Play all audios:
ABSTRACT Phospholipase D2 (PLD2), a signaling protein, plays a central role in cellular communication and various biological processes. Here, we show that PLD2 contributes to bone
homeostasis by regulating bone resorption through osteoclastic cell migration and microtubule-dependent cytoskeletal organization. _Pld2_-deficient mice exhibited a low bone mass attributed
to increased osteoclast function without altered osteoblast activity. While _Pld2_ deficiency did not affect osteoclast differentiation, its absence promoted the migration of osteoclast
lineage cells through a mechanism involving M-CSF-induced activation of the PI3K–Akt–GSK3β signaling pathway. The absence of _Pld2_ also boosted osteoclast spreading and actin ring
formation, resulting in elevated bone resorption. Furthermore, _Pld2_ deletion increased microtubule acetylation and stability, which were later restored by treatment with a specific
inhibitor of Akt, an essential molecule for microtubule stabilization and osteoclast bone resorption activity. Interestingly, PLD2 interacted with the M-CSF receptor (c-Fms) and PI3K, and
the association between PLD2 and c-Fms was reduced in response to M-CSF. Altogether, our findings indicate that PLD2 regulates bone homeostasis by modulating osteoclastic cell migration and
microtubule stability via the M-CSF-dependent PI3K–Akt–GSK3β axis. SIMILAR CONTENT BEING VIEWED BY OTHERS PHOSPHOLIPASE C Β4 PROMOTES RANKL-DEPENDENT OSTEOCLASTOGENESIS BY INTERACTING WITH
MKK3 AND P38 MAPK Article Open access 03 February 2025 SITE-1 PROTEASE CONTROLS OSTEOCLASTOGENESIS BY MEDIATING LC3 TRANSCRIPTION Article 19 January 2021 RAC1-DEPENDENT REGULATION OF
OSTEOCLAST AND OSTEOBLAST DIFFERENTIATION BY DEVELOPMENTALLY REGULATED GTP-BINDING 2 Article Open access 05 February 2025 INTRODUCTION Skeletal homeostasis is strongly controlled by the
dynamic coordination of bone-degrading osteoclasts and bone-synthesizing osteoblasts1,2,3,4. Osteoclasts are multinuclear cells responsible for resorbing the calcified bone matrix. The
development of these polykaryons is a multistage biological process that comprises the proliferation, differentiation, migration, and maturation of macrophage-monocyte lineage precursors.
The osteoclast differentiation process requires the activation of osteoclastogenic signaling cascades through the binding of receptor activator of nuclear factor-κB ligand (RANKL) to its
receptor, RANK5,6,7,8. On the other hand, precursor proliferation, migration, and cytoskeletal organization require the activation of signals through macrophage colony-stimulating factor
(M-CSF) and its receptor, c-Fms9,10,11,12. The degradation of bone matrix by osteoclasts depends on the organization of their actin cytoskeleton, resulting in polarization. When osteoclasts
contact bone, they become polarized, forming a unique adhesive structure, F-actin-rich ring or sealing zone, which surrounds the resorption area, and this event requires intact microtubule
integrity13,14. Microtubules are the major components of the eukaryotic cytoskeleton and comprise α- and β-tubulin protein subunits. These polymers play an important role in various cellular
events, including polarization, migration, vesicular trafficking, and the maintenance of cell shape. The importance of the tight modulation of microtubules in bone resorption has been well
documented in numerous studies15,16,17,18,19,20. Notably, acetylated tubulin, a marker of stable microtubules, plays a critical role in generating actin rings or sealing zones in mature
osteoclasts. The stabilization of the actin ring belt correlates with increased levels of tubulin acetylation, and this process is regulated by the Rho–mDia2–HDAC6 pathway15. The Akt–GSK3β
axis also governs osteoclast microtubule stability and bone resorption. The specific deletion of Akt1 and Akt2 in mature osteoclasts resulted in increased bone mass due to impaired actin
ring formation and bone resorption, revealing the essential role of Akt signaling in osteoclast bone-resorbing activity19. Members of the phospholipase D (PLD) family are enzymes that cleave
phosphatidylcholine, the major membrane phospholipid, into the bioactive lipid phosphatidic acid and choline21,22,23. Two main PLD isoforms (PLD1 and PLD2) share ~50% sequence homology and
have similar structures but differ in their subcellular localization. PLD1 is present in vesicular organelles, such as endosomes, autophagosomes, and lysosomes, whereas PLD2 is mainly
localized in the plasma membrane. PLD1 and PLD2 participate in several cellular functions, such as cell proliferation, migration, vesicle trafficking, and cytoskeletal organization.
Interestingly, a previous study using _Pld1_ or _Pld2_ knockout mice demonstrated that PLD1 and PLD2 play distinct roles in mast cell activation by regulating microtubule formation24. This
study further showed that PLD2 acts as a negative regulator of the organization of the mast cell cytoskeleton. In bone metabolism, several studies have revealed that PLD isoforms are
involved in modulating bone cells25,26,27,28. Our recent study reported that _Pld1_-deficient mice displayed reduced bone mass attributed to impaired osteoblastogenesis and increased
osteoclastogenesis26. Another study showed that PLD1, but not PLD2, enhances the osteoblast-mediated mineralization process25. Although PLD1 plays a role in bone metabolism, the function of
PLD2 in skeletal tissue homeostasis remains uncertain. In this study, employing _Pld2_-deficient mice, we demonstrate that PLD2 plays a vital role in bone homeostasis as a negative regulator
of osteoclastic bone resorption. Ablation of _Pld2_ accelerated cell migration and microtubule acetylation and increased the actin ring size, leading to an osteopenic phenotype in mice. We
further show that PLD2 functions in osteoclasts through the M-CSF-mediated PI3K–Akt–GSK3β signaling pathway. MATERIALS AND METHODS REAGENTS Recombinant human M-CSF and RANKL were acquired
from R&D Systems (Minneapolis, MN, USA). LY294002 and PD98059 were purchased from Cayman Chemical (Ann Arbor, MI, USA), and MK2206 was purchased from Selleck Chemicals (Selleck
Chemicals, TX, USA). Antibodies against Akt, ERK, JNK, p38, IκBα, PI3K, and PLD2 and phospho-specific antibodies for Akt, ERK, JNK, and p38 were obtained from Cell Signaling Technology
(Beverly, MA, USA). Antibodies against c-Fms, PLD2, GSK3β, and phospho-GSK3β were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-tubulin and anti-acetylated tubulin
antibodies were purchased from Sigma-Aldrich (St. Louis, MO, USA). MICE _Pld2_ knockout mice were produced as reported previously29 and kept on a C57BL/6 background. The animals were housed
in the animal facility at Kyungpook National University. All studies were approved by the Committee on the Ethics of Animal Experiments of Kyungpook National University (approval number:
KNU-2019-0038). MICROCOMPUTED TOMOGRAPHY (MICRO-CT) Mouse femurs were collected and fixed with 4% paraformaldehyde for 24 h. The microarchitectural properties of the femur were obtained
using a micro-CT system (eXplore Locus SP, GE Healthcare, Waukesha, WI, USA). A region between 0.7 and 2.3 mm below the growth plate was set as the region of interest. Next, the femurs were
scanned with a micro-CT calibrated to X-ray energy settings of 80 kV and 80 μA and an effective detector pixel size of 0.008 mm. Finally, the bone parameters were determined using eXplore
MicroView v.2.2 software provided with the micro-CT system. HISTOLOGY AND HISTOMORPHOMETRY For dynamic bone histomorphometric analysis, 7-week-old mice were injected intraperitoneally with
calcein green (10 mg/kg) and alizarin red (20 mg/kg) 6 and 2 days before sacrifice, respectively, as described in a previous study30. The tibiae were isolated, fixed in 4% paraformaldehyde,
and embedded in methyl methacrylate. The embedded tibiae tissues were cut into 6-μm-thick sections using a Leica RM2165 rotary microtome with a tungsten blade (Leica Microsystems, Germany)
and then imaged under a fluorescence microscope (Leica Microsystems, Germany). For von Kossa staining analysis, the lumbar vertebrae were fixed and embedded in methyl methacrylate. After
that, 6-µm-thick vertebrae sections were stained with von Kossa reagent. To evaluate osteoclast parameters, the tibiae were harvested and fixed in 4% paraformaldehyde, followed by
decalcification in 10% EDTA for 4 weeks at 4 °C. The specimens were dehydrated and embedded in paraffin. The paraffin sections were then stained for tartrate-resistant acid phosphatase
(TRAP) to identify osteoclasts. Bone histomorphometric analyses were conducted using the Bioquant OSTEO II program (Bio-Quant, Inc., Nashville, TN, USA). OSTEOCLAST CULTURE Murine bone
marrow-derived macrophages (BMMs) and osteoclasts were prepared as described previously. In brief, bone marrow was extracted from the long bones of 8–9-week-old mice, and red blood cells
were lysed for 2 min at room temperature. The cells were then plated in a Petri dish and cultured for 3–4 days in α-minimal essential medium (α-MEM) containing 10% fetal bovine serum and 10%
CMG 14-12 cell culture medium as the M-CSF source31. The attached cells were lifted and used as osteoclast precursor cells (BMMs). BMMs were then seeded at a density of 5 × 103 per well in
a 96-well cell culture plate and incubated in α-MEM supplemented with M-CSF and RANKL for osteoclast generation. Osteoclasts were fixed using 4% paraformaldehyde after 4–5 days in culture
and stained with TRAP solution containing 0.1 mg/ml naphthol AS-MX phosphate and 0.3 mg/ml Fast Red Violet. PROLIFERATION AND APOPTOSIS ASSAYS Proliferation assays were conducted using the
Cell Proliferation Biotrak ELISA kit (Amersham, GE Healthcare Life Sciences). BMMs were cultured in a 96-well plate at 5 × 103 cells per well in α-MEM supplemented with M-CSF at various
concentration for 3 days. After the cells had been incubated with 0.1% bromodeoxyuridine (BrdU) at 37 °C for 4 h, the amount of BrdU incorporation into the cellular DNA was determined by
measuring the absorbance at 450 nm. For the apoptosis assay, BMMs were plated at a concentration of 5 × 103 cells per well in a 96-well plate and cultured in α-MEM containing M-CSF (30
ng/ml) for 3 days. This assay was performed using the Cell Death Detection ELISA Plus kit (Roche, Mannheim, Germany) according to the manufacturer’s instructions. MIGRATION ASSAY Migration
assays were performed using a Transwell migration assay kit (Corning, Corning, NY, USA). First, BMMs (2 × 105 cells per well) or preosteoclasts (2 × 105 cells per well) were seeded in the
upper chamber in 100 μl of serum-free α-MEM, and M-CSF (50 ng/ml) was added to the lower chamber in 600 μl of serum-free α-MEM. After 16 h of incubation at 37 °C, the cells were fixed with
4% paraformaldehyde for 20 min and later stained with crystal violet. Finally, nonmigratory cells were removed from the upper surface of the Transwell membrane. RT–PCR Total RNA was prepared
from cultured cells using TRIzol (Invitrogen). cDNA was synthesized from 1 μg of RNA using the SuperScript synthesis system (Invitrogen). The primer sequences used for RT–PCR were as
follows: PLD2, 5′-CGAGAAGCTCCTGGTGGTAG-3′ and 5′-CCAGTCCTTGGTGATGAGGT-3′; TRAP, 5′-ACAGCCCCCCACTCCCACCCT-3′ and 5′-TCAGGGTCTGGGTCTCCTTGG-3′; and GAPDH, 5′-ACTTTGTCAAGCTCATTTCC-3′ and
5′-TGCAGCGAACTTTATTGATG-3′. QUANTITATIVE REAL-TIME PCR Quantitative PCR was performed with an ABI 7500 Real-Time PCR System and SYBR Green dye (Applied Biosystems, Foster City, CA). The
following primers were used: PLD2, 5′-CCAGCAAACAGAAATACTTGGAAA-3′ and 5′-GGCGTGGTAATTGCGATAGAA-3′; PLD1, 5′-TTGCTGATTTCATTGACAGGTACTC-3′ and 5′-CATGGACCACAGAGCCAATATC-3′; Atp6v0d2,
5′-GAGCTGTACTTCAATGTGGACCAT-3′ and 5′-CTGGCTTTGCATCCTCGAA-3′; DC-STAMP, 5′-CTTCCGTGGGCCAGAAGTT-3′ and 5′-AGGCCAGTGCTGACTAGGATGA-3′; c-Fos, 5′-AGGCCCAGTGGCTCAGAGA-3′ and
5′-GCTCCCAGTCTGCTGCATAGA-3′; NFATc1, 5′-ACCACCTTTCCGCAACCA-3′ and 5′-TTCCGTTTCCCGTTGCA-3′; TRAP, 5′-TCCCCAATGCCCCATTC-3′ and 5′-CGGTTCTGGCGATCTCTTTG-3′; and MMP-9,
5′-AAAGACCTGAAAACCTCCAACCT-3′ and 5′-GCCCGGGTGTAACCATAGC-3′. WESTERN BLOTTING AND IMMUNOPRECIPITATION BMMs or osteoclasts were washed with PBS and lysed with lysis buffer [50 mM Tris-HCl (pH
7.4), 150 mM NaCl, 1% NP-40, 1 mM EDTA] supplemented with Halt protease/phosphatase inhibitor cocktail (Thermo Scientific Inc., Rockford, IL, USA). First, the protein concentration of the
cell lysates was determined using a bicinchoninic acid kit (Pierce, Rockford, IL). Next, an aliquot of protein (40 μg) was subjected to 8% or 10% SDS-PAGE and transferred onto a
polyvinylidene difluoride membrane. The membrane was blocked in 5% skim milk or 1% BSA and immunoblotted with specific primary antibodies. Finally, immunoreactivity was quantified using an
ECL-Plus detection kit (Amersham Pharmacia Biotech, Piscataway, NJ, USA). For immunoprecipitation, cell lysates were incubated with anti-PLD2 antibody or control IgG followed by Sepharose A
beads (GE Healthcare). The immunoprecipitated proteins were separated by 8% SDS-PAGE and immunoblotted as described above. IMMUNOFLUORESCENCE AND ACTIN RING STAINING BMMs were seeded on
glass slides in 24-well plates and cultured with M-CSF and RANKL for 4–5 days. Then, the cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100 and blocked in 0.2%
BSA. The cells were then incubated with anti-tubulin or anti-acetylated tubulin primary antibodies. After washing with PBS, the samples were incubated with secondary antibodies and mounted
with 80% glycerol in PBS. For actin ring staining, F-actin was labeled with TRITC-conjugated phalloidin (Sigma) or Alexa Fluor 488 phalloidin (Invitrogen), and the nuclei were stained with
Hoechst 33258 (Sigma). The samples were visualized using a fluorescence microscope (Leica Microsystems, Germany). RESORPTION PIT ASSAY BMMs were cultured on bone slices in the presence of
M-CSF and RANKL. After 5 days, the cultured osteoclasts were removed from bone slices through mechanical agitation. The bone slices were then incubated with peroxidase-conjugated wheat germ
agglutinin (Sigma) and stained with 3,3′-diaminobenzidine (Sigma). The resorbed pit area and relative pit size were measured by a Java-based image analysis program (ImageJ). STATISTICS
Statistical analyses of all experiments were performed using Student’s _t_ test in Microsoft Excel 2016 (Microsoft, USA). Differences for which _p_ < 0.05 were considered statistically
significant, and the data are presented as the mean ± standard deviation (SD). RESULTS _PLD2-_DEFICIENT MICE EXHIBIT A LOW BONE MASS To evaluate the physiological role of PLD2 in bone
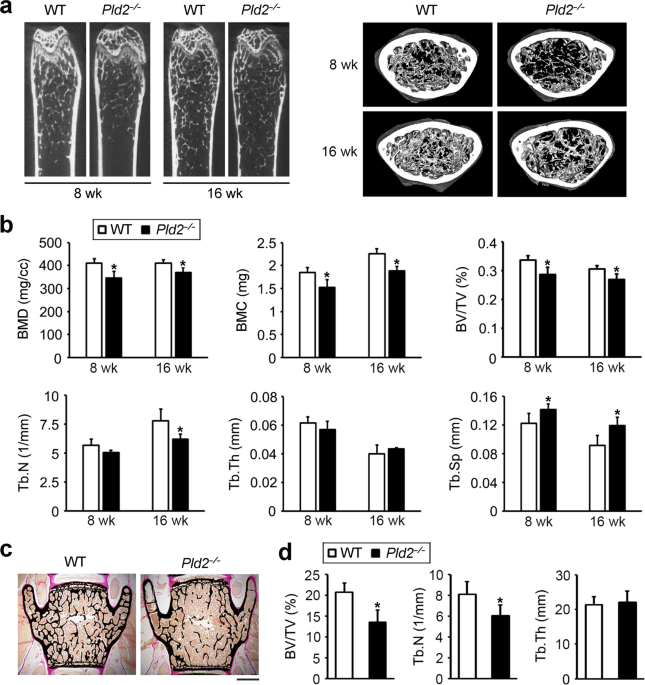
homeostasis, the femurs of 8- and 16-week-old _Pld2__−/−_ mice were analyzed with a micro-CT scanner. The _Pld2__−/−_ mice at both ages displayed a significant decrease in bone mineral
density, bone mineral content, and bone volume per tissue volume (BV/TV) compared to those of their wild-type (WT) littermates (Fig. 1a, b). Trabecular number (Tb.N) was decreased in the
_Pld2__−/−_ mice, with a related increase in trabecular separation (Th.Sp). No difference in trabecular thickness (Tb.Th) was observed between _Pld2__−/−_ mice and their WT counterparts
(Fig. 1b). Additionally, the low bone mass phenotype was further confirmed by nondecalcified bone histology. Von Kossa staining of lumbar vertebrae sections showed a significant reduction in
BV/TV and Tb.N without a change in Tb.Th in the _Pld2__−/−_ mice compared to WT littermates (Fig. 1c, d), as was the case upon analysis of the femurs. The osteopenic phenotype of the
_Pld2__−/−_ mice could be related to decreased bone formation and/or increased bone resorption. To address this issue, we also performed histological analysis of bone tissues. Dynamic
histomorphometry of calcein green- and alizarin red-labeled tibia sections showed no differences in the bone formation parameters such as mineral apposition rate (MAR) and bone formation
rate (BFR) between _Pld2__−/−_ mice and WT littermates (Fig. 2a, b). In contrast, the resorptive bone surface area was larger in _Pld2__−/−_ mice than in WT mice (Fig. 2c, d). Osteoclast
surface per bone surface (Oc.S/B.S) was significantly increased in _Pld2__−/−_ mice compared to WT controls, as indicated by TRAP staining. However, there was no difference in the osteoclast
number per bone surface (N.Oc/BS) (Fig. 2c, d). DELETION OF _PLD2_ INCREASES OSTEOCLAST SPREADING AND BONE RESORPTION Having established that _Pld2_ deficiency decreases bone mass due to
accelerated osteoclast development, we turned to in vitro osteoclast culture and first examined the expression pattern of PLD2 during osteoclastogenesis. PLD2 expression was abundant in
osteoclast precursors (BMMs), rapidly decreased within 1 day after the addition of RANKL and then increased until day 3 of culture (Fig. 3a, b). Next, we examined the impact of _Pld2_
deficiency on osteoclast formation. BMMs derived from _Pld2__−/−_ mice and WT littermates were cultured with RANKL and two different concentrations of M-CSF. Interestingly, the number of
well-spread osteoclasts was significantly higher in cultures isolated from _Pld2__−/−_ osteoclasts than in WT counterparts at both M-CSF concentrations (Fig. 3c, d). Additionally, the
relative osteoclast size was much greater in _Pld2__−/−_ cultures than in WT controls (Fig. 3e). To determine whether the osteoclast phenotype in the absence of _Pld2_ reflects improved
osteoclast fusion and/or differentiation, we assessed the expression of relevant osteoclastogenic indicators. _Pld2_ deletion had no impact on the mRNA expression of osteoclast fusion
markers (Atp6v0d2 and DC-STAMP) or various osteoclast differentiation markers (Fig. 3f). These observations were also confirmed at the protein level by immunoblotting (data not shown).
Furthermore, RANKL-mediated activation of signaling molecules required for osteoclastogenesis was unaltered in the absence of _Pld2_ (Fig. 3g). These results demonstrate that _Pld2_
deficiency does not affect osteoclast fusion or differentiation induced by RANKL. Given that _Pld2_ depletion does not alter osteoclastogenesis, we next investigated the impact of _Pld2_
deficiency on osteoclastic bone resorption. Mature osteoclasts from WT and _Pld2__−/−_ mice were generated on bone slices. After 5 days, the resorption pits were visualized with
peroxidase-conjugated wheat germ agglutinin staining. Consistent with their super spread morphology, _Pld2_-deficient osteoclasts significantly enhanced resorption lacuna formation (Fig. 3h,
i). In addition to the increased total resorption area, the average pit size was approximately three times larger in _Pld2__−/−_ osteoclasts (Fig. 3j). PLD2 REGULATES THE PROLIFERATION AND
MIGRATION OF OSTEOCLAST LINEAGE CELLS The proliferation, survival, and migration of osteoclast lineage cells are also key to osteoclast formation and function. Therefore, we performed a BrdU
incorporation assay and cell death ELISA to examine whether _Pld2_ deficiency affects the proliferation and/or apoptosis of osteoclast precursor cells (BMMs). The analysis of BrdU
incorporation showed that _Pld2_ deficiency slightly increased cell proliferation in response to M-CSF at various concentrations (Fig. 4a). In contrast, PLD2 had no impact on BMM survival,
as dictated by cell death detection via ELISA (Fig. 4b). We next investigated the role of PLD2 in cell migration using the Transwell culture system. The depletion of _Pld2_ strongly promoted
M-CSF-induced migration of both types of BMMs (Fig. 4c) and preosteoclasts at day 2 (Fig. 4d). M-CSF activates the PI3K/Akt and Grb2/ERK cascades in osteoclastic cells. Therefore, we
assessed whether Akt or ERK signaling is altered in the absence of _Pld2_. The phosphorylation levels of Akt and ERK were strongly increased in _Pld2_-deficient cells after M-CSF exposure
(Fig. 4e). Generally, PI3K–Akt regulates cell growth, migration, and survival through GSK3β. Activated Akt phosphorylates and inhibits GSK3β, thereby stimulating these cellular functions.
Therefore, we evaluated the phosphorylation level of GSK3β and found that GSK3β was hyperphosphorylated in _Pld2__−/−_ BMMs (Fig. 4f). The accelerated migration in _Pld2__−/−_ cells in
response to M-CSF reflects activation of the PI3K–Akt and/or Grb2–ERK pathway. To determine which signaling pathway is responsible for the M-CSF-induced increase in osteoclastic cell
migration, we used inhibitors of these pathways. Transwell migration assays showed that a PI3K inhibitor (LY294002), but not an ERK inhibitor (PD98059), strongly downregulated the capacity
of M-CSF to promote migration in WT precursor cells (Fig. 4g). Based on our observation that PI3K–Akt regulates osteoclastic cell migration, we then treated WT and _Pld2__−/−_ BMMs with a
PI3K inhibitor. The inactivation of PI3K in WT BMMs dose-dependently suppressed cell migration (Fig. 4h). Importantly, _Pld2_-deficient BMMs were more resistant to the suppressive effect of
the PI3K inhibitor on cell migration (Fig. 4h). Compared to WT BMMs, _Pld2__−/−_ BMMs migrated at normal levels in the presence of 5 μM LY294002. Thus, M-CSF-mediated activation of the
PI3K–Akt pathway is most likely responsible for the enhanced migration of _Pld2__−/−_ osteoclast lineage cells. _PLD2_ DEFICIENCY ACCELERATES CYTOSKELETAL ORGANIZATION IN OSTEOCLASTS The
tendency of _Pld2_-deficient osteoclasts to spread (Fig. 3c–e) suggests the accelerated organization of their actin cytoskeleton. At the beginning of osteoclastogenesis, the actin
cytoskeleton is organized into clusters, which are later arranged around the cell periphery to form F-actin rings in mature osteoclasts. The size of actin rings reflects the ability of
osteoclasts to resorb bone. To analyze the role of PLD2 in the osteoclast cytoskeleton, we generated WT and _Pld2__−/−_ osteoclasts on glass coverslips and stained the F-actin rings with
TRITC-conjugated phalloidin. Immunostaining data revealed that compared to WT cells, _Pld2_-deficient osteoclasts displayed larger actin ring formation (Fig. 5a). The relative size of the
actin rings was approximately twofold larger in _Pld2__−/−_ osteoclasts than in WT polykaryons (Fig. 5b). Actin ring formation in osteoclasts depends on an intact microtubule network. To
examine the impact of _Pld2_ depletion on microtubule organization, we applied nocodazole, a microtubule-depolymerizing agent. While nocodazole treatment resulted in significant disruption
of the microtubule cytoskeleton in WT osteoclasts, _Pld2_-deficient osteoclasts had a more nocodazole-resistant population of stable microtubules (Fig. 5c). The acetylation of microtubules
in mature osteoclasts represents microtubule stability, which led to our assessment of microtubule acetylation in _Pld2__−/−_ and WT osteoclasts. Immunofluorescence staining using an
anti-acetylated tubulin antibody showed that _Pld2__−/−_ osteoclasts possessed more abundant and highly acetylated tubulin than WT cells (Fig. 5d). These observations were further confirmed
by immunoblotting in the same cells (Fig. 5e). The Akt–GSK3β signaling axis is pivotal for microtubule stability in osteoclasts19. Therefore, we examined this key function and found that Akt
phosphorylation was strongly enhanced in _Pld2__−/−_ osteoclasts (Fig. 5f). Consistently, GSK3β phosphorylation was increased in _Pld2__−/−_ osteoclasts. Next, we applied the Akt-specific
inhibitor MK2206 to attenuate Akt activity in _Pld2__−/−_ osteoclasts. As shown in Fig. 5g, the increased microtubule acetylation in the absence of _Pld2_ was restored by the Akt inhibitor,
indicating that _Pld2_ deficiency promotes microtubule stabilization by increasing Akt activity. PLD2 FORMS A COMPLEX WITH C-FMS AND PI3K Given our data showing that PLD2 specifically
regulates the M-CSF-induced PI3K–Akt signaling pathway, we hypothesized that PLD2 can bind the M-CSF receptor, c-Fms, and/or PI3K. Therefore, endogenous PLD2 was immunoprecipitated using an
anti-PLD2 or mouse IgG control antibody to address this hypothesis. As shown in Fig. 6a, PLD2 interacted with both c-Fms and PI3K in BMMs. PI3K directly binds c-Fms after M-CSF
stimulation32,33, resulting in Akt activation. To further investigate the effect of M-CSF on PLD2-c-Fms-PI3K complex formation, cytokine-starved BMMs were treated with M-CSF and
immunoprecipitated with an anti-PLD2 antibody. Under basal conditions, PLD2 was associated with both c-Fms and PI3K (Fig. 6b). Interestingly, the binding of PLD2 to c-Fms was reduced by
M-CSF stimulation, whereas the interaction between PLD2 and PI3K increased following M-CSF exposure (Fig. 6b). Collectively, our data suggest that the enhanced interaction between PLD2 and
PI3K in response to M-CSF may be a key mechanism by which PLD2 controls the PI3K–Akt pathway. DISCUSSION PLD family proteins function as important signaling molecules in various tissues and
cellular processes. Numerous animal model studies involving targeted gene disruption and/or pharmacological approaches have suggested that PLD proteins could provide a therapeutic basis for
immunological, vascular, and neurological disorders21,22. In this study, we investigated the role of PLD2 in the skeleton using _Pld2__−/−_ mice and found that PLD2 plays a critical role in
bone metabolism and homeostasis. We also demonstrated that PLD2 negatively controls osteoclast bone resorption by regulating osteoclastic cell migration and microtubule stabilization via the
M-CSF-mediated PI3K–Akt–GSK3β axis and partly by modulating precursor cell proliferation via the Grb2–ERK pathway (Fig. 6c). Our in vivo data reveal that _Pld2__−/−_ mice display a decrease
in both trabecular and lumbar vertebral bone mass. The decrease in bone mass in the absence of _Pld2_ was associated with increased osteoclast function in vivo. Consistently, in vitro data
further confirmed the suppressive role of PLD2 in bone resorptive activity, as indicated by the increased resorption pit formation in _Pld2_-deficient osteoclasts. The accelerated bone
resorption was due to increased migration and cytoskeletal organization in _Pld2__−/−_ osteoclasts but not the differentiation and/or fusion of their precursors. Meanwhile, the osteoblast
parameters (MAR and BFR) were not significantly changed in the _Pld2__−/−_ mice, indicating that _Pld2_ deficiency does not affect bone formation in vivo. Consistent with our findings, a
recent study reported that in vitro pharmacological suppression of PLD2 with a chemical inhibitor had no profound effect on the osteoblast mineralization process25. Additionally, more
recently, our study showed that PLD2 was barely detected during osteoblast differentiation26. Thus, in addition to previous studies, our findings reveal that PLD2 does not significantly
affect osteoblast bone-forming activity. Cell migration is a highly active process that involves actin cytoskeleton remodeling34. In particular, the migration of osteoclastic cells is
crucial for their bone resorption function. Furthermore, PLD2 regulates the migration of diverse cell types, such as macrophages, leukocytes, and vascular smooth muscle cells35,36,37. Based
on these studies, we hypothesized that PLD2 can regulate the migration of osteoclast lineage cells. Supporting this hypothesis, we discovered that _Pld2_ deficiency increases osteoclastic
cell migration in response to M-CSF. The relevance of PLD2 to macrophage migration has also been reported in a previous study35. However, unlike our observations, a previous study showed
that silencing PLD2 using siRNA reduces M-CSF-mediated cell migration, whereas overexpression of PLD2 has the opposite effect. Furthermore, although the study suggested that PLD2 plays a
positive role in the migration of macrophage cells, the experiments were performed using the RAW/LR5 macrophage cell line, which often does not function identically to primary cells.
However, we used primary BMMs and preosteoclasts isolated from _Pld2__−/−_ mice in this study. This discrepancy may be due to the differences in cell type and off-target effects of knockdown
or the ectopic expression of PLD2. The activated PI3K–Akt pathway is a key signaling pathway for cell migration, proliferation, survival, and actin remodeling in various cell types. The
critical role of PI3K in osteoclast migration and function was well demonstrated by a genetic study38. Mice lacking the p85α subunit of PI3K exhibited increased bone mass with defective cell
migration and bone resorption, which were attributed to the reduced activation of Akt. Our results demonstrate that _Pld2_ deficiency increases Akt activation induced by M-CSF but not
RANKL. Thus, enhanced Akt activity by M-CSF contributes to the accelerated migration of osteoclast lineage cells in the absence of _Pld2_. In addition to cell migration, PLD2 regulates
osteoclast cytoskeletal organization. Its deficiency enhances osteoclast spreading, which generally reflects the ability of osteoclasts to form actin rings18,39,40,41,42. In fact,
_Pld2_-deficient osteoclasts possess enlarged actin rings and hyper-resorption. Actin ring organization in osteoclasts is governed by a microtubule network, specifically acetylated tubulin.
Consequently, failure to acetylate microtubules increases bone mass due to defective bone resorption16,19,20. The acetylation of microtubules, which stabilizes them, is regulated by several
factors, including PI3K–Akt. Thus, the PI3K–Akt cascade is critical for cell migration and a key factor for microtubule stabilization. Matsumoto et al. reported the importance of the Akt
signaling molecule in actin ring formation and microtubule stability19. Treatment with an Akt inhibitor in osteoclasts disrupted actin ring formation with abnormal regulation of acetylated
tubulin; however, the expression level of acetylated tubulin was enhanced when catalytically active Akt was overexpressed19. The effect of Akt on microtubule stabilization is regulated by
GSK3β, an effector signaling molecule downstream of Akt. The inhibition of GSK3β activity by Akt permits the activation of microtubule-associated proteins responsible for microtubule
stabilization19,43. These findings reveal that Akt promotes osteoclast bone-resorbing activity by regulating actin ring organization and microtubule stability. We demonstrated by
immunostaining and immunoblotting that _Pld2_ deficiency increases the expression of the acetylated form of tubulin. Moreover, the activation of Akt and consequent inactivation of GSK3β were
observed in _Pld2_-deficient osteoclasts. Furthermore, an Akt inhibitor restored tubulin acetylation and actin ring formation in the absence of _Pld2_. However, there was no significant
change in actin ring size. This may be due to the short treatment time and low concentration of the Akt inhibitor used in this study. Together, our results indicate that PLD2 controls
microtubule stability by regulating the Akt–GSK3β axis. Similar to our findings, a group working on mast cell function showed that PLD2 regulates dynamic microtubule rearrangement during
mast cell activation24. Importantly, microtubule formation and the FcεRI-mediated activation of Akt were increased in mast cells isolated from _Pld2__−/−_ mice, showing that PLD2 contributes
to the negative regulation of cytoskeletal organization in mast cells. In summary, we have identified PLD2 as a negative regulator of osteoclast function, specifically M-CSF signaling, and
is essential for osteoclastic cell migration and cytoskeletal organization. PLD2 binds the M-CSF receptor (c-Fms) and PI3K and antagonizes osteoclast migration and microtubule stabilization
by attenuating the Akt–GSK3β signaling axis, leading to impaired bone resorption. Therefore, the modulation of PLD2 may provide a novel strategy for treating skeletal diseases, including
osteoporosis and rheumatoid arthritis. REFERENCES * Boyle, W. J., Simonet, W. S. & Lacey, D. L. Osteoclast differentiation and activation. _Nature_ 423, 337–342 (2003). Article CAS
PubMed Google Scholar * Choi, J. Y. Healthy bone tissue homeostasis. _Exp. Mol. Med._ 52, 1165 (2020). Article CAS PubMed PubMed Central Google Scholar * Takayanagi, H.
Osteoimmunology: shared mechanisms and crosstalk between the immune and bone systems. _Nat. Rev. Immunol._ 7, 292–304 (2007). Article CAS PubMed Google Scholar * Teitelbaum, S. L. Bone
resorption by osteoclasts. _Science_ 289, 1504–1508 (2000). Article CAS PubMed Google Scholar * Kong, Y. Y. et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development
and lymph-node organogenesis. _Nature_ 397, 315–323 (1999). Article CAS PubMed Google Scholar * Lacey, D. L. et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast
differentiation and activation. _Cell_ 93, 165–176 (1998). Article CAS PubMed Google Scholar * Wong, B. R. et al. TRANCE (tumor necrosis factor [TNF]-related activation-induced
cytokine), a new TNF family member predominantly expressed in T cells, is a dendritic cell-specific survival factor. _J. Exp. Med._ 186, 2075–2080 (1997). Article CAS PubMed PubMed
Central Google Scholar * Yasuda, H. et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. _Proc.
Natl Acad. Sci. USA_ 95, 3597–3602 (1998). Article CAS PubMed PubMed Central Google Scholar * Feng, X. & Teitelbaum, S. L. Osteoclasts: new Insights. _Bone Res._ 1, 11–26 (2013).
Article PubMed CAS Google Scholar * Mun, S. H., Park, P. S. U. & Park-Min, K. H. The M-CSF receptor in osteoclasts and beyond. _Exp. Mol. Med._ 52, 1239–1254 (2020). Article CAS
PubMed PubMed Central Google Scholar * Teitelbaum, S. L. The osteoclast and its unique cytoskeleton. _Ann. N. Y. Acad. Sci._ 1240, 14–17 (2011). Article CAS PubMed Google Scholar *
Yoshida, H. et al. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. _Nature_ 345, 442–444 (1990). Article CAS PubMed Google
Scholar * Destaing, O., Saltel, F., Geminard, J. C., Jurdic, P. & Bard, F. Podosomes display actin turnover and dynamic self-organization in osteoclasts expressing actin-green
fluorescent protein. _Mol. Biol. Cell_ 14, 407–416 (2003). Article CAS PubMed PubMed Central Google Scholar * Jurdic, P., Saltel, F., Chabadel, A. & Destaing, O. Podosome and
sealing zone: specificity of the osteoclast model. _Eur. J. Cell Biol._ 85, 195–202 (2006). Article CAS PubMed Google Scholar * Destaing, O. et al. A novel Rho-mDia2-HDAC6 pathway
controls podosome patterning through microtubule acetylation in osteoclasts. _J. Cell Sci._ 118, 2901–2911 (2005). Article CAS PubMed Google Scholar * Gil-Henn, H. et al. Defective
microtubule-dependent podosome organization in osteoclasts leads to increased bone density in Pyk2(-/-) mice. _J. Cell Biol._ 178, 1053–1064 (2007). Article CAS PubMed PubMed Central
Google Scholar * Guimbal, S. et al. Dock5 is a new regulator of microtubule dynamic instability in osteoclasts. _Biol. Cell_ 111, 271–283 (2019). Article CAS PubMed Google Scholar *
Hong, J. M. et al. Calpain-6, a target molecule of glucocorticoids, regulates osteoclastic bone resorption via cytoskeletal organization and microtubule acetylation. _J. Bone Miner. Res._
26, 657–665 (2011). Article CAS PubMed Google Scholar * Matsumoto, T. et al. Regulation of bone resorption and sealing zone formation in osteoclasts occurs through protein kinase
B-mediated microtubule stabilization. _J. Bone Miner. Res._ 28, 1191–1202 (2013). Article CAS PubMed Google Scholar * Zalli, D. et al. The actin-binding protein cofilin and its
interaction with cortactin are required for podosome patterning in osteoclasts and bone resorption in vivo and in vitro. _J. Bone Miner. Res._ 31, 1701–1712 (2016). Article CAS PubMed
Google Scholar * Brown, H. A., Thomas, P. G. & Lindsley, C. W. Targeting phospholipase D in cancer, infection and neurodegenerative disorders. _Nat. Rev. Drug Discov._ 16, 351–367
(2017). Article CAS PubMed PubMed Central Google Scholar * Ghim, J., Chelakkot, C., Bae, Y. S., Suh, P. G. & Ryu, S. H. Accumulating insights into the role of phospholipase D2 in
human diseases. _Adv. Biol. Regul._ 61, 42–46 (2016). Article CAS PubMed Google Scholar * Frohman, M. A. The phospholipase D superfamily as therapeutic targets. _Trends Pharmacol. Sci._
36, 137–144 (2015). Article CAS PubMed PubMed Central Google Scholar * Zhu, M. et al. Differential roles of phospholipase D proteins in FcepsilonRI-mediated signaling and mast cell
function. _J. Immunol._ 195, 4492–4502 (2015). Article CAS PubMed Google Scholar * Abdallah, D. et al. Effects of phospholipase D during cultured osteoblast mineralization and bone
formation. _J. Cell. Biochem._ 120, 5923–5935 (2019). Article CAS PubMed Google Scholar * Kang, D. W. et al. Deletion of phospholipase D1 decreases bone mass and increases fat mass via
modulation of Runx2, beta-catenin-osteoprotegerin, PPAR-gamma and C/EBPalpha signaling axis. _Biochim. Biophys. Acta Mol. Basis Dis._ 1867, 166084 (2021). Article CAS PubMed Google
Scholar * Sylvia, V. L. et al. Regulation of phospholipase D (PLD) in growth plate chondrocytes by 24R,25-(OH)2D3 is dependent on cell maturation state (resting zone cells) and is specific
to the PLD2 isoform. _Biochim. Biophys. Acta_ 1499, 209–221 (2001). Article CAS PubMed Google Scholar * Tokuda, H. et al. Function of Ca2+ in phosphatidylcholine-hydrolyzing
phospholipase D activation in osteoblast-like cells. _Bone_ 19, 347–352 (1996). Article CAS PubMed Google Scholar * Ghim, J. et al. Endothelial deletion of phospholipase D2 reduces
hypoxic response and pathological angiogenesis. _Arterioscler. Thromb. Vasc. Biol._ 34, 1697–1703 (2014). Article CAS PubMed Google Scholar * Lim, K. E. et al. Core binding factor beta
of osteoblasts maintains cortical bone mass via stabilization of Runx2 in mice. _J. Bone Miner. Res._ 30, 715–722 (2015). Article CAS PubMed Google Scholar * Takeshita, S., Kaji, K.
& Kudo, A. Identification and characterization of the new osteoclast progenitor with macrophage phenotypes being able to differentiate into mature osteoclasts. _J. Bone Miner. Res._ 15,
1477–1488 (2000). Article CAS PubMed Google Scholar * Lee, A. W. & States, D. J. Both src-dependent and -independent mechanisms mediate phosphatidylinositol 3-kinase regulation of
colony-stimulating factor 1-activated mitogen-activated protein kinases in myeloid progenitors. _Mol. Cell. Biol._ 20, 6779–6798 (2000). Article CAS PubMed PubMed Central Google Scholar
* Xiong, Y. et al. A CSF-1 receptor phosphotyrosine 559 signaling pathway regulates receptor ubiquitination and tyrosine phosphorylation. _J. Biol. Chem._ 286, 952–960 (2011). Article CAS
PubMed Google Scholar * Ridley, A. J. et al. Cell migration: integrating signals from front to back. _Science_ 302, 1704–1709 (2003). Article CAS PubMed Google Scholar * Knapek, K.
et al. The molecular basis of phospholipase D2-induced chemotaxis: elucidation of differential pathways in macrophages and fibroblasts. _Mol. Cell. Biol._ 30, 4492–4506 (2010). Article CAS
PubMed PubMed Central Google Scholar * Lehman, N. et al. Phagocyte cell migration is mediated by phospholipases PLD1 and PLD2. _Blood_ 108, 3564–3572 (2006). Article CAS PubMed
PubMed Central Google Scholar * Wang, Z. et al. Phosphatidic acid generated by PLD2 promotes the plasma membrane recruitment of IQGAP1 and neointima formation. _FASEB J._ 33, 6713–6725
(2019). Article CAS PubMed PubMed Central Google Scholar * Munugalavadla, V. et al. The p85alpha subunit of class IA phosphatidylinositol 3-kinase regulates the expression of multiple
genes involved in osteoclast maturation and migration. _Mol. Cell. Biol._ 28, 7182–7198 (2008). Article CAS PubMed PubMed Central Google Scholar * Kim, H. J., Lee, D. K., Jin, X., Che,
X. & Choi, J. Y. Oleoylethanolamide exhibits GPR119-dependent inhibition of osteoclast function and GPR119-independent promotion of osteoclast apoptosis. _Mol. Cells_ 43, 340–349 (2020).
CAS PubMed PubMed Central Google Scholar * Kim, H. J. et al. G protein-coupled receptor 120 signaling negatively regulates osteoclast differentiation, survival, and function. _J. Cell.
Physiol._ 231, 844–851 (2016). Article CAS PubMed Google Scholar * Kim, H. J. et al. Glucocorticoids suppress bone formation via the osteoclast. _J. Clin. Invest._ 116, 2152–2160 (2006).
Article CAS PubMed PubMed Central Google Scholar * McHugh, K. P. et al. Mice lacking beta3 integrins are osteosclerotic because of dysfunctional osteoclasts. _J. Clin. Invest._ 105,
433–440 (2000). Article CAS PubMed PubMed Central Google Scholar * Hur, E. M. et al. GSK3 controls axon growth via CLASP-mediated regulation of growth cone microtubules. _Genes Dev._
25, 1968–1981 (2011). Article CAS PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS This work was supported by a National Research Foundation of Korea (NRF)
grant funded by the Korean Government (MSIT) (NRF-2018R1A2B6001298); the Basic Science Research Program through the NRF funded by the Ministry of Education (NRF-2021R1I1A1A01051983); and the
Korea Mouse Phenotyping Project of the Ministry of Science, ICT & Future Planning through the NRF (NRF-2014M3A9D5A01073658). AUTHOR INFORMATION Author notes * These authors contributed
equally: Hyun-Ju Kim, Dong-Kyo Lee. AUTHORS AND AFFILIATIONS * Department of Biochemistry and Cell Biology, Cell and Matrix Research Institute, BK21 Plus KNU Biomedical Convergence Program,
Korea Mouse Phenotyping Center, School of Medicine, Kyungpook National University, Daegu, 41944, Republic of Korea Hyun-Ju Kim, Dong-Kyo Lee, Xian Jin, Xiangguo Che & Je-Yong Choi *
Department of Life Sciences, Pohang University of Science and Technology, Pohang, 37673, Republic of Korea Sung Ho Ryu Authors * Hyun-Ju Kim View author publications You can also search for
this author inPubMed Google Scholar * Dong-Kyo Lee View author publications You can also search for this author inPubMed Google Scholar * Xian Jin View author publications You can also
search for this author inPubMed Google Scholar * Xiangguo Che View author publications You can also search for this author inPubMed Google Scholar * Sung Ho Ryu View author publications You
can also search for this author inPubMed Google Scholar * Je-Yong Choi View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS H.-J.K., S.H.R., and
J.-Y.C. designed the research; H.-J.K., D.-K.L., X.J., and X.C. performed the research; H.-J.K. and D.-K.L. analyzed the data; and H.-J.K. and J.-Y.C. wrote the paper. CORRESPONDING AUTHORS
Correspondence to Hyun-Ju Kim or Je-Yong Choi. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature
remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons
Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original
author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the
article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use
is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Kim, HJ., Lee, DK., Jin, X. _et al._ Phospholipase D2 controls bone homeostasis by
modulating M-CSF-dependent osteoclastic cell migration and microtubule stability. _Exp Mol Med_ 54, 1146–1155 (2022). https://doi.org/10.1038/s12276-022-00820-1 Download citation * Received:
03 March 2022 * Revised: 17 May 2022 * Accepted: 13 June 2022 * Published: 09 August 2022 * Issue Date: August 2022 * DOI: https://doi.org/10.1038/s12276-022-00820-1 SHARE THIS ARTICLE
Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided
by the Springer Nature SharedIt content-sharing initiative