Play all audios:
ABSTRACT Cytochrome _c_ and dATP/ATP induce oligomerization of Apaf-1 into two distinct apoptosome complexes: an ∼700 kDa complex, which recruits and activates caspases-9, -3 and -7, and an
∼1.4 MDa complex, which recruits and processes caspase-9, but does not efficiently activate effector caspases. While searching for potential inhibitors of the ∼1.4 MDa apoptosome complex, we
observed an ∼30 kDa Apaf-1 immunoreactive fragment that was associated exclusively with the inactive complex. We subsequently determined that caspase-3 cleaved Apaf-1 within its CED-4
domain (SVTD271↓S) in both dATP-activated lysates and apoptotic cells to form a prominent ∼30 kDa (p30) N-terminal fragment. Purified recombinant Apaf-1 p30 fragment weakly inhibited
dATP-dependent activation of caspase-3 _in vitro_. However, more importantly, prevention of endogenous formation of the p30 fragment did not stimulate latent effector caspase processing
activity in the large complex. Similarly, the possibility that XIAP, an inhibitor of apoptosis protein (IAP), was responsible for the inactivity of the ∼1.4 MDa complex was excluded as
immunodepletion of this caspase inhibitor failed to relieve the inhibition. However, selective proteolytic digestion of the ∼1.4 MDa and ∼700 kDa complexes showed that Apaf-1 was present in
conformationally distinct forms in these two complexes. Therefore, the inability of the ∼1.4 MDa apoptosome complex to process effector caspases most likely results from inappropriately
folded or oligomerized Apaf-1. SIMILAR CONTENT BEING VIEWED BY OTHERS MOLECULAR MECHANISMS UNDERLYING THE BIRC6-MEDIATED REGULATION OF APOPTOSIS AND AUTOPHAGY Article Open access 30 January
2024 EXPOSURE OF THE INNER MITOCHONDRIAL MEMBRANE TRIGGERS APOPTOTIC MITOPHAGY Article Open access 23 February 2024 HUMAN RIPK3 C-LOBE PHOSPHORYLATION IS ESSENTIAL FOR NECROPTOTIC SIGNALING
Article Open access 23 June 2022 INTRODUCTION Apoptosis is an evolutionarily conserved, biochemically and morphologically distinct form of cell death, characterized by an ordered dismantling
of the cell. In most cases, a highly selective class of cysteine proteases, known as caspases, carry out this process. Caspases are often categorized based on their function within the
cell, the length of their prodomains and their apparent substrate specificity (reviewed in1,2,3). Initiator caspases, such as caspases-8 and -9, contain long prodomains and are involved in
transducing receptor-mediated and stress-induced death signals, respectively.3,4 They activate effector caspases, such as caspase-3 and -7, which contain short prodomains and are responsible
for cleavage of many cellular proteins during the execution phase of apoptosis (reviewed in3,5). In stress-induced apoptosis, unknown cellular signals initiate the release of mitochondrial
cytochrome _c_, which interacts with Apaf-1.6,7 Apaf-1 is composed of three domains: an N-terminal caspase-recruitment domain (CARD), a CED-4 domain that contains Walker's A and B
boxes, and a series of C-terminal WD repeats (WDR).7 After associating with cytochrome _c_, Apaf-1 binds to dATP/ATP, undergoes a conformational change and self-oligomerizes _via_ its CED-4
domains.8,9,10 This process simultaneously exposes the CARD in Apaf-1, allowing it to recruit and facilitate processing of procaspase-9.9,10,11 This complex of cytochrome _c_, Apaf-1 and
caspase-9 is commonly referred to as the ‘apoptosome’. Interestingly, Apaf-1 proteins that lack WDR appear to spontaneously oligomerize in the absence of cytochrome _c_ and activate
caspase-9, but do not activate caspase-3. Thus, the WDR appear to negatively regulate Apaf-1 oligomerization and may be required for recruitment and activation of effector caspases.8,10,12
The precise binding site for cytochrome _c_ on Apaf-1 has not been identified, but an interaction between cytochrome _c_ and the WDR in Apaf-1 may induce a conformational change in Apaf-1
that exposes its CED-4 domain. In fact, WDR proteins are likely to form β-propeller structures, with the outer surfaces serving as stable platforms through which multiple proteins may
sequentially and/or simultaneously interact to form complexes.13 The apoptosome is a very large caspase-activating complex, containing multiple copies of Apaf-1 and caspase-9. Reconstitution
experiments with recombinant proteins indicate the apoptosome is ∼1.4 MDa in size, whereas those conducted in native cell lysates suggest it is ∼700 kDa.14,15,16 However, we have recently
described the formation of both these complexes in dATP-activated lysates. Significantly, however, only the ∼700 kDa complex was biologically active as assessed by its ability to process and
activate effector caspases.17 In the present study, we wished to determine why the ∼1.4 MDa Apaf-1 containing apoptosome complex was biologically inactive. We now report that in apoptotic
cells and dATP-activated cell lysates, Apaf-1 is cleaved by caspase-3 to yield an ∼30 kDa N-terminal fragment. This p30 fragment selectively associates with the ∼1.4 MDa apoptosome complex,
and addition of the purified recombinant protein to whole lysates inhibits dATP-activation of caspase-3. However, the p30 fragment is not responsible for the inactivity of the ∼1.4 MDa
complex. Therefore, the physiological function of the p30 fragment during apoptosis is currently unknown. We also exclude a potential role for the endogenous caspase inhibitor, XIAP, in
inhibiting the effector caspase processing activity of the ∼1.4 MDa complex. Instead, the conformational state of Apaf-1 within the ∼1.4 MDa and ∼700 kDa complexes appears to be
significantly different. Therefore, the inactive ∼1.4 MDa complex is likely formed as a result of inappropriate Apaf-1 oligomerization. RESULTS AN ∼30 KDA PROTEIN SELECTIVELY ASSOCIATES WITH
THE ∼1.4 MDA APOPTOSOME COMPLEX We recently described the formation of two Apaf-1 complexes in dATP-activated THP.1 lysates: a biologically active ∼700 kDa complex and an inactive ∼1.4 MDa
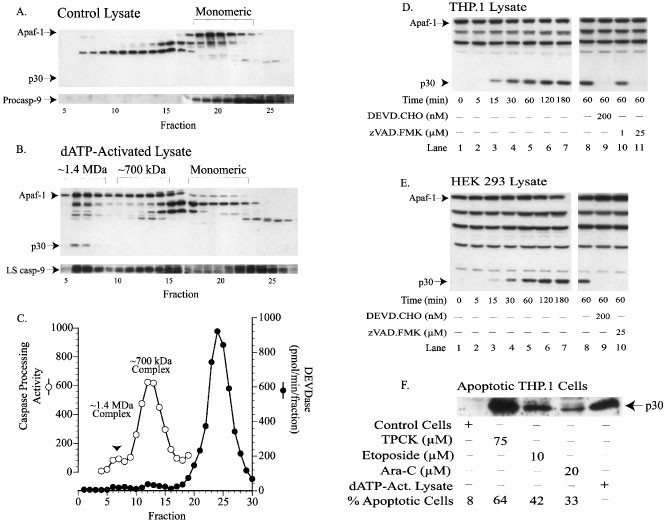
complex.17 In the current studies, in control lysates, Apaf-1 and procaspase-9 eluted off the Superose-6 column in fractions 18–24 (Mr∼158,000) and 18–26 (Mr∼60,000), respectively (Figure
1A). Following dATP activation of the lysate for 1 h, Apaf-1 oligomerized into ∼1.4 MDa and ∼700 kDa apoptosome complexes (Figure 1B, fractions 6–8 and fractions 10–16, respectively).
Procaspase-9 was processed to its catalytically active large subunits and was associated with both the ∼1.4 MDa and ∼700 kDa apoptosome complexes or was present as the free enzyme (Figure
1B). All DEVDase activity eluted in fractions 21–28 (Mr∼60,000) (Figure 1C), along with fully processed caspase-3 (data not shown). When both the ∼1.4 MDa and ∼700 kDa complexes were assayed
for their ability to process and activate exogenously added effector caspases, only the ∼700 kDa complex produced significant DEVDase activity (Figure 1C). These results were essentially
identical to those previously reported.17 However, upon dATP activation we also observed the formation of an Apaf-1 immunoreactive ∼30 kDa protein, which was selectively associated with the
inactive ∼1.4 MDa complex (Figure 1B, fractions 6–7). Consequently, we hypothesized that this ∼30 kDa protein (p30) may be a caspase-mediated cleavage product of Apaf-1 and may act as an
inhibitor of the ∼1.4 MDa complex. EFFECTOR CASPASES CLEAVE APAF-1 INTO AN ∼30 KDA FRAGMENT IN DATP-ACTIVATED LYSATES AND APOPTOTIC CELLS In order to test the hypothesis that Apaf-1 was
cleaved by caspases and to determine if the cleavage was cell-type specific, lysates from both THP.1 and HEK 293 cells were activated with dATP in the presence or absence of caspase
inhibitors. The p30 fragment was observed 15 min after activation and was inhibited by both DEVD.CHO and Z-VAD.FMK, indicating that cleavage of Apaf-1 was caspase-dependent (Figure 1D,E). In
addition, the p30 fragment was observed in THP.1 cells treated with either TPCK (75 μM), etoposide (10 μM), or Ara-C (20 μM), and the amount of the p30 formed was consistent with the degree
of apoptosis (Figure 1F). RECOMBINANT CASPASES-3 AND -7 CLEAVE PURIFIED APAF-1 AT SVTD260/271↓S Since DEVD.CHO potently inhibits the effector caspases-3 and -7, as well as the initiator
caspase-8,18 we next incubated purified unactivated Apaf-1 with either recombinant caspases-3, -7 or -8. Both caspases-3 and -7 processed Apaf-1 in a concentration-dependent manner into
several identifiable fragments, the most prominent of which was the p30 fragment previously observed in dATP-activated cell lysates (Figure 2A,B). Of note, ∼5–10 nM concentrations of active
caspases-3 and -7 were sufficient to process Apaf-1 and were below the concentration of active caspase-3 observed in dATP-activated THP.1 lysates. At the same concentrations, caspase-8
demonstrated no ability to process Apaf-1 (Figure 2C). Moreover, a role for caspase-6 was excluded because it did not process purified Apaf-1, and the caspase-6 selective inhibitor,
VEID.CHO, did not inhibit Apaf-1 cleavage in THP.1 lysates (data not shown). The p30 fragment was confirmed to be an N-terminal fragment of Apaf-1 because Flag-Apaf-1 expressed in HEK 293
produced a Flag-p30 fragment following incubation with active recombinant caspase-3 (Figure 2D). Since the p30 fragment was derived from the N-terminus of Apaf-1, we estimated the probable
caspase cleavage site motif to be SVTD260↓S in Apaf-1/Apaf-1L or SVTD271↓S in Apaf-1XL, which contains an additional eleven amino acids between the CARD and CED-4 domains (Figure 2E). To
confirm this cleavage site, we utilized a truncated form of Apaf-1 (residues 1–530; ∼62 kDa) which could be more easily mutated and _in vitro_ transcribed/translated. Caspase-3 cleaved
wild-type [35S]Apaf-530 into two fragments, one corresponding to the first 260 amino acids (p30 fragment) and the other corresponding to the remainder of the molecule, including the
C-terminal His6 tag (Figure 2E, lanes 2–4). The latter fragment appeared to undergo a second cleavage event to form a slightly smaller C-terminal fragment (Figure 2E, asterisk, lanes 3–4).
In contrast, the mutated [35S]Apaf-530 (D260A) was not cleaved at all by caspase-3 (Figure 2E, lanes 5–8). Thus, caspase-3 cleaves Apaf-1 within its CED-4 domain (SVTD↓S), downstream of the
Walker's B box (Figure 2E). RECOMBINANT P30 WEAKLY INHIBITS DATP-ACTIVATION OF CASPASE-3 In order to determine the biological effects of the p30 fragment, we cloned, expressed and
purified the protein for subsequent studies. We initially verified that recombinant p30 could associate with the ∼1.4 MDa complex when added to lysate that was subsequently dATP-activated
(data not shown). Next, we assessed the ability of purified Apaf-1L and p30 to activate caspase-9 in a pure recombinant system using _in vitro_ transcribed/translated [35S]procaspase-9,
cytochrome _c_ and dATP. Interestingly, while recombinant Apaf-1L readily activated caspase-9 (Figure 3A, lanes 1–4), the recombinant p30 was incapable of activating caspase-9 (Figure 3A,
lanes 5–7). Since the p30 fragment would be expected to bind procaspase-9 through a CARD-CARD interaction similar to Apaf-1L, we hypothesized that the p30 fragment might behave as a
competitive inhibitor of caspase-9 activation. Indeed, addition of purified p30 to THP.1 lysates slightly inhibited dATP-mediated activation of caspase-3 by caspase-9, as demonstrated by
inhibition of caspase-3 processing and decreased DEVDase activity (Figure 3B). Thus, the Apaf-1 p30 fragment appeared to compete weakly with endogenous full-length Apaf-1 for procaspase-9
and consequently decreased the number of available active Apaf-1/caspase-9 complexes. However, as the concentrations of p30 required to inhibit caspase activation were far above those
generated endogenously, the physiological significance of this result is presently unclear. IMMUNODEPLETION OF CASPASE-3 PREVENTS ASSOCIATION OF THE ∼30 KDA FRAGMENT WITH THE ∼1.4 MDA
COMPLEX Caspases-3 and -7 are frequently viewed as redundant caspases because they often cleave the same substrates _in vitro_. However, a number of studies suggest these caspases may play
distinct roles in dATP-activated cell lysates and apoptotic cells.15,19,20 Consequently, in order to determine the contribution of caspase-3 in producing the Apaf-1 p30 fragment and to
assess the effects of blocking its formation on apoptosome function, we immunodepleted caspase-3 from lysates prior to dATP activation. Following dATP activation and separation by gel
filtration, caspase-3 depleted lysate exhibited a >95% loss in DEVDase activity (Figure 3D). The remaining DEVDase activity co-eluted with processed caspase-7 in fractions 19–23 (Figure
3D and data not shown). Importantly, the p30 fragment was no longer associated with the ∼1.4 MDa apoptosome complex (Figures 3C and 1B, compare fractions 6–7). In contrast, immunodepletion
of caspase-7 did not significantly inhibit the association of the p30 fragment with the ∼1.4 MDa apoptosome complex (data not shown). This appeared somewhat surprising given that recombinant
caspase-7 was slightly more effective than caspase-3 at processing purified Apaf-1 _in vitro_ (Figure 2A,B). However, caspase-7 is present at significantly lower concentrations than
caspase-3 in our THP.1 cell lysates. Therefore, immunodepletion of caspase-3, but not caspase-7, effectively inhibited formation and association of the p30 fragment with the ∼1.4 MDa
complex. However, the ∼1.4 MDa apoptosome complex was still inactive when assayed for its ability to process effector caspases and generate DEVDase activity (Figure 3D, fractions 6–8). In
contrast, the ∼700 kDa complex displayed its normal capacity to process effector caspases (Figure 3D, fractions 10–16). Thus, caspase-3 appeared primarily responsible for the cleavage of
Apaf-1 and formation of the p30 fragment that associated with the large ∼1.4 MDa apoptosome complex. However, the immunodepletion experiments clearly demonstrated that the inability of the
∼1.4 MDa complex to activate effector caspases was not due to the presence of the N-terminal Apaf-1 p30 fragment or its corresponding C-terminal p105 fragment, nor was it due to caspase-3
inactivating the complex through cleavage of Apaf-1 or some other component of the apoptosome. XIAP IS NOT RESPONSIBLE FOR THE INACTIVITY OF THE ∼1.4 MDA APOPTOSOME COMPLEX In both our
present and previous studies, processed caspase-9 was present in both the ∼1.4 MDa and ∼700 kDa apoptosome complexes (Figures 1B and 3C), but only the latter activated effector caspases
(Figures 1C and 3D). Since IAPs, particularly XIAP, cIAP-1 and cIAP-2, are the primary endogenous inhibitors of caspase activity,21,22 it was possible they were responsible for inhibiting
the ∼1.4 MDa complex. However, only XIAP co-eluted with the ∼1.4 MDa apoptosome fractions following dATP activation of the lysate (data not shown). Therefore, THP.1 lysates were
immunodepleted of XIAP (Figure 4A), activated with dATP, and fractionated as already described. XIAP was not responsible for the inactivity of the ∼1.4 MDa apoptosome complex as the complex
remained incapable of activating effector caspases (Figure 4B, fractions 6–8.). Interestingly, removal of XIAP also had little effect on the activity of the ∼700 kDa complex (Figures 1C and
4B, compare fractions 10–16), suggesting that the concentration of XIAP in our THP.1 lysate preparation was either insufficient to effectively inhibit effector caspase activation or that
XIAP was rendered ineffective _via_ the action of the recently discovered IAP inhibitor, Smac/Diablo.23,24 APAF-1 EXHIBITS ALTERNATIVE CONFORMATIONS IN THE ∼700 KDA AND ∼1.4 MDA APOPTOSOME
COMPLEXES Since the ∼1.4 MDa apoptosome complex was (i) not active at any stage during its formation,17 (ii) approximately twice the size of the active ∼700 kDa apoptosome complex, (iii) not
directly inactivated by caspase-3 or the endogenously derived p30 fragment, and (iv) not inactivated by the presence of IAPs, we hypothesized that the ∼1.4 MDa apoptosome complex might
simply be inappropriately formed. We speculated that Apaf-1 might be present in a conformational state that prevents normal recruitment/processing of effector caspases. Therefore, we
isolated both ∼700 kDa and ∼1.4 MDa apoptosome complexes from dATP-activated caspase-3 immunodepleted lysates and subjected them to partial proteolysis with trypsin, chymotrypsin or
staphylococcal V8 protease. Apaf-1 in the ∼1.4 MDa complex was generally more susceptible to proteolytic cleavage by all of the proteases compared to Apaf-1 in the ∼700 kDa complex (Figure
4C). In addition, the pattern of protein fragments was strikingly different. Trypsin produced at least three low molecular weight fragments (<50 kDa) from the ∼1.4 MDa complex that were
not detected following proteolysis of the ∼700 kDa complex, and both trypsin and chymotrypsin produced numerous larger molecular weight fragments (∼65–125 kDa) that were primarily generated
from the larger complex. The apparent sensitivity of the ∼1.4 MDa complex to proteolytic cleavage led to us to question if this complex might likewise demonstrate greater sensitivity to
caspase-3 mediated cleavage. Indeed, Apaf-1 within the ∼1.4 MDa complex (Figure 4C, fractions 6–8) was clearly more susceptible to cleavage by caspase-3 than Apaf-1 within the ∼700 kDa
complex (Figure 4C, fractions 11–13). These data were compatible with the hypothesis that the ∼700 kDa apoptosome complex initially activated caspase-3, which in turn cleaved Apaf-1 within
the ∼1.4 MDa apoptosome complex to form the ∼30 kDa fragment. Caspase-3 was capable of cleaving purified monomeric Apaf-1 into the ∼30 kDa fragment (Figure 2A) and recombinant p30 associated
with the ∼1.4 MDa complex when added to dATP-activated lysates (data not shown). Thus, it appears that both monomeric and inappropriately oligomerized Apaf-1 are sensitive to cleavage by
caspase-3 and that in either case the resulting Apaf-1 p30 fragment associates with the ∼1.4 MDa apoptosome complex. However, since the vast majority of Apaf-1 undergoes oligomerization in
dATP-activated lysates prior to activation of effector caspases,17 it appears more likely that caspase-3 targets the inappropriately oligomerized Apaf-1 in the ∼1.4 MDa complex rather than
unactivated Apaf-1. DISCUSSION Cytochrome _c_/dATP-dependent oligomerization of Apaf-1 is required for formation of a functional apoptosome that processes caspase-9, followed by caspases-3
and -7.9,10,14,15,16 However, this process is still somewhat ill defined. In reconstitution experiments using purified Apaf-1, caspase-9 and cytochrome _c_, Apaf-1 apparently oligomerizes
into an ∼1.4 MDa complex.14,16 Nevertheless, we find that in cell lysates, this complex is relatively inactive compared with the ∼700 kDa apoptosome complex.17 However, the ∼1.4 MDa
apoptosome complex can be observed in apoptotic cells and appears to be dependent upon the apoptotic stimulus and the degree of apoptosis.17 In the present studies we have attempted to
explain why the ∼1.4 MDa complex is inactive. In doing so, we discovered that in dATP-activated lysates and apoptotic cells, caspase-3 can cleave Apaf-1 at SVTD260/271↓S to generate an
Apaf-1 fragment that contains an intact CARD domain and a truncated CED-4 region (Figures 1 and 2). Purified p30 fragment was capable of weakly inhibiting dATP-mediated activation of
caspase-3 by caspase-9 in lysates, but only did so at concentrations that are likely to exceed those produced endogenously. Consistent with these findings, immunodepletion of caspase-3 from
lysates prevented formation and association of the N-terminal Apaf-1 p30 fragment and its corresponding C-terminal p105 fragment with the ∼1.4 MDa apoptosome by preventing cleavage of
Apaf-1. However, the ∼1.4 MDa complex was formed to the same extent in both caspase-3 depleted and normal dATP-activated lysates, indicating that the p30 fragment did not ‘convert’ an active
∼700 kDa apoptosome complex into an inactive ∼1.4 MDa apoptosome complex. In addition, the ∼1.4 MDa apoptosome complexes remained incapable of activating effector caspases, even in the
absence of any Apaf-1 p30 fragment (Figure 3). Thus, although the biological consequences of Apaf-1 cleavage remain unknown, neither cleavage of Apaf-1 nor the resulting p30 fragment were
responsible for producing or inhibiting the ∼1.4 MDa complex. Interestingly, processed caspase-9 was associated with the inactive ∼1.4 MDa apoptosome complex, implying that the large complex
might be capable of promoting trans-activation of procaspase-9 or alternatively, might simply recruit processed caspase-9 released from the active ∼700 kDa apoptosome complex. If the former
were in fact true, then the presence of a caspase-9 inhibitor, for example a member of the inhibitor of apoptosis (IAP) family of proteins such as XIAP,21,22 might explain the inability of
the ∼1.4 MDa complex to activate effector caspases. However, XIAP was not responsible for the inactivity of the ∼1.4 MDa complex as its removal did not restore the activity of the large
complex (Figure 4). Rather, partial proteolysis of both the ∼700 kDa and ∼1.4 MDa complexes revealed markedly different proteolytic digestion profiles and suggested that the ∼1.4 MDa
apoptosome complex was inappropriately formed (Figure 4). Indeed, all of the proteases tested, including caspase-3, appeared to cleave Apaf-1 in the ∼1.4 MDa complex far more efficiently
than in the ∼700 kDa complex. It is interesting to note that the WD repeats present in Gβ-subunits form β-propeller structures, which are very resistant to trypsin, unless they are misfolded
or disturbed by particular amino acid substitutions.25 Apaf-1 contains a large C-terminal WD40 repeat region, which has been suggested to form β-propellers and which has recently been
implicated in the recruitment of effector caspases-3 and -7 to the apoptosome.7,12 In our studies, both trypsin and chymotrypsin produced numerous large N-terminal Apaf-1 fragments in the
inactive ∼1.4 MDa apoptosome complex, but not in the ∼700 kDa complex (Figure 4). Thus, inappropriately oligomerized Apaf-1 in the ∼1.4 MDa apoptosome complex may contain misfolded WD
repeats, which are sensitive to trypsin degradation and are incapable of effectively recruiting and processing effector caspases-3 and -7. However, the specific factors that dictate
inappropriate formation of the ∼1.4 MDa apoptosome complex in apoptotic cells are currently unclear, but the focus of future research. In conclusion, we have demonstrated that active
effector caspase-3 cleaves Apaf-1 within the ∼1.4 MDa apoptosome complex into a prominent ∼30 kDa N-terminal fragment, which remains associated with the complex. However, the inability of
this complex to efficiently activate effector caspases is not due to degradation and inactivation of the complex by active caspase-3 or to the presence of an Apaf-1 fragment. Moreover, IAPs
do not appear responsible for its lack of activity. Instead, the inability of the ∼1.4 MDa apoptosome complex to process effector caspases is likely because the complex contains
inappropriately oligomerized Apaf-1. MATERIALS AND METHODS PREPARATION AND ACTIVATION OF CELL LYSATES/APOPTOTIC CELLS Human monocytic THP.1 cells and human embryonic kidney (HEK) 293 cells
were grown in RPMI 1640 and DMEM, respectively, supplemented with 10% heat inactivated FBS in 5% CO2 at 37°C. Cell lysates were prepared as previously described.15 _In vitro_ activation of
caspases was initiated by incubating lysates (10 mg/ml) with dATP/MgCl2 (2 mM) for various times at 37°C. Exogenous cytochrome _c_ was not required as it was released from mitochondria
during lysate preparation. In some experiments, THP.1 cells were treated with either _N_-tosyl-_l_-phenylalanyl chloromethyl ketone (TPCK; 75 μM), etoposide (10 μM), or
1-β-D-arabinofuranosylcytosine hydrochloride (Ara-C; 20 μM) for varying times, and apoptosis was assessed by phosphatidylserine exposure as previously described.26 CASPASE CLEAVAGE OF
ENDOGENOUS AND RECOMBINANT APAF-1 THP.1 and HEK293 lysates (25 μg protein) were activated with dATP in the presence or absence of the caspase inhibitors, benzyloxycarbonyl-Val-Ala-Asp(OMe)
fluoromethyl-ketone (Z-VAD.FMK) or Ac-Asp-Glu-Val-Asp aldehyde (DEVD.CHO). Z-VAD.FMK inhibits all known caspases, whereas DEVD.CHO is more selective for caspases-3, -7 and -8.18 Purified
recombinant unactivated Apaf-1 (∼200 ng) was incubated with active caspases-3, -6, -7 or -8 for 1 h at 37°C. All protein samples were mixed with 2× SDS loading buffer, separated by SDS–PAGE,
and immunoblotted for Apaf-1 cleavage products using an antibody generously provided by Dr. Xiaodong Wang (University of Texas Southwestern Medical Center, Dallas, TX, USA). This antibody
was raised against the N-terminus of Apaf-1 (amino acids 10–254).7 APAF-1 MUTAGENESIS/DELETIONS The caspase cleavage site in Apaf-1 was determined using a truncated form of Apaf-1
(pET28a-Apaf-530; generously provided by Dr. Emad S. Alnemri).10 Asp-260 was mutated to an Ala using Stratagene's Quik-Change kit with the following primers (mutation underlined)
(5′→CAAGAGTGTTACAGCTTCAGTAATGG→3′; 5′→CCATTACTGAAGCTGTAACACTCTTG→3′) and confirmed by DNA sequencing. The wild-type Apaf-530 and Apaf-530 (D260→A) plasmids were transcribed/translated using
the T7 TNT system (Promega), and the 35S-labeled proteins were incubated with recombinant caspase-3 for 2 h and analyzed by SDS–PAGE/autoradiography. This mutation corresponds to D271→A in
Apaf-1XL/Apaf-1WD13,12,16 which contains an additional 11 amino acids between the CARD and CED-4 domains. While conducting these studies, Apaf-1XL was shown to be the primary form of Apaf-1
present in cells.27 Consequently, we PCR amplified the sequence corresponding to the N-terminal cleavage product of Apaf-1XL (residues 1–271), using an Apaf-1XL construct (kindly provided by
Dr. Gabriel Nuñez) as a template and the following primers which contained the necessary restriction sites (5′→CGGGATCCATGGATGCAAAAGCTCGAAATTG→3′ and
5′→GGGGTACCAGCTGTAACACTCTTGTCTCTGGTTG→3′). The PCR product was cloned into pTriEX-1 (Novagen) at the _Bam_HI/_Kpn_I sites and incorporated C-terminal HSV and His-6 tags. In addition, the
D271→A mutation was also included in the fragment to ensure that the HSV and His-6 tags would not be removed by caspase-3 during dATP activation experiments. EXPRESSION AND PURIFICATION OF
HUMAN RECOMBINANT APAF-1 (AND ITS P30 CLEAVAGE FRAGMENT) AND CASPASES Sf-9 insect cells were infected with high titer baculoviral stocks encoding full-length Apaf-1.16 Cells were harvested
48 h later and His-tagged Apaf-1 was purified on a Ni2+ column essentially as previously described.16 Similarly, the p30 Apaf-1 fragment was bacterially expressed, isolated on Ni2+ beads,
and further purified by DEAE-chromatography. Active caspases-3, -6, -7 and -8 were expressed, purified and active site titrated as previously described.28,29 ANALYSIS OF APOPTOSOME
COMPLEXES/IMMUNODEPLETION OF CASPASE-3 AND XIAP Control and dATP-activated lysates were fractionated by size-exclusion chromatography, using an FPLC protein purification system with a
Superose 6 HR 10/30 column (Amersham Pharmacia Biotech, Herts, UK). Apoptosome complexes (∼700 kDa and ∼1.4 MDa) were eluted using column buffer supplemented with 50 mM NaCl, assayed for
their ability to process exogenously added caspases-9, -3 and -7, and immunoblotted for Apaf-1 and caspase-9 (D.R. Green, La Jolla Institute for Allergy and Immunology, San Diego, CA, USA)
as previously described.17 In some experiments, lysates were immunodepleted of caspase-3 or XIAP prior to dATP-activation. Briefly, protein G sepharose beads (500 μl of 50% slurry; Amersham
Pharmacia Biotech, Herts, UK) were blocked with 3% BSA, preincubated for 2 h with anti-caspase-3 or XIAP antibodies (1 : 50; Transduction Laboratories/Pharmingen, San Diego, CA, USA) and
thoroughly washed to remove unbound antibody. Lysates (15 mg/ml) were then incubated with antibody coated beads for 2 h. Partially immunodepleted supernatants were removed by centrifugation
and subjected to a second round of immunodepletion in order to completely deplete caspase-3 or XIAP. PARTIAL PROTEOLYSIS OF ∼1.4 MDA AND ∼700 KDA APOPTOSOME COMPLEXES Following dATP
activation of caspase-3 immunodepleted lysates, apoptosome complexes were isolated by gel fitration as described above. A portion of each column fraction (30 μl) was incubated with either
100 nM caspase-3 for 2 h or 1 ng/ml of sequencing grade trypsin, chymotrypsin or staphylococcal V8 protease (Boehringer Mannheim, Lewes, UK) for 15 min at 37°C. Reactions were terminated
with an equal volume of 2×SDS loading buffer and analyzed for Apaf-1 cleavage products. ABBREVIATIONS * CARD: caspase recruitment domain * Z-VAD.FMK: benzyloxycarbonyl-Val-Ala-Asp-(OMe)
fluoromethyl ketone * DEVD.CHO: acetyl-Asp-Glu-Val-Asp aldehyde * HEK 293 cells: human embryonic kidney cells REFERENCES * Cohen GM . 1997 Caspases: the executioners of apoptosis _Biochem.
J._ 326: 1–16 Article CAS Google Scholar * Bratton SB, MacFarlane M, Cain K, Cohen GM . 2000 Protein complexes activate distinct caspase cascades in death receptor and stress-induced
apoptosis _Exp. Cell Res._ 256: 27–33 Article CAS Google Scholar * Earnshaw WC, Martins LM, Kaufmann SH . 1999 Mammalian caspases: structure, activation, substrates, and functions during
apoptosis _Annu. Rev. Biochem._ 68: 383–424 Article CAS Google Scholar * Sun XM, MacFarlane M, Zhuang J, Wolf BB, Green DR, Cohen GM . 1999 Distinct caspase cascades are initiated in
receptor-mediated and chemical-induced apoptosis _J. Biol. Chem._ 274: 5053–5060 Article CAS Google Scholar * Nicholson DW . 1999 Caspase structure, proteolytic substrates, and function
during apoptotic cell death _Cell Death Differ._ 6: 1028–1042 Article CAS Google Scholar * Green DR, Reed JC . 1998 Mitochondria and apoptosis _Science_ 281: 1309–1312 Article CAS
Google Scholar * Zou H, Henzel WJ, Liu X, Lutschg A, Wang X . 1997 Apaf-1, a human protein homologous to C. elegans CED-4, participants in cytochrome c-dependent activation of caspase-3
_Cell_ 90: 405–413 Article CAS Google Scholar * Hu Y, Ding L, Spencer DM, Nunez G . 1998 WD-40 repeat region regulates Apaf-1 self-association and procaspase-9 activation _J. Biol. Chem._
273: 33489–33494 Article CAS Google Scholar * Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X . 1997 Cytochrome c and dATP-dependent formation of
Apaf-1/caspase-9 complex initiates an apoptotic protease cascade _Cell_ 91: 479–489 Article CAS Google Scholar * Srinivasula SM, Ahmad M, Fernandes-Alnemri T, Alnemri ES . 1998
Autoactivation of procaspase-9 by Apaf-1-mediated oligomerization _Mol. Cell_ 1: 949–957 Article CAS Google Scholar * Qin H, Srinivasula SM, Wu G, Fernandes-Alnemri T, Alnemri ES, Shi Y .
1999 Structural basis of procaspase-9 recruitment by the apoptotic protease-activating factor 1 _Nature_ 399: 549–557 Article CAS Google Scholar * Hu Y, Benedict MA, Ding L, Nunez G .
1999 Role of cytochrome _c_ and dATP/ATP hydrolysis in Apaf-1-mediated caspase-9 activation and apoptosis. EMBO J _18:_ 18: 3586–3595 CAS Google Scholar * Smith TF, Gaitatzes C, Saxena K,
Neer EJ . 1999 The WD repeat: a common architecture for diverse functions _Trends Biochem. Sci._ 24: 181–185 Article CAS Google Scholar * Saleh A, Srinivasula SM, Acharya S, Fishel R,
Alnemri ES . 1999 Cytochrome c and dATP-mediated oligomerization of Apaf-1 is a prerequisite for procaspase-9 activation _J. Biol. Chem._ 274: 17941–17945 Article CAS Google Scholar *
Cain K, Brown DG, Langlais C, Cohen GM . 1999 Caspase activation involves the formation of the aposome, a large (∼700 kDa) caspase-activating complex _J. Biol. Chem._ 274: 22686–22692
Article CAS Google Scholar * Zou H, Li Y, Liu X, Wang X . 1999 An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9 _J. Biol. Chem._ 274:
11549–11556 Article CAS Google Scholar * Cain K, Bratton SB, Langlais C, Walker G, Brown DG, Sun XM, Cohen GM . 2000 Apaf-1 oligomerizes into biologically active ∼700 kDa and inactive
∼1.4 MDa apoptosome complexes _J. Biol. Chem._ 275: 6067–6070 Article CAS Google Scholar * Garcia-Calvo M, Peterson EP, Leiting B, Ruel R, Nicholson DW, Thornberry NA . 1998 Inhibition of
human caspases by peptide-based and macromolecular inhibitors _J. Biol. Chem._ 273: 32608–32613 Article CAS Google Scholar * Chandler JM, Cohen GM, MacFarlane M . 1998 Different
subcellular distribution of caspase-3 and caspase-7 following Fas-induced apoptosis in mouse liver _J. Biol. Chem._ 273: 10815–10818 Article CAS Google Scholar * Germain M, Affar EB,
D'Amours D, Dixit VM, Salvesen GS, Poirier GG . 1999 Cleavage of automodified poly(ADP-ribose)polymerase during apoptosis. Evidence for involvement of caspase-7 _J. Biol. Chem._ 274:
28379–28384 Article CAS Google Scholar * Deveraux QL, Takahashi R, Salvesen GS, Reed JC . 1997 X-linked IAP is a direct inhibitor of cell-death proteases _Nature_ 388: 300–304 Article
CAS Google Scholar * Deveraux QL, Roy N, Stennicke HR, Van Arsdale T, Zhou Q, Srinivasula SM, Alnemri ES, Salvesen GS, Reed JC . 1998 IAPs block apoptotic events induced by caspase-8 and
cytochrome c by direct inhibition of distinct caspases _EMBO J._ 17: 2215–2223 Article CAS Google Scholar * Du C, Fang M, Li Y, Li L, Wang X . 2000 Smac, a mitochondrial protein that
promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition _Cell_ 102: 33–42 Article CAS Google Scholar * Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM,
Reid GE, Moritz RL, Simpson RJ, Vaux DL . 2000 Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins _Cell_ 102: 43–53 Article
CAS Google Scholar * Garcia-Higuera I, Gaitatzes C, Smith TF, Neer EJ . 1998 Folding a WD repeat propeller. Role of highly conserved aspartic acid residues in the G protein beta subunit
and Sec13 _J. Biol. Chem._ 273: 9041–9049 Article CAS Google Scholar * Zhuang J, Ren Y, Snowden RT, Zhu H, Gogvadze V, Savill JS, Cohen GM . 1998 Dissociation of phagocyte recognition of
cells undergoing apoptosis from other features of the apoptotic program _J. Biol. Chem._ 273: 15628–15632 Article CAS Google Scholar * Benedict MA, Hu Y, Inohara N, Nunez G . 2000
Expression and functional analysis of Apaf-1 isoforms. Extra WD-40 repeat is required for cytochrome c binding and regulated activation of procaspase-9 _J. Biol. Chem._ 275: 8461–8468
Article CAS Google Scholar * Srinivasula SM, Ahmad M, Fernandes-Alnemri T, Litwack G, Alnemri ES . 1996 Molecular ordering of the Fas-apoptotic pathway: the Fas/APO-1 protease Mch5 is a
CrmA-inhibitable protease that activates multiple Ced-3/ICE-like cysteine proteases _Proc. Natl. Acad. Sci. USA_ 93: 14486–14491 Article CAS Google Scholar * Garcia-Calvo M, Peterson EP,
Rasper DM, Vaillancourt JP, Zamboni R, Nicholson DW, Thornberry NA . 1999 Purification and catalytic properties of human caspase family members _Cell Death Differ._ 6: 362–369 Article CAS
Google Scholar Download references ACKNOWLEDGEMENTS The authors thank Dr. Xiaodong Wang (University of Texas Southwestern Medical Center, Dallas, TX) for the baculoviral Apaf-1 stock and
anti-Apaf-1 antibody, Dr. Emad S. Alnemri for the Apaf-1-530 construct, Dr. Douglas R. Green (La Jolla Institute for Allergy and Immunology, San Diego, CA) for anti-caspase-9 antibody, and
Dr. D.W. Nicholson (Merck Frosst, Canada) for anti-caspase-3 antibody. The authors also thank X-M Sun, C Langlais and D Brown for their technical expertise and helpful discussions during the
preparation of this manuscript. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * MRC Toxicology Unit, Hodgkin Building, University of Leicester, P.O. Box 138, Lancaster Road, Leicester, LE1
9HN, UK S B Bratton, G Walker, D L Roberts, K Cain & G M Cohen Authors * S B Bratton View author publications You can also search for this author inPubMed Google Scholar * G Walker View
author publications You can also search for this author inPubMed Google Scholar * D L Roberts View author publications You can also search for this author inPubMed Google Scholar * K Cain
View author publications You can also search for this author inPubMed Google Scholar * G M Cohen View author publications You can also search for this author inPubMed Google Scholar
CORRESPONDING AUTHOR Correspondence to G M Cohen. ADDITIONAL INFORMATION Edited by G Melino RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Bratton, S.,
Walker, G., Roberts, D. _et al._ Caspase-3 cleaves Apaf-1 into an ∼30 kDa fragment that associates with an inappropriately oligomerized and biologically inactive ∼1.4 MDa apoptosome complex.
_Cell Death Differ_ 8, 425–433 (2001). https://doi.org/10.1038/sj.cdd.4400834 Download citation * Received: 08 November 2000 * Revised: 04 December 2000 * Accepted: 14 December 2000 *
Published: 01 May 2001 * Issue Date: 01 April 2001 * DOI: https://doi.org/10.1038/sj.cdd.4400834 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this
content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative
KEYWORDS * Apaf-1 * apoptosome * effector caspases