Play all audios:
ABSTRACT BACKGROUND: Autophagy allows recycling of cellular components and may facilitate cell survival after chemotherapy. Pantoprazole inhibits proton pumps and is reported to inhibit
autophagy. Here we evaluate the effects of pantoprazole to modify cytotoxicity of the anticancer drug docetaxel, and underlying mechanisms. METHODS: Effects of docetaxel±pantoprazole were
studied against wild-type and autophagy-deficient PC3 cells and against four human xenografts. Effects of pantoprazole on autophagy were evaluated by quantifying LC3-I, LC3-II and p62
proteins in western blots, and by fluorescent microscopy of cells transfected with RFP-GFP-LC3. The distribution of drug effects and of autophagy was quantified in tumour sections in
relation to blood vessels and hypoxia by immunohistochemistry using _γ_H2AX, cleaved caspase-3, Ki67 and LC3/ p62. RESULTS: Pantoprazole increased the toxicity of docetaxel _in vitro_,
increased docetaxel-induced expression of _γ_H2AX and cleaved caspase-3, and decreased Ki67 in tumour sections. Pantoprazole increased growth delay of four human xenografts of low, moderate
and high sensitivity to docetaxel, with minimal increase in toxicity. Docetaxel led to increased autophagy throughout tumour sections. Pantoprazole inhibited autophagy, and effects of
pantoprazole were reduced against genetically modified cells with decreased ability to undergo autophagy. CONCLUSIONS: Autophagy is a mechanism of resistance to docetaxel chemotherapy that
may be modified by pantoprazole to improve therapeutic index. SIMILAR CONTENT BEING VIEWED BY OTHERS AUTOPHAGY AS A THERAPEUTIC TARGET IN PANCREATIC CANCER Article Open access 15 September
2020 3-BROMOPYRUVATE OVERCOMES CETUXIMAB RESISTANCE IN HUMAN COLORECTAL CANCER CELLS BY INDUCING AUTOPHAGY-DEPENDENT FERROPTOSIS Article Open access 09 August 2023 PEVONEDISTAT (MLN4924):
MECHANISM OF CELL DEATH INDUCTION AND THERAPEUTIC POTENTIAL IN COLORECTAL CANCER Article Open access 21 July 2020 MAIN Causes of resistance to chemotherapy have focused on molecular changes
in individual tumour cells, including expression of drug export pumps such as P-glycoprotein and altered expression of tubulin isotypes, which convey resistance to taxanes (Bradley and Ling,
1994; Terry et al, 2009; Ploussard et al, 2010). Other mechanisms depend on the solid tumour microenvironment including problems of limited delivery of anticancer drugs to tumour cells that
are distal to functional blood vessels (Lankelma et al, 1999; Tannock et al, 2002; Huxham et al, 2004; Tredan et al, 2007), and resistance of these slowly proliferating, poorly nourished
(often hypoxic) cells to cycle-dependent chemotherapy. We have shown that limited distribution of activity in poorly nourished or hypoxic regions of solid tumours is common to many
anticancer drugs, including docetaxel, and is an important cause of therapeutic resistance (Saggar et al, 2013). Autophagy is a cellular process of self-consumption characterised by
sequestration of bulk cytoplasm, long-lived proteins and cellular organelles into double-membrane vesicles called autophagosomes, which are delivered to and degraded in lysosomes (White,
2012). Markers of autophagy co-localise in hypoxic and poorly nourished regions of tumours (Hoyer-Hansen and Jaattela, 2007; Rouschop et al, 2010). Autophagy is prognostic of poor outcome in
multiple tumour types (Sivridis et al, 2010; Karpathiou et al, 2011; Sivridis et al, 2011), and high levels of autophagy have been associated with resistance to systemic therapy in several
preclinical and clinical models, presumably because autophagy facilitates survival of stressed or damaged cells through recycling of cellular breakdown products (Yang et al, 2011). Hence,
targeting of autophagy with pharmacological agents may be a mechanism to improve the effectiveness of anticancer drugs for solid tumours. Current clinical strategies for inhibiting autophagy
include the use of hydroxychloroquine (HCQ) and proton pump inhibitors (PPIs) that disrupt lysosomal pH regulation and thus prevent autolysosome formation and degradation of captured
cytoplasmic content. Several clinical trials of HCQ in combination with cytotoxic (docetaxel, temozolomide) and targeted (gefitinib) agents are underway, based on the premise that inhibition
of autophagy by HCQ should enhance the efficacy of these drugs (Poklepovic and Gewirtz, 2014; Rosenfeld et al, 2014). Inhibitors of autophagy with greater specificity than HCQ are also
being developed (McAfee et al, 2012; Deng et al, 2013). Pantoprazole is a PPI that inhibits the gastric H+, K+-ATPase proton pump; but at higher concentration, PPIs inhibit other proton
pumps, including those that acidify endosomes; they have been reported to inhibit autophagy possibly through inhibiting acidification of endosomes or their fusion with autophagosomes
(Udelnow et al, 2011). Proton pump inhibitors have been reported to sensitise cancer cells and solid tumours to different chemotherapeutic agents (Luciani et al, 2004). Multiple mechanisms
are probably involved, but appear to relate to changes in acidity in intra- and extracellular compartments of tumour cells. Several studies have shown that PPIs such as omeprazole,
esomeprazole and pantoprazole have activity against human hematopoietic and solid tumours; they may revert chemo-resistance in drug-resistant tumours and directly induce killing of tumour
cells (Yeo et al, 2004; De Milito et al, 2007; de Milito et al, 2010). Growing evidence suggests that the major mechanism may be inhibition of autophagy (Levy et al, 2014; Pan et al, 2014;
Wang and Wu, 2014; Yang et al, 2014a, 2014b). In the present study, we report that pantoprazole enhances the _in vitro_ and _in vivo_ activity of docetaxel, a drug in wide clinical use, and
provide evidence that the major underlying mechanism is the inhibition of autophagy. MATERIALS AND METHODS CELL LINES, DRUGS AND REAGENTS Human breast carcinoma MCF-7 cells, human vulvar
epidermoid carcinoma A-431 cells, and human prostate cancer PC3 and LNCap cells were used. All cell lines were purchased from the American Type Culture Collection in 2011. MCF-7, A-431 and
cells have been maintained in our laboratory and were grown in _α_-minimum essential medium supplemented with 10% FBS (Hyclone, Logan, UT, USA). The PC3 and LNCaP cells were grown in Ham’s
F-12K medium (Life Technologies Inc., Carlsbad, CA, USA) supplemented with 10% FBS. All cells were grown in a humidified atmosphere of 95% air/5% CO2 at 37 °C and experiments were performed
on 4th and 5th passages generated from the frozen stock. Routine tests to exclude Mycoplasma in all cell lines were conducted several times each year. Short tandem repeat analysis was
conducted to ensure cells (MCF-7, A-431, PC3 and LNCaP) were of human origin in May 2013. To generate tumours, 4- to 6-week-old male athymic nude mice (Jackson Laboratory, Bar Harbor, ME,
USA) were injected subcutaneously in both flanks with 2 × 106 PC3 or LNCaP cells, and 4- to 6-week-old female athymic nude mice (Harlan Sprague–Dawley) with implanted 17_β_-estradiol tablets
(60-day release; Innovative Research of America) were injected subcutaneously with 5 × 106 MCF-7 cells per side; non-estradiol-implanted female athymic nude mice were injected with 1 × 106
A-431 cells. There were six mice per treatment group (12 tumours) and each experiment was repeated three times. Docetaxel was obtained from Sanofi Inc (Laval, Quebec). Pantoprazole was
purchased from the hospital pharmacy as a lyophilised powder and dissolved in 0.9% saline. EF5 was provided by the National Cancer Institute as a powder and then dissolved in distilled water
supplemented with 2.4% ethanol and 5% dextrose to make a 10-mM stock solution that was stored at room temperature. Cy5-conjugated mouse anti-EF5 antibody was purchased from Dr Cameron Koch,
University of Pennsylvania, Philadelphia, PA, USA. DiOC7 was purchased from AnaSpec (San Jose, CA, USA) and a stock solution (2.5 mg ml−1) was made by dissolving in dimethyl sulphoxide;
this stock was diluted 1 : 10 in phosphate-buffered saline and 10% Solutol HS 15 (Sigma-Aldrich, Oakville, ON, Canada). _γ_H2aX was recognised with a rabbit anti-human _γ_H2aX primary
antibody (Cell Signaling, Danvers, MA, USA). Cleaved caspase-3 was recognised with primary rabbit anti-human cleaved caspase-3 antibody (Cell Signaling, Danvers, MA, USA). Ki67 was
identified with primary rabbit anti-human Ki67 antibody (NovusBiologicals, Oakville, ON, Canada). LC3 was recognised with a rabbit anti-human LC3 primary antibody and p62 was recognised with
a rabbit anti-human p62 primary antibody (ABGENT, San Diego, CA, USA). Application of all primary antibodies was followed by Cy3-conjugated goat anti-rabbit IgG secondary antibody and
visualised using the Olympus fluorescent upright microscope. _IN VITRO_ CYTOTOXICITY Single-cell suspensions were treated at 37 °C in 95% air+5% CO2 in stirred glass polyshell vials with or
without 50 nM docetaxel in the presence or absence of pantoprazole at various concentrations. Cells were counted and placed into a 13 ml tube at a concentration of 105cells ml−1. Serial
dilutions were made to 104 and 103 cells ml−1 and each concentration was plated in triplicate into six-well plates. Colonies generated 8–14 days later were stained with methylene blue and
counted. The average colony count for each concentration was recorded and surviving fraction was calculated using the following formula: GROWTH DELAY Mice bearing MCF-7, A-431, PC3 and LNCap
tumours were divided into groups of six mice and treated weekly for 3 weeks with saline, docetaxel (15 mg kg−1 i.p.), pantoprazole (200 mg kg−1 i.p.) or pantoprazole 2 h before docetaxel.
These were maximum tolerated doses that caused minimal weight loss (Patel et al, 2013). All mice were ear-tagged and randomised to avoid bias. Two perpendicular diameters of tumours growing
in the flanks of mice were measured with a caliper every 2–3 days and treatment began once tumours reached a diameter of 5–8 mm. Measurements were taken until tumours reached a maximum
diameter of 1.5 cm or began to ulcerate, when mice were killed humanely. To minimise bias, we only continued measurements if at least 10 tumours were available for assessment. Tumour volume
was estimated using the formula: 0.5(ab2), where a is the longest diameter and b is the shortest diameter. EFFECT OF ANTICANCER DRUGS ON BIOMARKERS Mice bearing tumours of mean
cross-sectional area 0.7–0.8 cm2 were treated with docetaxel (15 mg kg−1 i.p.), or pantoprazole (200 mg kg−1 i.p.) alone or 2 h before docetaxel. To detect hypoxia and functional blood
vessels, EF5 was injected i.p. ∼2 h before killing the mice (0.2 ml of a 10 mM stock per mouse) and the perfusion marker DiOC7 (1 mg kg−1) was injected i.v.1 min before sacrifice. Mice were
killed 10 min or 24 h after docetaxel injection and tumours were excised, embedded in OCT compound, frozen in liquid nitrogen and stored at −70 °C. Whole cryostat sections (10 _μ_m thick)
were analysed and artifacts and regions of necrosis excluded. Tumour sections were first imaged for DiOC7 using a FITC filter set. Sections were then stained for hypoxic regions using a
Cy5-conjugated mouse anti-EF5 antibody (1 : 50) and with appropriate antibodies to one of the following biomarkers: _γ_H2aX cleaved caspase-3, Ki67, LC3 and p62; the sections were imaged
using Cy3 filter set (530–560 nm excitation/573–746 nm emission). Image analysis and quantification of biomarker distribution in relation to blood vessels and regions of hypoxia were
performed as described previously (Fung et al, 2012). EVALUATION OF AUTOPHAGY Microtubule-associated protein 1 light chain 3 (LC3), a specific marker for autophagosome formation, has two
forms, LC3-I and its proteolytic derivative LC3-II (molecular weight, 18 kDa and 16 kDa, respectively)(Xie and Klionsky, 2007). LC3-I is localised in the cytoplasm, whereas LC3-II binds to
autophagosomes. Various stresses, such as hypoxia and starvation, stimulate the conversion of LC3-I to LC3-II, and upregulation of LC3 expression(Mizushima et al, 2010). Relative autophagic
flux can be measured by the levels of LC3-II degraded in autolysosomes in which lysosomal hydrolases are functional (Klionsky et al, 2008). In addition to LC3, we also used p62/SQSTM1 as a
marker of autophagy. The p62 protein serves as a link between LC3 and ubiquitinated substrates but, unlike LC3, is degraded within the mature autolysosome (Bjorkoy et al, 2005). Thus,
observation of increased p62 is indicative of a build-up of the protein due to inhibition of lysosomal fusion to the autophagosome. For western blot analysis, PC3 cells treated with or
without pantoprazole in the presence or absence of bafilomycin A1 were lysed in RIPA buffer and centrifuged at 13 000 G at 4 °C for 30 min. Protein concentration in the supernatant was
determined using a Bio-Rad Protein Assay (Bio-Rad Laboratories, Hercules, CA, USA) to quantify LC3-I and LC3-II and p62. The PC3 cells were also transfected with a plasmid containing LC3-II
tagged at the N terminus with green (GFP) and red fluorescent protein (RFP); this probe allows distinction of autophagosomes (GFP+RFP+ yellow puncta) and autolysosomes (GFP-RFP+ red puncta),
as GFP fluorescence is quenched in the acidic autolysosomes(Kimura et al, 2007). Cells showing red fluorescence have increased autophagy whereas control cells show yellow fluorescence.
Images were examined under a × 20 lens on an Olympus fluorescence microscope using standard filter sets for GFP and RFP. The western blots and the number and spatial distribution of punctae
were quantified using Image Pro software (Version premier 9). RNA INTERFERENCE OF AUTOPHAGY GENES The PC3 cells with knockdown of _ATG7_ and _BECLIN1_ (or both) were generated. Lentiviral
shRNA (_ATG7_ and _BECLIN1_) constructs were purchased from Open Biosystems (RMM4534_019584 and RMM4534_028835). The _ATG7_ and _BECLIN1_ shRNAs were transfected into PC3 cells, either alone
or together with packaging plasmids following the manufacturer’s protocol (Invitrogen ViraPower Lentiviral Expression Systems kit, Carlsbad, CA, USA). The silencing efficacy of the various
shRNA was assessed by WB analysis of ATG7 and BECLIN1 proteins using polyclonal antibodies. ANIMAL STUDIES AND ETHICAL TREATMENT Animal experiments described in this paper were carried out
using Animal Use Protocol (AUP1232.15, 09/05/14) approved by Princess Margaret Cancer Center, University Health Network (UHN) Animal Care Committee under the guidelines of the Canadian
Council on Animal Care. STATISTICAL ANALYSIS One-way ANOVA, followed by Tukey’s _post hoc_ test, determined statistical differences between treatment groups. _P_<0.05 was used to indicate
statistical significance; all tests were two-sided and no corrections were applied for multiple significance testing. All the experiments were repeated at least three times. Drug and
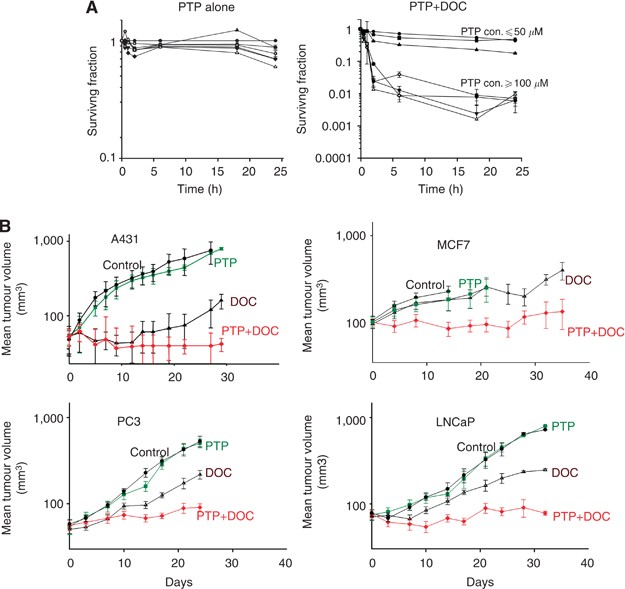
biomarker distributions are represented as mean values±s.e.m. RESULTS PANTOPRAZOLE INCREASES TOXICITY OF DOCETAXEL We pretreated the human breast cancer cell line (MCF-7) with pantoprazole
(10–1000 _μ_ M) for 2 h followed by docetaxel (50 nM) and evaluated cell survival by a colony-formation assay (Figure 1A). Pantoprazole alone was not toxic to MCF-7 cells but pretreatment
with concentrations at or above 100 _μ_ M increased the cytotoxicity of docetaxel. Similar effects were seen for human prostate cancer PC3 cells (see Figure 4A). Mice bearing subcutaneous
A-431, MCF-7, PC3 or LNCaP xenografts were treated i.p. weekly for three consecutive weeks with pantoprazole, docetaxel or pantoprazole before docetaxel. Pantoprazole had no significant
effect on tumour growth; docetaxel had variable effects to cause growth delay, whereas combined treatment increased growth delay of each type of xenograft (Figure 1B). To study antitumour
effects in relation to the tumour microenvironment, we used IHC to quantify the distribution of biomarkers following treatment in relation to functional blood vessels of PC3 xenografts.
_γ_H2AX was induced within 10 min by docetaxel, with greater activity close to blood vessels, and increased activity following combined treatment (Figure 2A). At 24 h, there was some
activation of cleaved caspase-3, a marker of apoptosis, by pantoprazole, greater induction by docetaxel and marked induction by the combination, again with decreasing activity with
increasing distance from blood vessels (Figure 2B). Reduction in Ki67, a marker of cell proliferation, was also much greater for the combination of pantoprazole and docetaxel than for either
drug alone (Figure 2C). Photomicrographs of Ki67 (magenta) in relation to blood vessels (red) of PC3 xenografts are illustrated for controls, for pantoprazole and following docetaxel with
and without pretreatment with pantoprazole (Figure 2D). PANTOPRAZOLE INCREASES THE EFFECTIVENESS OF DOCETAXEL BY INHIBITION OF AUTOPHAGY We used western blotting to examine the conversion of
LC3-I to LC3-II in control and pantoprazole-treated PC3 cells in the presence or absence of the specific lysosomal H+-ATPase inhibitor bafilomycin A1 (Baf). We used Baf to differentiate
autophagosome formation from turnover and to identify the accumulation of LC3-II that occurs due to inhibition of autophagy flux. We observed that pantoprazole increased accumulation of
LC3-II substantially (median 2.25±0.21-fold) compared with control in the absence of Baf, but with a non-significant effect (median 1.15±0.18-fold) in the presence of Baf, (Figure 3A)
Pantoprazole-induced alteration of the autophagic flux is confirmed by accumulation of p62 (median 3.33±0.26-fold) compared with control, again with non-significant effects in the presence
of Baf (Figure 3B). Thus pantoprazole appears to inhibit autophagy through a similar mechanism to Baf. PC3 cells transfected with the plasmid containing LC3-II tagged at the N terminus with
GFP and RFP were exposed to 100 _μ_ M pantoprazole, 5 nM docetaxel either alone or in combination, saline as control, starvation and 50 _μ_ M HCQ as positive control. Starvation induces
autophagy (red fluorescence), as does docetaxel, consistent with increased survival due to autophagy being a mechanism of resistance. HCQ, pantoprazole and combined treatment showed
increased yellow fluorescence (Figure 3C), suggesting that pantoprazole leads to accumulation of autophagosomes due to blocking of the fusion of autophagosomes with lysosomes, thereby
inhibiting autophagy. We generated PC3 cells deficient in the autophagy-associated proteins BECLIN1 and ATG7 by transfecting specific shRNAs, either alone or in combination. There were
minimal differences in the sensitivity of wild-type (WT) and single knockdown cells to docetaxel, with greater enhancement of toxicity of pantoprazole in the single knockdown cells (Figure
4A–C); however, the double knockdown cell line was more sensitive to docetaxel with minimal added effects of pantoprazole (Figure 4D). The silencing efficacy of the various siRNAs was
assessed by western blot analysis of ATG7 and BECLIN1 proteins using polyclonal antibodies (Figure 4E). We quantified the markers of autophagy LC3 and p62 in relation to functional blood
vessels (recognised by DiOC7) and region of hypoxia (recognised by EF5) using IHC in two human prostate cancer xenografts, PC3 and LNCaP, treated with docetaxel, with and without
pantoprazole pretreatment. Results showed that (i) LC3 expression was increased in tumour regions distal to functional blood vessels and proximal to hypoxic regions in wild-type PC3 and
LNCaP tumours (Figure 5A–F), (ii) LC3 expression was increased by docetaxel but was reduced by pantoprazole pretreatment, with lowest levels after treatment with both pantoprazole and
docetaxel (Figure 5A,C,D and F) and (iii) p62 expression was decreased by docetaxel and increased by pantoprazple alone and by combined treatment (Figure 5B and E). Photomicrographs of LC3
(green) in relation to blood vessels (red) and hypoxic regions (cyan) are illustrated for control, and docetaxel-treated PC3 xenografts with and without pretreatment with pantoprazole
(Figure 5G). DISCUSSION Proton pump inhibitors have been reported to sensitise cancer cells and solid tumours to different chemotherapeutic agents (Luciani et al, 2004). Multiple mechanisms
might be involved, but probably relate to changes in acidity in both intra-and extracellular compartments of tumour cells. In initial studies, we administered pantoprazole before doxorubicin
(a weak base that concentrates in acidic endosomes of cells); we reasoned that sequestration of doxorubicin in acidic endosomes might decrease drug available to bind to DNA (and cause
cytotoxicity) and decrease drug available to diffuse to more distal cells. We showed that high-dose pantoprazole increases endosomal pH in cultured cells and decreases sequestration of
doxorubicin within them (Lee and Tannock, 2006; Tredan et al, 2007; Patel et al, 2013). Pantoprazole increased toxicity of doxorubicin for cultured tumour cells, improved the distribution of
doxorubicin in tumour tissue, and increased growth delay when doxorubicin was used to treat experimental tumours (Patel et al, 2013). In the present study, we showed similar or greater
effects when pantoprazole was given before docetaxel, a drug that is not basic and not known to be sequestered in acidic endosomes, and we sought alternative mechanisms to explain these
effects. It has been reported that inhibition of autophagy increases cytotoxicity of several anticancer drugs in preclinical models (Sotelo et al, 2006; Carew et al, 2007; Firat et al,
2012). Chloroquine (CQ) and hydroxychloroquine (HCQ) are known to inhibit autophagy and have been investigated in preclinical studies and in more than 30 clinical trials; however, the ocular
toxicities and minimal single-agent anticancer efficacy of CQ or HCQ have restricted their clinical application. In a recently published phase I/II trial of HCQ with temozolomide for
glioblastoma multiforme, pharmacokinetic–pharmacodynamic studies indicated that the high micromolar concentrations of HCQ required to inhibit autophagy _in vitro_ are inconsistently achieved
in patients (Rosenfeld et al, 2014). New and safer inhibitors of autophagy are worthy of investigation. There is evidence that PPIs inhibit autophagy, probably because fusion of
autophagosomes with acidic endosomes is central to the process (Marino et al, 2010). Autophagy can be evaluated with an RFP-GFP-LC3-II tandem construct, a method that depends on the
acidification and capacity for degradation of the lysosome (Kimura et al, 2007). Our results suggest that pantoprazole inhibits autophagy by raising lysosomal pH and/or by inhibiting fusion
of autophagosomes with lysomes, leading to the accumulation of autophagosomes. This mechanism is supported by results of western blots, which showed that pantoprazole increased the
accumulation of both LC3-II and p62, effects that were reduced or absent in the presence of bafilomycin, indicating similar mechanisms of action. A recent study shows that steady state
levels of the p62 protein reflect the autophagic status, and that p62 levels increase when autophagy is blocked (Bardag-Gorce et al, 2005; Bjorkoy et al, 2005). It has been reported that
deletion of ATG7 and BECLIN1 inhibits autophagy in cervical cancer cells induced by nutrient deprivation and increases cell death (Yu et al, 2004). Stable knockdown of ATG7 in human breast
cancer cells inhibited cell growth in soft agar and tumour formation in nude mice (Kim et al, 2011). Several studies have suggested that autophagy may also function as a survival mechanism
adopted by cancer cells facing hostile microenvironment conditions (such as hypoxia) and the resultant metabolic stress. We have compared therapeutic effects _in vitro_ and _in vivo_ of
wild-type prostate cancer PC3 cells with autophagy-deficient PC3 cells where shRNA has been used to knock down BECLIN1, ATG7 or both. Knockdown of BECLIN1 or ATG7 increased
pantoprazole-induced cytotoxicity of docetaxel, presumably because of combined effects to abrogate autophagy, whereas the double knockdown cell line with absent autophagy is already more
sensitive to docetaxel and toxicity was not increased further by pantoprazole (Figure 4). Our data confirm that autophagy is upregulated in poorly nourished regions of PC3 and LNCaP
xenografts, consistent with the findings of others (Rouschop et al, 2010), suggesting its role in cell survival under stressed microenvironmental conditions. Docetaxel leads to substantial
upregulation of autophagy, throughout the tumour as indicated by an increase in LC3 and a decrease in p62, consistent with an effect of autophagy to promote cell survival by recycling
cellular components in cells damaged by chemotherapy. Pantoprazole has a marked effect to inhibit docetaxel-induced autophagy throughout PC3 and LNCaP tumours (Figure 5). Pantoprazole given
2 h before docetaxel led to consistent and substantial augmentation of growth delay due to docetaxel in four different xenografts, although pantoprazole alone had no effect. This occurred in
xenografts with no (MCF-7), modest (PC3 and LNCaP) and substantial (A-431) sensitivity to docetaxel alone (Figure 1B) and with minimal increase in toxicity. The distribution of activity of
docetaxel (evaluated by the biomarkers _γ_H2AX, cleaved caspase-3 and Ki67) in PC3 xenografts was increased substantially by pantoprazole (Figure 2). Figure 2B showed slightly increased
apoptosis by pantoprazole alone, and this is most likely due to the role of autophagy in the degradation of toxic proteins (Li et al, 2008). Autophagy is also linked to the unfolded-protein
response (UPR), which is important for dealing with cell stress and if inhibited may trigger apoptosis (Fribley et al, 2009). However, combined treatment shows an increase in apoptosis that
is synergistic rather than additive. Another group also reported that proton pump inhibitors have caspase- and pH-dependent antineoplastic activity in human melanoma (Marino et al, 2010).
Our group has undertaken a phase I trial of pantoprazole given every 3 weeks before doxorubicin, which indicated a maximum tolerated dose of pantoprazole of 240 mg in patients (Brana et al,
2014). The mean serum concentration 1–2 h after injection of pantoprazole 240 mg was 84 _μ_ M with some patients exceeding 100 _μ_ M – a concentration that leads to antitumour effects in
mice (Patel et al, 2013)–and the mean half-life in serum was 7.5 h. The present results and those of the phase I trial led to initiation of a phase II study of pantoprazole and docetaxel as
first-line chemotherapy for men with castration-resistant prostate cancer (docetaxel is the standard treatment for such men). Clinical interaction with docetaxel will be checked in our
ongoing phase 2 trial for prostate cancer but an interaction seems unlikely in view of lack of added toxicity, and our phase I trial showed that there was no interaction between pantoprazole
and doxorubicin. Our present findings, together with data suggesting that markers of autophagy are associated with poor prognosis and poor response to treatment in several types of cancer
(Hu et al, 2012), suggest that autophagy might be a general mechanism of drug resistance in solid tumours. Inhibitors of autophagy should be evaluated further for their effects to modify
treatments with drugs and radiotherapy. CHANGE HISTORY * _ 03 MARCH 2015 This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at
publication _ * _ 20 MARCH 2024 This article has been retracted. Please see the Retraction Notice for more detail: https://doi.org/10.1038/s41416-024-02660-4 _ REFERENCES * Bardag-Gorce F,
Francis T, Nan L, Li J, He Lue Y, French BA, French SW (2005) Modifications in P62 occur due to proteasome inhibition in alcoholic liver disease. _Life Sci_ 77: 2594–2602. Article CAS
PubMed Google Scholar * Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T (2005) p62/SQSTM1 forms protein aggregates degraded by autophagy and has a
protective effect on huntingtin-induced cell death. _J Cell Biol_ 171: 603–614. Article PubMed PubMed Central Google Scholar * Bradley G, Ling V (1994) P-glycoprotein, multidrug
resistance and tumor progression. _Cancer Metastasis Rev_ 13: 223–233. Article CAS PubMed Google Scholar * Brana I, Ocana A, Chen EX, Razak AR, Haines C, Lee C, Douglas S, Wang L, Siu
LL, Tannock IF, Bedard PL (2014) A phase I trial of pantoprazole in combination with doxorubicin in patients with advanced solid tumors: evaluation of pharmacokinetics of both drugs and
tissue penetration of doxorubicin. _Invest New Drugs_ 32: 1269–1277. Article CAS PubMed Google Scholar * Carew JS, Nawrocki ST, Kahue CN, Zhang H, Yang C, Chung L, Houghton JA, Huang P,
Giles FJ, Cleveland JL (2007) Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistance. _Blood_ 110:
313–322. Article CAS PubMed PubMed Central Google Scholar * De Milito A, Canese R, Marino ML, Borghi M, Iero M, Villa A, Venturi G, Lozupone F, Iessi E, Logozzi M, Della Mina P,
Santinami M, Rodolfo M, Podo F, Rivoltini L, Fais S (2010) pH-dependent antitumor activity of proton pump inhibitors against human melanoma is mediated by inhibition of tumor acidity. _Int J
Cancer_ 127: 207–219. Article CAS PubMed Google Scholar * De Milito A, Iessi E, Logozzi M, Lozupone F, Spada M, Marino ML, Federici C, Perdicchio M, Matarrese P, Lugini L, Nilsson A,
Fais S (2007) Proton pump inhibitors induce apoptosis of human B-cell tumors through a caspase-independent mechanism involving reactive oxygen species. _Cancer Res_ 67: 5408–5417. Article
CAS PubMed Google Scholar * Deng L, Lei Y, Liu R, Li J, Yuan K, Li Y, Chen Y, Liu Y, Lu Y, Edwards CK 3rd, Huang C, Wei Y (2013) Pyrvinium targets autophagy addiction to promote cancer
cell death. _Cell Death Dis_ 4: e614. Article CAS PubMed PubMed Central Google Scholar * Firat E, Weyerbrock A, Gaedicke S, Grosu AL, Niedermann G (2012) Chloroquine or
chloroquine-PI3K/Akt pathway inhibitor combinations strongly promote gamma-irradiation-induced cell death in primary stem-like glioma cells. _PLoS One_ 7: e47357. Article CAS PubMed
PubMed Central Google Scholar * Fribley A, Zhang K, Kaufman RJ (2009) Regulation of apoptosis by the unfolded protein response. _Methods Mol Biol_ 559: 191–204. Article CAS PubMed
PubMed Central Google Scholar * Fung AS, Jonkman J, Tannock IF (2012) Quantitative immunohistochemistry for evaluating the distribution of Ki67 and other biomarkers in tumor sections and
use of the method to study repopulation in xenografts after treatment with paclitaxel. _Neoplasia_ 14: 324–334. Article CAS PubMed PubMed Central Google Scholar * Hoyer-Hansen M,
Jaattela M (2007) Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. _Cell Death Differ_ 14: 1576–1582. Article CAS PubMed Google Scholar * Hu
YL, Jahangiri A, Delay M, Aghi MK (2012) Tumor cell autophagy as an adaptive response mediating resistance to treatments such as antiangiogenic therapy. _Cancer Res_ 72: 4294–4299. Article
CAS PubMed PubMed Central Google Scholar * Huxham LA, Kyle AH, Baker JH, Nykilchuk LK, Minchinton AI (2004) Microregional effects of gemcitabine in HCT-116 xenografts. _Cancer Res_ 64:
6537–6541. Article CAS PubMed Google Scholar * Karpathiou G, Sivridis E, Koukourakis MI, Mikroulis D, Bouros D, Froudarakis ME, Giatromanolaki A (2011) Light-chain 3A autophagic
activity and prognostic significance in non-small cell lung carcinomas. _Chest_ 140: 127–134. Article PubMed Google Scholar * Kim MJ, Woo SJ, Yoon CH, Lee JS, An S, Choi YH, Hwang SG,
Yoon G, Lee SJ (2011) Involvement of autophagy in oncogenic K-Ras-induced malignant cell transformation. _J Biol Chem_ 286: 12924–12932. Article CAS PubMed PubMed Central Google Scholar
* Kimura S, Noda T, Yoshimori T (2007) Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. _Autophagy_ 3: 452–460. Article CAS
PubMed Google Scholar * Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, Bamber BA, Bassham DC, Bergamini E, Bi X,
Biard-Piechaczyk M, Blum JS, Bredesen DE, Brodsky JL, Brumell JH, Brunk UT, Bursch W, Camougrand N, Cebollero E, Cecconi F, Chen Y, Chin LS, Choi A, Chu CT, Chung J, Clarke PG, Clark RS,
Clarke SG, Clave C, Cleveland JL, Codogno P, Colombo MI, Coto-Montes A, Cregg JM, Cuervo AM, Debnath J, Demarchi F, Dennis PB, Dennis PA, Deretic V, Devenish RJ, di Sano F, Dice JF, Difiglia
M, Dinesh-Kumar S, Distelhorst CW, Djavaheri-Mergny M, Dorsey FC, Droge W, Dron M, Dunn WA Jr, Duszenko M, Eissa NT, Elazar Z, Esclatine A, Eskelinen EL, Fesus L, Finley KD, Fuentes JM,
Fueyo J, Fujisaki K, Galliot B, Gao FB, Gewirtz DA, Gibson SB, Gohla A, Goldberg AL, Gonzalez R, Gonzalez-Estevez C, Gorski S, Gottlieb RA, Haussinger D, He YW, Heidenreich K, Hill JA,
Hoyer-Hansen M, Hu X, Huang WP, Iwasaki A, Jaattela M, Jackson WT, Jiang X, Jin S, Johansen T, Jung JU, Kadowaki M, Kang C, Kelekar A, Kessel DH, Kiel JA, Kim HP, Kimchi A, Kinsella TJ,
Kiselyov K, Kitamoto K, Knecht E, Komatsu M, Kominami E, Kondo S, Kovács AL, Kroemer G, Kuan CY, Kumar R, Kundu M, Landry J, Laporte M, Le W, Lei HY, Lenardo MJ, Levine B, Lieberman A, Lim
KL, Lin FC, Liou W, Liu LF, Lopez-Berestein G, López-Otín C, Lu B, Macleod KF, Malorni W, Martinet W, Matsuoka K, Mautner J, Meijer AJ, Meléndez A, Michels P, Miotto G, Mistiaen WP,
Mizushima N, Mograbi B, Monastyrska I, Moore MN, Moreira PI, Moriyasu Y, Motyl T, Münz C, Murphy LO, Naqvi NI, Neufeld TP, Nishino I, Nixon RA, Noda T, Nürnberg B, Ogawa M, Oleinick NL,
Olsen LJ, Ozpolat B, Paglin S, Palmer GE, Papassideri I, Parkes M, Perlmutter DH, Perry G, Piacentini M, Pinkas-Kramarski R, Prescott M, Proikas-Cezanne T, Raben N, Rami A, Reggiori F,
Rohrer B, Rubinsztein DC, Ryan KM, Sadoshima J, Sakagami H, Sakai Y, Sandri M, Sasakawa C, Sass M, Schneider C, Seglen PO, Seleverstov O, Settleman J, Shacka JJ, Shapiro IM, Sibirny A,
Silva-Zacarin EC, Simon HU, Simone C, Simonsen A, Smith MA, Spanel-Borowski K, Srinivas V, Steeves M, Stenmark H, Stromhaug PE, Subauste CS, Sugimoto S, Sulzer D, Suzuki T, Swanson MS, Tabas
I, Takeshita F, Talbot NJ, Tallóczy Z, Tanaka K, Tanaka K, Tanida I, Taylor GS, Taylor JP, Terman A, Tettamanti G, Thompson CB, Thumm M, Tolkovsky AM, Tooze SA, Truant R, Tumanovska LV,
Uchiyama Y, Ueno T, Uzcátegui NL, van der Klei I, Vaquero EC, Vellai T, Vogel MW, Wang HG, Webster P, Wiley JW, Xi Z, Xiao G, Yahalom J, Yang JM, Yap G, Yin XM, Yoshimori T, Yu L, Yue Z,
Yuzaki M, Zabirnyk O, Zheng X, Zhu X, Deter RL (2008) Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. _Autophagy_ 4: 151–175. Article CAS
PubMed Google Scholar * Lankelma J, Dekker H, Luque FR, Luykx S, Hoekman K, van der Valk P, van Diest PJ, Pinedo HM (1999) Doxorubicin gradients in human breast cancer. _Clin Cancer Res_
5: 1703–1707. CAS PubMed Google Scholar * Lee CM, Tannock IF (2006) Inhibition of endosomal sequestration of basic anticancer drugs: influence on cytotoxicity and tissue penetration. _Br
J Cancer_ 94: 863–869. Article CAS PubMed PubMed Central Google Scholar * Levy JM, Thompson JC, Griesinger AM, Amani V, Donson AM, Birks DK, Morgan MJ, Mirsky DM, Handler MH, Foreman
NK, Thorburn A (2014) Autophagy inhibition improves chemosensitivity in BRAFV600E brain tumors. _Cancer Discov_ 4: 773–780. Article CAS PubMed PubMed Central Google Scholar * Li J, Ni
M, Lee B, Barron E, Hinton DR, Lee AS (2008) The unfolded protein response regulator GRP78/BiP is required for endoplasmic reticulum integrity and stress-induced autophagy in mammalian
cells. _Cell Death Differ_ 15: 1460–1471. Article CAS PubMed Google Scholar * Luciani F, Spada M, De Milito A, Molinari A, Rivoltini L, Montinaro A, Marra M, Lugini L, Logozzi M,
Lozupone F, Federici C, Iessi E, Parmiani G, Arancia G, Belardelli F, Fais S (2004) Effect of proton pump inhibitor pretreatment on resistance of solid tumors to cytotoxic drugs. _J Natl
Cancer Inst_ 96: 1702–1713. Article CAS PubMed Google Scholar * Marino ML, Fais S, Djavaheri-Mergny M, Villa A, Meschini S, Lozupone F, Venturi G, Della Mina P, Pattingre S, Rivoltini L,
Codogno P, De Milito A (2010) Proton pump inhibition induces autophagy as a survival mechanism following oxidative stress in human melanoma cells. _Cell Death Dis_ 1: e87. Article CAS
PubMed PubMed Central Google Scholar * McAfee Q, Zhang Z, Samanta A, Levi SM, Ma XH, Piao S, Lynch JP, Uehara T, Sepulveda AR, Davis LE, Winkler JD, Amaravadi RK (2012) Autophagy
inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. _Proc Natl Acad Sci USA_ 109: 8253–8258. Article CAS PubMed PubMed
Central Google Scholar * Mizushima N, Yoshimori T, Levine B (2010) Methods in mammalian autophagy research. _Cell_ 140: 313–326. Article CAS PubMed PubMed Central Google Scholar * Pan
B, Chen D, Huang J, Wang R, Feng B, Song H, Chen L (2014) HMGB1-mediated autophagy promotes docetaxel resistance in human lung adenocarcinoma. _Mol Cancer_ 13: 165. Article PubMed PubMed
Central Google Scholar * Patel KJ, Lee C, Tan Q, Tannock IF (2013) Use of the proton pump inhibitor pantoprazole to modify the distribution and activity of doxorubicin: a potential
strategy to improve the therapy of solid tumors. _Clin Cancer Res_ 19: 6766–6776. Article CAS PubMed Google Scholar * Ploussard G, Terry S, Maille P, Allory Y, Sirab N, Kheuang L, Soyeux
P, Nicolaiew N, Coppolani E, Paule B, Salomon L, Culine S, Buttyan R, Vacherot F, de la Taille A (2010) Class III beta-tubulin expression predicts prostate tumor aggressiveness and patient
response to docetaxel-based chemotherapy. _Cancer Res_ 70: 9253–9264. Article CAS PubMed PubMed Central Google Scholar * Poklepovic A, Gewirtz DA (2014) Outcome of early clinical trials
of the combination of hydroxychloroquine with chemotherapy in cancer. _Autophagy_ 10: 1478–1480. Article CAS PubMed PubMed Central Google Scholar * Rosenfeld MR, Ye X, Supko JG,
Desideri S, Grossman SA, Brem S, Mikkelson T, Wang D, Chang YC, Hu J, McAfee Q, Fisher J, Troxel AB, Piao S, Heitjan DF, Tan KS, Pontiggia L, O’Dwyer PJ, Davis LE, Amaravadi RK (2014) A
phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. _Autophagy_
10: 1359–1368. Article CAS PubMed PubMed Central Google Scholar * Rouschop KM, van den Beucken T, Dubois L, Niessen H, Bussink J, Savelkouls K, Keulers T, Mujcic H, Landuyt W, Voncken
JW, Lambin P, van der Kogel AJ, Koritzinsky M, Wouters BG (2010) The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B
and ATG5. _J Clin Invest_ 120: 127–141. Article CAS PubMed Google Scholar * Saggar JK, Fung AS, Patel KJ, Tannock IF (2013) Use of molecular biomarkers to quantify the spatial
distribution of effects of anticancer drugs in solid tumors. _Mol Cancer Ther_ 12: 542–552. Article CAS PubMed Google Scholar * Sivridis E, Koukourakis MI, Mendrinos SE, Karpouzis A,
Fiska A, Kouskoukis C, Giatromanolaki A (2011) Beclin-1 and LC3A expression in cutaneous malignant melanomas: a biphasic survival pattern for beclin-1. _Melanoma Res_ 21: 188–195. Article
CAS PubMed Google Scholar * Sivridis E, Koukourakis MI, Zois CE, Ledaki I, Ferguson DJ, Harris AL, Gatter KC, Giatromanolaki A (2010) LC3A-positive light microscopy detected patterns of
autophagy and prognosis in operable breast carcinomas. _Am J Pathol_ 176: 2477–2489. Article CAS PubMed PubMed Central Google Scholar * Sotelo J, Briceno E, Lopez-Gonzalez MA (2006)
Adding chloroquine to conventional treatment for glioblastoma multiforme: a randomized, double-blind, placebo-controlled trial. _Ann Intern Med_ 144: 337–343. Article CAS PubMed Google
Scholar * Tannock IF, Lee CM, Tunggal JK, Cowan DS, Egorin MJ (2002) Limited penetration of anticancer drugs through tumor tissue: a potential cause of resistance of solid tumors to
chemotherapy. _Clin Cancer Res_ 8: 878–884. CAS PubMed Google Scholar * Terry S, Ploussard G, Allory Y, Nicolaiew N, Boissiere-Michot F, Maille P, Kheuang L, Coppolani E, Ali A, Bibeau F,
Culine S, Buttyan R, de la Taille A, Vacherot F (2009) Increased expression of class III beta-tubulin in castration-resistant human prostate cancer. _Br J Cancer_ 101: 951–956. Article CAS
PubMed PubMed Central Google Scholar * Tredan O, Galmarini CM, Patel K, Tannock IF (2007) Drug resistance and the solid tumor microenvironment. _J Natl Cancer Inst_ 99: 1441–1454.
Article CAS PubMed Google Scholar * Udelnow A, Kreyes A, Ellinger S, Landfester K, Walther P, Klapperstueck T, Wohlrab J, Henne-Bruns D, Knippschild U, Wurl P (2011) Omeprazole inhibits
proliferation and modulates autophagy in pancreatic cancer cells. _PLoS One_ 6: e20143. Article CAS PubMed PubMed Central Google Scholar * Wang J, Wu GS (2014) Role of autophagy in
cisplatin resistance in ovarian cancer cells. _J Biol Chem_ 289: 17163–17173. Article CAS PubMed PubMed Central Google Scholar * White E (2012) Deconvoluting the context-dependent role
for autophagy in cancer. _Nat Rev Cancer_ 12: 401–410. Article CAS PubMed PubMed Central Google Scholar * Xie Z, Klionsky DJ (2007) Autophagosome formation: core machinery and
adaptations. _Nat Cell Biol_ 9: 1102–1109. Article CAS PubMed Google Scholar * Yang A, Rajeshkumar NV, Wang X, Yabuuchi S, Alexander BM, Chu GC, von Hoff DD, Maitra A, Kimmelman AC
(2014a) Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. _Cancer Discov_ 4: 905–913. Article CAS PubMed PubMed Central Google Scholar *
Yang M, Zeng P, Kang R, Yu Y, Yang L, Tang D, Cao L (2014b) S100A8 contributes to drug resistance by promoting autophagy in leukemia cells. _PLoS One_ 9: e97242. Article PubMed PubMed
Central Google Scholar * Yang ZJ, Chee CE, Huang S, Sinicrope FA (2011) The role of autophagy in cancer: therapeutic implications. _Mol Cancer Ther_ 10: 1533–1541. Article CAS PubMed
PubMed Central Google Scholar * Yeo M, Kim DK, Kim YB, Oh TY, Lee JE, Cho SW, Kim HC, Hahm KB (2004) Selective induction of apoptosis with proton pump inhibitor in gastric cancer cells.
_Clin Cancer Res_ 10: 8687–8696. Article CAS PubMed Google Scholar * Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, Baehrecke EH, Lenardo MJ (2004) Regulation of an ATG7-beclin 1
program of autophagic cell death by caspase-8. _Science_ 304: 1500–1502. Article CAS PubMed Google Scholar Download references ACKNOWLEDGEMENTS We thank all members of the Pathology
Research Program (PRP) and the Advanced Optical Microscopy Facility (AOMF). Supported by grant KG100252 from the Komen Foundation and by a grant from the Canadian Institutes of Health
Research. AUTHOR INFORMATION Author notes * Q Tan and A M Joshua: These authors contributed equally to this work. AUTHORS AND AFFILIATIONS * Department of Medical Biophysics, University
Health Network, University of Toronto, Toronto, M5G2M9, ON, Canada Q Tan, J K Saggar, M Yu, M Wang, N Kanga, J Y Zhang, B G Wouters & I F Tannock * Division of Medical Oncology and
Hematology, Princess Margaret Cancer Center and University Health Network, University of Toronto, Toronto, M5G2M9, ON, Canada A M Joshua, X Chen & I F Tannock * Institute of Medical
Science, University Health Network, University of Toronto, Toronto, M5G2M9, ON, Canada A M Joshua & I F Tannock Authors * Q Tan View author publications You can also search for this
author inPubMed Google Scholar * A M Joshua View author publications You can also search for this author inPubMed Google Scholar * J K Saggar View author publications You can also search for
this author inPubMed Google Scholar * M Yu View author publications You can also search for this author inPubMed Google Scholar * M Wang View author publications You can also search for
this author inPubMed Google Scholar * N Kanga View author publications You can also search for this author inPubMed Google Scholar * J Y Zhang View author publications You can also search
for this author inPubMed Google Scholar * X Chen View author publications You can also search for this author inPubMed Google Scholar * B G Wouters View author publications You can also
search for this author inPubMed Google Scholar * I F Tannock View author publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to I F
Tannock. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no conflict of interest. ADDITIONAL INFORMATION This work is published under the standard license to publish agreement.
After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. RIGHTS AND
PERMISSIONS From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of
this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/ ABOUT THIS ARTICLE CITE THIS ARTICLE Tan, Q., Joshua, A., Saggar, J. _et al._ RETRACTED ARTICLE: Effect of pantoprazole
to enhance activity of docetaxel against human tumour xenografts by inhibiting autophagy. _Br J Cancer_ 112, 832–840 (2015). https://doi.org/10.1038/bjc.2015.17 Download citation * Received:
17 December 2014 * Revised: 24 December 2014 * Accepted: 07 January 2015 * Published: 03 February 2015 * Issue Date: 03 March 2015 * DOI: https://doi.org/10.1038/bjc.2015.17 SHARE THIS
ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard
Provided by the Springer Nature SharedIt content-sharing initiative KEYWORDS * autophagy * proton pump inhibitor * drug distribution * pharmacodynamic markers * tumour microenvironment