Play all audios:
ABSTRACT The physiological role of the pro-survival BCL-2 family member A1 has been debated for a long time. Strong mRNA induction in T cells on T cell receptor (TCR)-engagement suggested a
major role of A1 in the survival of activated T cells. However, the investigation of the physiological roles of A1 was complicated by the quadruplication of the _A1_ gene locus in mice,
making _A1_ gene targeting very difficult. Here, we used the recently generated _A1__−/−_ mouse model to examine the role of A1 in T cell immunity. We confirmed rapid and strong induction of
A1 protein in response to TCR/CD3 stimulation in CD4+ as well as CD8+ T cells. Surprisingly, on infection with the acute influenza HKx31 or the lymphocytic choriomeningitis virus docile
strains mice lacking A1 did not show any impairment in the expansion, survival, or effector function of cytotoxic T cells. Furthermore, the ability of _A1__−/−_ mice to generate
antigen-specific memory T cells or to provide adequate CD4-dependent help to B cells was not impaired. These results suggest functional redundancy of A1 with other pro-survival BCL-2 family
members in the control of T cell-dependent immune responses. SIMILAR CONTENT BEING VIEWED BY OTHERS TRAF2 REGULATES T CELL IMMUNITY BY MAINTAINING A TPL2-ERK SURVIVAL SIGNALING AXIS IN
EFFECTOR AND MEMORY CD8 T CELLS Article 17 November 2020 TIM-3 DRIVES TEMPORAL DIFFERENCES IN RESTIMULATION-INDUCED CELL DEATH SENSITIVITY IN EFFECTOR CD8+ T CELLS IN CONJUNCTION WITH
CEACAM1 Article Open access 14 April 2021 THEMIS SUPPRESSES THE EFFECTOR FUNCTION OF CD8+ T CELLS IN ACUTE VIRAL INFECTION Article 28 March 2023 MAIN On antigenic challenge, T lymphocytes
need to rapidly switch from their IL-7/IL-7R-regulated naive, quiescent state1, 2 to a T cell antigen-receptor (TCR/CD3) stimulation-induced activation state.3 In case of inappropriate
stimulation of the TCR, for example, in the absence of co-receptor stimulation, this shift in the survival programme is not induced and leads to rapid T cell death.4 Conversely,
appropriately stimulated T cells expand rapidly, allowing accumulation of T cell clones expressing TCRs of high affinity for specific antigens. During this clonal expansion, BCL-2 family
regulated apoptosis acts as a mechanism to remove low-affinity T cells, thereby ensuring the generation of a highly effective immune response.5 On infection clearance, most of the activated
T cells are removed by apoptosis,6 leaving only some T cells with antigen-specific high-affinity TCRs in reserve as long-lived memory T cells.7 The BCL-2 family of proteins regulate
apoptotic cell death, with the balance between pro-survival and pro-apoptotic family members determining whether a cell lives or dies. The expression of pro-survival BCL-2 family members is
dynamically regulated during T cell activation.8 TCR/CD3 ligation leads to the downregulation of BCL-2 and induction of BCL-XL.3 Accordingly, _Bcl-2__−/−_ mice display a loss of mature,
unstimulated T cells, and the death of these cells can be prevented by TCR/CD3 stimulation.9 Interestingly, although BCL-XL is substantially upregulated on TCR/CD3 stimulation, its loss did
not increase apoptosis or impair proliferation of T cells stimulated with mitogenic antibodies.10 In contrast, MCL-1 has been shown to be a crucial pro-survival factor after T cell
activation.11, 12 A1 is a pro-survival BCL-2 family protein that has been proposed to be important for activated T cell survival based on its expression status in different T cell subsets.
In naive T cells, A1 protein is hardly detectable, but its expression is strongly and rapidly induced on TCR/CD3 stimulation.3, 13 Addressing the physiological role of A1 in mice has been
difficult due to a quadruplication of the _Bcl2a1_ locus in the mouse genome.14 _A1_-knockdown studies using _in vivo_ expression of shRNAs in the haematopoietic system suggested a role for
A1 in mast cell maturation,15 mature B cell survival8 and early T cell development,16 although not all of these phenotypes were found across the different mouse models analysed. To
unambiguously determine the functions of A1, we developed an _A1_ knockout mouse strain in which all three functional paralogues of _A1_ (_A1-a_, _A1-b_ and _A1-d_) had been deleted.17
Interestingly, these _A1_-deficient mice show little to no abnormalities under steady state conditions. However, these observations might not be totally surprising, given that A1 protein
levels are very low in unstimulated cells.18 Therefore, we investigated a role for A1 in mice that had been challenged with different viral infections. These studies clearly demonstrate that
loss of A1 alone does not impair T cell-driven immune responses. It therefore appears likely that A1 has critical overlapping functions with other pro-survival BCL-2 proteins in T cell
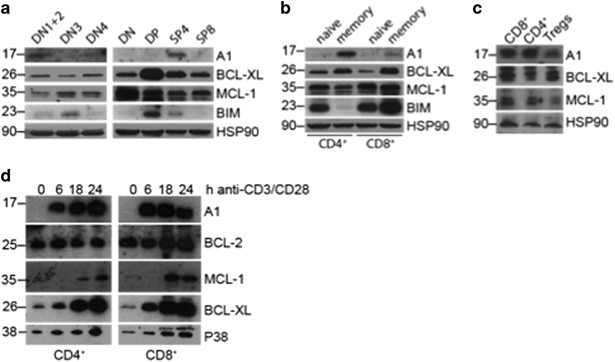
immune responses. RESULTS _A1_ IS PREDOMINANTLY EXPRESSED IN CD4-SINGLE POSITIVE THYMOCYTES AND MEMORY T CELLS AND IS RAPIDLY UPREGULATED ON TCR/CD3 STIMULATION A1 expression was reported on
pre-TCR signalling19 as well as in double-positive (DP) thymocytes,20 and is massively induced on T cell activation.13 Owing to the absence of a reliable antibody detecting murine A1 at
that time, these observations were based mainly on mRNA expression analysis. To test A1 protein levels in thymocyte subsets, we isolated them according to their expression of cell subset
markers and performed western blot analysis on these extracts. We could detect robust A1 protein expression only in DN1 and 2 and in SP4 thymocytes and to a lesser extent in DN4 stage
thymocytes (Figure 1a). This was consistent with our quantitative real-time PCR (qRT-PCR) results (Supplementary Figure 1A). As described previously, BCL-XL expression was highest in DP
thymocytes and MCL-1 was expressed throughout all T cell developmental stages. In mature CD4+ and CD8+ splenic T cells, A1 was predominantly expressed in CD62L−CD44+ memory-like T cells but
was barely detectable in naive T cells (Figure 1b), agreeing with our qRT-PCR data (Supplementary Figure 1B). Interestingly, less A1 protein was detected in FoxP3+ Tregs compared with
conventional T cells (Figure 1c). Furthermore, we isolated CD4+ and CD8+ T cells from the spleens of wild-type mice and stimulated them in culture with anti-CD3 and anti-CD28 to mimic
antigenic activation. We confirmed rapid A1 induction at the protein (Figure 1d) as well as the mRNA level (Supplementary Figure 1C). Regarding the other pro-survival BCL-2 family members,
BCL-2 protein and mRNA levels did not change after TCR/CD3 stimulation, while the amount of MCL-1 protein increased. BCL-XL mRNA and protein levels both increased over the time of TCR/CD3
stimulation, although at a slower rate compared with A1 (Figure 1d and Supplementary Figure 1C). These findings reveal that A1 is weakly expressed throughout T cell development in the thymus
and in resting peripheral T cells but is upregulated on antigen-receptor stimulation. _A1_−/− T CELLS SHOW NORMAL RESPONSES ON ACUTE INFECTION WITH INFLUENZA VIRUS IN A HAEMATOPOIETIC
CHIMAERIC SETTING As A1 expression was rapidly induced in T cells on activation, we investigated whether the lack of A1 would impair T cell immune responses in a viral infection model. To
avoid possible premature death of _A1_−/− mice due to impaired anti-viral immunity, we generated mixed bone-marrow chimaeric mice (C57BL/6-Ly5.1 wild-type: C57BL/6-Ly5.2 _A1_−/− 1:1) and
infected them intra-nasally with the influenza strain HKx31 (Figure 2a). Another advantage of mixed bone-marrow chimaeras is that we can detect putative competitive disadvantages of
_A1__−/−_ lymphoid cells on activation and can therefore directly compare wild-type and _A1__−/−_ T cells responding to viral challenge within the same animal. After 8 days of infection,
lungs, mediastinal LN (mLN), and spleens were isolated and analysed by flow cytometry. First, we compared the Ly5.1 to Ly5.2 ratio among CD4+ T cells, CD8+ T cells, and B cells in the spleen
to the respective cell types in the blood 5 weeks after reconstitution. _A1__−/−_ haematopoietic stem cells did not show any disadvantage in reconstituting the above mentioned cell lineages
and the ratios between the _A1__−/−_ cells and their competitors remained stable over the observation time (Supplementary Figure 2A). Leukocyte infiltrates were isolated from the lung by
gradient centrifugation and analysed for CD8+ cytotoxic T cells (CTLs). Ly5.1/Ly5.2 DP T cells originating from the host were gated out to allow visualisation only of the _A1__−/−_ and
wild-type competitor donor-derived cells. No differences were observed in the frequencies of CTLs of wild-type _versus A1__−/−_ origin (Figure 2b). Furthermore, antigen-specific CD8+NP+ CTLs
of wild-type _versus A1__−/−_ origin did not differ in their numbers and frequencies (Figure 2b). _A1__−/−_ derived short-lived effector CTLs were found to be slightly increased (Figure
2b). However, a similar trend was observed when WT-Ly5.1:WT-Ly5.2 mixed bone-marrow chimaeras were analysed. This might suggest an advantage of Ly5.2+ cells in this T cell subset
(Supplementary Figure 2B). A1-deficient lung infiltrating CTLs contained fewer central memory-like T cells (TCM) compared with those of wild-type origin. However, a similar tendency was seen
in WT-Ly5.2+:WT-Ly5.1+ reconstituted mice (Supplementary Figure 2A). Importantly, the numbers of the most abundant T cell subset in the lungs, the effector-like memory T cells (TEM), were
comparable between _A1__−/−_ and wild-type origin (Figure 2c). Moreover, the frequencies of total CTLs in the spleen was comparable between the wild-type and _A1__−/−_ compartments (Figure
2d). Approximately 5% of CTLs were NP-specific for both the T cells of _A1__−/−_ and wild-type origin (Figure 2d). In addition, the frequencies of TEM and TCM CTLs were comparable between
those of _A1__−/−_ and wild-type origin (Figure 2d). Similarly, _A1__−/−_ and wild-type haematopoietic stem cell derived CD4+ T cells showed comparable frequencies of TCM and TEM (Figure
2e). Furthermore, no differences in regulatory T cells (Tregs), Type-1 helper cells (Th1) and follicular T helper cells (Tfh) were observed for T-lymphoid cells derived from _A1__−/−_ or
wild-type in the spleen (Figure 2f). Moreover, GC B cell and PC formation appeared normal in the _A1__−/−_ compartment when compared with their wild-type counterparts (Figure 2g).
Collectively, these findings show that _A1__−/−_ lymphocytes are able to mount a normal immune response to infection with influenza even in competition with wild-type T cells within the same
animal. Apart from a slight reduction in lung infiltrating TCM in the _A1__−/−_ population compared with their wild-type counterparts, no impact of loss of A1 could be observed in any
haematopoietic cell type analysed. _A1__−/−_ MICE SHOW NORMAL T CELL IMMUNE RESPONSES TO CHRONIC INFECTION WITH LYMPHOCYTIC CHORIOMENINGITIS VIRUS DOCILE Next, we examined the impact of
A1-deficiency on T cell immune responses in a chronic virus infection model. _A1__−/−_ and wild-type mice were infected with lymphocytic choriomeningitis virus (LCMV) docile, which cannot be
cleared and eventually leads to exhaustion of CTLs.21 Eight days post infection, mice were killed and their spleens analysed. The total cellularity of the spleens was comparable between
wild-type and _A1__−/−_ mice (Supplementary Figure 3A), and we clearly observed an enrichment of CTLs in mice of both genotypes (Figure 3a and Supplementary Figure 3B). Next, we analysed the
activation profile of T cells from the infected animals. TEM-like cells were most abundant within the CTL pool. Loss of A1 had no impact on their numbers (Figure 3b) or frequencies
(Supplementary Figure 3C). Moreover, _A1__−/−_ CD4+ T cells did not show any alterations in their memory compartment compared with infected wild-type mice (Supplementary Figure 3D).
_A1__−/−_ mice displayed a slight reduction in B cells on infection and a tendency towards a compensatory increase in granulocytes and macrophages (Supplementary Figure 3B). CTLs can be
further characterised based on their expression of KLRG1 and the IL-7 receptor alpha chain (CD127) into short-lived effector cells (SLECs, CD8+KLRG1+CD127−) and memory precursor effector
cells (MPECs, CD8+KLRG1−CD127+). Although A1 is mainly expressed in memory T cells (Figure 1), _A1__−/−_ and wild-type CTLs contained similar frequencies and numbers of SLECs and MPECs
(Figure 3c). Next, we analysed antigen-specific CTLs by staining with class I MHC tetramers loaded with GP33 or NP396 peptides; allowing discrimination between low-affinity and high-affinity
antigen-specific T cells, respectively. No differences in the numbers and frequencies of low- or high-affinity CTLs could be observed between infected _A1__−/−_ and wild-type mice (Figure
3d and Supplementary Figure 3E). To assess whether A1-deficient antigen-specific T cells were capable of producing IFN-_γ_ in an _in vitro_ recall response, total splenocytes from infected
animals were stimulated overnight with GP33 or NP396 peptides before cytokine production was analysed by intracellular flow cytometry. No differences in the frequencies and numbers of
IFN-_γ_-producing T cells was observed between splenocytes from infected _A1__−/−_ _versus_ wild-type mice (Figure 3e and Supplementary Figure 3F). Finally, we analysed the load of LCMV in
the spleen, liver, kidney, lung and brain of infected mice. No differences in viral titres were noted between wild-type and _A1__−/−_ mice (Figure 3f and Supplementary Figure 3G). On the
basis of these findings we conclude that loss of A1 does not impair the activation or expansion of CTLs in a chronic infection model of LCMV Docile. A1-DEFICIENT T CELLS SHOW NORMAL MEMORY
RESPONSES AND EXPAND NORMALLY ON REPEATED CHALLENGE WITH INFLUENZA VIRUS _IN VIVO_ In a separate study, we found that unchallenged _A1__−/−_ mice have slightly reduced numbers of memory T
cells compared with their wild-type counterparts.17 Therefore, we analysed the impact of A1-deficiency in memory T cell formation. Hence, mice were infected intra-nasally with influenza
HKx31 virus and memory T cells were enumerated 41 days after infection. First, we analysed the intrinsic ability of _A1__−/−_ CTLs to generate long-lived memory T cells by infecting BM
chimaeras (Figure 4 and Supplementary Figure 4). After 41 days, comparable frequencies of antigen-specific memory CTLs could be found in the wild-type and _A1__−/−_ compartments (Figure 4a).
Furthermore, the frequencies of _A1__−/−_ TCM and TEM CTLs were comparable (Figure 4a), as were the frequencies of Tregs (Figure 4b). To test for possible extrinsic effects on CD8+ memory T
cell formation, intact _A1__−/−_ and wild-type mice were infected with influenza virus. No differences in the memory profile of CTLs in the spleen (Figure 4c) were observed. Moreover, the
numbers of NP-specific CTLs were equivalent between the infected wild-type and _A1__−/−_ mice (Figure 4d). Finally, loss of A1 did not appear to impair the development of CD4+ memory T cells
(Figure 4e). Next, we investigated whether A1 has a role in the reactivation of memory T cells. C57BL/6-Ly5.2+ wild-type and _A1__−/−_ mice were infected with influenza virus and killed
after 41 days. CTLs were enriched by magnetic bead purification and 2 × 104 NP-specific A1-deficient or wild-type T cells were transferred into naive C57BL/6-Ly5.1+ (wild-type) recipient
mice (Figure 5a). These mice were challenged with influenza virus one day later and analysed 8 days post infection. We readily identified Ly5.2+ donor-derived T cells in the spleen, mLN and
lungs. No differences were observed between mice transplanted with either wild-type or _A1__−/−_ memory T cells (Figure 5b). In the lungs ~20% of the infiltrating leukocytes were Ly5.2+
donor-derived CTLs (Figure 5b), and this frequency was comparable to the Ly5.1+ infiltrates that were derived from a primary infection (Ly5.1+host, Supplementary Figure 5A). Approximately
30% of Ly5.2+ wild-type and Ly5.2 _A1__−/−_ CTLs were antigen-specific (Figure 5c), indicating a strong memory response. The expansion of total NP+CTLs was calculated by comparing the number
of injected NP+CTLs to the number recovered from spleen, mLN and lung. No differences were observed between the two genotypes (Figure 5d). To rule out that other pro-survival BCL-2 family
members were upregulated to compensate for the loss of A1, BCL-2 and MCL-1 protein levels were determined by intracellular flow cytometry in Ly5.2+wild-type, Ly5.2+_A1__−/−_ donor-derived
CTLs and Ly5.1+wild-type host CTLs. No evidence of upregulation of BCL-2 or MCL-1 in _A1__−/−_ CTLs was observed at the time points tested (Figure 5e). We were unable to assess BCL-XL by
flow cytometry due to the relatively poor quality of these antibodies. Moreover, these cells could not be isolated in sufficient numbers to perform Western blot analysis. Interestingly,
Ly5.2 wild-type CTLs showed a slight increase in MCL-1 protein levels, which was not observed in _A1__−/−_ CTLs. BCL-2 expression was reduced in both Ly5.2+ wild-type and _A1__−/−_ CTLs.
Collectively, these findings demonstrate that A1 is dispensable for CD8+ memory T cell formation, expansion and function. T-HELPER CELL FUNCTION AND B CELL RESPONSES ARE NOT IMPAIRED IN
_A1__−/−_ MICE IMMUNISED WITH NP-KLH/ALUM Influenza and LCMV infection primarily activate CTLs, but CD4+ T-helper cells and B cells also participate in the clearance of viruses and other
pathogens.22 To address whether A1 might have an impact on Tfh cells or the formation of GC B cells and long-lived PC, _A1__−/−_ and wild-type mice were immunised with NP-KLH/Alum and
analysed 14 days later by flow cytometry. Total spleen cellularity was similar between wild-type and _A1__−/−_ mice (Figure 6a). The total numbers of Tfh were slightly although not
significantly reduced in _A1__−/−_ mice (Figure 6b). The frequencies and numbers of antigen-specific Ig class-switched GC B cells were comparable between immunised _A1__−/−_ and wild-type
mice (Figure 6c). This indicates that the slight reduction of Tfh cells observed in _A1__−/−_ mice might not be physiologically relevant. The total numbers of PC were slightly but not
significantly reduced in the immunised A1-deficient mice compared with their wild-type counterparts (Figure 6d). To enumerate the antigen-specific PC in the spleen and bone marrow, ELISpot
assays were performed. No differences were observed in the production of low-affinity (NP16 binding) or high-affinity (NP2 binding) antibody secreting cells between the two genotypes (Figure
6e). These results show that A1 on its own is not critical for the development of Tfh as well as GC B cells and PC during a T cell-dependent humoral immune response. DISCUSSION We show that
loss of A1 does not impair T cell immunity in a diverse set of experimental systems. We therefore conclude that A1 does not exert a unique role in T cells or B cells during immune
responses. It remains possible that A1 has overlapping roles with other pro-survival BCL-2 family members controlling lymphocyte homoeostasis and activation. The lack of an obvious phenotype
was unexpected, as the levels of _A1_ mRNA and protein were both rapidly increased in T cells on TCR/CD3 ligation or stimulation with mitogens17, 18, 23 A1 induction has been defined as a
central step in rewiring the cell survival machinery in T cells during the transition from a resting to an activated state. This is associated with a change from a cytokine receptor (mainly
IL-7R) to a TCR-driven survival programme and includes the induction of BCL-XL and A1, accompanied by a downregulation of BCL-2.3 Therefore, it was surprising that _A1__−/−_ mice had no
difficulties in clearance of an acute viral infection. Interestingly, T cell-specific loss of BCL-XL, which is also strongly induced on TCR/CD28 stimulation,24 does not impair T cell
responses to _Listeria monocytogenes_ infection and humoral immune responses.10 This may suggest functional redundancy between these two BCL-2-like proteins. Besides exhaustion and
subsequent inactivation of CTLs, BID- and BIM-dependent apoptosis occurs during persistent viral infection to regulate T cell shutdown.25 Therefore, we examined whether A1 might be involved
in the response to chronic LCMV infection. Surprisingly, we found neither a role for A1 in the overall immune response nor in the survival of high- as well as low-affinity T cell
populations. These findings demonstrate that A1 is not essential for the development and survival of CTLs during persistent viral infection or for the preferential survival of high-affinity
TCR bearing T cell clones. Our data further strengthen the notion that MCL-1 appears to be the only pro-survival BCL-2 family member that cannot be substituted by other BCL-2 pro-survival
family members during T cell activation.12 A1 is readily detectable in memory T cells, and unchallenged _A1__−/−_ mice display a slight reduction in CD4+ memory T cells.17 These observations
suggested a role for A1 as a survival factor for long-lived memory T cells that are still present after the termination of an anti-viral immune response. Although it has been shown that the
BIM/BCL-2 axis is a main regulator of the formation and maintenance of memory T cells,26, 27 NOXA has also been proposed to have a role in regulating the size and clonal diversity of memory
T cell populations.5, 28 This is of particular interest as NOXA, like A1, is induced transcriptionally after TCR ligation.5 NOXA is believed to antagonise MCL-1 to set a threshold for the
survival of those CTLs with high-affinity TCRs.5 Accordingly, _Noxa__−/−_ mice have a higher number of antigen-specific CTLs and an increased memory compartment with higher clonal diversity
on infection with influenza.28 A1 is the closest pro-survival relative to MCL-1 and the only other described high-affinity binding partner of NOXA.29 Although we could not observe any
defects in the generation of antigen-specific memory T cells in response to influenza infection in _A1__−/−_ mice, we cannot exclude that the clonal repertoire of _A1__−/−_ T cells differs
from that of wild-type T cells. It will be interesting to test whether A1 loss can enhance the gene dosage-dependent phenotypes noted in mice lacking one allele of _Mcl-1._30, 31 Along this
line it is worth mentioning that such unilateral functional redundancy was also reported in mice that lack the BH3-only protein BMF on a BIM-deficient background. _Bmf__−/−_lymphocytes, such
as pre-B cells, showed no or only minor resistance to apoptotic stimuli, but this resistance was massively increased on additional loss of even only one allele of _Bim,_ exceeding
resistance observed in _Bim__−/−_ cells.32 Given that Tregs require continuous TCR signalling for their maintenance and function,33 it was surprising that we did not observe higher A1
protein levels in Tregs when compared with conventional T cells (Tcons). It has been reported that _A1_ mRNA levels are high in splenic Tregs but even higher in thymic Tregs when compared
with the expression observed in CD4+ Tcons.34 However, this was not demonstrated at the protein level in this study. Our results nonetheless support similar or even lower protein expression
levels of A1 in Tregs compared with Tcons. Therefore, we can only speculate that tonic TCR signalling as received by Tregs leads to the posttranslational downregulation of A1, which is only
visible at the protein level. Indeed, it has been shown before that A1 is tightly regulated by ubiquitin-dependent proteasomal degradation.18, 35 Future experiments of A1 protein stability
in Tregs will show whether this hypothesis holds true. Interestingly, non-challenged A1_−/−_ mice displayed a slight reduction in Tregs compared with wild-type mice.17 However, this small
difference was abolished on viral infection. This may imply a role for A1 in the survival of Tregs under homoeostatic conditions. Alternatively, after TCR/CD3 stimulation, Treg cell survival
might depend exclusively on MCL-1.36 Using a conditional RNAi mouse model to knockdown A1 expression, Carrington _et al._37 showed that conventional DCs (cDCs), which are critical for
certain immune responses, rely on A1 for their survival. Schenk _et al._17 also demonstrated that A1-deficient mice have reduced numbers of cDCs. However, the normal immune response of
_A1__−/−_ mice to viral infections observed here leads us to the question whether A1 may only be an auxiliary to MCL-1 in cDC survival or whether there are A1-independent antigen-presenting
cells that can take over the control of CTL-mediated immune responses in _A1__−/−_ mice. This question remains to be further investigated. It is also noteworthy that the above mentioned and
other RNAi-based A1-knockdown mouse models did not show any reductions in the numbers of mature, naive T cells or defects in T cell activation in culture. The lack of a T cell defect was
originally ascribed to incomplete knockdown of A1 in activated T cells,8, 15 but our analysis here points towards functional redundancy between A1 and other pro-survival BCL-2 family members
in the survival of quiescent as well as activated T cells. In conclusion, we showed that A1 ablation is not sufficient to impair viral T cell immune responses in mice. It is, however,
interesting that although A1 and BCL-XL are both highly induced in T cells in response to TCR and co-stimulatory signals that contribute to T cell survival,38, 39, 40 the loss of neither
impairs T cell function. This leaves the possibility that A1 and BCL-XL act in a redundant manner or are only of minor importance compared with the highly potent survival protein MCL-1.
These questions can now be addressed by the generation of compound mutant mice lacking A1 and/or BCL-XL, BCL-2 and/or MCL-1 in mature T cells. MATERIALS AND METHODS MICE All experiments with
mice were conducted according to the guidelines of The Walter and Eliza Hall Institute of Medical Research Animal Ethics Committee. _A1__−/−_ mice were generated on a C57BL/6 background.17
Mice used for _in vivo_ experiments were all 6–10 weeks of age. Bone-marrow (BM) chimaeras were generated by reconstitution of lethally irradiated recipient mice (two doses of 5.5 Gy, 3 h
apart) with a mixture of _A1__−/−_ or wild-type bone marrow (Ly5.2) and isogenic wild-type bone marrow (Ly5.1). Mice were analysed 8-12 weeks post reconstitution. _Foxp3__YFP-cre_ mice41
were used for sorting wild-type Treg cells based on YFP expression. _IN VITRO_ T CELL ACTIVATION CD4+ and CD8+ T cells were FACS-sorted from spleens of wild-type mice and cultured in
RPMI1640 medium supplemented with 10% foetal calf serum (FCS, PAA, Pasching, Austria, FBS Gold #A15-151), 2 mM l-Glutamine, 1% Penicillin plus Streptomycin (10.000 U/ml Penicillin, 10 mg/ml
Streptomycin in 0.9% NaCl) and 50 _μ_g/ml Gentamicin. T cells were activated with 5 _μ_g/ml plate-bound anti-CD3 (BioLegend, San Diego, CA, USA, clone 145-2C11) and 1 _μ_g/mL anti-CD28
(BioLegend, clone 37.51) antibodies for the indicated time points. LCMV INFECTION LCMV infections were performed by injection of 2 × 106 pfu LCMV docile into the tail vein of mice. The
origin and biology of LCMV docile has been described.21 This virus was propagated in L929 cells. To analyse the cytokine production in response to restimulation of LCMV-specific cells, 1 ×
106 total splenocytes were cultured overnight in the presence of 10-8 M LCMV GP33 (sequence KAVYNFATM), LCMV NP396 (sequence FQPQNGQFI), or non-stimulatory adenovirus (SGPSNTPPEI) in the
presence of the protein transport inhibitor Monensin (BD Biosciences, Franklin Lakes, NJ, USA) and stained thereafter for cytokine production as described below. Viral titres were analysed
using the LCMV focus forming assay as described before.42 In short, brain, liver, lung, spleen and kidney were collected in MEM 2% FCS and frozen at −80 °C before further analysed. Organs
were homogenised and supernatant dilutions were used to infect monolayers of MC57 cells (8 × 105 per ml), Cells were overlayed with 2% methyl cellulose (Fluka #64620) in DME and plates were
incubated at 37 °C, 5% CO2 for 48 h. Viral plaque staining was performed after fixation of the cells with 4% Formalin-PBS and permeabilization with 0.1% Triton X (FLUKA #93418) with VL-4 rat
anti-LCMV monoclonal antibody (WEHI Antibody Facility, Melbourne, VIC, Australia) followed by Peroxidase-conjugated AffiniPure goat anti-rat IgG (H+L) antibodies (Jackson ImmunoResearch
Laboratories, West Grove, PA, USA #112-035-003). A colour-reaction of ortho-phenylendiamin (Sigma-Aldrich, Pty. Ltd. Sydney, Australia, #P3888) substrate was used for detection of focus
forming units. The focus forming unit was used to calculate the viral titre in the original supernatant. INFLUENZA HKX31 INFECTION Mice were lightly anaesthetised by inhalation of
methoxyflurane, and infected i.n. with 3000 pfu of HKx31 (H3N2) influenza virus43 in 25 _μ_l PBS. Virus stocks were grown in the allantoic cavity of 10 day old embryonated chicken eggs, from
which the viral titre was determined by plaque assay on monolayers of Madin Derby canine kidney (MDCK) cells. Infiltrates from the lung were isolated as follows: lungs were mashed through
70 _μ_m cell strainers (BD Biosciences, Cat# 352350) and washed with PBS. Pellets were resuspended in 5 ml PBS supplemented with 2% FCS and underlayed with 3 ml Histopaque 1083 (Sigma, Cat#
1083-1) followed by 30 min centrifugation at 400x_g_ at room temperature with the brakes set off. Mononuclear cells were isolated from the opaque interface and washed extensively with PBS.
Staining for cell surface and intracellular markers was performed as described below. ENRICHMENT OF CD8+ CTLS FOR INFLUENZA RECHALLENGING MODEL For the enrichment of CD8+ CTLs from spleens
of infected mice, splenocytes were incubated with supernatants from hybridoma clones producing the following mABs (WEHI, Antibody Facility, Melbourne, VIC, Australia): rat anti-B220, rat
anti-CD11b, rat anti-GR-1, rat anti-CD4, and rat anti-MHCII. Spleen cells expressing these surface markers were depleted using goat anti-rat IgG antibody conjugated magnetic beads (NEB,
Ipswich, MA, USA, Cat# S1433S). Enrichment efficiency was assessed by flow cytometry and 2 × 104 NP-specific CTLs were injected in 200 _μ_l PBS i.v. into Ly5.1 wild-type recipient mice. T
LYMPHOCYTE-DEPENDENT B CELL IMMUNE RESPONSES Mice were immunised by i.p. injection of 100 _μ_g/20 g body weight of 4-hydroxy-3-nitrophenylacetyl (NP) coupled to Keyhole Limpet Hemocyanin
(KLH) at a ratio of 27 : 1 and precipitated onto alum.44 Mice were sacrificed 14 days post immunisation and spleen and bone marrow were isolated for analysis. ENZYME-LINKED IMMUNOSPOT ASSAY
The frequency of ASC was determined as described.44 Briefly, cells were incubated O/N at 37 °C on pre-coated 96-well MultiScreen-HA filter plates (Merck Millipore, Bayswater, VIC,
Australia). Spots were visualised with goat anti-mouse IgG1 antibodies conjugated to horseradish peroxidase (Southern Biotechnology Associates, Birmingham, AL, USA), and staining was
performed with 3-amino-9-ethyl carbazole (Sigma-Aldrich). Plates were washed extensively, and spots were counted using an AID ELIspot reader system (Autoimmun Diagnostika, Strassberg,
Germany). WESTERN BLOT ANALYSIS Cells were lysed in lysis buffer (50 mM Tris pH 8, 150 mM NaCl, 0.5% NP-40, 50 mM NaF, 1 mM Na3VO4, 1 mM PMSF, one tablet protease inhibitors (EDTA free,
Roche Austria, Vienna, Austria) per 10 ml and 30 _μ_g/ml DNaseI (Sigma-Aldrich, St. Louis, MO, USA) and protein was quantified with Bradford reagent (500-0006, Bio-Red, Munich, Germany).
Overall, 30 _μ_g total protein was loaded on 12% Bis-Tris acryl-amide gels and blotted on AmershamTM HybondTM-ECL nitrocellulose membranes (GE Healthcare, Little Chalfont, UK). The following
antibodies were used for protein detection: rat anti-mouse A1 (6D6, 2 _μ_g/ml),18 rat anti-BIM (WEHI Antibody Facility, Melbourne, VIC, Australia, clone 3C5, 2.6 _μ_g/ml), mouse anti-HSP90
(F8, Santa Cruz, Dallas, TX, USA, Cat# sc-13119, 0.2 _μ_g/ml), rabbit anti-MCL-1 (polyclonal, Rockland, Limerick, PA, USA, Cat# 600-401-394, 2.2 _μ_g/ml), rabbit anti-BCL-XL (54H6, Cell
Signaling, Danvers, MA, USA, Cat# CS2764, 1:1000), rabbit anti-P38 MAPK (polyclonal, Cell Signaling Cat# 9212, 1:1000). All primary antibodies were diluted in 5% BSA in PBST and blots were
incubated overnight at 4 °C. MRNA PURIFICATION AND QRT-PCR ANALYSIS RNA from approximately 1 × 105 cells was isolated, using the Quick-RNA MicroPrep (Zymo Research, Irvine, CA, USA, #R1051)
and up to 200 ng RNA was used to generate cDNA using the iScript cDNA Synthesis Kit (Bio-Rad #170-8891). qRT-PCR was performed using Platinum SYBR Green qPCR SuperMix-UDG reagent
(Invitrogen, Waltham, MA, USA) and following primers: A1 fwd 5′CCTGGCTGAGCACTACCTTC3′; A1 rev 5′TCCACGTGAAAGTCATCCAA3′; Mcl-1 fwd 5′TAACAAACTGGGGCAGGATT3′ Mcl-1 rev 5′GTCCCGTTTCGTCCTTACAA3′;
Bcl-xL fwd 5′TTCGGCATGGAGTAAACTGG3′ Bcl-xL rev 5′TGGATCCAAGGCTCTAGGTG3′; Bcl-2 fwd 5′CTGGCATCTTCTCCTTCCAG3′ Bcl-2 rev 5′GACGGTAGCGACGAGAGAAG3′; and actin fwd 5′ACTGGGACGACATGGAGAAG3′ actin
rev 5′GGGGTGTTGAAGGTCTCAAA3′. PCR conditions: 7 min 95 °C; 40 cycles × (10 s 95 °C; 20 s 60 °C; 20 s 72 °C); 15 s on 95 °C; 15 s 60 °C; melting curve 20 min; 15 s 95 °C. Relative
quantification was calculated using the ΔΔCT method. ANTIBODIES AND FLOW CYTOMETRY Fluorochrome-conjugated rat monoclonal antibodies directed against the following cell subset specific
surface markers were used for flow cytometric analysis: CD4 (RM 4-5), CD62L (MEL-14), Ly5.2 (104), CD44 (IM7), Ly6C (AL-21), CD11b (M1/70), IgG1 (RMG1-1), GR1 (Ly6G/C), CD69 (H1.2F3), CD19
(1D3) from BD Pharmingen (Franklin Lakes, NJ, USA); CCR2 (#475301) from R&D (Minneapolis, MN, USA); ICOS (7E-17G9), PD-1 (J43), CD103 (2E7), KLRG1 (2F1), CD25 (PC61.5), Tigit (GIGD7),
TCR_β_ (H57-597), CXCR3 (CXCR3-173), Ly5.1 (A20), ST2 (RMST2-2), Ly6C (HK1.4), GATA-3 (TWAJ), CD11c (N418), F4/80 (BM8), CD19 (MB19-1), CD127 (A7R34), CD8 (53-6.7) from eBioscience (San
Diego, CA, USA); CXCR5 (L138D7), B220 (RA3-6B2), Syndecan (281-2) from BioLegend (San Diego, CA, USA); NP-PE from Biosearch Technologies (Steinach-Thur, Germany, #N-5070-1). Intracellular
staining of FoxP3 (eBioscience, clone FJK-16 s, 0.1 _μ_g/ml) was performed using the eBioscience FOXP3 staining kit (Cat# 00-5523-00) according to the manufacturer's protocol.
Intracellular staining for fluorochrome-conjugated IFN-_γ_ (eBioscience, clone XMG1.2, 0.1 _μ_g/ml), MCL-1 (clone 19C4-15, 0.5 _μ_g/ml)45 and BCL-2 (BD biosciences, clone 3F11, 1:200) was
performed using the BD Cytofix/Cytoperm Fixation/Permeabilization kit (Cat# 554714) according to the manufacturer’s protocol. Influenza virus (HKx31)-specific responses were quantified by
staining with phycoerythrin-labelled of major histocompatibility complex (MHC) class I tetrameric complexes specific for the H-2b-restricted immunodominant epitope of influenza virus: NP366
(H-2Db). LCMV-specific CD8+ T cells were quantified with allophycocyanin-coupled tetramers MHC class I (H-2Db) in complex with LCMV GP33 (sequence KAVYNFATM), or NP396 (sequence FQPQNGQFI)
(Baylor College Medicine, Houston, TX, USA). Antibody-stained cells were analysed in a BD Fortessa1 or BD Fortessa X20. Cell sorting was performed using a BD FacsAriaTMIII Cell sorter (all
BD Biosciences). STATISTICAL ANALYSIS If not indicated differently, statistical analyses were conducted using a two-tailed Student’s _t_ test with Prism v.5.03 software (GraphPad, La Jolla,
CA, USA). Data are shown as mean±S.E. with _P_-values of <0.05 considered statistically significant.46, 47, 48, 49, 50 REFERENCES * Tan JT, Dudl E, LeRoy E, Murray R, Sprent J, Weinberg
KI _et al_. IL-7 is critical for homeostatic proliferation and survival of naive T cells. _Proc Natl Acad Sci USA_ 2001; 98: 8732–8737. Article CAS Google Scholar * Opferman JT, Letai A,
Beard C, Sorcinelli MD, Ong CC, Korsmeyer SJ . Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. _Nature_ 2003; 426: 671–676. Article CAS Google Scholar *
Koenen P, Heinzel S, Carrington EM, Happo L, Alexander WS, Zhang JG _et al_. Mutually exclusive regulation of T cell survival by IL-7 R and antigen receptor-induced signals. _Nat Commun_
2013; 4: 1735. Article Google Scholar * Sandalova E, Wei CH, Masucci MG, Levitsky V . Regulation of expression of Bcl-2 protein family member Bim by T cell receptor triggering. _Proc Natl
Acad Sci USA_ 2004; 101: 3011–3016. Article CAS Google Scholar * Wensveen FM, van Gisbergen KP, Derks IA, Gerlach C, Schumacher TN, van Lier RA _et al_. Apoptosis threshold set by Noxa
and Mcl-1 after T cell activation regulates competitive selection of high-affinity clones. _Immunity_ 2010; 32: 754–765. Article CAS Google Scholar * Hildeman DA, Zhu Y, Mitchell TC,
Bouillet P, Strasser A, Kappler J _et al_. Activated T cell death _in vivo_ mediated by proapoptotic bcl-2 family member bim. _Immunity_ 2002; 16: 759–767. Article CAS Google Scholar *
Best JA, Blair DA, Knell J, Yang E, Mayya V, Doedens A _et al_. Transcriptional insights into the CD8(+) T cell response to infection and memory T cell formation. _Nat Immunol_ 2013; 14:
404–412. Article CAS Google Scholar * Sochalska M, Ottina E, Tuzlak S, Herzog S, Herold M, Villunger A . Conditional knockdown of BCL2A1 reveals rate-limiting roles in BCR-dependent B
cell survival. _Cell Death Differ_ 2016; 23: 628–639. Article CAS Google Scholar * Nakayama K, Nakayama K, Negishi I, Kuida K, Shinkai Y, Louie MC _et al_. Disappearance of the lymphoid
system in Bcl-2 homozygous mutant chimeric mice. _Science_ 1993; 261: 1584–1588. Article CAS Google Scholar * Zhang N, He YW . The antiapoptotic protein Bcl-xL is dispensable for the
development of effector and memory T lymphocytes. _J Immunol_ 2005; 174: 6967–6973. Article CAS Google Scholar * Dzhagalov I, Dunkle A, He YW . The anti-apoptotic Bcl-2 family member
Mcl-1 promotes T lymphocyte survival at multiple stages. _J Immunol_ 2008; 181: 521–528. Article CAS Google Scholar * Tripathi P, Koss B, Opferman JT, Hildeman DA . Mcl-1 antagonizes
Bax/Bak to promote effector CD4(+) and CD8(+) T cell responses. _Cell Death Differ_ 2013; 20: 998–1007. Article CAS Google Scholar * Verschelde C, Walzer T, Galia P, Biemont MC, Quemeneur
L, Revillard JP _et al_. A1/Bfl-1 expression is restricted to TCR engagement in T lymphocytes. _Cell Death Differ_ 2003; 10: 1059–1067. Article CAS Google Scholar * Hatakeyama S,
Hamasaki A, Negishi I, Loh DY, Sendo F, Nakayama K _et al_. Multiple gene duplication and expression of mouse bcl-2-related genes, A1. _Int Immunol_ 1998; 10: 631–637. Article CAS Google
Scholar * Ottina E, Lyberg K, Sochalska M, Villunger A, Nilsson GP . Knockdown of the antiapoptotic Bcl-2 family member A1/Bfl-1 protects mice from anaphylaxis. _J Immunol_ 2015; 194:
1316–1322. Article CAS Google Scholar * Ottina E, Grespi F, Tischner D, Soratroi C, Geley S, Ploner A _et al_. Targeting antiapoptotic A1/Bfl-1 by _in vivo_ RNAi reveals multiple roles in
leukocyte development in mice. _Blood_ 2012; 119: 6032–6042. Article CAS Google Scholar * Schenk RL, Tuzlak S, Carrington EM, Zhan Y, Heinzel S, Teh CE _et al_. Characterisation of mice
lacking all functional isoforms of the pro-survival BCL-2 family member A1 reveals minor defects in the haematopoietic compartment. _Cell Death Differ_ 2016 (in press;
doi:10.1038/cdd.2016.156). Article CAS Google Scholar * Lang MJ, Brennan MS, O'Reilly LA, Ottina E, Czabotar PE, Whitlock E _et al_. Characterisation of a novel A1-specific
monoclonal antibody. _Cell Death Dis_ 2014; 5: e1553. Article CAS Google Scholar * Mandal M, Borowski C, Palomero T, Ferrando AA, Oberdoerffer P, Meng F _et al_. The BCL2A1 gene as a
pre-T cell receptor-induced regulator of thymocyte survival. _J Exp Med_ 2005; 201: 603–614. Article CAS Google Scholar * Tomayko MM, Punt JA, Bolcavage JM, Levy SL, Allman DM, Cancro MP
. Expression of the Bcl-2 family member A1 is developmentally regulated in T cells. _Int Immunol_ 1999; 11: 1753–1761. Article CAS Google Scholar * Moskophidis D, Lechner F, Pircher H,
Zinkernagel RM . Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. _Nature_ 1993; 362: 758–761. Article CAS Google Scholar
* Swain SL, McKinstry KK, Strutt TM . Expanding roles for CD4+ T cells in immunity to viruses. _Nat Rev Immunol_ 2012; 12: 136–148. Article CAS Google Scholar * Grumont RJ, Rourke IJ,
Gerondakis S . Rel-dependent induction of A1 transcription is required to protect B cells from antigen receptor ligation-induced apoptosis. _Genes Dev_ 1999; 13: 400–411. Article CAS
Google Scholar * Groux H, Monte D, Plouvier B, Capron A, Ameisen JC . CD3-mediated apoptosis of human medullary thymocytes and activated peripheral T cells: respective roles of
interleukin-1, interleukin-2, interferon-gamma and accessory cells. _Eur J Immunol_ 1993; 23: 1623–1629. Article CAS Google Scholar * Masson F, Kupresanin F, Mount A, Strasser A, Belz GT
. Bid and Bim collaborate during induction of T cell death in persistent infection. _J Immunol_ 2011; 186: 4059–4066. Article CAS Google Scholar * Wojciechowski S, Tripathi P, Bourdeau T,
Acero L, Grimes HL, Katz JD _et al_. Bim/Bcl-2 balance is critical for maintaining naive and memory T cell homeostasis. _J Exp Med_ 2007; 204: 1665–1675. Article CAS Google Scholar *
Kurtulus S, Tripathi P, Moreno-Fernandez ME, Sholl A, Katz JD, Grimes HL _et al_. Bcl-2 allows effector and memory CD8+ T cells to tolerate higher expression of Bim. _J Immunol_ 2011; 186:
5729–5737. Article CAS Google Scholar * Wensveen FM, Klarenbeek PL, van Gisbergen KP, Pascutti MF, Derks IA, van Schaik BD _et al_. Pro-apoptotic protein Noxa regulates memory T cell
population size and protects against lethal immunopathology. _J Immunol_ 2013; 190: 1180–1191. Article CAS Google Scholar * Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG _et
al_. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. _Mol Cell_ 2005; 17: 393–403. Article CAS Google Scholar *
Delbridge AR, Opferman JT, Grabow S, Strasser A . Antagonism between MCL-1 and PUMA governs stem/progenitor cell survival during hematopoietic recovery from stress. _Blood_ 2015; 125:
3273–3280. Article CAS Google Scholar * Grabow S, Delbridge AR, Valente LJ, Strasser A . MCL-1 but not BCL-XL is critical for the development and sustained expansion of thymic lymphoma in
p53-deficient mice. _Blood_ 2014; 124: 3939–3946. Article CAS Google Scholar * Labi V, Woess C, Tuzlak S, Erlacher M, Bouillet P, Strasser A _et al_. Deregulated cell death and
lymphocyte homeostasis cause premature lethality in mice lacking the BH3-only proteins Bim and Bmf. _Blood_ 2014; 123: 2652–2662. Article CAS Google Scholar * Vahl JC, Drees C, Heger K,
Heink S, Fischer Julius C, Nedjic J _et al_. Continuous T cell receptor signals maintain a functional regulatory T cell pool. _Immunity_ 2014; 41: 722–736. Article CAS Google Scholar *
Tischner D, Gaggl I, Peschel I, Kaufmann M, Tuzlak S, Drach M _et al_. Defective cell death signalling along the Bcl-2 regulated apoptosis pathway compromises Treg cell development and
limits their functionality in mice. _J Autoimmun_ 2012; 38: 59–69. Article CAS Google Scholar * Herold MJ, Zeitz J, Pelzer C, Kraus C, Peters A, Wohlleben G _et al_. The stability and
anti-apoptotic function of A1 are controlled by its C terminus. _J Biol Chem_ 2006; 281: 13663–13671. Article CAS Google Scholar * Pierson W, Cauwe B, Policheni A, Schlenner SM,
Franckaert D, Berges J _et al_. Antiapoptotic Mcl-1 is critical for the survival and niche-filling capacity of Foxp3(+) regulatory T cells. _Nat Immunol_ 2013; 14: 959–965. Article CAS
Google Scholar * Carrington EM, Zhang JG, Sutherland RM, Vikstrom IB, Brady JL, Soo P _et al_. Prosurvival Bcl-2 family members reveal a distinct apoptotic identity between conventional and
plasmacytoid dendritic cells. _Proc Natl Acad Sci USA_ 2015; 112: 4044–4049. Article CAS Google Scholar * Rogers PR, Song J, Gramaglia I, Killeen N, Croft M . OX40 promotes Bcl-xL and
Bcl-2 expression and is essential for long-term survival of CD4 T cells. _Immunity_ 2001; 15: 445–455. Article CAS Google Scholar * Song J, Salek-Ardakani S, Rogers PR, Cheng M, Van
Parijs L, Croft M . The costimulation-regulated duration of PKB activation controls T cell longevity. _Nat Immunol_ 2004; 5: 150–158. Article CAS Google Scholar * Lei F, Song J, Haque R,
Haque M, Xiong X, Fang D _et al_. Regulation of A1 by OX40 contributes to CD8(+) T cell survival and anti-tumor activity. _PLoS ONE_ 2013; 8: e70635. Article CAS Google Scholar * Rubtsov
YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X _et al_. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. _Immunity_ 2008; 28: 546–558.
Article CAS Google Scholar * Wang C, McPherson AJ, Jones RB, Kawamura KS, Lin GH, Lang PA _et al_. Loss of the signaling adaptor TRAF1 causes CD8+ T cell dysregulation during human and
murine chronic infection. _J Exp Med_ 2012; 209: 77–91. Article CAS Google Scholar * Flynn KJ, Belz GT, Altman JD, Ahmed R, Woodland DL, Doherty PC . Virus-specific CD8+ T cells in
primary and secondary influenza pneumonia. _Immunity_ 1998; 8: 683–691. Article CAS Google Scholar * Smith KG, Light A, Nossal GJ, Tarlinton DM . The extent of affinity maturation differs
between the memory and antibody-forming cell compartments in the primary immune response. _EMBO J_ 1997; 16: 2996–3006. Article CAS Google Scholar * Campbell KJ, Bath ML, Turner ML,
Vandenberg CJ, Bouillet P, Metcalf D _et al_. Elevated Mcl-1 perturbs lymphopoiesis, promotes transformation of hematopoietic stem/progenitor cells, and enhances drug resistance. _Blood_
2010; 116: 3197–3207. Article CAS Google Scholar * Czabotar PE, Lessene G, Strasser A, Adams JM . Control of apoptosis by the BCL-2 protein family: implications for physiology and
therapy. _Nat Rev Mol Cell Biol_ 2014; 15: 49–63. Article CAS Google Scholar * Llambi F, Wang YM, Victor B, Yang M, Schneider DM, Gingras S _et al_. BOK Is a non-canonical BCL-2 family
effector of apoptosis regulated by ER-associated degradation. _Cell_ 2016; 165: 421–433. Article CAS Google Scholar * Ke F, Voss A, Kerr JB, O'Reilly LA, Tai L, Echeverry N _et al_.
BCL-2 family member BOK is widely expressed but its loss has only minimal impact in mice. _Cell Death Differ_ 2012; 19: 915–925. Article CAS Google Scholar * Chipuk JE, Green DR . How do
BCL-2 proteins induce mitochondrial outer membrane permeabilization? _Trends Cell Biol_ 2008; 18: 157–164. Article CAS Google Scholar * Thomas LW, Lam C, Edwards SW . Mcl-1; the molecular
regulation of protein function. _FEBS Lett_ 2010; 584: 2981–2989. Article CAS Google Scholar Download references ACKNOWLEDGEMENTS We thank all members of the Herold laboratory for their
support and advice. G Siciliano, H Johnsons and their team for animal husbandry; S Monard and his team for help with flow cytometry; and D Tischner for help with RNA analysis. This work was
supported by a Leukemia Foundation National Research Program PhD Scholarship (to RS), National Health and Medical Research Council, Australia (programme grant 1016701 and fellowship 1020363,
to AS), project grant APP1049720 (to MJH). This work was made possible through Victorian State Government Operational Infrastructure Support and Australian Government National Health and
Medical Research Council Independent Research Institutes Infrastructure Support Scheme. This work was supported by grants from the Austrian Science Fund (FWF), Grant I1298 (FOR-2036), the
MCBO Doctoral College ‘Molecular Cell Biology and Oncology’ (W1101) and the ‘Österreichische Krebshilfe Tirol’. MDH and ST are supported by a DOC-fellowship from the Austrian Academy of
Science (ÖAW). AUTHOR CONTRIBUTIONS ST performed experiments, analysed data, wrote paper and prepared figures; RS performed experiments, analysed data and prepared figures; AV, MDH, SP, and
DZ performed experiments and analysed data; AV and AS planned study design and edited paper, MJH conceived and planned study, and wrote paper. AUTHOR INFORMATION Author notes * Selma Tuzlak
and Robyn L Schenk: These authors contributed equally to this work. AUTHORS AND AFFILIATIONS * Division of Developmental Immunology, BIOCENTER, Medical University Innsbruck, Innsbruck,
Austria Selma Tuzlak, Manuel D Haschka & Andreas Villunger * The Walter & Eliza Hall Institute for Medical Research, Parkville, Melbourne, 3052, VIC, Australia Selma Tuzlak, Robyn L
Schenk, Ajithkumar Vasanthakumar, Simon P Preston, Dimitra Zotos, Axel Kallies, Andreas Strasser & Marco J Herold * Department of Medical Biology, University of Melbourne, Parkville,
Melbourne, 3050, VIC, Australia Robyn L Schenk, Ajithkumar Vasanthakumar, Simon P Preston, Dimitra Zotos, Axel Kallies, Andreas Strasser & Marco J Herold * Tyrolean Cancer Research
Institute, Innsbruck, Austria Andreas Villunger Authors * Selma Tuzlak View author publications You can also search for this author inPubMed Google Scholar * Robyn L Schenk View author
publications You can also search for this author inPubMed Google Scholar * Ajithkumar Vasanthakumar View author publications You can also search for this author inPubMed Google Scholar *
Simon P Preston View author publications You can also search for this author inPubMed Google Scholar * Manuel D Haschka View author publications You can also search for this author inPubMed
Google Scholar * Dimitra Zotos View author publications You can also search for this author inPubMed Google Scholar * Axel Kallies View author publications You can also search for this
author inPubMed Google Scholar * Andreas Strasser View author publications You can also search for this author inPubMed Google Scholar * Andreas Villunger View author publications You can
also search for this author inPubMed Google Scholar * Marco J Herold View author publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence
to Marco J Herold. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no conflict of interest. ADDITIONAL INFORMATION Edited by C Borner Supplementary Information accompanies this
paper on _Cell Death and Differentiation_ website . SUPPLEMENTARY INFORMATION SUPPLEMENTARY FIGURE S1 (JPG 197 KB) SUPPLEMENTARY FIGURE S2 (JPG 1293 KB) SUPPLEMENTARY FIGURE S3 (JPG 104 KB)
SUPPLEMENTARY FIGURE S4 (JPG 59 KB) SUPPLEMENTARY FIGURE S5 (JPG 38 KB) SUPPLEMENTARY LEGENDS (DOCX 19 KB) RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS
ARTICLE Tuzlak, S., Schenk, R., Vasanthakumar, A. _et al._ The BCL-2 pro-survival protein A1 is dispensable for T cell homeostasis on viral infection. _Cell Death Differ_ 24, 523–533 (2017).
https://doi.org/10.1038/cdd.2016.155 Download citation * Received: 14 July 2016 * Revised: 08 November 2016 * Accepted: 01 December 2016 * Published: 13 January 2017 * Issue Date: March
2017 * DOI: https://doi.org/10.1038/cdd.2016.155 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is
not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative