Play all audios:
Bcl-2 E1B 19-KDa interacting protein 3 (BNIP3) is a mitochondrial death and mitophagy marker, which is involved in inducing cardiac remodeling post myocardial infarction. In this study, we
show that BNIP3 expression increases in stressed cardiomyocytes in vitro and in response to pressure overload in vivo, and that its transcription is directly related to JNK activity. BNIP3
expression gradually increased in the first weeks after pressure overload and peaked at the heart failure stage. Ultrastructurally, the mitochondrial area was inversely proportional to BNIP3
expression. Both JNK and AKT activities increased with pressure overload; however, JNK signaling dominated over AKT signaling for the activation of the transcription factor FOXO3a and for
the transcription of its effector, BNIP3. 3-methyladenine attenuated JNK signaling and significantly decreased BNIP3 expression and reversed cardiac remodeling in heart failure.
Ultrastructurally, the mitochondrial area was significantly increased in the 3-methyladenine group compared with placebo. Moreover, adenoviral gene delivery of dominant negative JNK in a rat
model of pressure overload hypertrophy abolished the increase in BNIP3 expression in response to pressure overload. These results suggest that JNK signaling is a critical modulator of the
transcription factor FOXO3a driving the expression of its effector, BNIP3, in heart failure and that JNK, through BNIP3, induces mitochondrial apoptosis and mitophagy.
Heart failure is a clinical syndrome characterized by the activation of the neurohormonal and renin angiotensin aldosterone system followed by remodeling of the left ventricle (LV) and
alterations in the LV geometry.1 The integrity of the endoplasmic reticulum (ER) and the juxtaposed mitochondria is pivotal for the proper function of the cardiomyocyte. Ultrastructurally,
these two organelles are located at very close proximity and crosstalk with each other via calcium signaling.2, 3, 4, 5, 6 In heart failure, both the ER and the mitochondria, and each on its
own, execute death signals that take the form of programmed apoptotic and autophagic cell death.7 The decline in cardiac function of heart failure patients is in part due to the loss of the
diseased cardiac myocytes in the form of necrotic, apoptotic and autophagic cell death. The Bcl-2 family proteins serve as a critical death regulators that reside immediately upstream of
the mitochondria. They consist of anti-apoptotic members such as Bcl-2 protein and pro-apoptotic members. The pro-apoptotic Bcl-2 members are subdivided into ‘multidomain’ and ‘Bcl-2
Homology (BH3)’ proteins. Multidomain pro-apoptotic proteins such as Bax and Bak display sequence conservation in the BH domains 1–3 and their expression is directly regulated by the
anti-apoptotic Bcl-2 protein. On the other hand, the BH3-only members display sequence conservation only in the α-helical BH3 region, which constitutes the critical death domain.8 Of the BH3
members, the Bcl-2 E1B 19-KDa interacting protein 3 (BNIP3) is unique in the sense that it induces mitochondrial apoptosis as well as mitochondrial autophagy (mitophagy).9 In the initial
phase of apoptosis, BNIP3 inserts into the outer mitochondrial membrane with the N terminus oriented into the cytoplasm and the C terminus inside the mitochondria. It induces
mitochondria-mediated apoptosis and fragmentation by driving mitochondrial permeability transition pore opening, cytochrome C release and the destruction of the mitochondrial cristae.10, 11
On the other hand, BNIP3 is an autophagy receptor that activates mitophagy in a non-canonical order leading to their sequestration and subsequent removal.9, 12, 13 What makes BNIP3 more
interesting is that, unlike the other Bcl-2 members, it is the effector of the transcription factor FOXO3a in post-mitotic skeletal muscles and cardiomyocytes.14 Many studies have suggested
that BNIP3 expression increases under ischemic condition in cardiac myocytes and that cardiac remodeling is directly related to BNIP3 expression.15, 16, 17, 18, 19 In this study, we show
that BNIP3 is expressed in response to cardiomyocyte stressors, such as phenylephrine (PE) or calcium, in vitro and to pressure overload in vivo. Moreover, we show how the interplay between
AKT and JNK signaling modulates FOXO3a for the transcription of its effector BNIP3. Moreover, we show that 3-methyladenine (3 MA), by interfering with JNK signaling, modulates the expression
of t
he mitochondrial death and mitophagy marker BNIP3 in vivo, and reverses cardiac remodeling in a rat model of pressure overload-induced heart failure. This signaling pathway was further
validated via the adenoviral gene delivery of dominant negative JNK (Ad-DN-JNK) in a rat model of pressure overload hypertrophy (POH).
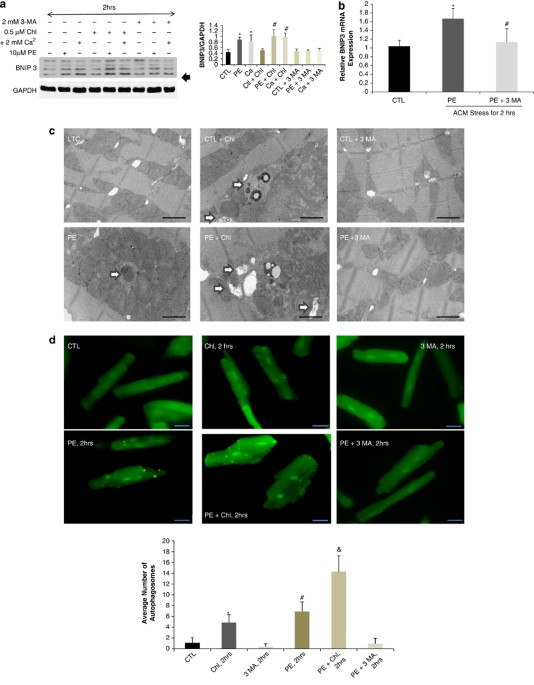
BNIP3 expression is increased by two-fold 2 h after cardiomyocyte stress with PE or calcium in vitro. BNIP3 expression was also increased with the addition of chloroquine, a well-known
autophagolysosome fusion inhibitor, in stressed cardiomyocytes, whereas 3 MA, an autophagy induction inhibitor, inhibited the increase in BNIP3 expression Figure 1a. The relative BNIP3 mRNA
expression was significantly increased in PE-stressed cardiomyocytes for 2 h and its increase was significantly inhibited by 3 MA Figure 1b. Autophagosomes, containing mitochondria with
different stages of vacuolar degeneration, were observed in chloroquine, PE and PE plus chloroquine-stressed cardiomyocytes Figure 1c and Supplementary Figure 1. The number of autophagosomes
increased with the addition of chloroquine and in PE-stressed cardiomyocytes for 2 h with the highest number of autophagosomes observed in the PE plus chloroquine-treated cardiomyocytes. 3
MA inhibited the formation of autophagosomes in PE-stressed cardiomyocytes Figure 1d. This suggests that the increase in autophagosomes with PE treatment is due to the increase in
autophagosomes formation rather than the consequence of a degradation removal problem. The overexpression of FOXO3a using an adenovirus containing constitutively active FOXO3a (Ad-FOXO3a)
increased the expression of BNIP3 in cardiac myocytes in vitro compared with adenovirus green fluorescent protein (Ad-GFP) and dominant negative FOXO3a (Ad-DN-FOXO3a), respectively Figure
1e. Moreover, the overexpression of BNIP3 in eGFP-LC3 expressing cardiomyocytes by simultaneous infection with an adenovirus containing BNIP3 (Ad-BNIP3) and another with eGFP-LC3
(Ad-eGFP-LC3), robustly increased the number of autophagosomes compared with adenovirus Null (Ad-Null) and adenovirus Sh BNIP3 (Ad-Sh BNIP3)-infected cardiomyocytes, respectively Figure 1f.
Western blot data shown in Supplementary Figure 2. Ultrastructurally, BNIP3 overexpression in cultured cardiac myocytes was associated with a marked increase in autophagosomes and robust
decrease in mitochondrial area compared with Ad-Null and Ad-Sh BNIP3-infected cardiac myocytes, respectively Figure 1g and Supplementary Figure 3.
BNIP3 is upregulated 2 h after cardiomyocyte stress with PE or calcium and is inhibited by 3 MA. (a) Western blotting analysis of protein lysates from adult cardiomyocytes (ACM) stressed
with PE or Calcium. BNIP3 expression was significantly upregulated 2 h after ACM stress with PE or calcium and with chloroquine (Chl) treatment, *P