Play all audios:
ABSTRACT The class I selective inhibitor of the histone deacetylases, mocetinostat, has promising antitumor activities in both preclinical studies and the clinical trials. To understand how
mocetinostat induces apoptosis, we examined the effects of mocetinostat on miR-31, a proapoptotic microRNA that was previously found to be epigenetically silenced in prostate cancer. We
found that miR-31 was significantly upregulated by mocetinostat in prostate cancer cells. Antiapoptotic protein E2F6, the target of miR-31, was decreased by mocetinostat treatment. When
miR-31 was blocked with an inhibitor, the ability of mocetinostat to induce apoptosis was reduced. We further demonstrated that mocetinostat enhanced the activity of docetaxel in apoptosis
induction. While siRNA knockdown of E2F6 sensitized cancer cells to mocetinostat-induced apoptosis, overexpression of E2F6 blocked mocetinostat-induced apoptosis. In an orthotopic xenograft
model, we demonstrated that mocetinostat activated miR-31, decreased E2F6, induced apoptosis, and significantly reduced prostate cancer growth. Importantly, we found that mocetinostat also
increased miR-31 expression, decreased E2F6, and induced apoptosis in the primary prostate cancer stem cells. Thus, activation of miR-31 and downregulation of E2F6 constitute an important
mechanism in mocetinostat-induced apoptosis in prostate cancer. SIMILAR CONTENT BEING VIEWED BY OTHERS MIR-93 SUPPRESSES TUMORIGENESIS AND ENHANCES CHEMOSENSITIVITY OF BREAST CANCER VIA DUAL
TARGETING E2F1 AND CCND1 Article Open access 14 August 2020 THE MICRORNA MIR-454 AND THE MEDIATOR COMPLEX COMPONENT MED12 ARE REGULATORS OF THE ANDROGEN RECEPTOR PATHWAY IN PROSTATE CANCER
Article Open access 25 March 2025 SYNERGISTIC APOPTOTIC EFFECT OF MIR-183-5P AND POLO-LIKE KINASE 1 INHIBITOR NMS-P937 IN BREAST CANCER CELLS Article Open access 24 September 2021 Histone
deacetylases (HDACs) are important epigenetic regulators of gene expression.1 These enzymes deacetylate lysines of the core histone tail and result in transcription repression.2 HDACs also
deacetylate lysines of other proteins and regulate cellular functions ranging from proliferation and apoptosis to metastasis and angiogenesis.3,4 There are 18 mammalian HDACs and they are
classified into four groups:5 class I HDACs (HDAC1, -2, -3, and -8); class II HDACs (HDAC4, -5, -6, -7, -9, and -10); class III HDACs (sirt1–7); and class IV HDAC (HDAC11). The class I HDACs
are often overexpressed in various types of cancers comparing with the corresponding normal tissues and their overexpression is correlated with a poor prognosis.6–8 HDAC inhibitors have
been developed and extensively tested in phase I–III clinical trials. However, these new agents showed minimal clinical activity in patients with solid tumors while undesired side effects
are also a problem.9 One of the reasons for the poor performance of the early generation of the HDAC inhibitors may be because they inhibit multiple classes of HDACs, which results in
toxicity and limits the achievable therapeutic doses. Mocetinostat, also known as MGCD0103, is one of the new class I selective HDAC inhibitors.10 Preclinical studies have demonstrated broad
spectrum antitumor activities of mocetinostat against various types of cultured cell lines and tumor xenografts in nude mice.10,11 It has been shown that mocetinostat inhibits colon cancer
cell growth by upregulating WNT ligand DKK-1 expression.12 In B-cell chronic lymphocytic leukemia cells, mocetinostat has been shown to induce apoptosis through decreasing antiapoptotic
Mcl-1 protein and inducing Bax translocation to the mitochondria.13 While mocetinostat has been shown to effectively kill prostate cancer cells,10 the mechanisms of apoptosis induction
remain poorly understood. microRNAs (miRNAs) are small single-stranded non-coding RNAs that suppress gene expression by cleaving the target mRNAs or by inhibiting their translation.14 The
miRNAs are initially transcribed from their encoding genes as long pri-miRNAs. The pri-miRNAs are further processed into ~60–70 nucleotide-long pre-miRNAs by Drosha RNase III endonuclease.15
The RNase III endonuclease Dicer then processes the pre-miRNAs to form the mature single-strand miRNAs, which are incorporated into the RNA-induced silencing complex to bind to and silence
the target mRNAs. Recent studies have demonstrated important roles of miRNAs in cancer, regulating cell cycle,16 differentiation,17,18 metabolism,19 invasion and metastasis,20 as well as
apoptosis.21 Recently, we have reported that miR-31 was significantly downregulated in prostate cancer cells and its downregulation resulted in overexpression of antiapoptotic protein E2F6
and resistance to apoptosis.22 Studies by Lin _et al._23 have shown that miR-31 is epigenetically silenced in prostate cancer by promoter hypermethylation and histone H3K27 trimethylation.
Since the HDACs are often involved in DNA methylation-mediated gene silencing,24 we tested whether mocetinostat can activate miR-31 expression. We found that mocetinostat activated miR-31
expression and downregulated its target, the antiapoptotic protein E2F6 both _in vitro_ and _in vivo_ in an orthotopic xenograft model. These findings identified a novel mechanism that
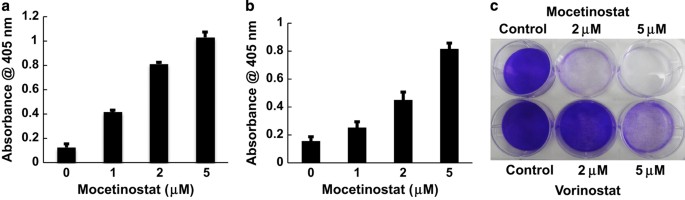
contributes to mocetinostat-induced apoptosis in prostate cancer. RESULTS MOCETINOSTAT INDUCES APOPTOSIS IN PROSTATE CANCER CELLS We determined whether mocetinostat can induce apoptosis in
prostate cancer cells. As shown in Figure 1a, mocetinostat induced significant levels of apoptosis in DU-145 cells in a dose-dependent manner. Induction of apoptosis was determined by the
cell death ELISA assay measuring mono- and oligonucleosomes in the lysates of apoptotic cells. Similarly, mocetinostat also induced significant levels of apoptosis in PC-3 cells (Figure 1b).
We compared the antitumor activities of mocetinostat and vorinostat (suberanilohydroxamic acid),25 an FDA-approved non-selective HDAC inhibitor. Mocetinostat was significantly more potent
than vorinostat in prostate cancer suppression (Figure 1c). MOCETINOSTAT INDUCES MIR-31 EXPRESSION AND DOWNREGULATES E2F6 We recently demonstrated that the downregulation of miR-31
contributes to apoptosis resistance in prostate cancer cells.22 Since miR-31 was shown to be repressed by epigenetic mechanisms in prostate cancer,23 we hypothesized that mocetinostat may
activate miR-31 expression. The effects of mocetinostat on miR-31 expression were determined by real-time PCR. As shown in Figures 2a and c, mocetinostat significantly induced miR-31
expression in both DU-145 and PC-3 cells. We have previously shown that miR-31 targets E2F6,22 which is a potent antiapoptotic protein that can inhibit UV- and hypoxia-induced
apoptosis.26,27 As a result of miR-31 induction, mocetinostat significantly decreased E2F6 protein in DU-145 and PC-3 cells (Figures 2b and d). To determine whether HDAC inhibition by
mocetinostat is responsible for the activation of miR-31, we used siRNA to specifically knock down HDAC1. As shown in Figures 2e and f, siRNA knockdown of HDAC1 activated miR-31 expression
and decreased E2F6 protein. To further understand the mechanism of mocetinostat-induced apoptosis, we determined the effects of mocetinostat on the expression of the proapoptotic members of
the Bcl-2 family proteins.28 Interestingly, Bad was significantly increased by mocetinostat treatment while the expression levels of the other proteins were either reduced (Puma, Bid, and
Bax) or unchanged (Bak) (Figure 3a). Since Bad is a key proapoptotic protein that triggers the intrinsic pathway of apoptosis,29,30 we examined the effects of mocetinostat on the caspases.
As shown in Figure 3b, mocetinostat treatment significantly increased the levels of activated (cleaved) caspase-9 and caspase-3. The cleaved products of PARP, substrates of the caspases,
were also increased by mocetinostat. MIR-31 PLAYS A CRITICAL ROLE IN MOCETINOSTAT-INDUCED APOPTOSIS AND MOCETINOSTAT ENHANCES THE ACTIVITY OF DOCETAXEL To determine the role of miR-31 in
mocetinostat-induced apoptosis, we used an miR-31 inhibitor to block the activity of miR-31 in DU-145 cells. As shown in Figure 3c, the ability of mocetinostat to induce apoptosis was
reduced when miR-31 was inhibited. In metastatic castration-resistant prostate cancer, docetaxel is the only approved treatment and it only has limited efficacy. To determine whether
mocetinostat can enhance the antitumor activity of docetaxel, we treated DU-145 cells with a single agent of mocetinostat and docetaxel, or the combination of the two agents. As shown in
Figure 3d, mocetinostat significantly enhanced apoptosis induction by docetaxel. E2F6 REGULATES MOCETINOSTAT-INDUCED APOPTOSIS IN PROSTATE CANCER CELLS To determine the role of E2F6 in
mocetinostat-induced apoptosis, we used siRNA to knock down E2F6 in DU-145 cells. As shown in Figure 4a, E2F6 protein expression was significantly reduced by siRNA treatment. Knockdown of
E2F6 sensitized DU-145 cells to apoptosis induced by mocetinostat (Figure 4b). We expressed E2F6 exogenously to determine if E2F6 can contribute to apoptosis resistance in prostate cancer
cells. Overexpression of E2F6 was confirmed by western blotting (Figure 4c). When treated with mocetinostat, DU-145 cells overexpressing E2F6 were significantly more resistant to
drug-induced apoptosis, comparing with empty vector-transfected cells (Figure 4d). MOCETINOSTAT INDUCES MIR-31 EXPRESSION AND ACTIVATES APOPTOSIS _IN VIVO_ IN AN ORTHOTOPIC TUMOR XENOGRAFT
MODEL To determine whether miR-31 activation plays an important role in mocetinostat-induced apoptosis _in vivo_, we established an orthotopic xenograft model in which the prostate cancer
was implanted into the prostate of nude mice. After treatment with mocetinostat, we observed _in vivo_ activation of miR-31, downregulation of E2F6, increased expression of Bad, and
activation of caspase-3 (Figures 5a and b). As a result, significant level of apoptosis was induced in the tumors and orthotopic tumor growth was suppressed by nearly 50% (Figures 5c and d).
We did not observe any toxicity in the mice (such as loss of body weight, etc). MOCETINOSTAT INDUCES MIR-31 EXPRESSION AND ACTIVATES APOPTOSIS IN PRIMARY PROSTATE CANCER STEM CELLS It is
postulated that cancer stem cells mediate tumor formation, metastasis, and resistance to chemotherapy.31 Thus, it is critical to identify drugs that can eliminate cancer stem cells.32 We
tested if mocetinostat is effective against prostate cancer stem cells. As shown in Figure 6a, mocetinostat induced significant levels of apoptosis in patient-derived primary prostate cancer
stem cells. Furthermore, mocetinostat increased miR-31 expression (Figure 6b), and decreased E2F6 protein (Figure 6c) in the prostate cancer stem cells. The level of proapoptotic protein
Bad was increased by mocetinostat (Figure 6c). DISCUSSION Mocetinostat is a class I selective HDAC inhibitor and has promising antitumor activities in preclinical studies.10,11 More
importantly, recent clinical trials have demonstrated excellent response of mocetinostat against myelodysplastic syndrome and relapsed Hodgkin's lymphoma, with acceptable safety
profiles.33,34 However, a phase II study also found that mocetinostat has limited efficacy as a single agent in relapsed and refractory chronic lymphocytic leukemia.35 Thus, it is important
to understand the mechanism of mocetinostat-induced apoptosis in order to improve its efficacy in cancer therapy and overcome drug resistance. Our findings in this report serve for this
purpose by helping to understand the molecular basis of mocetinostat-induced apoptosis. We have previously shown that in prostate cancer cell lines derived from advanced metastatic cancers,
miR-31 is downregulated to contribute to the resistance to chemotherapy-induced apoptosis.22 Here, we demonstrated that mocetinostat activates the expression of miR-31, which in turn
decreases the antiapoptotic protein E2F6. We also found that mocetinostat increases the expression of proapoptotic protein Bad. Our data have established a mechanistic model to explain how
mocetinostat induces apoptosis in prostate cancer (Figure 6d). By inducing miR-31 to decrease E2F6 while in the meantime increasing Bad, mocetinostat tipped the balance between the
antiapoptotic and proapoptotic proteins,36 resulting in increased apoptosis. Evidences are emerging that the prostate cancer stem cells may play important roles in resistance to castration37
and chemotherapy.31 Recently, it has been shown that mocetinostat induces cell cycle arrest and apoptosis in colon-cancer-initiating cells.12 Our data suggest that mocetinostat can
efficiently induce apoptosis in the primary prostate cancer stem cells through a mechanism involving miR-31 and E2F6 (Figure 6). Thus, mocetinostat is an effective drug for eliminating the
prostate cancer stem cells. Azacitidine can activate the expression of epigenetically silenced genes by inhibiting DNA promoter methylation.38 Moreover, recent clinical trials have
demonstrated excellent results when azacitidine and mocetinostat are used together in cancer therapy.33 It will be interesting to test if azacitidine can act with mocetinostat
synergistically to activate miR-31 and induce apoptosis in prostate cancer cells, since the two drugs target different mechanisms of miR-31 silencing. In current clinical trials of
mocetinostat and other HDAC inhibitors, HDAC activity inhibition and histone acetylation are used as biomarkers to measure pharmacodynamics responses. Our findings in this study have
identified new parameters (miR-31, E2F6, Bad) that may be used to monitor tumor response to mocetinostat. These apoptosis regulators may be useful to predict how efficiently mocetinostat can
kill the prostate cancer cells. MATERIALS AND METHODS CELLS AND TRANSFECTION The cell lines PC-3 and DU-145 were purchased from American Type Culture Collection (Manassas, VA, USA). Cells
were cultured in RPMI1640 media containing 10% FBS. Patient-derived human prostate cancer stem cells were purchased from CELPROGEN (San Pedro, CA, USA) and cultured following the
manufacturer’s protocol. For transient transfection, plasmids were transfected into cells using Lipofectamine Plus Reagent (Invitrogen, Grand Island, NY, USA) following the manufacturer’s
protocol. E2F6 siRNA (Ambion, Grand Island, NY, USA; Cat # 4185), HDAC1 siRNA (Ambion; Cat # s73) and anti-miR-31 inhibitor (Ambion, Cat # AM11465) were transfected into cells using X-treme
GENE siRNA transfection reagent (Roche, Indianapolis, IN, USA) following the manufacturer’s protocol. DRUGS AND CHEMICALS Mocetinostat was purchased from Selleckchem (Houston, TX, USA).
Vorinostat was purchased from Biovision (Mountain View, CA, USA). PLASMID CONSTRUCTION The full-length _E2F6_ cDNA was obtained by PCR using an EST clone as a template and constructed into a
pcDNA3-HA vector. WESTERN BLOT ANALYSIS Cells were lysed in RIPA buffer (1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS in PBS). Complete protease inhibitor cocktail (Roche) was added to
lysis buffer before use. Protein concentration was determined by Bio-Rad DC protein assay (Bio-Rad). Protein samples were subjected to SDS-PAGE and transferred onto nitrocellulose membrane.
The membrane was blocked in 5% non-fat milk in PBS overnight and incubated with primary antibody and subsequently with appropriate horse radish peroxidase-conjugated secondary antibody.
Signals were developed with ECL reagents (Pierce, Rockford, IL, USA) and exposure to X-ray films. Anti-β-tubulin and anti-E2F6 antibodies were purchased from Santa Cruz Biotechnology
(Dallas, TX, USA). Cleaved caspase-9, cleaved caspase-3, cleaved PARP, HDAC1, Bad, Bid, Bak, Puma, and Bax antibodies were purchased from Cell Signaling (Danvers, MA, USA). REAL-TIME PCR The
miRNA expression was measured by real-time PCR using TaqMan MicroRNA assays (Cat # TM1100 for miR-31) from Applied Biosystems (Foster city, CA, USA). Total RNA was isolated from cells using
_mir_Vana miRNA Isolation Kit (Ambion). Five micrograms of total RNA was used in reverse transcription reaction. The cDNAs were used as templates to perform PCR on an Applied Biosystems
7500 Real-time PCR System following the manufacturer’s protocol. Relative miRNA expression levels were calculated using 18S RNA as reference. DETECTION OF APOPTOSIS For _in vitro_ apoptosis
detection, the Cell Death Detection ElisaPLUS kit (Roche) was used to detect apoptosis following the manufacturer’s protocol. This assay determines apoptosis by measuring mono- and
oligonucleosomes in the lysates of apoptotic cells. The cell lysates were placed into a streptavidin-coated microplate and incubated with a mixture of anti-histone-biotin and
anti-DNA-peroxidase. The amount of peroxidase retained in the immunocomplex was photometrically determined with ABTS as the substrate. Absorbance was measured at 405 nm. For _in vivo_
apoptosis detection, the _in situ_ BrdU-Red DNA Fragmentation (TUNEL) Assay Kit (Abcam, Cambridge, MA, USA) was used following the manufacturer’s protocol. This assay utilizes Br-dUTP
(bromolated deoxyuridine triphosphate nucleotides), which is readily incorporated into DNA strand breaks. The Br-dUTP sites are identified by a red fluorescence labeled anti-BrdU monoclonal
antibody. CRYSTAL VIOLET STAINING The cells were treated with various doses of mocetinostat or the same doses of vorinostat for 72 h. The colonies were fixed with paraformaldehyde and
stained with 0.05% crystal violet. ORTHOTOPIC TUMOR XENOGRAFT IN NUDE MICE Six- to eight-week-old male nude mice (Nu/Nu) were purchased from Charles River (Wilmington, MA, USA). The mice
were maintained in sterile conditions using the Innovive IVC System from Innovive (San Diego, CA, USA), following the protocol approved by the Institutional Animal Care and Use Committee of
North Dakota State University. Orthotopic tumor xenografts were established by injection of 3×105 cancer cells in 50 μl serum-free media (containing 50% Matrigel) into the prostate of the
mice, after an incision was made 3 mm above the pubic symphysis and the bladder and seminal vesicles were carefully lifted to expose the dorsal prostate. Mocetinostat was dissolved in PBS
acidified with 0.1 N HCl and dosed p.o. as solutions daily. PBS was given p.o. to the control group. STATISTICAL ANALYSIS Differences between the mean values were analyzed for significance
using the unpaired two-tailed Student’s test for independent samples; _P_⩽0.05 was considered to be statistically significant. REFERENCES * Gray SG, Ekstrom TJ . The human histone
deacetylase family. _Exp Cell Res_ 2001; 262: 75–83. Article CAS Google Scholar * Strahl BD, Allis CD . The language of covalent histone modifications. _Nature_ 2000; 403: 41–45. Article
CAS Google Scholar * Minucci S, Pelicci PG . Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. _Nat Rev Cancer_ 2006; 6: 38–51. Article CAS
Google Scholar * Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. _Science_
2009; 325: 834–840. Article CAS Google Scholar * Haberland M, Montgomery RL, Olson EN . The many roles of histone deacetylases in development and physiology: implications for disease and
therapy. _Nat Rev Genet_ 2009; 10: 32–42. Article CAS Google Scholar * Weichert W, Roske A, Gekeler V, Beckers T, Ebert MP, Pross M et al. Association of patterns of class I histone
deacetylase expression with patient prognosis in gastric cancer: a retrospective analysis. _Lancet Oncol_ 2008; 9: 139–148. Article CAS Google Scholar * Weichert W, Roske A, Gekeler V,
Beckers T, Stephan C, Jung K et al. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical
prostatectomy. _Br J Cancer_ 2008; 98: 604–610. Article CAS Google Scholar * Weichert W, Roske A, Niesporek S, Noske A, Buckendahl AC, Dietel M et al. Class I histone deacetylase
expression has independent prognostic impact in human colorectal cancer: specific role of class I histone deacetylases _in vitro_ and _in vivo_. _Clin Cancer Res_ 2008; 14: 1669–1677.
Article CAS Google Scholar * Slingerland M, Guchelaar HJ, Gelderblom H . Histone deacetylase inhibitors: an overview of the clinical studies in solid tumors. _Anti-cancer Drugs_ 2014; 25:
140–149. Article CAS Google Scholar * Fournel M, Bonfils C, Hou Y, Yan PT, Trachy-Bourget MC, Kalita A et al. MGCD0103, a novel isotype-selective histone deacetylase inhibitor, has broad
spectrum antitumor activity _in vitro_ and _in vivo_. _Mol Cancer Ther_ 2008; 7: 759–768. Article CAS Google Scholar * Bonfils C, Kalita A, Dubay M, Siu LL, Carducci MA, Reid G et al.
Evaluation of the pharmacodynamic effects of MGCD0103 from preclinical models to human using a novel HDAC enzyme assay. _Clin Cancer Res_ 2008; 14: 3441–3449. Article CAS Google Scholar *
Sikandar S, Dizon D, Shen X, Li Z, Besterman J, Lipkin SM . The class I HDAC inhibitor MGCD0103 induces cell cycle arrest and apoptosis in colon cancer initiating cells by upregulating
Dickkopf-1 and non-canonical Wnt signaling. _Oncotarget_ 2010; 1: 596–605. PubMed PubMed Central Google Scholar * El-Khoury V, Moussay E, Janji B, Palissot V, Aouali N, Brons NH et al.
The histone deacetylase inhibitor MGCD0103 induces apoptosis in B-cell chronic lymphocytic leukemia cells through a mitochondria-mediated caspase activation cascade. _Mol Cancer Ther_ 2010;
9: 1349–1360. Article CAS Google Scholar * Bartel DP . MicroRNAs: target recognition and regulatory functions. _Cell_ 2009; 136: 215–233. Article CAS Google Scholar * Bartel DP .
MicroRNAs: genomics, biogenesis, mechanism, and function. _Cell_ 2004; 116: 281–297. Article CAS Google Scholar * Kota J, Chivukula RR, O'Donnell KA, Wentzel EA, Montgomery CL, Hwang
HW et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. _Cell_ 2009; 137: 1005–1017. Article CAS Google Scholar * Chen CZ, Li L, Lodish HF,
Bartel DP . MicroRNAs modulate hematopoietic lineage differentiation. _Science_ 2004; 303: 83–86. Article CAS Google Scholar * Zhang J, Jima DD, Jacobs C, Fischer R, Gottwein E, Huang G
et al. Patterns of microRNA expression characterize stages of human B-cell differentiation. _Blood_ 2009; 113: 4586–4594. Article CAS Google Scholar * Poy MN, Eliasson L, Krutzfeldt J,
Kuwajima S, Ma X, Macdonald PE et al. A pancreatic islet-specific microRNA regulates insulin secretion. _Nature_ 2004; 432: 226–230. Article CAS Google Scholar * Ma L, Teruya-Feldstein J,
Weinberg RA . Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. _Nature_ 2007; 449: 682–688. Article CAS Google Scholar * Hermeking H . The miR-34 family in
cancer and apoptosis. _Cell Death Differ_ 2009; 17: 193–199. Article Google Scholar * Bhatnagar N, Li X, Padi SK, Zhang Q, Tang MS, Guo B . Downregulation of miR-205 and miR-31 confers
resistance to chemotherapy-induced apoptosis in prostate cancer cells. _Cell Death Dis_ 2010; 1: e105. Article CAS Google Scholar * Lin PC, Chiu YL, Banerjee S, Park K, Mosquera JM,
Giannopoulou E et al. Epigenetic repression of miR-31 disrupts androgen receptor homeostasis and contributes to prostate cancer progression. _Cancer Res_ 2013; 73: 1232–1244. Article CAS
Google Scholar * McCabe MT, Brandes JC, Vertino PM . Cancer DNA methylation: molecular mechanisms and clinical implications. _Clin Cancer Res_ 2009; 15: 3927–3937. Article CAS Google
Scholar * Marks PA . Discovery and development of SAHA as an anticancer agent. _Oncogene_ 2007; 26: 1351–1356. Article CAS Google Scholar * Yang WW, Wang ZH, Zhu Y, Yang HT . E2F6
negatively regulates ultraviolet-induced apoptosis via modulation of BRCA1. _Cell Death Differ_ 2007; 14: 807–817. Article CAS Google Scholar * Yang WW, Shu B, Zhu Y, Yang HT . E2F6
inhibits cobalt chloride-mimetic hypoxia-induced apoptosis through E2F1. _Mol Biol Cell_ 2008; 19: 3691–3700. Article CAS Google Scholar * Danial NN, Korsmeyer SJ . Cell death: critical
control points. _Cell_ 2004; 116: 205–219. Article CAS Google Scholar * Jiang P, Du W, Wu M . p53 and Bad: remote strangers become close friends. _Cell research_ 2007; 17: 283–285.
Article CAS Google Scholar * Danial NN . BAD: undertaker by night, candyman by day. _Oncogene_ 2008; 27: S53–S70. Article CAS Google Scholar * Sharpe B, Beresford M, Bowen R, Mitchard
J, Chalmers AD . Searching for prostate cancer stem cells: markers and methods. _Stem Cell Rev_ 2013; 9: 721–730. Article CAS Google Scholar * Ajani JA, Song S, Hochster HS, Steinberg IB
. Cancer stem cells: the promise and the potential. _Semin Oncol_ 2015; 42: S3–S17. Article CAS Google Scholar * Luger SM, O'Connell CL, LKlimek V, Cooper MA, Besa EC, Rossetti JM et
al. A phase II study of mocetinostat, an oral isotype-selective histone deacetylase (HDAC) inhibitor, in combination with 5-azacitidine in patients with myelodysplastic syndrome (MDS). _J
Clin Oncol_ 2013; 31(suppl): abstr 7116. * Younes A, Oki Y, Bociek RG, Kuruvilla J, Fanale M, Neelapu S et al. Mocetinostat for relapsed classical Hodgkin's lymphoma: an open-label,
single-arm, phase 2 trial. _Lancet Oncol_ 2011; 12: 1222–1228. Article CAS Google Scholar * Blum KA, Advani A, Fernandez L, Van Der Jagt R, Brandwein J, Kambhampati S et al. Phase II
study of the histone deacetylase inhibitor MGCD0103 in patients with previously treated chronic lymphocytic leukaemia. _Br J Haematol_ 2009; 147: 507–514. Article CAS Google Scholar *
Willis SN, Adams JM . Life in the balance: how BH3-only proteins induce apoptosis. _Curr Opin Cell Biol_ 2005; 17: 617–625. Article CAS Google Scholar * Germann M, Wetterwald A,
Guzman-Ramirez N, van der Pluijm G, Culig Z, Cecchini MG et al. Stem-like cells with luminal progenitor phenotype survive castration in human prostate cancer. _Stem Cells_ 2012; 30:
1076–1086. Article CAS Google Scholar * O'Dwyer K, Maslak P . Azacitidine and the beginnings of therapeutic epigenetic modulation. _Expert Opin Pharmacother_ 2008; 9: 1981–1986.
Article CAS Google Scholar Download references ACKNOWLEDGEMENTS We thank Dr. Tao Wang for providing help with real-time PCR studies. This research was supported by NIH Grants CA186100,
GM103332, and GM114080. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Pharmaceutical Sciences, School of Pharmacy, North Dakota State University, Fargo, 58108, ND, USA Q Zhang,
M Sun, S Zhou & B Guo Authors * Q Zhang View author publications You can also search for this author inPubMed Google Scholar * M Sun View author publications You can also search for
this author inPubMed Google Scholar * S Zhou View author publications You can also search for this author inPubMed Google Scholar * B Guo View author publications You can also search for
this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to B Guo. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no conflict of interest. ADDITIONAL INFORMATION
Edited by I Harris RIGHTS AND PERMISSIONS This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are
included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to
obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/ Reprints and permissions ABOUT THIS
ARTICLE CITE THIS ARTICLE Zhang, Q., Sun, M., Zhou, S. _et al._ Class I HDAC inhibitor mocetinostat induces apoptosis by activation of miR-31 expression and suppression of E2F6. _Cell Death
Discovery_ 2, 16036 (2016). https://doi.org/10.1038/cddiscovery.2016.36 Download citation * Received: 07 April 2016 * Accepted: 28 April 2016 * Published: 06 June 2016 * DOI:
https://doi.org/10.1038/cddiscovery.2016.36 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative