Play all audios:
ABSTRACT Rett syndrome (RTT) is a disorder that affects patients’ ability to communicate, move and behave. RTT patients are characterized by impaired language, stereotypic behaviors,
frequent seizures, ataxia and sleep disturbances, with the onset of symptoms occurring after a period of seemingly normal development. RTT is caused by mutations in methyl-CpG binding
protein 2 (_MECP2_), an X-chromosome gene encoding for MeCP2, a protein that regulates gene expression. _MECP2_ generates two alternative splice variants encoding two protein isoforms that
differ only in the N-terminus. Although no functional differences have been identified for these splice variants, it has been suggested that the RTT phenotype may occur in the presence of a
functional MeCP2-e2 protein. This suggests that the two isoforms might be functionally distinct. Supporting this notion, the two variants show regional and age-related differences in
transcript abundance. Here, we show that transgenic expression of either the MeCP2-e1 or MeCP2-e2 splice variant results in prevention of development of RTT-like phenotypic manifestations in
a mouse model lacking _Mecp2_. Our results indicate that the two MeCP2 splice variants can substitute for each other and fulfill the basic functions of MeCP2 in the mouse brain. SIMILAR
CONTENT BEING VIEWED BY OTHERS INTEGRATED GENE EXPRESSION AND ALTERNATIVE SPLICING ANALYSIS IN HUMAN AND MOUSE MODELS OF RETT SYNDROME Article Open access 22 January 2025 THE MOLECULAR
GENETICS OF NELAVL IN BRAIN DEVELOPMENT AND DISEASE Article Open access 12 September 2023 COPY NUMBER VARIATION IN TRNA ISODECODER GENES IMPAIRS MAMMALIAN DEVELOPMENT AND BALANCED
TRANSLATION Article Open access 18 April 2023 INTRODUCTION Rett syndrome (RTT, MIM 312750) is a progressive neurodevelopmental disorder that affects predominantly females. It is one of the
leading causes of intellectual disability and autistic features in females.1 Clinical features of RTT include psychomotor regression, intellectual disability, communication dysfunction,
seizures, postural hypotonia, stereotypic hand movements, tremors, autonomic dysfunction and growth failure.2 The vast majority of RTT cases are caused by mutations in the X-linked gene
_MECP2_,3 encoding for methyl-CpG binding protein 2 (MeCP2). MeCP2 is a nuclear protein that binds to methylated CpGs and regulates gene expression.4, 5 In addition, mutations in _CDKL5_ and
_FOXG1_ have been also associated with RTT.1, 6 In mammals, _MECP2_ generates two alternative splice variants, encoding protein isoforms that differ only in the N-terminus.7, 8 The MeCP2-e1
mRNA splice variant, in which exon 2 is spliced out, produces a 496-amino acid polypeptide with an acidic N-terminus translated from an ATG initiation codon in exon 1. The second variant,
MeCP2-e2, encodes a slightly shorter protein (486-aminoacids) translated from an ATG in exon 2. Splicing variants often encode functionally diverse protein isoforms.9 Evidence that this
could also be the case for MeCP2 splice variants comes from the findings that MeCP2 variants show regional and age-related differences in transcript abundance in the mouse brain – MECP2-e1
is the predominant form in most adult brain structures10 – and that _Mecp2-e1_ and _Mecp2-e2_ transcripts appear to show different preferences for alternative polyadenylation sites within
the long 3′-UTR.10 In addition, research into the relationship between genotype and phenotype in RTT provides further support to the notion that MeCP2-e1 and MeCP2-e2 could be functionally
distinct; several mutations in _MECP2_ exon 1 have been identified in classic RTT patients,8, 11, 12, 13, 14, 15, 16, 17 including point mutations that allegedly do not to affect
transcription or translation of MeCP2-e2,16, 17 suggesting that endogenously expressed MeCP2-e2 is unable to compensate for the lack of MeCP2-e1. On the other hand, it was reported that the
sole expression of MeCP2-e2 was able to rescue the phenotype of _Mecp2__−/Y_ mice, leading to the conclusion that expression of _MeCP2-_e2 – in the absence of MeCP2-e1 – is sufficient for
attaining normal neuronal function.18, 19, 20 These seemingly contradictory results could stem from mouse–human differences in MeCP2 requirements, but could also be reconciled by the
hypothesis that MeCP2-e1 and MeCP2-e2 could perform similar functions in brain cells, if they were expressed at comparative levels. Thus, MeCP2-e2 cannot compensate for the lack of MeCP2-e1
in the reported patients carrying mutations that specifically affect MeCP2-e1, because there is not enough MeCP2-e2 in these cells. However, in mice engineered to express MeCP2-e2 from a
heterologous promoter, the expression of MeCP2-e2 could suffice to compensate for the lack of MeCP2-e1. Supporting this hypothesis, both MeCP2 isoforms show similar intranuclear localization
and chromatin binding kinetics in transfected cells.21 Mutations that affect specifically the expression of MeCP2-e2 have not been described, precluding a definitive answer to the
functional significance of the two differing MeCP2 splice variants. We reasoned that if the two MeCP2 splice variants subserved different functions, then the extent of phenotypic rescue
displayed by mice lacking endogenous _Mecp2_, but expressing either MeCP2-e1 or MeCP2-e2 cDNAs, should be distinct. Thus, we generated transgenic mice expressing either MeCP2-e1 or MeCP2-e2
cDNAs in brain cells, crossed them with mice lacking _Mecp2_, and compared the ability of each of the variants to compensate for the lack of the endogenous gene. We observed that expression
of either of the isoforms mitigated the phenotypic consequences from the lack of _Mecp2_ and allowed _Mecp2_−/Y mice to survive to adulthood, indicating a significant degree of functional
overlap. MATERIALS AND METHODS ANIMALS MECP2 transgenes were generated by linearizing pcDNA3.1A-_MECP2_B-myc and pEGFP-_MECP2_ (kindly provided by B Minassian and S Kudo, respectively) to
release vector sequences and microinjected into the pronuclei of B6CBF2 zygotes. MeCP2-e1-myc and EGFP-MeCP2-e2 transgenic mice were crossed with 129/SvJ _Mecp2__−/_+ mice.22 Animals were
kept in an animal room under SPF conditions, in a 12/12 h light/dark cycle with free access to food and water. All the experiments were approved by the Centro de Estudios Científicos Animal
Care and Use Committee. FLUORESCENT IMMUNOHISTOCHEMISTRY Floating cryostat sections (40 _μ_m) were incubated overnight with primary antibodies anti-MeCP2 1:100 (Upstate/Millipore, Billerica,
MA, USA), anti-Myc 1:1000 (Sigma, St Louis, MO, USA) and anti-GFP 1:500 (Molecular Probes, Carlsbad, CA, USA). Secondary antibodies conjugated to Alexa Fluor 488 (Molecular Probes) or Cy3
(Jackson Immunoresearch Laboratories, West Grove, PA, USA) were used at a 1:500 dilution. Images were captured using a Zeiss Axiovert 100 M confocal microscope equipped with a QImaging 3.3
RTV cooled CCD camera (Zeiss, Göttingen, Germany) and Adobe Photoshop 7.0 (Adobe Systems, San Jose, CA, USA). WESTERN BLOT ANALYSIS Brain regions were dissected from 1 mm coronal sections
and homogenized in lysis buffer containing 125 mM Tris (pH 6.8) and 2% SDS supplemented with 1 × protease inhibitor cocktail (Sigma, P8340). Thirty microgram of protein were electrophoresed
and transferred on PVDF membranes (Bio-Rad, Hercules, CA, USA). Membranes were incubated with anti-MeCP2 1:2500 (Upstate) or anti-b-Tubulin 1:1000 (Santa Cruz Biotechnology, Santa Cruz, CA,
USA), and anti-rabbit HRP-conjugated IgG 1:40 000 (Pierce, Biotechnology, Rockford, IL, USA). Densitometry of immunoreactive bands was quantitated with Quantity One software (Bio-Rad).
PHENOTYPIC CHARACTERIZATION Starting from 3 weeks old, mice were tested on a weekly basis for survival, body weight determination, and presence and severity of stereotypic hand movements and
clasping. Severity of clasping was determined according to an arbitrary scaling (from 0 to 3), based on how fast mice clasp their feet together when picked by the tail and also on their
capability of releasing the posture: 0 (no clasping), 1 (reversible clasping in which only hind-limbs press into the stomach), 2 (delayed, but irreversible) and 3 (immediate and
irreversible). At 7 weeks of age, each mouse was subjected to a battery of behavioral tests performed always in the same order: elevated plus maze, open field, elevated beam test and hanging
wire test. For all experiments, the data are presented as mean±SEM. The data were analyzed using the Student’s _t_-test. Statistical significance was set at a minimum of _P_<0.05.
Survival analysis was conducted by means of a Kaplan–Meier survival analysis. ELEVATED PLUS MAZE Mice were placed in the center of a cross-shaped maze elevated 45 cm from the floor with two
open and two closed arms. The behavior of the mice was observed and the time spent in either the closed, open or at the center of the maze was recorded. ELEVATED BEAM TEST The elevated beam
test consists of two elevated platforms connected through a 70 cm long dowel of 0.7 cm radius. Before testing, mice were placed on the dowel 10 cm away from one of the platforms. Only those
mice that reached the platform in the first 60 s were further assessed. Next, mice were placed in the middle of the dowel and the time of first arrival, the total number of arrivals and the
number of falls were recorded for 2 min. HANGING WIRE TEST The hanging wire test was performed by hanging the mice by its forepaws from a suspended wire and recording the number of falls in
2 min. RESULTS GENERATION OF TRANSGENIC MICE FOR EITHER MECP2-E1 OR MECP2-E2 We generated transgenic mice carrying either the cDNA for MeCP2-e1 or MeCP2-e2 under the control of the human
cytomegalovirus immediate-early promoter/enhancer region (CMV promoter), which drives consistently strong expression of genes in both cultured cells and in differentiated neurons. In order
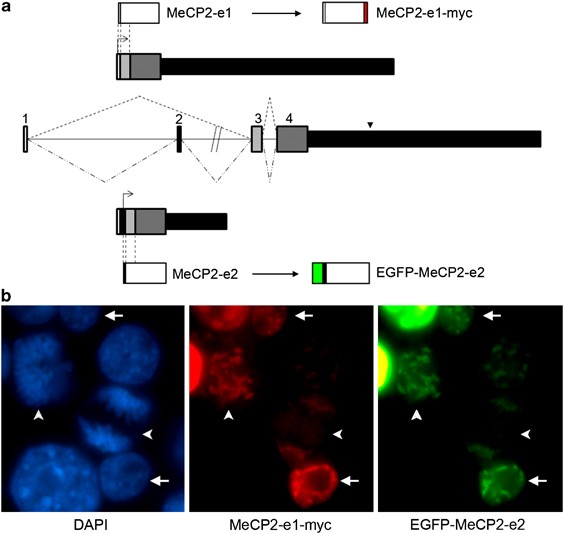
to easily differentiate between the two isoforms, MeCP2-e1 was fused C terminally to a myc tag, whereas EGFP was added to the N-terminus of MeCP2-e2 (Figure 1a). The functionality of these
constructs was confirmed by testing the ability to repress an _in vitro_ methylated reported construct, to modulate alternative splicing of reporter constructs and to correctly localize to
heterochromatic foci (Supplementary Figure 1). As previously reported, both constructs completely colocalize in N2A-transfected cells in interphase, as well as in cells undergoing active
replication (Figure 1b). These data suggest that the addition of the tags does not alter functionality of the isoforms. Several founder mice were obtained for both constructs. Two
independent transgenic lines expressing different amounts of MeCP2-e1-myc in the brain, e1-TgH and e1-TgL, were selected for this study. As it was already shown that _MeCP2-e2_ expression
rescued lethality, normalized body weight regulation and restored motor activity of _Mecp2_−/Y mice to wild-type levels,18, 19, 20 we decided to study only one MeCP2-e2 transgenic line that
exhibited widespread neuronal expression of the transgene to confirm these previous observations (e2-Tg). EXPRESSION OF ISOFORM-SPECIFIC CDNAS IN TRANSGENIC MICE The expression pattern of
the transgenes on the selected transgenic lines is shown in Figure 2, determined by indirect immunofluorescence for the Myc tag and EGFP in the MeCP2-e1 and MeCP2-e2 transgenics,
respectively. Transgene expression was widely distributed in the brain for all three lines. There are qualitative and quantitative differences in the expression of MeCP2-e1 in e1-TgH and
e1-TgL lines. In line e1-TgH, the intensity of the fluorescent signal was similar across all brain regions, with the exception of the cerebellum, in which the intensity was higher, and the
lateral nucleus of the thalamus and the CA1 area of the hippocampus, which showed decreased labeling (Figure 2a). Myc labeling in e1-TgL was also widespread, but exhibited lower levels of
expression than e1-TgH in general and more cell to cell heterogeneity, with highest expression in cortical layer 2 and lowest expression in the cerebellum (Figure 2a). Line e2-Tg showed
brain-wide EGFP–MeCP2-e2 expression at comparable levels throughout the brain, excluding the dentate gyrus of the hippocampus where the expression was undetectable (Figure 2a). Co-staining
of transgenic MeCP2 and the intermediate filament marker glial fibrillary acidic protein (GFAP) indicated that none of the transgenic lines express MeCP2 in astrocytes at detectable levels
(Supplementary Figure 2a). However, as it has been shown that endogenous MeCP2 expression in glial cells is not easily revealed due to its low levels,23 we analyzed the expression of the
transgenes in primary cultures of mixed cortical cells and found expression of MeCP2 transgenes in some GFAP-positive glial cells in the three transgenic lines studied (Supplementary Figure
2b). To determine the percentage of neurons expressing transgenic MeCP2-e1 or MeCP2-e2 in the different brain regions, we co-stained for MeCP2, which labels neurons expressing both
endogenous MeCP2, as well as the transgenic MeCP2-e1 or MeCP2-e2, depending on the transgenic mice analyzed (Figures 2b–d). Co-immunofluorescence analysis using anti-MeCP2 and anti-myc
indicated that transgenic line e1-TgH expressed MeCP2-e1 in 16 to 98% of neurons, depending on the brain region (Table 1). Line e1-TgL expressed transgenic MeCP2-e1 in 7–96% of neurons, and
MeCP2-e2 was expressed in 9–88% in the e2-Tg transgenic line (Table 1 and Supplementary Figure 3). It has been demonstrated that neurons are sensitive to inappropriate MeCP2 expression
levels.24, 25 Thus, we determined the level of expression of transgenic MeCP2 by comparing the immunoreactivity for MeCP2 in protein extracts obtained from a variety of brain regions of
transgenic and wild-type mice. Analysis of the western blot results indicated that expression of MeCP2-e1 in e1-TgH and e1-TgL varied between brain regions, averaging at 40 and 15% of MeCP2
wild-type expression, respectively (data not shown). A higher level of expression was observed in cerebellum of line e1-TgH, in which transgenic MeCP2-e1 was approximately 75% of the
wild-type MeCP2 expression (data not shown). Line e2-Tg expressed 30–40% of wild-type MeCP2 in regions such as the brain stem, cerebral cortex and hippocampus, but expressed close to 100% in
the caudate/putamen areas, hypothalamus and thalamus (Supplementary Figure 4). These results were confirmed by quantitating the amount of MeCP2 expressed by transgenic mice crossed into a
_Mecp2_ null background (_Mecp2_−/Y;e1-TgH, _Mecp2_−/Y;e1-TgL and _Mecp2_−/Y;e2-Tg (Figures 3a and b). Thus, considering the percentage of cells expressing the transgene in the different
brain regions (Table 1) and the total relative amount expressed per region (Figure 3b), the estimated expression of transgenic proteins per cell amounts to approximately 30–80% in e1-TgH,
35–95% in e1-TgL (higher than 40% only in cerebellum, with approximate expression of MeCP2-e1 of 95%) and 40–140% in e2-Tg, compared with the expression of endogenous MeCP2. Interestingly,
we did not observe phenotypic abnormalities in e1-TgL mice. However, e1-TgH mice, modestly overexpressing MeCP2-e1 developed a recognizable late onset phenotype that includes hypoactivity
and premature death at 12–16 months of age (Abrams _et al_, unpublished). TRANSGENIC MECP2-E1 IS ENOUGH TO PREVENT RETT-LIKE PHENOTYPES IN MECP2-DEFICIENT MICE The ability of MeCP2-e1 to
compensate for loss of endogenous MeCP2 was tested in male mice derived from matings of _Mecp2_−/+ females crossed with e1-TgH, e1-TgL or e2-Tg (as positive controls). Two independent blind
observers were able to easily and accurately sort out _Mecp2_−/Y mice from mice of all other genotypes after 7 weeks of age, by simply looking at the mice in their home cage. The main
differentiating parameters were activity and body posture. _Mecp2_−/Y mice appeared less active, hesitant and usually adopted a crouched posture, as compared with the rest of the mice in the
cage, suggesting that both transgenes had a modifier effect on the neurobehavioral phenotype of the _Mecp2_−/Y mice. We then performed a more systematic phenotypic evaluation by determining
life span, measuring body weight, testing motor skills, evaluating anxiety-related behaviors and documenting seizures. LIFE SPAN Fifty percent of _Mecp2_−/Y mice did not survive past 10
weeks and all mice with this genotype died before 30 weeks (Figure 3c). Notably, more than 80% of _Mecp2_−/Y;e1-TgH were still alive at 43 weeks, with some mice surviving past 16 months.
Thus, MeCP2-e1 was able to fully prevent the early death seen in Mecp2 null mice. BODY WEIGHT _Mecp2__−/y_ mice show a characteristic weight gain pattern: weight escalates around week 8 and
then starts declining significantly at 14 weeks (Figure 3d and data not shown). The weight gain curve of the _Mecp2_−/Y;e1-TgH mice was indistinguishable from wild-type littermates,
demonstrating that the presence of this isoform by itself may prevent the development of this weight gain phenotype. HINDPAW CLASPING To determine whether the neurobehavioral decline shown
by _Mecp2_−/Y mice was modified by expression of the MeCP2-e1 variant, we quantitatively assessed paw clasping. At 3 weeks of age, _Mecp2__−/y_ mice displayed hindpaw clasping of a severity
level of 1, and by 8 weeks, it progressed to 2. By 12 weeks of age, all _Mecp2__−/y_ mice studied presented grade 2–3 clasping. Expression of MeCP2-e1 significantly improved (_P_<0.01)
the average clasping score of the mutant mice (Figure 4a). No clasping was observed in _Mecp2__−/y_;e1-TgH mice until 9 weeks of age, when some began to show a subtle clasping that by 18
weeks was classified as of level 1. ANXIETY As it has been previously reported, _Mecp2_−/Y mice exhibited a behavior that correlates with decreased anxiety when tested in the plus maze, they
did not show a preference for the closed (commonly interpreted as safe) arm of the maze, as wild-type mice did (Figure 4b). _Mecp2_−/Y mice spent a similar amount of time in both, open and
closed arms of the maze, a behavior that was significantly different than that exhibited by wild-type and _Mecp2__−/y_;e1-TgH mice. MOTOR CONTROL We subjected the mice to the elevated beam
test, in which a mouse is induced to walk on a thin wooden dowel to reach the safety of a platform. _Mecp2_−/Y mice performed significantly worse than their wild-type littermates (Figure 4).
The performance of _Mecp2__−/y_;e1-TgH mice was similar to the wild-type littermates (Figures 4c–f). To further characterize the motor phenotype of the transgenic mice, we measured their
ability to remain suspended from a horizontal wire. Wild-type and _Mecp2__−/y_;e1-TgH mice outperformed the _Mecp2__−/y_ mice in this test (Figure 4g). Collectively, these results indicate
that MeCP2-e1 was able to normalize the motor phenotype caused by lack of endogenous MeCP2. THE PHENOTYPIC RESCUE EXERTED BY TRANSGENIC MECP2-E1 IS DOSAGE-DEPENDENT Transgenic expression of
MeCP2-e2 is sufficient to prevent the RTT-like phenotype in MeCP2 null mice;18, 19, 20 however, the fact that MeCP2-e2 seems to be unable to compensate for the absence of the MeCP2-e1
variant in human RTT patients carrying mutations that exclusively affect MeCP2-e116, 17 raises the possibility of a MeCP2 dose-dependency in the extent of rescue. We explored this
possibility by comparing the extent of rescue elicited by the e1-TgH and e1-TgL MeCP2-e1 transgenic lines, which showed different levels of transgene expression. Although initial home cage
observation suggested phenotypic rescue in _Mecp2__−/y_;e1-TgL mice (see above), a more detailed analysis showed that this rescue was only partial. The presence of the e1-TgL transgene
extended the median life span (0.5 probability survival rate) of the _Mecp2_−/Y mice from 10 weeks in _Mecp2_−/Y to 25 weeks in _Mecp2_−/Y;e1-TgL mice, but this effect is noticeably less
significant than the one observed in the _Mecp2_−/Y;e1-TgH mice. In addition, _Mecp2_−/Y;e1-TgL mice showed partial amelioration in clasping severity (Figure 4a) and only a trend for rescue
in anxiety and motor performance (Figure 4), suggesting a gene dose-dependent effect on severity of these phenotypes. TRANSGENIC MECP2-E2 ALSO PREVENTS RETT-LIKE PHENOTYPES To compare the
rescuing capacity of MeCP2-e1 and MeCP2-e2, we characterized the phenotype of the _Mecp2__−/y_;e2-Tg mice. _Mecp2_−/Y;e2-Tg mice have a longer life expectancy than _Mecp2_−/Y, with a median
life span of 42 weeks and some animals surviving for more than 14 months (Figure 3a). Body weight and behavior on the plus maze were also normalized (Figure 3b). Thus, both MeCP2 isoforms
were able to prevent the early death and the phenotypes seen in Mecp2 null mice to comparable extents. However, hindlimb clasping and motor abnormalities were only partially rescued in
_Mecp2__−/y_;e2-Tg, raising the possibility of minor differences in brain function of both isoforms (Figure 4). IN VIVO COLOCALIZATION OF TRANSGENIC MECP2 ISOFORMS Transfected MeCP2-e1 and
MeCP2-e2 show an apparent similar intracellular localization when overexpressed in cultured cells. However, it is possible that _in vivo_, both isoforms have distinct intracellular
localization and the observed complete colocalization could be an artifact of _in vitro_ transient overexpression. Our mice expressing transgenic MeCP2 isoforms at levels comparable to the
endogenous proteins allows for intracellular colocalization studies of the transgenic MeCP2 isoforms in intact brain cells. We crossbred our transgenic mice into a _Mecp2_ null background to
generate mice expressing both e1-TgH and e2-Tg in the absence of endogenous MeCP2. Confocal analysis of tissue sections obtained from _Mecp2__−/y_;e1-TgH;e2-Tg mice indicate that transgenic
MeCP2-e1 and MeCP2-e2 appear to colocalize completely in cells in the brain, skeletal muscle, heart, eye and kidney (Supplementary Figure 5). DISCUSSION This study was designed to determine
whether the two MeCP2 isoforms derived from alternative splicing have diverse or completely redundant functions. MeCP2-e1 and MeCP2-e2 differ only in the N-terminus; MeCP2-e1-specific
N-terminus contains the sequence MAAAAAAAPSGGGGGGEEERL, which in MeCP2-e2 is changed to MVAGMLGLR. The presence of polyA and polyG repeats in the MeCP2-e1 N-terminus is suggestive of a
differential function of the isoforms, as there is accumulating evidence that tandem repeats may serve functional roles within coding sequences, in addition to be regulatory elements or
mutational hotspots.26 However, most _in vitro_ studies failed to detect any functional difference between the two isoforms.21 In spite of the limited information about MeCP2-e2 expression
in human brain, the existence of Rett-causing mutations in _MECP2_ that apparently do not interfere with MeCP2-e2 expression, but affect MeCP2-e1, suggests that the sole presence of MeCP2-e2
is not enough to provide the full functional role. Although, as with most cases of neurological disorders, integrating and interpreting human and mouse data is not completely
straightforward, the available mouse data suggest that expression of MeCP2-e2 is sufficient to prevent the development of disease signs in mice lacking endogenous MeCP2 expression.18, 19, 20
Our data show for the first time that the sole expression of MeCP2-e1 was able to compensate for the lack of MeCP2 in mice. The rescuing effect of transgene expression seemed to be dose
dependent, as transgenics expressing lower levels of MeCP2-e1 were only partially rescued, suggesting that the hypothetical inability of MeCP2-e2 to compensate for the absence of MeCP2-e1 in
patients carrying mutations affecting only MeCP2-e1 is most probably due to insufficient endogenous expression levels. We have previously reported that transgenic mice expressing MeCP2-e2
under the NSE or CamKIIa promoters, when crossed to _Mecp2__−/y_ elicited no significant behavioral or lifespan improvements.27 Contrasting this previous study with the current data, in
which we observe significant phenotypic prevention with widespread brain expression of MeCP2-e2, suggests that not only level, but also location of expression is important for MeCP2's
overall activity in brain function. In addition, the expression of transgenic MeCP2 in glial cells in the transgenic mice described in this report could contribute to the behavioral
amelioration and might explain the different results observed.23, 28 Do our results mean that MeCP2-e1 and MeCP2-e2 are practically interchangeable? Life span, body weight control and
anxiety were equally rescued by both isoforms, suggesting equal function at similar levels of expression. However, the rescue exhibited by both isoforms was not 100% concordant; the degree
of rescue of the clasping and motor phenotypes was significantly higher for MeCP2-e1. This might suggest differential relevance of the two isoforms for the development of motor and clasping
phenotypes, but the possibility of differences due to variable expression of the transgenes in different brain tissues cannot be ruled out. Further, the study of phenotypes not analyzed in
this study could potentially reveal unidentified functional differences between the two isoforms. Notably, the approximate expression of MeCP2-e1 in the transgenic line that showed a
significant normalization of the Rett-like phenotypes presented by _Mecp2__−/y_ mice was only 40–60% of endogenous, suggesting that equaling endogenous level of expression is not strictly
necessary for a significant phenotypic rescue. Even more remarkable is the observation that _Mecp2__−/y_;e1-TgL mice showed rescue effects that, although partial, were in most cases
equivalent or more significant than what was seen in the _Mecp2__−/y_;e2-Tg mice. This implies that only minor amounts of MeCP2-e1 are sufficient to elicit dramatic behavioral recovery, and
that the influence of the two forms on phenotypic function might not be completely equivalent. We and others have shown that mice carrying a _Mecp2_ floxed allele and expressing
approximately 40% less MeCP2 than wild-type mice exhibited discernible phenotypes, suggesting that even mild reductions in MeCP2 protein might cause neuronal dysfunction.24, 29 This apparent
inconsistency could be due to small differences in patterns of transgene expression, or to the different time point in which the mice were tested (7 weeks of age (this study) _versus_ 12
weeks). Also, subtle phenotypic manifestations not analyzed in this study, such as social behavior, could still be present in _Mecp2__−/y_ mice expressing transgenic MeCP2-e1. The finding
that expression of MeCP2-e1 by itself was sufficient to significantly extend the life span of _Mecp2__−/y_ mice, to prevent the development of common manifestations of neurological
dysfunction in mice, such as clasping, and to normalize anxiety-related and motor phenotypes, is noteworthy and relevant to the design of efficient strategies aiming to restore MeCP2
activity, including gene therapy30 and protein administration approaches. REFERENCES * Weaving LS, Ellaway CJ, Gécz J, Christodoulou J : Rett syndrome: clinical review and genetic update. _J
Med Genet_ 2005; 42: 1–7. Article CAS Google Scholar * Chahrour M, Zoghbi HY : The story of Rett syndrome: from clinic to neurobiology. _Neuron_ 2007; 56: 422–437. Article CAS Google
Scholar * Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY : Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. _Nat Genet_ 1999;
23: 185–188. Article CAS Google Scholar * Jones PL, Veenstra GJ, Wade PA _et al_: Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. _Nat Genet_ 1998; 19:
187–191. Article CAS Google Scholar * Chahrour M, Jung SY, Shaw C _et al_: MeCP2, a key contributor to neurological disease, activates and represses transcription. _Science_ 2008; 320:
1224–1229. Article CAS Google Scholar * Philippe C, Amsallem D, Francannet C _et al_: Phenotypic variability in Rett syndrome associated with FOXG1 mutations in females. _J Med Genet_
2010; 47: 59–65. Article CAS Google Scholar * Kriaucionis S, Bird A : The major form of MeCP2 has a novel N-terminus generated by alternative splicing. _Nucleic Acids Res_ 2004; 32:
1818–1823. Article CAS Google Scholar * Mnatzakanian GN, Lohi H, Munteanu I _et al_: A previously unidentified MECP2 open reading frame defines a new protein isoform relevant to Rett
syndrome. _Nat Genet_ 2004; 36: 339–341. Article CAS Google Scholar * Tress ML, Martelli PL, Frankish A _et al_: The implications of alternative splicing in the ENCODE protein complement.
_Proc Natl Acad Sci USA_ 2007; 104: 5495–5500. Article CAS Google Scholar * Dragich JM, Kim YH, Arnold AP, Schanen NC : Differential distribution of the MeCP2 splice variants in the
postnatal mouse brain. _J Comp Neurol_ 2007; 501: 526–542. Article Google Scholar * Amir RE, Fang P, Yu Z _et al_: Mutations in exon 1 of MECP2 are a rare cause of Rett syndrome. _J Med
Genet_ 2005; 42: e15. Article CAS Google Scholar * Bartholdi D, Klein A, Weissert M _et al_: Clinical profiles of four patients with Rett syndrome carrying a novel exon 1 mutation or
genomic rearrangement in the MECP2 gene. _Clin Genet_ 2006; 69: 319–326. Article CAS Google Scholar * Chunshu Y, Endoh K, Soutome M, Kawamura R, Kubota T : A patient with classic Rett
syndrome with a novel mutation in MECP2 exon 1. _Clin Genet_ 2006; 70: 530–531. Article CAS Google Scholar * Quenard A, Yilmaz S, Fontaine H _et al_: Deleterious mutations in exon 1 of
MECP2 in Rett syndrome. _Eur J Med Genet_ 2006; 49: 313–322. Article Google Scholar * Ravn K, Nielsen JB, Schwartz M : Mutations found within exon 1 of MECP2 in Danish patients with Rett
syndrome. _Clin Genet_ 2005; 67: 532–533. Article CAS Google Scholar * Fichou Y, Nectoux J, Bahi-Buisson N _et al_: The first missense mutation causing Rett syndrome specifically
affecting the MeCP2_e1 isoform. _Neurogenetics_ 2009; 10: 127–133. Article CAS Google Scholar * Saunders CJ, Minassian BE, Chow EW, Zhao W, Vincent JB : Novel exon 1 mutations in MECP2
implicate isoform MeCP2_e1 in classical Rett syndrome. _Am J Med Genet A_ 2009; 149A: 1019–1023. Article CAS Google Scholar * Luikenhuis S, Giacometti E, Beard CF, Jaenisch R : Expression
of MeCP2 in postmitotic neurons rescues Rett syndrome in mice. _Proc Natl Acad Sci USA_ 2004; 101: 6033–6038. Article CAS Google Scholar * Giacometti E, Luikenhuis S, Beard C, Jaenisch R
: Partial rescue of MeCP2 deficiency by postnatal activation of MeCP2. _Proc Natl Acad Sci USA_ 2007; 104: 1931–1936. Article CAS Google Scholar * Jugloff DG, Vandamme K, Logan R,
Visanji NP, Brotchie JM, Eubanks JH : Targeted delivery of an Mecp2 transgene to forebrain neurons improves the behavior of female Mecp2-deficient mice. _Hum Mol Genet_ 2008; 17: 1386–1396.
Article CAS Google Scholar * Kumar A, Kamboj S, Malone BM _et al_: Analysis of protein domains and Rett syndrome mutations indicate that multiple regions influence chromatin-binding
dynamics of the chromatin-associated protein MECP2 _in vivo_. _J Cell Sci_ 2008; 121: 1128–1137. Article CAS Google Scholar * Guy J, Hendrich B, Holmes M, Martin JE, Bird A : A mouse
Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. _Nat Genetic_ 2001; 27: 322–326. Article CAS Google Scholar * Ballas N, Lioy DT, Grunseich C, Mandel G :
Non-cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. _Nat Neurosci_ 2009; 12: 311–317. Article CAS Google Scholar * Kerr B, Alvarez-Saavedra M, Saez MA,
Saona A, Young JI : Defective body-weight regulation, motor control and abnormal social interactions in Mecp2 hypomorphic mice. _Hum Mol Genet_ 2008; 17: 1707–1717. Article CAS Google
Scholar * Collins AL, Levenson JM, Vilaythong AP _et al_: Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. _Hum Mol Genet_ 2004; 13: 2679–2689. Article CAS
Google Scholar * Kashi Y, King D, Soller M : Simple sequence repeats as a source of quantitative genetic variation. _Trends Genet_ 1997; 13: 74–78. Article CAS Google Scholar *
Alvarez-Saavedra M, Saez MA, Kang D, Zoghbi HY, Young JI : Cell-specific expression of wild-type MeCP2 in mouse models of Rett syndrome yields insight about pathogenesis. _Hum Mol Genet_
2007; 16: 2315–2325. Article CAS Google Scholar * Maezawa I, Swanberg S, Harvey D, LaSalle JM, Jin LW : Rett syndrome astrocytes are abnormal and spread MeCP2 deficiency through gap
junctions. _J Neurosci_ 2009; 29: 5051–5061. Article CAS Google Scholar * Samaco R, Fryer J, Fyffe S, Chao HT, Zoghbi H, Neul JL : A partial loss of function allele of _Methyl-CpG-Binding
Protein 2_ predicts a human neurodevelopmental syndrome. _Hum Mol Genet_ 2008; 17 (12): 1718–1727. Article CAS Google Scholar * Rastegar M, Hotta A, Pasceri P _et al_: MECP2
isoform-specific vectors with regulated expression for Rett syndrome gene therapy. _PLoS One_ 2009; 4: e6810. Article Google Scholar Download references ACKNOWLEDGEMENTS We greatly
appreciate the gift of MeCP2-e1-myc and EGFP-MeCP2-e2 cDNAs from Drs Berge Minassian (The Hospital for Sick Children, Canada) and Shinichi Kudo (Hokkaido Institute for Public Health, Japan),
respectively. We thank the CECS mouse facility for animal transgenesis and care. This work was supported by FONDECYT (Grants 1061067, 1051079 and 11070237). CECS is funded by the Chilean
Government through the Centers of Excellence Base Financing Program of Conicyt. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Biology, Centro de Estudios Científicos, Valdivia,
Chile Bredford Kerr, Jessica Soto C, Mauricio Saez, Katherina Walz & Juan I Young * Department of Biochemistry, Universidad Austral de Chile, Valdivia, Chile Jessica Soto C &
Mauricio Saez * Department of Human Genetics, John P Hussman Institute for Human Genomics, Miller School of Medicine, University of Miami, Miami, Fl, USA Alexander Abrams, Katherina Walz
& Juan I Young Authors * Bredford Kerr View author publications You can also search for this author inPubMed Google Scholar * Jessica Soto C View author publications You can also search
for this author inPubMed Google Scholar * Mauricio Saez View author publications You can also search for this author inPubMed Google Scholar * Alexander Abrams View author publications You
can also search for this author inPubMed Google Scholar * Katherina Walz View author publications You can also search for this author inPubMed Google Scholar * Juan I Young View author
publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to Juan I Young. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no
conflict of interest. ADDITIONAL INFORMATION Supplementary Information accompanies the paper on European Journal of Human Genetics website SUPPLEMENTARY INFORMATION SUPPLEMENTARY FIGURES
(DOC 28109 KB) RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Kerr, B., Soto C, J., Saez, M. _et al._ Transgenic complementation of MeCP2 deficiency:
phenotypic rescue of _Mecp2_-null mice by isoform-specific transgenes. _Eur J Hum Genet_ 20, 69–76 (2012). https://doi.org/10.1038/ejhg.2011.145 Download citation * Received: 29 September
2010 * Revised: 12 May 2011 * Accepted: 03 June 2011 * Published: 10 August 2011 * Issue Date: January 2012 * DOI: https://doi.org/10.1038/ejhg.2011.145 SHARE THIS ARTICLE Anyone you share
the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer
Nature SharedIt content-sharing initiative KEYWORDS * Rett syndrome * MeCP2 * alternative splicing * mouse model