Play all audios:
ABSTRACT Members of the genus _Candidatus_ Accumulibacter are important in many wastewater treatment systems performing enhanced biological phosphorus removal (EBPR). The Accumulibacter
lineage can be subdivided phylogenetically into multiple clades, and previous work showed that these clades are ecologically distinct. The complete genome of _Candidatus_ Accumulibacter
phosphatis strain UW-1, a member of Clade IIA, was previously sequenced. Here, we report a draft genome sequence of _Candidatus_ Accumulibacter spp. strain UW-2, a member of Clade IA,
assembled following shotgun metagenomic sequencing of laboratory-scale bioreactor sludge. We estimate the genome to be 80–90% complete. Although the two clades share 16S rRNA sequence
identity of >98.0%, we observed a remarkable lack of synteny between the two genomes. We identified 2317 genes shared between the two genomes, with an average nucleotide identity (ANI) of
78.3%, and accounting for 49% of genes in the UW-1 genome. Unlike UW-1, the UW-2 genome seemed to lack genes for nitrogen fixation and carbon fixation. Despite these differences, metabolic
genes essential for denitrification and EBPR, including carbon storage polymer and polyphosphate metabolism, were conserved in both genomes. The ANI from genes associated with EBPR was
statistically higher than that from genes not associated with EBPR, indicating a high selective pressure in EBPR systems. Further, we identified genomic islands of foreign origins including
a near-complete lysogenic phage in the Clade IA genome. Interestingly, Clade IA appeared to be more phage susceptible based on it containing only a single Clustered Regularly Interspaced
Short Palindromic Repeats locus as compared with the two found in Clade IIA. Overall, the comparative analysis provided a genetic basis to understand physiological differences and ecological
niches of Accumulibacter populations, and highlights the importance of diversity in maintaining system functional resilience. SIMILAR CONTENT BEING VIEWED BY OTHERS “_CANDIDATUS_
DECHLOROMONAS PHOSPHORITROPHA” AND “_CA_. D. PHOSPHORIVORANS”, NOVEL POLYPHOSPHATE ACCUMULATING ORGANISMS ABUNDANT IN WASTEWATER TREATMENT SYSTEMS Article Open access 21 June 2021 THE NOVEL
GENUS, ‘_CANDIDATUS_ PHOSPHORIBACTER’, PREVIOUSLY IDENTIFIED AS _TETRASPHAERA_, IS THE DOMINANT POLYPHOSPHATE ACCUMULATING LINEAGE IN EBPR WASTEWATER TREATMENT PLANTS WORLDWIDE Article Open
access 25 February 2022 ANOXYGENIC PHOTOSYNTHESIS AND IRON–SULFUR METABOLIC POTENTIAL OF _CHLOROBIA_ POPULATIONS FROM SEASONALLY ANOXIC BOREAL SHIELD LAKES Article 03 August 2020
INTRODUCTION Enhanced biological phosphorus removal (EBPR) is an activated sludge process used worldwide to remove phosphorus from wastewaters. Bacteria conducting EBPR do so by accumulating
large quantities of polyphosphate inside their cells, presumably in response to alternating anaerobic and aerobic conditions in the activated sludge tank (Mino et al., 1998). An archetypal
EBPR organism is readily enriched with acetate as a primary carbon source in laboratory-scale sequencing batch reactors (Oehmen et al., 2007; McMahon et al., 2010), and is named
‘_Candidatus_ Accumulibacter phosphatis’ (Hesselmann et al., 1999) (henceforth referred to as Accumulibacter). The Accumulibacter lineage is phylogenetically subdivided into two types (Types
I and II) based on comparative sequence analysis of the gene encoding polyphosphate kinase (_ppk1_) (McMahon et al., 2002). Each type is comprised of several coherent clades exhibiting
unique distribution patterns in wastewater treatment systems (He et al., 2007; He et al., 2010a) and natural aquatic environments (Peterson et al., 2008). Comparative genome analysis can
reveal factors that identify genes or pathways that relate to niche dimensions. Differential gene content can at least partly explain the segregation of _Prochlorococcus_ ecotypes between
high- and low-light conditions in the ocean (Kettler et al., 2007). A recent paper that compared the genomes of _Salinispora_ species discovered several genes required for marine life
adaption (Penn and Jensen, 2012). Comparisons among strains or populations within a species often point to evolutionary-scale forces shaping genomes, such as selection and recombination. A
comparison of seven isolated _Sulfolobus islandicas_ strains isolated from three geographically sites revealed extensive gene loss and gain from recombination within these populations using
mobile elements to maintain genetic diversity (Reno et al., 2009). More recently, metagenomics has enabled such comparisons among uncultured organisms (Gilbert and Dupont, 2011).
Accumulibacter-enriched sludge was the subject of a metagenomic sequence analysis (Garcia Martin et al., 2006) that eventually resulted in the completion of the genome for Accumulibacter
Clade IIA strain UW-1 (GenBank CP001715, Goldstamp Gc01096, hereafter referred to as ‘Clade IIA UW-1’). The resulting genome consisted of a 5.1-Mbp chromosome and three plasmids. The genome
sequencing clarified several features of EBPR metabolism that had been contentious (Garcia Martin et al., 2006; Oehmen et al., 2007). Most notably, it confirmed that the
Embden–Meyerhof–Parnas pathway for glycolysis was fully present and also revealed that Accumulibacter had genes for nitrogen and carbon fixation. The latter was unexpected considering the
carbon- and nitrogen-rich characteristics of wastewater. In addition, a novel cytochrome that consisted of a fusion of cytochrome _b_/_b_6, several transmembrane domains and a nicotinamide
adenine dinucleotide/flavin adenine dinucleotide (NAD/FAD)-binding site was identified and proposed to allow for anaerobic tricarboxylic acid cycle (TCA cycle) operation. Recent work has
revealed ecophysiological differences among Accumulibacter clades. Two morphologically distinct Accumulibacter populations were enriched using different carbon sources and found to have
different nitrate reduction abilities (Carvalho et al., 2007). Subsequently, Flowers et al. (2009) observed that Clade IA-enriched sludge could couple phosphorus uptake with nitrate
reduction, whereas Clade IIA could not. Wexler et al. (2009) investigated the protein expression of two EBPR bioreactors enriched with different Accumulibacter clade composition using
radio-labeled proteomics. One bioreactor revealed enhanced TCA cycle gene expression aerobically, whereas the other showed enhanced synthesis anaerobically, suggesting that these two
Accumulibacter populations had distinct anaerobic and aerobic metabolisms. Another study explored the impact of polyphosphate content on anaerobic performance and stoichiometry, and found
that when polyphosphate content decreased in the cells, Clade IIA appeared to switch to a glycogen-accumulating metabolism, which no longer assisted in removing phosphate from the system
(Acevedo et al., 2012). These findings suggest that distinct Accumulibacter clades inhabit different niches in EBPR ecosystems, each providing an important role in ecosystem function.
Several recent studies have assessed Accumulibacter gene expression using metaproteomics, metatranscriptomics or reverse transcription quantitative real-time polymerase chain reaction (qPCR)
(Burow et al., 2008; Wilmes et al., 2008; Wexler et al., 2009; He et al., 2010b; He and McMahon, 2011). However, such analyses have proven difficult because multiple strains of
Accumulibacter usually coexist in activated sludges, whereas only the Clade IIA UW-1 genome is available as a reference. Therefore, we sequenced the metagenome of a Clade IA-enriched
community to generate a Clade IA reference genome and to better understand ecophysiological and genomic differences between Accumulibacter clades. By developing a greater understanding of
the genomic basis underlying hypothesized distinct niches for the clades, we will also be able to provide more accurate models that help to predict the overall performance of EBPR systems
(Oehmen et al., 2010c). MATERIALS AND METHODS SAMPLE COLLECTION AND PROCESSING The operation of the lab-scale acetate-fed sequencing batch reactor was described as the ‘US sludge’ in Garcia
Martin et al. (2006), except that the pH was controlled at 7.0–7.5, and 4 mg l−1 of allylthiourea was added to the reactor to inhibit nitrification (for more details see Supplementary Online
Material). A biomass sample was collected from the sequencing batch reactor on 21 December 2007 for metagenomic sequencing. Community genomic DNA was extracted from ∼0.2-g cell pellet using
an enzymatic digestion method described previously (Garcia Martin et al., 2006). At the same time, fluorescence _in situ_ hybridization using the PAOMIX probes (Crocetti et al., 2000) was
performed as described previously (He et al., 2008) to estimate the total Accumulibacter abundance. Also, _ppk1_-targeted qPCRs were conducted, as described previously (He et al., 2007), to
determine relative proportions of the two Accumulibacter clades. METAGENOMIC SEQUENCING The extracted genomic DNA was used for sequencing with three different technologies: (1) two whole
genome shotgun libraries with 3- (plasmid) and 40-kb (fosmid) inserts in pUC18 and pCC1FOS, respectively, were end-sequenced with the Sanger technology as described previously (Garcia Martin
et al., 2006); (2) two runs of pyrosequencing with the Roche 454 GS-FLX system and Titanium chemistry, including one using a 15-kbp paired-end library; and (3) one lane of paired-end (2 ×
76 bp) Illumina GA II (San Diego, CA, USA). METAGENOME ASSEMBLY To avoid a mosaic assembly between Clades IA and IIA, sequences generated from the paired-end Illumina run were screened to
remove reads that shared high sequence identities (i.e. ⩾97%) to Clade IIA UW-1 using the runMapping module of Newbler version 2.4 (454 Life Sciences, Branford, CT, USA). Following
screening, raw reads were assembled using Velvet assembler (v.1.0.10) (Zerbino and Birney, 2008) with a hash length of 57, a minimum contig length of 200 bp and a paired-end insert size of
300 bp. The hash length of 57 was chosen over lengths of 4151, and 61 based on contig N50, total contig size, total number of contigs and maximum contig size. The Velvet-assembled contigs
were then shredded into 1800-bp fragments that overlapped by 900 bp. For 454 reads, dinucleotide repeats were removed using Newbler, and redundant reads were removed by Cd-hit versions
2007-0131 (Li and Godzik, 2006) with a requirement of 100% sequence identity on the initial 10 bp and 90% identity on the entire read. Both 454 and Sanger reads were trimmed to an accuracy
of 97% using Lucy v.1.19p (Chou and Holmes, 2001), followed by a screen using the runMapping module of Newbler v.2.4 to remove reads that were ⩾97% identical to Clade IIA UW-1. The final set
of 454 reads, Sanger reads and fragmented Velvet contigs were assembled using Newbler v.2.4 with a minimum identity of 98% and a minimum word length of 80 bp. ACCUMULIBACTER CLADE IA
SCAFFOLD BINNING AND ANNOTATION To identify scaffolds derived from Accumulibacter Clade IA, tetranucleotide frequency analysis was performed (Teeling et al., 2004). Briefly, the frequencies
of all 256 possible tetranucleotides were determined for each DNA scaffold using a custom Perl script. Because the scaffolds were assembled from contigs based on paired-end reads, large
regions within scaffolds contained no sequence data. To ensure that the scaffolds were not constructed from contigs from multiple organisms, the scaffolds were broken into pieces where Ns
were present and analyzed as separate fragments. In addition, these fragments were screened to remove those that were smaller than 20 kbp. In a previous study using self-organizing maps, it
was found that 10-kbp fragments were required for accurate phylogenetic binning (Abe et al., 2003); therefore, 20 kbp was chosen to improve accuracy. To provide a reference for phylogenetic
binning, 11 complete genomes from organisms related to those detected in previous 16S rRNA gene clone libraries, as well as the Accumulibacter Clade IIA UW-1 genome, were shredded into 50-kb
fragments and analyzed for tetranucleotide frequency separately. The resulting 66 unknown fragments from the metagenome and the 3086 fragments from representative organisms were then
analyzed and plotted using correspondence analysis in the R software package (R Development Core Team, 2009). Based on the ordination patterns from correspondence analysis, fragments
clustered with Accumulibacter Clade IIA UW-1, but distinct from other organisms, were considered putative Clade IA fragments. For fragmented scaffolds, at least half of the fragments had to
be classified as Clade IA for the scaffold to be considered as being derived from Clade IA. A total of seven putative Accumulibacter Clade IA scaffolds were identified and annotated using
the RAST server (Aziz et al., 2008). The resulting annotation had 3877 protein coding sequences and 42 tRNA genes. ORTHOLOG IDENTIFICATION Genes from Clade IA were compared with the genes of
Clade IIA UW-1 and _vice versa_, using blastn (Altschul et al., 1997), to identify orthologs between the two genomes with the following blast parameters modified from Rusch et al. (2007) to
detect sequences with up to 45% divergence: blastall −p blastn −F ‘m l’ −m 8 −r 8 −q −8 −X 150 −e 1e−5. The blast results were then screened to remove any alignment that was <40% of the
gene length. From the resulting blast output, putative orthologs were identified by examining reciprocal (or bi-directional) best blast hits. CALCULATION OF SYNONYMOUS AND NON-SYNONYMOUS
SUBSTITUTION RATIO Each pair of amino-acid sequences for all orthologs identified during reciprocal best blast-hit analysis was aligned using MUSCLE (v.3.8.31 default parameters) (Edgar,
2004). The resulting alignments and the nucleotide sequences for each pair were then used to create a codon-based alignment using PAL2NAL (v.14 default parameters) (Suyama et al., 2006). The
resulting codon-based DNA alignment for each pair was then analyzed using the codeml package from PAML (v.4) (Yang, 2007) to estimate the synonymous and non-synonymous substitution ratio
based on maximum likelihood. ANALYSIS OF GENOME SYNTENY To evaluate the differences in genomic structure between Clade IIA UW-1 genome and the Clade IA scaffolds, these sequences were
aligned using the Artemis Comparison Tool (Carver et al 2005). The default parameters were used for the megablast alignment as the input for Artemis Comparison Tool including a maximum
expected value of 1e−4. To increase our ability to detect syntenic regions, adjacent syntenic regions that were <1 kbp apart (∼1 gene) were joined to allow for a single gene insertion.
The total length of syntenic regions was determined by summing the lengths of all of these regions, which were at least 2 kbp long. GENOMIC ISLANDS Clade IA genomic islands (GIs) were
identified by blasting all genes from Clade IIA UW-1 genome against the Clade IA scaffolds with the following parameters: blastall −p blastn −m 8 −e 1e−4 −r 8 −q −8 −X 150 −F ‘m l’. Blast
results were then screened to remove any alignment that was <40% of the gene length, and for each gene the blast result with the highest bit score was selected. Once the blast results
were mapped onto the Clade IA scaffolds, GIs were identified as regions at least 20 kbp in length without any blast alignments. The process was repeated to find Clade IIA UW-1 GIs by
blasting all genes from Clade IA scaffolds against the Clade IIA UW-1 genome. For each identified GI, the Codon Usage Deviation, defined as the sum of difference of codon use for each
triplet between each island and the scaffolds/genome, was determined using the European Molecular Biology Open Software Suite (EMBOSS) package CUSP and CODCMP (Rice et al., 2000). RESULTS
SEQUENCING, ASSEMBLY AND BINNING OF CLADE IA SCAFFOLDS A biomass sample was collected from a laboratory-scale bioreactor that was continuously maintaining the same activated sludge biomass
that was previously sampled for metagenomic sequencing in 2004 (Garcia Martin et al., 2006). However, in contrast to the 2004 sample, the community was enriched in Clade IA rather than Clade
IIA (Table 1). We will use the nomenclature in Table 1 to discuss the two samples hereafter. Sample R107-IA was subjected to metagenomic sequencing using a combination of technologies:
traditional Sanger 3-kb plasmid end sequencing (109 Mbp), Sanger 40-kbp fosmid end sequencing (8 Mbp), 454 Titanium Shotgun (238 Mbp), 454 Titanium Paired End (178 Mbp) and Illumina Paired
End (2392 Mbp) (Supplementary Table S1). This effort generated a total of 2827 Mbp of sequence. Before assembly, we removed reads sharing ⩾97% nucleotide identity with the finished Clade IIA
UW-1 genome to prevent false mosaic assemblies of Clade IIA reads into Clade IA contigs, as Clade IIA was still present in the community (Table 1). After this preassembly screen, 95% of the
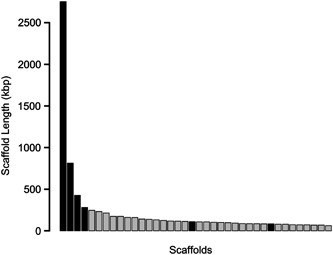
reads remained (Supplementary Table S1), and were assembled into 18 776 contigs (25.1 Mbp), with a largest contig of length 85 kbp. The contigs were then joined into 1466 scaffolds (173
Mbp), with a largest scaffold of length 2.75 Mbp (Figure 1). We used tetranucleotide frequency analysis to identify scaffolds that were putatively derived from Accumulibacter Clade IA
(Figure 2). A large number of the R107-IA scaffold fragments clustered with the Clade IIA UW-1 fragments, and were distinct from fragments from other reference genomes. Based on these
results, a total of seven putative Accumulibacter Clade IA scaffolds were identified, amounting to 4.5 Mbp of sequence (4.2 Mbp when excluding Ns) with an average GC content of 64%. These
seven Clade IA scaffolds (Supplementary Table S2) were annotated using the RAST server, yielding 3923 predicted genes (Table 2). In comparison, the Accumulibacter Clade IIA UW-1 chromosome
and three plasmids totaled 5.3 Mbp of sequence and had 4792 predicted genes. We also identified a _ppk1_ homolog in the largest scaffold (Scaffold01135) sharing 98% nucleotide identity with
a Clade IA _ppk1_ identified previously (GenBank AF502200.1; McMahon et al., 2002). As this locus was originally used to define the Accumulibacter clade phylogeny, the presence of a Clade IA
_ppk1_ homolog within this putative Clade IA scaffold provides confidence that the scaffolds were assembled and binned properly. We examined the seven putative Clade IA scaffolds to
determine genome completeness. Forty-two of the 49 tRNAs annotated in the finished Clade IIA UW-1 genome were identified by blastn. All of the 52 ribosomal proteins and 16 of 20 tRNA
synthetases were identified by tblast. We also conducted a Cluster of Orthologous Genes (COG)-based analysis by first determining all of the COG functions shared among the four neighboring
Rhodocyclaceae sequenced genomes besides Clade IIA UW-1 (_Aromatoleum aromaticum_ EbN1, _Dechloromonas aromatica_ RCB, _Azoarcus_ sp. BH72 and _Thauera_ sp. MZ1T), yielding 1273 COG
functions that were at least present once in all of the four neighbors. This COG list was then compared with a similar list for the Accumulibacter Clade IA scaffolds, and it was determined
that 119 (∼10%) of the COG functions were missing from Clade IA scaffolds. The average copy number of each COG function was then determined for these four neighbor genomes, and the average
abundance was compared with Clade IA scaffolds. From this method, it was determined that Clade IA lacked roughly 20% of the COG functions. Therefore, the genome was estimated to be roughly
80–90% complete. GENOME ALIGNMENT ANALYSIS To visualize the structural differences between the two genomes, the Accumulibacter Clade IA scaffolds were aligned against the Clade IIA UW-1
chromosome and three plasmids (Supplementary Figure S1). The order of the Clade IA scaffolds was determined by using the contig reordering function in Mauve v.2.3.1 (Darling et al., 2010)
using the Clade IIA UW-1 genome as a reference. The alignments clearly show that the two genomes have significantly different structure. Considering that the two strains share 98.7% identity
in their 16S rRNA genes, the amount of difference in genome structure is surprising. The largest syntenous region between the two genomes was 10.5 kbp long; however, only 206 regions were
>2000 bp (∼2 genes long), which totaled 623 kbp. If syntenous regions that were <1000 bp apart (∼<1 gene) were merged and any region <2000 bp when combined was removed, a total
of 236 syntenic regions that total just under 1 Mbp in length remained. This accounts for∼25% of the Clade IA genome. It is possible that this number is underestimated because of the removal
of potentially highly conserved regions during the preassembly sequence screening that removed sequences <97% similar to Clade IIA, which likely resulted in gaps in the Clade IA
scaffolds. Therefore, the Clade IA genome should be completed to verify these results. SHARED AND DIFFERENTIAL GENE CONTENT We compared the genes shared between the two clades using
Reciprocal Best Hit Analysis. A total of 2317 genes and 2410 proteins were identified as shared between the two clades based on nucleotide and protein sequence, respectively (Table 2),
although this is a conservative estimate because the Clade IA genome is not finished. These genes share an average nucleotide identity (ANI) of 78.3% (Figure 3) and constitute 49% of the
total Clade IIA UW-1 genes. Surprisingly, scaffolds from Clade IA matched to genes on the plasmids from Clade IIA UW-1. Specifically, Scaffold00037 contained both conjugation genes to allow
for genetic exchange between two bacteria from one of the plasmids as well as genes from the Clade IIA UW-1 chromosome including genes for development of precursors for Vitamin B12
synthesis, which is an important cofactor in many metabolic reactions. Also, Scaffold01134 contained genes from all three Clade IIA UW-1 plasmids as well as a few from the Clade IIA UW-1
chromosome. In contrast, Scaffold000028, which is the smallest Clade IA scaffold (35 kbp), only had a single Reciprocal Best Hit Analysis with Clade IIA UW-1. Although the majority of the
genes (∼80%) on Scaffold000028 were hypothetical, a few genes were phage-related, which suggests that these genes may be remnants of a lysogenic phage (discussed below) that carried these
genes into the genome. Genes from both clades were functionally classified according to RAST Functional Categories (Aziz et al., 2008) and the frequency of each category was compared in both
genomes (Supplementary Figure S2). We found minimal differences in RAST Functional Category distribution, despite the fact that ∼50% of the genes are non-orthologous based on the Clade IIA
UW-1 gene count. To understand some of the functional differences between Clade IA and IIA, the distribution of RAST Functional Category for genes unique (non-orthologous) to both clades was
analyzed (Figure 4). Most categories were comparably represented within the two genomes, but several categories appeared to be enriched in Clade IA, including ‘Stress Response’,
‘Respiration’, ‘Cell Wall and Capsule’ and ‘Protein, Nucleoprotein transport’. Several categories were enriched in Clade IIA UW-1, including ‘Fatty Acid, Lipids, and Isoprenoids’, ‘Membrane
Transport’, ‘Cofactors, Vitamins, Prosthetic Groups, Pigments’ and ‘Phosphorus Metabolism.’ Considering these latter categories are related to basic cell physiology, it is possible that
these highly conserved sequences were removed during the preassembly screen and not incorporated into the final Clade IA genome. GENOMIC ISLANDS We used the finished Clade IIA UW-1 genome to
recruit genes from the Clade IA scaffolds to identify relatively large regions of the Clade IIA UW-1 genome that may be absent in the Clade IA genome (GIs). Twenty-eight putative Clade IIA
UW-1 GIs were identified that contained 575 genes (Table 3). The largest GI was 41 kbp. Of these genes, 270 (∼46%) were hypothetical proteins. Of the 28 GIs, 16 contained some elements of
foreign DNA based on the presence of phage, transposase or integrase genes, which likely explains why these regions contained unique genes. Another GI contained several genes affiliated with
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPRs), which have been linked to phage immunity (Barrangou et al., 2007) (also discussed later). Surprisingly, GI 21, which had
the largest codon usage deviation, did not contain any evidence of a foreign DNA source, such as phage, transposase or integrase genes. Despite having apparent remnants of foreign DNA, the
16 GIs with phage, transposase or integrase genes had comparable codon usage deviation to GIs without these genes. This suggests that the genes within these 16 GIs were either present in an
ancestral lineage from which both clades were derived and subsequently lost in Clade IA or transferred into Clade IIA UW-1 long enough to allow for codon usage to converge. In addition, some
of these GIs may have been a result of predation and integration into the Clade IIA genome just before collecting the sample R104-IIA. We note that it is possible that some of the
identified GIs were missing in the Clade IA scaffolds because of the removal of reads sharing high identity with Clade IIA UW-1 during the preassembly screening for sample R107-IA, although
the presence of genes involved in horizontal transfer provides support for the likelihood that they are indeed GIs. Similarly, we searched for GIs in the Clade IA scaffolds by recruiting
genes from the finished Clade IIA UW-1 genome. We identified five Clade IA GIs that contained 129 genes (Table 4). Of these, 93 genes (72%) were annotated as hypothetical genes.
Interestingly, GI 4, which appears to be derived from foreign DNA based on having the largest codon usage deviation, is entirely comprised of hypothetical genes. When these genes were
compared using blast against the NCBI non-redundant database (nr), no significant blast hits (e-value <1) were obtained for any of these genes. The largest GI in Clade IA was over 50 kbp
long and contained a mix of hypothetical and phage-related genes. Despite the large fraction of phage genes, this GI had the smallest codon usage deviation, implying that the island was due
to lysogenic phage integration less recently than in other islands. GI 1 contained genes for several membrane proteins. With the codon usage similar to the average value, it is likely that
this is simply a region of unique gene functions and not recently horizontally transferred. Similar to GI 4, very few of these genes had significant blast hits with sequences in the nr
database. Surprisingly, Scaffold00028, which is entirely a GI 2, is nearly identical (>98% similar over 92% of its length) to a podovirus (EPV 1) sequenced in a viral metagenome
(Skennerton et al., 2011) generated from the same bioreactor 7 months after the R104-IIA sample and roughly 2.5 years earlier when the R107-IA sample was collected. Based on having similar
coverage and tetranucleotide frequencies to Clade IA scaffolds, we believe that this is a lysogenic phage in the Clade IA genome. The only differences between the EPV 1 scaffold and GI 2 are
two non-contiguous ∼1000-bp regions that contain hypothetical genes. GENETIC BASIS FOR ECOPHYSIOLOGICAL DIFFERENTIATION Previous studies using Accumulibacter-enriched bioreactors have
suggested that Clade IA can reduce nitrate, whereas Clade IIA cannot (Carvalho et al., 2007; Flowers et al., 2009; Guisasola et al., 2009; Oehmen et al., 2010b). Clade IIA UW-1’s inability
to reduce nitrate was also suggested as its genome lacked all of the subunits for the respiratory nitrate reductase gene (_narGHI_). Interestingly, no _nar_ genes could be identified in the
Clade IA scaffolds presented here. The Clade IIA UW-1 genome did contain identifiable periplasmic nitrate reductase (_napDAGHBF_) in a single gene cluster. There was also a _napC/nirT_
homolog that has been shown in other organisms to be used with _nap_ genes in another locus to perform nitrate reduction while providing enough proton-motive force for energy production
(Gonzalez et al., 2006). These same genes with nearly the same organization were also found in the Clade IA genome (Scaffold01135), except that the _napF_ gene was missing; however, based on
conserved operon structure in model organisms, the _napF_ gene should have been located where a gap exists in the Clade IA scaffold. It is still possible that the gap exists as a result of
the preassembly screening, which removed the _napF_ gene because of having sequence homology >97%, but this is unlikely given that the ANI of shared genes in this gene neighborhood was
83% (across 28 kbp). If Clade IA does use these _nap_ genes for nitrate respiration, the observed differences in nitrate-reducing capabilities between these two clades might be related to
gene regulation rather than gene content. The metagenome assembly from sample R107-IA did contain a cluster of _narGHIJ_ genes on Scaffold00577, which is ∼13 kbp long (6 kbp when excluding
Ns), but the mean coverage for this scaffold (5 × ) is very low as compared with the Clade IA scaffolds (16.2 X). The best hits to the scaffold obtained using NCBI’s blastx against nr were
to the genera _Intrasposrangium_, _Serinicoccus_ and _Janibacter_ (members of the _Actinobacteria_ phylum), suggesting a different origin. Still, given the fact that the Accumulibacter Clade
IA genome is not complete, the absence of _narGHIJ_ genes within the Accumulibacter Clade IA genome cannot be confirmed currently. Clade IIA UW-1 had genes required for nitrogen fixation
and carbon fixation, but most of these genes appear to be absent in the Clade IA genome because they were not found in the Clade IA scaffolds or at significantly high read coverage in the
metagenome. Specifically, Clade IA lacked two of the important genes required for the Calvin cycle, ribulose-1,5-bisphosphate carboxylase oxygenase (_rubisco_) and ribulose-phosphate
3-epimerase (_Rpe_), which were present in Clade IIA UW-1 (JGI-IMG/M gene OID: 2 014 613 882 and 2 014 614 231, respectively). Also, Clade IA contained none of the nitrogenase subunits,
_nifDHK_, present in the Clade IIA UW-1 genome. The absences of these genes might be explained by a loss over time because of being present in our bioreactors for multiple years or may
indicate that Clade IA originated from natural environments where nitrogen and carbon fixation are not required. Of course, it is also possible that the draft Clade IA genome lacked these
genes because of incomplete genome coverage or assembly. To assess this likelihood, we searched for these genes in other scaffolds and among the raw metagenomic reads. However, we found no
definitive evidence for the presence of these genes in the Clade IA genome (see Supplementary Online Material). Additional sequencing and further assembly will need to be done to confirm
these gene-level differences between Clade IA and IIA. COMPARISON OF CRISPR LOCI CRISPR elements are thought to provide resistance to invasion of phage or other foreign DNA by storing
segments of previously confronted phage or other foreign DNA as spacers that are surrounded by short palindromic-like repeat sequences adjacent to a series of CRISPR-associated genes (CAS)
(Barrangou et al., 2007; Marraffini and Sontheimer, 2010). The detection of CRISPRs in a genome is significant because the presence or absence of these loci can determine which populations
remain after a viral predation event. In a previous study, Accumulibacter Clade IIA UW-1 was thought to contain three CRISPR loci, whereas the genome was still in a draft form (Kunin et al.,
2008); however, the finished genome contained only two. Here, we found that the Clade IA scaffolds only contained one CRISPR locus (Table 5). Interestingly, the CRISPR repeat sequences and
CAS gene structure in the two Clade IIA UW-1 loci were different. The CRISPR locus in Clade IA did not share any repeat sequences with the loci in Clade IIA UW-1; however, the CAS gene order
and predicted function in Clade IA (CR3) were identical to those present in one of the Clade IIA UW-1 loci (CR2). Despite the similar gene order and predicted function, the genes appear to
have different origins based on low sequence similarities. Comparison of the spacers in the three loci using blastn determined that there were no spacers shared among them. Interestingly,
blast analysis of the spacer sequences from all three loci with the viral metagenome (Skennerton et al., 2011), generated from the same bioreactor 7 months after the R104-IIA sample and
roughly 2.5 years before the R107-IA sample, yielded only perfect matches with the Clade IA CRISPR Spacers (Table 5). In total, nine Clade IA CRISPR spacers matched perfectly with the EPV1
phage that was found to be a likely lysogen in the Clade IA genome (Scaffold00028 corresponding to GI 2). In contrast, neither of the Clade IIA CRISPR regions matched any viral metagenomic
sequences completely. Skennerton _et al._ (2011) predicted that this phage was specific for Accumulibacter and they also noted the presence of a histone-like nucleoid-structuring (H-NS)
protein that might make the phage resistant to CRISPR activity. The repeated presence of phage DNA in CRISPR spacers, particularly near the leader region (which represent the most current
phage insertion events), suggest that the histone-like nucleoid structuring allows for EPV1 to infect continually Clade IA. This suggests that these same phage populations or similar types
were still active 2.5 years later during the time of collecting the R107-IA sample, and that only Clade IA is susceptible to predation by the EPV1 phage in this system. METABOLIC
RECONSTRUCTION OF ACCUMULIBACTER CLADE IA Some elements of EBPR metabolism have been under contention for several decades now. The main area of disagreement is the source and balancing of
anaerobic-reducing equivalents for polyhydroxyalkanoate production. Researchers disagree about the pathway used for anaerobic operation of glycolysis (Embden–Meyerhof–Parnas versus
Entner–Douderoff (ED) pathway) and the direction of the TCA cycle (reductive, split, full or through glyoxylate shunt) (Oehmen et al., 2007). The finished genome of Accumulibacter Clade IIA
UW-1 revealed that it only contained genes for the Embden–Meyerhof–Parnas pathway and also showed the presence of all genes necessary for the varied modes of the TCA cycle previously
suggested for EBPR (Garcia Martin et al., 2006). Not too surprisingly, the draft Clade IA genome also contained nearly all of the important genes associated with EBPR metabolism, and also
lacked ED pathway genes. The only important EBPR-related gene that seemed to be absent in the Clade IA genome encodes the E1 subunit of the pyruvate dehydrogenase complex; however, the other
subunits for the pyruvate dehydrogenase complex were present and there is a gap in the scaffold where the E1 subunit should be located. Therefore, it is likely that this missing gene will
be found in the finished genome. Although the overall gene content was nearly identical, there were differences in gene counts between the two organisms. For example, Clade IA contained two
copies of the fumarase (fig|759355.3.peg.2281 and fig|759355.3.peg.1714), whereas Clade IIA only contains one (JGI-IMG/M gene OID: 645 011 516). Whether the additional fumarase in Clade IA
is expressed anaerobically needs to be determined by future gene expression analysis. One interesting discovery was the existence of three Na(+)-translocating NADH-quinone reductase
(Rnf/Nqr) encoding gene clusters in Clade IIA (IMG/M gene OID: 645 012 180–645 012 185 and 645 010 733–645 010 738 and 645 012 144–645 012 149) as compared with a single such cluster in the
Clade IA genome (fig|759355.3.peg.2984–2978). One of the Clade IIA clusters (IMG/M gene OID: 645 010 733–645 010 738) was relatively homologous (>81% amino acid similarity) with Clade IA,
but the remaining two Clade IIA operons (IMG/M gene OID: 645 012 144–645 012 149 and 645 012 180–645 012 185) were much more divergent (<68% similar) from the Clade IA operon. Also, one
of those operons had only three of the six subunits with any nucleotide similarity (<63%) to _Clade IA_ genes. Although the function of these unique gene clusters in Clade IIA is unknown,
it as well as the other clusters share nearly identical structure (i.e. gene order) with _rnf_ gene clusters that have been shown to catalyze the transfer or electron from reduced
ferredoxin to NAD+ coupled with Na+ translocation (Muller et al., 2008). If either of these gene clusters are expressed anaerobically, the Rnf/Nqr might assist in anaerobic operation of the
TCA cycle since any reduced ferredoxin from pyruvate ferredoxin:oxidoreductase or α-ketoglutarate oxidoreductase can be used to produce NADH as well as a proton motive force for ATP
production anaerobically. In the original report of the Accumulibacter Clade IIA UW-1-enriched metagenome, several interesting findings were discussed including the presence of a novel
fusion protein consisting of a cytochrome _b/b6_ domain with several transmembrane helices as well as a NAD(P)- and flavin-binding domain. The fusion protein is proposed to allow for
oxidation of reduced quinone from succinate dehydrogenase in the absence of oxygen by transferring electrons to NAD+ and FAD (Garcia Martin et al., 2006). Since then, the genomes of two
strains of _Alicycliphilus denitrificans_ (BC and K601), which were isolated from a wastewater treatment plant, were found to have an protein with homologous structure (Mechichi et al.,
2003; Weelink et al., 2008; Oosterkamp et al., 2011). We determined that the wrong gene id (JGI-IMG/M gene OID: 2 001 028 710 or 645 009 129 in finished genome) was provided for the novel
cytochrome in that report rather than the actual protein with these domains (JGI-IMG/M gene OID: 2 001 028 680 or 645 009 126 on finished genome), so recent studies exploring its expression
targeted the wrong gene (Burow et al., 2008; He et al., 2010b). Thus, its role in anaerobic metabolism is still unproven. We discovered that these two genes (JGI-IMG/M gene OID: 645 009 129
and 645 009 126) have orthologs in Clade IA (fig|759355.3.peg.563 and fig|759355.3.peg.561) with 81% and 86% nucleotide identity, respectively, that appear to be part of a conserved operon
(Supplementary Figure S3). In addition, Clade IA region shares a larger nine-gene cluster (fig|759355.3.peg.561–fig|759355.3.peg.570) in that region with both strains of _Alicycliphilus
denitrificans_. The additional sequences are all related to cytochrome _c_ biosynthesis (Supplementary Figure S3). Despite having similar synteny to _Alicycliphilus_, the Clade IA novel
fusion proteins are more similar to Clade IIA than to their homologs in _Alicycliphilus_, suggesting that Clades IA and IIA are more likely to have a common origin despite having different
operon structure. What impact these different operon structures have on metabolism is yet to be determined. CONSERVATION OF _EBPR_ GENES We postulated that genes associated with the hallmark
carbon and phosphorus cycling pathways in EBPR metabolism were under significant selective pressure because of their importance to the fitness of the bacteria in the EBPR ecosystems. To
test this, we evaluated the level of conservation of EBPR genes as compared with genes not associated with EBPR (NON-EBPR genes) (Supplementary Table S3). Using the Student’s _t_-test, we
determined that the average nucleotide identities between these two clades for the EBPR and NON-EBPR genes, 82% and 78%,respectively, were statistically different (_P_<0.00001). In
contrast to observed structural and gene content differences between the clades, the higher level of conservation for EBPR genes probably suggests that these important genes are under the
same selective pressure. This idea is further supported by EBPR genes having a synonymous and non-synonymous substitution ratio of 0.05 that is also significantly different (_P_<0.005)
than the synonymous and non-synonymous substitution ratio for NON-EBPR genes (0.07). These results suggest that mutations in EBPR genes are under stronger negative selection since mutations
that cause amino-acid sequences changes are selected against more than in NON-EBPR genes. DISCUSSION Before this work, several lines of evidence pointed to the existence of multiple distinct
Accumulibacter clades in EBPR systems, despite the high 16S rRNA sequence identity within the Accumulibacter lineage (McMahon et al., 2010). Although the R104-IIA sludge sample was enriched
in Clade IIA, reads from closely related co-occurring species and strains were detected in its metagenome (Garcia Martin et al., 2006). At that time, Accumulibacter lineage diversity was
poorly characterized. Subsequently, a rigorous phylogenetic analysis based on the _ppk_1 locus was used to partition the lineage into two Types (I and II), which could be further subdivided
into five and seven clades, respectively (He et al., 2007; Peterson et al., 2008). In this study, we compared the gene content between representatives of Accumulibacter Clades IA and IIA.
Although the complete genome for Clade IIA UW-1 is available, the genome for Clade IA was constructed by first removing reads, which were highly similar to Clade IIA UW-1 (⩾ 97%) from the
metagenomic sequencing reads of sample R107-IA and assembling the remaining reads. The removal of these reads helped in preventing the development of a mosaic genome comprised of both Clade
IA- and IIA-derived reads, but it also likely caused gaps in the Clade IA scaffolds in regions where there was high sequence conservation between Clades IA and IIA. From all of the assembled
scaffolds, the Clade IA fragments were identified based on having tetranucleotide frequencies similar to Clade IIA UW-1. The limitation of this method to identify accurately fragments below
20 kbp likely reduced the number of Clade IA fragments that could be identified. As a result, the assembly of this genome is considered as a draft and comparisons between Clade IA and IIA
populations are considered preliminary. Despite these limitations, this novel approach allowed for assembly of large genomic fragments of our target organism from a mixed community that
contained an organism of similar phylogeny and function. The assembly was considered effective as indicated by the high level of estimated completeness (80–90%) of the genome based on the
presence of essential COG functions, and specifically tRNAs, tRNA synthetases and ribosomal proteins. Comparison of genes related to EBPR metabolism (including those involved in central
carbon metabolism) did not reveal any marked differences between the two clades. Previous studies suggested that these two clades used the glycolytic and TCA cycle differently under
anaerobic conditions (Wexler et al., 2009; Oehmen et al., 2010a; Acevedo et al., 2012). Although we did not detect any apparent genome-level differences, we did note instances where the gene
copy number for a particular enzyme important in the TCA cycle varied, which could influence expression levels. Specifically, we noticed a higher abundance of a protein complex
(Na(+)-translocating NADH-quinone reductase) in Clade IIA, which may allow for it to operate either a reductive or complete TCA cycle anaerobically. Although Clade IA did contain a single
gene cluster encoding this complex, it is possible that these genes are not expressed or have another function for Clade IA that is yet to be determined. With highly conserved 16S rRNA
sequences (98.5%) and similar functions in EBPR processes, the genomes of these two clades were expected to have similar genomic structure and gene content originating from a common
ancestor. However, our results suggest that these two clades are significantly different in both characteristics. Accumulibacter Clades IA and IIA only have 25% of their genomes exhibiting
any synteny and they only share an estimated 63% and 48% of their gene content, respectively, at an ANI of 78%. The sequencing of a sludge sample enriched in Clade IA has revealed
significant genome-level differentiation between the two clades, which could arguably be considered as two ‘species’ of Accumulibacter. Several GIs were identified in both clades. We
identified 28 putative GIs in Clade IIA UW-1 that contained a variety of genes including phage-related genes. Contrastingly, Clade IA only had five putative GIs in its scaffolds with
inferred characteristics ranging from phage-related to hypothetical. When considering the functions of the genes and the codon usage deviations in the GIs, it is likely that many of these
differences are associated with foreign DNA integration into each genome. Because the Clade IA genome is not complete, several of the putative GIs in Clade IIA UW-1 are probably associated
with regions currently missing in the Clade IA scaffolds; however, it was surprising that only five GIs existed in Clade IA totaling 150 kbp (∼3% of the scaffold length) considering the fact
that such a large fraction of the total genes lacked orthologs in Clade IIA UW-1. This indicates that while there are large genomic differences, the location of these differences are
sprinkled throughout the genome and not restricted to specific regions. This idea is supported by viewing the genome alignments, which revealed a spider web of matching regions between the
genomes (Supplementary Figure S1). The genes associated with EBPR metabolism appear to be the exception to the general interspecies differences, as they all are present in both clades and
share an ANI of 81%, which is higher than the average for the two genomes. It is possible that these genes, which are thought to be essential for EBPR performance, are diverging less. This
idea is supported by the EBPR genes having an average synonymous and non-synonymous substitution ratio of 0.05, which is statistically lower than that of the NON-EBPR genes (0.07). A
previous long-term studies on our bioreactors have shown extended periods of both Clade IA and Clade IIA dominance (He et al., 2010a). It is possible that changes in the community structure
were associated with phage predation events that specifically targeted each clade. In theory, the additional CRISPR locus in the Clade IIA genome should provide it with more resistance to
phage predation because of the presence of more phage spacer sequences (221 versus 100) and lessen the likelihood of phage predation, but recent papers have shed light on possible mechanisms
by which phages counteract the CRISPR activity including a podovirus identified in the viral metagenome from our bioreactors(Skennerton et al., 2011; Bondy-Denomy et al., 2013). The
persistence of viral populations despite CRISPR spacer matches present in the bacterial genomes is supported by the detection of spacers located near the leader region, which have high
identity to a partially assembled phage from a viral metagenome (Skennerton et al., 2011) sampled 2.5 years earlier as well as the apparent presence of the same lysogenic phage in the Clade
IA genome. One of the more interesting findings in this study was the apparent conservation of some pieces of the Accumulibacter Clade IIA UW-1 genome and loss of others. Specifically, there
is evidence that either the plasmids or plasmid-associated genes are retained within the Clade IA genome. It is currently unclear if these scaffolds are derived from a new plasmid or from
chromosomal fragments, but the large number of genes associated with conjugation suggested plasmid origin. The plasmids from Accumulibacter Clade IIA UW-1 mostly contained genes associated
with conjugation, transposases and heavy metal resistance, but one of the Clade IA scaffolds that may be plasmid derived (Scaffold01134) contains numerous genes associated with important
functions including cytochromes, ubiquinol-cytochrome _C_ reductase and PHA synthase. One surprising finding during the sequencing of Clade IIA UW-1 was the presence of carbon and nitrogen
fixation genes, considering the fact that wastewater is a carbon- and nitrogen-rich environment. The Clade IA scaffolds lack most of the genes associated with these processes; however, there
is some weak evidence that these genes were simply removed during the preassembly screen of the R107-IA raw reads. Whether this gene loss reflects the genomic structure of ‘wild’ Clade IA
populations, is a result of natural gene loss over time spent in the bioreactor or an artifact due to removal of reads during the preassembly screen remains to be determined. CONCLUSION
Through metagenomic sequencing of a Clade IA-enriched lab-scale bioreactor, a large fraction of the genome for Clade IA was assembled. Some of the results from this study are still
preliminary because of the required treatment to Clade IIA UW-1-like sequence reads to avoid a mosaic Clade IA and IIA genome. Nevertheless, the study does reveal that while Clades IA and
IIA have all of the necessary genes for EBPR metabolism and nitrite reduction, there are marked genomic differences between the two clades. Specifically, there is little genome synteny
between the two clades and there appears to be differences in their ability to fix carbon and nitrogen. In addition, Accumulibacter Clade IIA UW-1 may have better defenses against phage
predation based on the presence of an additional CRISPR locus as compared with Clade IA. Overall, these findings provide a greater understanding of the differences between these two clades
and will assist in further exploration of metabolic differences through future gene expression studies. REFERENCES * Abe T, Kanaya S, Kinouchi M, Ichiba Y, Kozuki T, Ikemura T . (2003).
Informatics for unveiling hidden genome signatures. _Genome Res_ 13: 693–702. Article CAS PubMed PubMed Central Google Scholar * Acevedo B, Oehmen A, Carvalho G, Seco A, Borras L, Barat
R . (2012). Metabolic shift of polyphosphate-accumulating organisms with different levels of polyphosphate storage. _Water Res_ 46: 1889–1900. Article CAS PubMed Google Scholar *
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W _et al_ (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. _Nucleic Acids Res_ 25:
3389–3402. Article CAS PubMed PubMed Central Google Scholar * Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA _et al_ (2008). The RAST server: rapid annotations using
subsystems technology. _BMC Genom_ 9: 75. Article Google Scholar * Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S _et al_ (2007). CRISPR provides acquired resistance
against viruses in prokaryotes. _Science_ 315: 1709–1712. Article CAS PubMed Google Scholar * Bondy-Denomy J, Pawluk A, Maxwell KL, Davidson AR . (2013). Bacteriophage genes that
inactivate the CRISPR/Cas bacterial immune system. _Nature_ 493: 429–432. Article CAS PubMed Google Scholar * Burow LC, Mabbett AN, Blackall LL . (2008). Anaerobic glyoxylate cycle
activity during simultaneous utilization of glycogen and acetate in uncultured Accumulibacter enriched in enhanced biological phosphorus removal communities. _ISME J_ 2: 1040–1051. Article
CAS PubMed Google Scholar * Carvalho G, Lemos PC, Oehmen A, Reis MAM . (2007). Denitrifying phosphorus removal: linking the process performance with the microbial community structure.
_Water Res_ 41: 4383–4396. Article CAS PubMed Google Scholar * Carver TJ, Rutherford KM, Berriman M, Rajandream M-A, Barrell BG, Parkhill J . (2005). ACT: the Artemis comparison tool.
_Bioinformatics_ 21: 3422–3423. CAS PubMed Google Scholar * Chou H-H, Holmes MH . (2001). DNA sequence quality trimming and vector removal. _Bioinformatics_ 17: 1093–1104. Article CAS
PubMed Google Scholar * Crocetti GR, Hugenholtz P, Bond PL, Schuler A, Keller J, Jenkins D _et al_ (2000). Identification of polyphosphate accumulating organisms and the design of 16S
rRNA-directed probes for their detection and quantitation. _Appl Environ Microbiol_ 66: 1175–1182. Article CAS PubMed PubMed Central Google Scholar * Darling AE, Mau B, Perna NT .
(2010). ProgressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. _PLoS One_ 5: e11147. Article PubMed PubMed Central Google Scholar * Edgar RC . (2004).
MUSCLE: multiple sequence alignment with high accuracy and high throughput. _Nucleic Acids Res_ 32: 1792–1797. Article CAS PubMed PubMed Central Google Scholar * Flowers JJ, He S,
Yilmaz S, Noguera DR, McMahon KD . (2009). Denitrification capabilities of two biological phosphorus removal sludges dominated by different ‘_Candidatus Accumulibacter_’ clades. _Environ
Microbiol Rep_ 1: 583–588. Article CAS PubMed PubMed Central Google Scholar * Garcia Martin H, Ivanova N, Kunin V, Warnecke F, Barry KW, McHardy AC _et al_ (2006). Metagenomic analysis
of two enhanced biological phosphorus removal (EBPR) sludge communities. _Nat Biotechnol_ 24: 1263–1269. Article PubMed Google Scholar * Gilbert JA, Dupont CL . (2011). Microbial
metagenomics: beyond the genome. In: Carlson CA, Giovannoni SJ (eds) _Annual Review of Marine Science_ VOL. 3. Annual Reviews: Palo Alto, pp 347–371. Google Scholar * Gonzalez PJ, Correia
C, Moura I, Brondino CD, Moura JJG . (2006). Bacterial nitrate reductases: molecular and biological aspects of nitrate reduction. _J Inorg Biochem_ 100: 1015–1023. Article CAS PubMed
Google Scholar * Guisasola A, Qurie M, MdM Vargas, Casas C, Baeza JA . (2009). Failure of an enriched nitrite-DPAO population to use nitrate as an electron acceptor. _Process Biochem_ 44:
689–695. Article CAS Google Scholar * He S, Bishop FI, McMahon KD . (2010a). Bacterial community and Accumulibacter population dynamics in biological phosphorus removal sludge. _Appl
Environ Microbiol_ 76: 5479–5487. Article CAS PubMed PubMed Central Google Scholar * He S, Gall DL, McMahon KD . (2007). ‘_Candidatus Accumulibacter_’ population structure in enhanced
biological phosphorus removal sludges as revealed by polyphosphate kinase genes. _Appl Environ Microbiol_ 73: 5865–5874. Article CAS PubMed PubMed Central Google Scholar * He S, Gu AZ,
McMahon KD . (2008). Progress towards understanding the distribution of Accumulibacter among full-scale enhanced biological phosphorus removal systems. _Microb Ecol_ 55: 229–236. Article
PubMed Google Scholar * He S, Kunin V, Haynes M, Garcia Martin H, Ivanova N, Kyrpides N _et al_ (2010b). Metatranscriptomic analysis of ‘Candidatus Accumulibacter’-enriched enhanced
biological phosphorus removal sludge. _Environ Microbiol_ 12: 1205–1217. Article CAS PubMed Google Scholar * He S, McMahon KD . (2011). ‘Candidatus Accumulibacter’ gene expression in
response to dynamic EBPR conditions. _ISME J_ 5: 329–340. Article CAS PubMed Google Scholar * Hesselmann RPX, Werlen C, Hahn D, van der Meer JR, Zehnder AJB . (1999). Enrichment,
phylogenetic analysis and detection of a bacterium that performs enhanced biological phosphate removal in activated sludge. _System Appl Microbiol_ 22: 454–465. Article CAS Google Scholar
* Kettler GC, Martiny AC, Huang K, Zucker J, Coleman ML, Rodrigue S _et al_ (2007). Patterns and implications of gene gain and loss in the evolution of Prochlorococcus. _PLoS Genet_ 3:
2515–2528. Article CAS Google Scholar * Kunin V, He S, Warnecke F, Peterson SB, Martin HG, Haynes M _et al_ (2008). A bacterial metapopulation adapts locally to phage predation despite
global dispersal. _Genome Res_ 18: 293–297. Article CAS PubMed PubMed Central Google Scholar * Li W, Godzik A . (2006). Cd-hit: a fast program for clustering and comparing large sets of
protein or nucleotide sequences. _Bioinformatics_ 22: 1658–1659. Article CAS PubMed Google Scholar * Marraffini LA, Sontheimer EJ . (2010). CRISPR interference: RNA-directed adaptive
immunity in bacteria and archaea. _Nat Rev Genet_ 11: 181–190. Article CAS PubMed PubMed Central Google Scholar * McMahon KD, Dojka MA, Pace NR, Jenkins D, Keasling JD . (2002).
Polyphosphate kinase from activated sludge performing enhanced biological phosphorus removal. _Appl Environ Microbiol_ 68: 4971–4978. Article CAS PubMed PubMed Central Google Scholar *
McMahon KD, He S, Oehmen A . (2010) _The microbiology of phoshorus removal_ In: Seviour RJ, Nielsen PH (eds) Microbial Ecology of Activated Sludge IWA Publishing: London, pp 281–320. Google
Scholar * Mechichi T, Stackebrandt E, Fuchs G . (2003). _Alicycliphilus denitrificans_ gen. nov., sp. nov., a cyclohexanol-degrading, nitrate-reducing β-proteobacterium. _Int J Syst Evol
Microbiol_ 53: 147–152. Article CAS PubMed Google Scholar * Mino T, Van Loosdrecht MCM, Heijnen JJ . (1998). Microbiology and biochemistry of the enhanced biological phosphate removal
process. _Water Res_ 32: 3193–3207. Article CAS Google Scholar * Muller V, Imkamp F, Biegel E, Schmidt S, Dilling S . (2008). Discovery of a ferredoxin: NAD(+)-oxidoreductase (Rnf) in
acetobacterium woodii—a novel potential coupling site in acetogens. In: Wiegel J, Maier RJ, Adams MWW (eds) _Incredible Anaerobes: From Physiology to Genomics to Fuels_. Blackwell
Publishing: Oxford, pp 137–146. Google Scholar * Oehmen A, Carvalho G, Freitas F, Reis MAM . (2010a). Assessing the abundance and activity of denitrifying polyphosphate accumulating
organisms through molecular and chemical techniques. _Water Sci Technol_ 61: 2061–2068. Article CAS PubMed Google Scholar * Oehmen A, Carvalho G, Freitas F, Reis MAM . (2010b). Assessing
the abundance and activity of denitrifying polyphosphate accumulating organisms through molecular and chemical techniques. _Water Sci Technol_ 61: 2061–2068. Article CAS PubMed Google
Scholar * Oehmen A, Carvalho G, Lopez-Vazquez CM, van Loosdrecht MCM, Reis MAM . (2010c). Incorporating microbial ecology into the metabolic modelling of polyphosphate accumulating
organisms and glycogen accumulating organisms. _Water Res_ 44: 4992–5004. Article CAS PubMed Google Scholar * Oehmen A, Lemos PC, Carvalho G, Yuan Z, Keller J, Blackall LL _et al_
(2007). Advances in enhanced biological phosphorus removal: from micro to macro scale. _Water Res_ 41: 2271–2300. Article CAS PubMed Google Scholar * Oosterkamp MJ, Veuskens T, Plugge
CM, Langenhoff AAM, Gerritse J, van Berkel WJH _et al_ (2011). Genome sequences of _Alicycliphilus denitrificans_ strains BC and K601T. _J Bacteriol_ 193: 5028–5029. Article CAS PubMed
PubMed Central Google Scholar * Penn K, Jensen PR . (2012). Comparative genomics reveals evidence of marine adaptation in _Salinispora_ species. _BMC Genom_ 13: 86. Article Google Scholar
* Peterson SB, Warnecke F, Madejska J, McMahon KD, Hugenholtz P . (2008). Environmental distribution and population biology of the genus Accumulibacter, a primary agent of biological
phosphorus removal in activated sludge. _Environ Microbiol_ 10: 2692–2703. Article CAS PubMed PubMed Central Google Scholar * R Development Core Team (2009) _R: A Language and
Environment for Statistical Computing_. R Foundation for Statistical Computing _Vienna, Austria_. * Reno ML, Held NL, Fields CJ, Burke PV, Whitaker RJ . (2009). Biogeography of the
_Sulfolobus islandicus_ pan-genome. _Proc Natl Acad Sci_ 106: 8605–8610. Article CAS PubMed PubMed Central Google Scholar * Rice P, Longden I, Bleasby A . (2000). EMBOSS: The European
Molecular Biology Open Software Suite. _Trends Genet_ 16: 276–277. Article CAS PubMed Google Scholar * Rusch DB, Halpern AL, Sutton G, Heidelberg KB, Williamson S, Yooseph S _et al_
(2007). The Sorcerer II Global Ocean Sampling expedition: Northwest Atlantic through Eastern Tropical Pacific. _PLoS Biol_ 5: 398–431. Article CAS Google Scholar * Skennerton C, Angly F,
Breitbart M, Bragg J, He S, Hugenholtz P _et al_ (2011). Phage encoded H-NS: a potential Achilles heel in the bacterial defence system. _PLoS One_ 6: e20095. Article CAS PubMed PubMed
Central Google Scholar * Suyama M, Torrents D, Bork P . (2006). PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. _Nucleic Acids Res_ 34:
W609–W612. Article CAS PubMed PubMed Central Google Scholar * Teeling H, Meyerdierks A, Bauer M, Amann R, Glockner FO . (2004). Application of tetranucleotide frequencies for the
assignment of genomic fragments. _Environ Microbiol_ 6: 938–947. Article CAS PubMed Google Scholar * Weelink SAB, Tan NCG, ten Broeke H, van den Kieboom C, van Doesburg W, Langenhoff AAM
_et al_ (2008). Isolation and characterization of _Alicycliphilus denitrificans_ strain BC, which grows on benzene with chlorate as the electron acceptor. _Appl Environ Microbiol_ 74:
6672–6681. Article CAS PubMed PubMed Central Google Scholar * Wexler M, Richardson DJ, Bond PL . (2009). Radiolabelled proteomics to determine differential functioning of
<i>Accumulibacter</i> during the anaerobic and aerobic phases of a bioreactor operating for enhanced biological phosphorus removal. _Environ Microbiol_ 11:
3029–3044. Article CAS PubMed Google Scholar * Wilmes P, Andersson AF, Lefsrud MG, Wexler M, Shah M, Zhang B _et al_ (2008). Community proteogenomics highlights microbial strain-variant
protein expression within activated sludge performing enhanced biological phosphorus removal. _ISME J_ 2: 853–864. Article CAS PubMed Google Scholar * Yang ZH . (2007). PAML 4:
Phylogenetic analysis by maximum likelihood. _Mol Biol Evol_ 24: 1586–1591. Article CAS PubMed Google Scholar * Zerbino DR, Birney E . (2008). Velvet: Algorithms for _de novo_ short read
assembly using de Bruijn graphs. _Genome Res_ 18: 821–829. Article CAS PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS We thank Francisco Rodriguez-Valera,
Rohit Ghai and Ana Belén Martín Cuadrado at the Universidad Miguel Hernandez in Alicante, Spain, for insightful discussion, ideas for analysis and access to custom perl scripts, and Forest
Bishop, Alisha Truman and Nick Bartolerio for assistance with running bioreactors. We also thank Ben Oyserman for thoughtful comments on early drafts of the manuscript. KDM acknowledges
funding from the US National Science Foundation (CBET-0967646) and the UW-Madison Graduate School. The work conducted by the US Department of Energy Joint Genome Institute is supported by
the Office of Science of the US Department of Energy under Contract No. DE-AC02–05CH11231. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Departments of Civil and Environmental Engineering,
and Bacteriology, University of Wisconsin at Madison, Madison, WI, USA Jason J Flowers & Katherine D McMahon * US Department of Energy Joint Genome Institute, Walnut Creek, CA, USA
Shaomei He, Stephanie Malfatti, Tijana Glavina del Rio & Susannah G Tringe * Australian Centre for Ecogenomics, University of Queensland, St Lucia, Queensland, Australia Philip
Hugenholtz Authors * Jason J Flowers View author publications You can also search for this author inPubMed Google Scholar * Shaomei He View author publications You can also search for this
author inPubMed Google Scholar * Stephanie Malfatti View author publications You can also search for this author inPubMed Google Scholar * Tijana Glavina del Rio View author publications You
can also search for this author inPubMed Google Scholar * Susannah G Tringe View author publications You can also search for this author inPubMed Google Scholar * Philip Hugenholtz View
author publications You can also search for this author inPubMed Google Scholar * Katherine D McMahon View author publications You can also search for this author inPubMed Google Scholar
CORRESPONDING AUTHOR Correspondence to Katherine D McMahon. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no conflict of interest. ADDITIONAL INFORMATION Supplementary
Information accompanies this paper on The ISME Journal website SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION (DOC 713 KB) RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS
ARTICLE CITE THIS ARTICLE Flowers, J., He, S., Malfatti, S. _et al._ Comparative genomics of two ‘_Candidatus_ Accumulibacter’ clades performing biological phosphorus removal. _ISME J_ 7,
2301–2314 (2013). https://doi.org/10.1038/ismej.2013.117 Download citation * Received: 06 December 2012 * Revised: 14 May 2013 * Accepted: 11 June 2013 * Published: 25 July 2013 * Issue
Date: December 2013 * DOI: https://doi.org/10.1038/ismej.2013.117 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a
shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative KEYWORDS * enhanced biological phosphorus
removal * ‘_Candidatus_ Accumulibacter phosphatis’ * activated sludge