Play all audios:
ABSTRACT Dubin-Johnson syndrome (DJS) is an autosomal recessive disorder characterized by conjugated hyperbilirubinemia and caused by mutations of the ATP-binding cassette (ABC) transporter
encoding gene_ MRP2/cMOAT/ABCC2_. Previous studies reported on mutations in DJS patients and polymorphisms in healthy human individuals. The genomic DNA sequence of a female Caucasian DJS
patient was analyzed by DNA sequencing and revealed the identification of a homozygous missense mutation C2302T. This DJS-causing alteration results in an amino acid exchange Arg768Trp.
INTRODUCTION Dubin-Johnson syndrome (DJS, MIM 237500) is a rare, benign, and autosomal recessive disorder, which is characterized by a defect in the transfer of endogenous and exogenous
anionic conjugates from hepatocytes into the bile, causing conjugated hyperbilirubinemia and deposition of brown pigments in the liver (Dubin and Johnson 1954). The molecular basis of this
impaired hepatobiliary transport in DJS patients is the absence of functional ABCC2 protein. The ATP-binding cassette (ABC) transporter ABCC2, also known as canalicular multispecific organic
anion transporter (cMOAT) or multidrug resistance protein 2 (MRP2), is an integral membrane glycoprotein of about 190 kDa localized to the apical domain of polarized cells, such as the
canalicular membrane of hepatocytes or the apical membrane of renal proximal tubules (König et al. 1999). In the liver, the ABCC2 protein mediates the multispecific efflux of various types
of organic anions, including glucuronate, sulfate, and glutathione conjugates, across the canalicular hepatocyte membrane into the bile, dependent on ATP-hydrolysis (Suzuki and Sugiyama
2002). So far, several mutations and polymorphisms of the human_ ABCC2_ gene have been reported (Wada et al. 1998; Toh et al. 1999; Ito et al. 2001; Mor-Cohen et al. 2001; Itoda et al. 2002;
Saito et al. 2002). In this study, we report a mutation of the_ ABCC2_ gene in a patient with DJS, which was also described to be a single nucleotide polymorphism (SNP) in healthy specimens
by other authors (Ito et al. 2001; Saito et al. 2002). Additionally, the SNP C-24T (NCBI SNP ID rs717620) of the 5'-untranslated region (5'-UTR) in a set of healthy European
individuals and a panel of human tumor cell lines was analyzed. PATIENT AND METHODS A female Caucasian DJS patient of Turkish origin, aged 37, was examined by a needle biopsy performed for
diagnostic reasons. Anamnesis of the family showed no additional proven case of DJS. The serum total and conjugated bilirubin levels were elevated (2.4 mg/dl and 1.9 mg/dl, respectively).
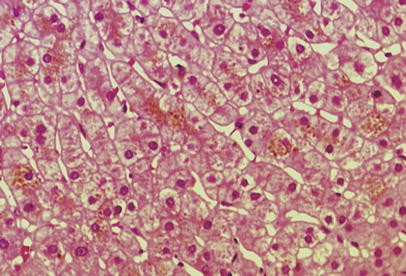
Histopathologic examination of the liver biopsy revealed golden-brown pigmented granules in the cytoplasm of the liver cells, typical for DJS (Fig. 1). Control subjects were healthy patients
in terms of DJS at the University Hospital Charité (Berlin, Germany). The DJS patient and control subjects gave their informed consent prior to their inclusion in the study. Blood samples
of the DJS patient and 20 control subjects were used for genetic analysis. The following human tumor cell lines were used: the melanoma cell line MeWo (Fogh et al. 1978), the gastric
carcinoma cell line EPG85–257 (Dietel et al. 1990), the pancreatic carcinoma cell line EPP85–181 (Lage und Dietel 2002), the ovarian carcinoma cell line A2780 (Eva et al. 1982), the colon
carcinoma cell line HT-29 (Thomas et al. 1974), and the adrenocortical carcinoma cell line D43/86 (not published). Cell lines were grown using standard procedures as described previously
(Dietel et al. 1990; Lage and Dietel 2002). Genomic DNA was prepared from peripheral blood leukocytes and cultured tumor cell lines by standard methods as described previously (Lage and
Dietel 2002). Polymerase chain reaction (PCR) amplification was performed using 150 ng genomic DNA, 0.2 µM of each oligonucleotide primer (Table 1), 0.2 mM of each dNTP, 50 mM KCl, 10 mM
Tris-HCl, pH 8.3, 1.5 mM MgCl2, 0.001% gelatin, and 1 unit Ampli_Taq-_Gold DNA-polymerase (Perkin Elmer) in a final volume of 25 µl. Cycling conditions: initial denaturation at 94°C for 12
min followed 36 cycles of amplification (denaturation at 94°C, 1 min; annealing at 55°C, 90 s; extension at 72°C, 90 s). The last cycle contained a 5-min extension step at 72°C. The PCR
products were sequenced directly by using dye terminator sequencing and an ABI-373 sequencer (Perkin Elmer). The sequencing was repeated independently, and mutation analysis was performed by
comparison with the_ ABCC2_ reference sequence NM_000392 (NCBI). RESULTS AND DISCUSSION The genomic DNA of a Caucasian DJS patient was analyzed with PCR methods using primer pairs (Table 1)
that cover the entire ABCC2 cDNA and exon-intron junction regions and screened for mutations. The only alteration found was the missense mutation C2302T causing an amino acid exchange
Arg768Trp indicating to be the molecular basis for DJS in this Turkish patient. This mutation was not found in any of the 26 control specimens tested. The nucleotide exchange C2302T located
in the highly conserved domain, the Walker C motif, was previously described by other authors (Wada et al. 1998; Toh et al. 1999; Ito et al. 2001) in DJS patients as well as in healthy
specimens. Wada et al. (1998) and Toh et al. (1999) reported the homozygous mutation C2302T as genetic alteration causing DJS in patients. Ito et al. (2001) examined a panel of healthy
Japanese specimens to detect mutations in the_ ABCC2_ gene and found the mutation C2302T to be heterozygous in one of 48 specimens. In accordance with our results, Itoda et al. (2002) could
not detect the alteration C2302T in any of 72 cell lines derived from various tissues of healthy Japanese individuals for the purpose of detecting polymorphisms in the_ ABCC2_ gene. It was
reported that the amino acid exchange Arg768Trp causes a deficient maturation of the ABCC2 protein leading to a diminished glycosylation, impaired sorting to the apical membrane, and
degradation via the proteasome pathway (Hashimoto et al. 2002). These results support our findings that the missense mutation Arg768Trp alone is responsible for the defect in transporting
bilirubin conjugates, leading to hyperbilirubinemia in the context of DJS in the examined patient. The observation that this homozygous mutation was found in Japanese patients (Wada et al.
1998; Toh et al. 1999) and in a Caucasian DJS patient with Turkish descent (present study) suggests that this dysfunctional allel is relatively common in the human genetic pool. Further
analyzes with more healthy specimens and DJS patients are needed to determine whether this allel is also present in other ethnic populations, and to compare allel frequencies among them.
With data of other genetic loci, these data could give new insights in population founding, migration, and development. In the_ ABCC2_ gene mutation analysis of the DJS patient we used
genomic DNA of 20 healthy individuals and six human tumor cell lines as control. The DJS-causing base substitution C2302T was not detected in any of the control specimens. In order to
demonstrate that the used control subjects and cell lines are representative for the population, one of the most common polymorphisms of the ABCC2 encoding gene (Ito et al. 2001; Mor-Cohen
et al. 2001; Itoda et al. 2002), SNP C-24T, was analyzed. In four of 20 control individuals the SNP C-24T was detected, whereas the DJS patient did not show this polymorphism. Among the six
human tumor cell lines analyzed, the melanoma cell line MeWo and the pancreatic carcinoma cell line EPP85–181 contained this SNP, whereas the other four cell lines did not (Table 2). All six
control specimen with the SNP C-24T were heterozygous for this polymorphism. The allel frequency of 0.889 for C and 0.111 for T at this locus was similar to other studies, i.e., C=0.813 and
T=0.188 (Ito et al., 2001), or C=0.819 and T=0.181 (Itoda et al. 2002) in Japanese healthy individuals. Mor-Cohen et al. (2001) examined three Jewish populations and reported allel
frequencies between 0.739 and 0.877 for C and between 0.122 and 0.261 for T for the SNP C-24T. These data indicate a comparable allel frequency for the SNP C-24T in all analyzed populations.
CONCLUSIONS The homozygous missense mutation C2302T causing the amino acid exchange Arg768Trp is the molecular reason for DJS in a female Caucasian patient examined. Since this DJS-causing
mutation was reported in a Japanese population previously, the data indicate that C2302T is a common mutation in DJS patients in different populations. Analysis of the_ ABCC2_ gene in DJS
patients and molecular proof of the various mutations will improve the understanding of this ABC transporter in liver function. REFERENCES * Dietel M, Arps H, Lage H, Niendorf A (1990)
Membrane vesicle formation due to acquired mitoxantrone resistance in human gastric carcinoma cell line EPG85–257. Cancer Res 50:6100–6106 CAS PubMed Google Scholar * Dubin IN, Johnson FB
(1954) Chronic idiopathic jaundice with unidentified pigment in liver cells: a new clinical entity with a report of 12 cases. Medicine 33:952–961 Article Google Scholar * Eva A, Robbins
KC, Andersen PR, Srinivasan A, Tronick SR, Reddy EP, Ellmore NW, Galen AT, Lautenberger JA, Papas TS, Westin EH, Wong-Staal F, Gallo RC, Aaronson SA (1982) Cellular genes analogues to
retroviral onco genes are transcribed in human tumour cells. Nature 295:116–119 Article CAS Google Scholar * Fogh J, Bean MA, Brüggen J, Fogh H, Fogh JM, Hammar SP, Kodera Y, Loveless JD,
Sorg C, Wright WC (1978) Comparison of human tumor cell line before and after growth in the nude mouse. In: Fogh J, Giovanella B, (eds). The nude mouse in experimental and clinical
research. Academic Press, New York: 215–234 * Hashimoto K, Uchiumi T, Konno T, Ebihara T, Nakamura T, Wada M, Sakisaka S, Maniwa F, Amachi T, Ueda K, Kuwano M (2002) Trafficking and
functional defects by mutations of the ATP-binding domains in MRP2 in patients with Dubin-Johnson syndrome. Hepatology 36:1236–1245 Article CAS Google Scholar * Ito S, Ieiri I, Tanabe M,
Suzuki A, Higuchi S, Otsubo K (2001) Polymorphism of the ABC transporter genes_ MDR1_,_ MRP1_ and_ MRP2/cMOAT_, in healthy Japanese subjects. Pharmacogenetics 11:175–184 Article CAS Google
Scholar * Itoda M, Saito Y, Soyama A, Saeki M, Murayama N, Ishida S, Sai K, Nagano M, Suzuki H, Sugiyama Y, Ozawa S, Sawada J (2002) Polymorphisms in the_ ABCC2_ (_cMOAT/MRP2_) gene found
in 72 established cell lines derived from Japanese individuals: an association between single nucleotide polymorphisms in the 5'-untranslated region and exon 28. Drug Metab Dispos
30:363–364 Article CAS Google Scholar * König J, Nies AT, Cui Y, Leier I, Keppler D (1999) Conjugate export pumps of the multidrug resistance protein (MRP) family: localization, substrate
specificity, and MRP2-mediated drug resistance. Biochim Biophys Acta 1461:377–394 Article Google Scholar * Lage H, Dietel M (2002) Multiple mechanisms confer different drug-resistant
phenotypes in pancreatic carcinoma cells. J Cancer Res Clin Oncol 128:349–357 Article CAS Google Scholar * Mor-Cohen R, Zivelin A, Rosenberg N, Shani M, Muallem S, Seligsohn U (2001)
Identification and functional analysis of two novel mutations in the multidrug resistance protein 2 gene in Israeli patients with Dubin-Johnson syndrome. J Biol Chem 276:36923–36930 Article
CAS Google Scholar * Saito S, Iida A, Sekine A, Miura Y, Ogawa C, Kawauchi S, Higuchi S, Nakamura Y (2002) Identification of 779 genetic variations in eight genes encoding members of
ATP-binding cassette, subfamily C (ABCC/MRP/CFTR). J Hum Genet 47:147–171 Article CAS Google Scholar * Suzuki H, Sugiyama Y (2002) Single nucleotide polymorphisms in multidrug resistance
associated protein 2 (MRP2/ABCC2): its impact on drug disposition. Adv Drug Deliv Rev 54:1311–1331 Article CAS Google Scholar * Toh S, Wada M, Uchiumi T, Inokuchi A, Makino Y, Horie Y,
Adachi Y, Sakisaka S, Kuwano M (1999) Genomic structure of the canalicular multispecific organic anion-transporter gene (_MRP2/cMOAT_) and mutations in the ATP-binding-cassette region in
Dubin-Johnson syndrome. Am J Hum Genet 64:739–746 Article CAS Google Scholar * Thomas DR, Philpott GW, Jaffe BM (1974) Prostaglandin E (PGE) control of cell proliferation in vitro:
characteristics of HAT-29. J Surg Res 16:463–465 Article CAS Google Scholar * Wada M, Toh S, Taniguchi K, Nakamura T, Uchiumi T, Kohno K, Yoshida I, Kimura A, Sakisaka S, Adachi Y, Kuwano
M (1998) Mutations in the canalicular multispecific organic anion transporter (cMOAT) gene, a novel ABC transporter, in patients with hyperbilirubinemia II/Dubin-Johnson syndrome. Human Mol
Genet 7:203–207 Article CAS Google Scholar Download references ACKNOWLEDGEMENTS This work was supported by grants LA1039/1-3 and LA1039/2-1 of the Deutsche Forschungsgemeinschaft (DFG).
The authors thank Dr. Christiane Otto (Berlin) for her support and for providing clinical data. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Institute of Pathology, Charité Campus Mitte,
Humboldt University Berlin, Schumannstr. 20/21, 10117, Berlin, Germany Verena Materna & Hermann Lage Authors * Verena Materna View author publications You can also search for this author
inPubMed Google Scholar * Hermann Lage View author publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to Hermann Lage. RIGHTS AND
PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Materna, V., Lage, H. Homozygous mutation Arg768Trp in the ABC-transporter encoding gene_ MRP2/cMOAT/ABCC2_ causes
Dubin-Johnson syndrome in a Caucasian patient. _J Hum Genet_ 48, 484–486 (2003). https://doi.org/10.1007/s10038-003-0057-8 Download citation * Received: 15 May 2003 * Accepted: 01 July 2003
* Published: 27 August 2003 * Issue Date: September 2003 * DOI: https://doi.org/10.1007/s10038-003-0057-8 SHARE THIS ARTICLE Anyone you share the following link with will be able to read
this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative
KEYWORDS * MRP2 * cMOAT * ABCC2 * DJS * Mutation * Polymorphism