Play all audios:
ABSTRACT During inflammation, host- and microbial-derived proteases trigger the activation of protease-activated receptors (PARs), a family of G-protein-coupled receptors. We report here
that activation of Toll-like receptors (TLRs) by fungi unmasks an essential and divergent role for PAR1 and PAR2 in downstream signaling and inflammation. TLRs activated PARs and triggered
distinct signal transduction pathways involved in inflammation and immunity to _Candida albicans_ and _Aspergillus fumigatus._ Inflammation was promoted by PAR1 and PAR2 activation in
response to _Candida_ and by PAR2 inhibition in response to _Aspergillus_. This occurred by TLR regulation of PAR signaling, with TLR2 promoting PAR1 activity, and TLR4 suppressing PAR2
activity. Thus, tissue injury and pathogens induce signals that are integrated at the level of distinct TLR/PAR-dependent pathways, the exploitation or subversion of which contributes to
divergence in microbial promotion of inflammatory response. SIMILAR CONTENT BEING VIEWED BY OTHERS DIFFERENTIAL SIGNALLING REQUIREMENTS FOR RIPK1-DEPENDENT PYROPTOSIS IN NEUTROPHILS AND
MACROPHAGES Article Open access 04 July 2024 ACTIVATION OF LEUKOTRIENE B4 RECEPTOR 1 IS A PREREQUISITE FOR COMPLEMENT RECEPTOR 3-MEDIATED ANTIFUNGAL RESPONSES OF NEUTROPHILS Article 31
January 2024 PHAGOSOMAL SIGNALLING OF THE C-TYPE LECTIN RECEPTOR DECTIN-1 IS TERMINATED BY INTRAMEMBRANE PROTEOLYSIS Article Open access 06 April 2022 INTRODUCTION Toll-like receptors (TLRs)
control the inflammatory response to fungal infection, which is characteristic of innate immunity.1 However, cooperation between innate immune receptors is of critical importance to
regulating and shaping antimicrobial immunity.2, 3, 4 During inflammation, host- or fungal-derived proteases are released into the extracellular environment.5, 6, 7 Certain extracellular
proteases can specifically cleave and trigger protease-activated receptors (PARs), a family of four G-protein-coupled receptors.5, 6 Thus, PARs are viewed as an integral component of the
host antimicrobial alarm system capable of affecting host defense and immunity.8 Four PARs have been cloned and they all share the same basic mechanism of activation: the proteolytic
unmasking of a tethered peptide ligand resides in the receptor's N-terminal exodomain, and synthetic peptides that mimic this sequence function as agonists that activate PARs
independent of receptor cleavage. Through their unique ability to sense serine proteases, such as thrombin, trypsin, and mast cell tryptase, PARs act as “sensors” of extracellular protease
gradients.5 ,6 Thrombin activates PAR1, PAR3, and PAR4, whereas trypsin and mast cell tryptase activate PAR2. However, certain microbial proteases can also activate mammalian PARs.9
Activated PARs couple to signaling cascades that affect, among others, coagulation and inflammatory responses.10 The role of PARs in inflammation is complex, as individual PARs have both
proinflammatory and protective roles in the airway11 and the gastrointestinal tract6, 12, 13, 14, 15 as well as in the brain,16 depending on disease context and cellular type. Several
observations suggest that PARs contribute to the host responses to fungal infections. First, serine proteases are activated when mannan lectins bind to fungi.17 Second, fungal proteases, in
common with PAR agonists (PAR-APs), initiate inflammatory responses.18, 19 Third, fungi are able to interact with cells and pathways of the coagulation cascade, as evidenced by the
stimulation of tissue factor activity by _Aspergillus_ conidia and the massive intravascular thrombosis at foci of _Aspergillus_ infection.20 However, the involvement of PARs in fungal
infections has not been directly examined. In the present study, we used PAR-APs, PAR antagonists (PAR-ANTs), and mice lacking or overexpressing PARs and TLRs to assess the PAR/TLR cross
talk in infections caused by _Candida albicans_ and _Aspergillus fumigatus,_ two major fungal pathogens.1 We found that PAR1 and PAR2 have opposing roles in governing the inflammatory
response and pathology to _Aspergillus_ or _Candida_ and this occurred by delegation of different TLRs. Thus, the study identifies a previously unknown cross talk between PARs and TLRs in
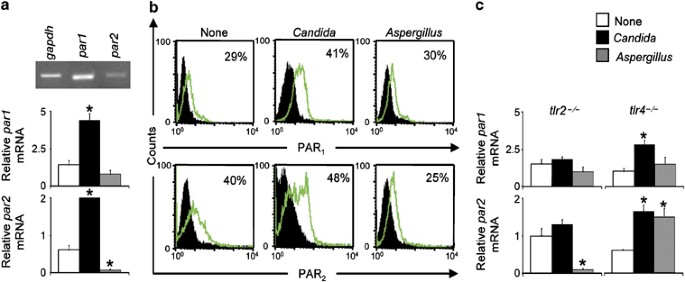
fungal infections. RESULTS FUNGI REGULATE EXPRESSION OF PAR1 AND PAR2 IN PMNS _IN VITRO_ To establish whether fungal infections affect PAR expression, we studied polymorphonuclear
neutrophils (PMNs) because they are essential for the initiation and execution of the acute inflammatory response to fungi and human PMNs express functional PAR2.21 We found that murine PMNs
expressed both _par1_ and _par2_ mRNA and PAR1 and PAR2 proteins (Figure 1a,b). Exposure of PMNs to fungi differentially regulated PAR1 and PAR2 expression. _Candida_ yeasts increased the
expression of _par1_ and _par2_ mRNA and immunoreactivity, whereas _Aspergillus_ conidia decreased _par2_ expression (Figure 1a,b). FUNGI REGULATE PAR EXPRESSION THROUGH TLR-DEPENDENT
MECHANISMS In mice, the fungicidal activity and inflammatory pathology of PMNs are strictly dependent upon distinct TLR signaling pathways.22 As TLR2 and TLR4 exert distinct effects on the
inflammatory responses of mice with candidiasis or aspergillosis,23 we assessed the involvement of TLR2 and TLR4 in the modulation of _par_ expression by fungi. _Candida_ failed to increase
_par1_ and _par2_ gene expression in _tlr2_−/− PMNs; _Aspergillus_ instead increased _par2_ expression in _tlr4_−/− PMNs (Figure 1c). These data demonstrate that fungi regulate expression of
_par1_ and _par2_ in a manner that is differentially dependent upon TLR2 and TLR4. FUNGI REGULATE EXPRESSION OF _PAR1_ AND _PAR2 IN VIVO_ To assess whether modulation of _par_ expression
occur also _in vivo_ during infection, we evaluated _par1_ and _par2_ expression in the stomach of mice with gastrointestinal candidiasis or the lungs of mice with pulmonary aspergillosis.
Consistent with our _in vitro_ findings, _par1_ expression was increased in the stomach of mice with candidiasis, whereas _par2_ expression was decreased in the lungs of mice with
aspergillosis (Figure 2). However, _par2_ was not upregulated in the stomach of _Candida_-infected mice as it was in isolated PMNs. The expression of _par1_ was not increased in _tlr2_−/−
mice with candidiasis, and _par2_ expression was upregulated in the lungs of _tlr4_−/− mice with aspergillosis (Figure 2). Thus, TLR signaling, in response to fungi, modulates _par_
expression both _in vitro_ and _in vivo_, even though the expression _in vivo_ likely reflects higher levels of complexity. HOST PROTEASES REGULATE THE EXPRESSION AND FUNCTION OF PAR Serine
proteases from PMNs, such as elastase and cathepsin G, which are stored in and released from azurophilic granules, can cleave PAR1 and PAR2 at sites that disable these receptors, and may
thereby act in an autocrine and paracrine fashion to downregulate PAR signaling.24 Fungi control secretory responses of PMNs, including protease activity, through TLR signaling.22, 23
Therefore, we hypothesized that fungi, through TLR activation, stimulate the release of proteases from PMNs that cleave PAR1 and PAR2 and modulate their activity. Because _par1_ was
upregulated by _Candida_ and _par2_ downregulated by _Aspergillus_, we analyzed PAR1 or PAR2 activation in response to _Candida_ or _Aspergillus_, respectively. To test the hypothesis, we
analyzed calcium mobilization in HEK293 cells, which naturally express both PAR1 and PAR2,25 exposed to the following stimuli: (i) elastase and cathepsin G; (ii) supernatants from wild type
(WT) PMNs exposed to _Candida_ or _Aspergillus_ (each containing 20 ng ml−1 protease activity); and (iii) supernatants from _tlr2_−/− or _tlr4_−/− PMNs challenged with the above fungi. We
subsequently stimulated HEK cells with thrombin or trypsin and measured [Ca2+]i and receptor surface expression to assess the activation of PAR1 or PAR2, respectively.We found that (i)
thrombin and trypsin stimulated a prompt and transient increase in [Ca2+]i in untreated HEK293 cells, which suggests activation of PAR1 and PAR2 (Figure 2a); (ii) the combination of elastase
and cathepsin G also increased [Ca2+]i and this treatment had no effect on responses to thrombin (Figure 3a, upper panel) but abolished responses to trypsin (Figure 3a, lower panel), a
finding in line with the PAR2 disarming ability of neutrophil serine proteases;24 (iii) supernatants from WT, _tlr2_−/−, and _tlr4_−/− PMNs also increased [Ca2+]i, although we do not know
whether this response is mediated by PARs or other mechanisms; and (iv) pre-exposure to supernatants from _Candida_-exposed PMNs, either WT or TLR-deficient, did not affect thrombin
signaling (Figure 3a, upper panel), but supernatants from _Aspergillus_-stimulated WT and _tlr2_−/−, but not _tlr4_−/−, PMNs, prevented trypsin signaling (Figure 3a, lower panel).
Cytofluorimetric analysis confirmed the disparate activity of the different supernatants on PAR activation. Exposure to thrombin or trypsin reduced the levels of immunoreactive PAR1 and PAR2
at the cell surface, which indicates cleavage, activation, and internalization of these receptors (Figure 3b). The supernatant alone from _Candida_-exposed PMNs increased the surface
expression of PAR1, a finding suggestive of receptor exocytosis (Figure 3b, top panel). Subsequent exposure to thrombin decreased the immunoreactivity of PAR1 at the cell surface, indicating
an intact mechanism of receptor activation. In contrast, subsequent exposure to trypsin did not cause a decrease in immunoreactivity of PAR2 at the cell surface after treatment with
supernatant from _Aspergillus_-exposed PMNs, suggesting that the supernatant had prevented PAR2 activation (Figure 3b, lower panel). Together, these results indicate that fungi activate PMNs
in a TLR-dependent fashion to release factors that modulate the activity of PAR1 and PAR2. FUNGAL PROTEASES CONTRIBUTE TO PAR ACTIVATION As both fungi secrete a variety of proteases _in
vivo_ and _in vitro_,7 and microbial proteases are known to activate PAR receptors,9 we assessed whether the ability of _Aspergillus_ culture supernatant to inhibit trypsin signaling was
sensitive to protease inhibition. For this purpose, supernatants from both _Candida_ and _Aspergillus_ were treated with protease inhibitors and used to assess [Ca2+]i mobilization in HEK293
in response to thrombin and trypsin, respectively. The results show that, while treatment with protease inhibitors did not modify the activity of _Candida_ culture supernatants, it
abolished the ability of supernatants from _Aspergillus_ cultures to inhibit trypsin signaling (Figure 3c). These findings suggest that _Aspergillus_ proteases are involved in PAR2
deactivation. Recombinant fungal proteases (the _Aspergillus_ serine protease Aspf18 and _Candida_ aspartyl protease SAP2) failed to show any activity (data not shown). Further studies in
HEK293 cells exposed to _Candida_ supernatant or SAP2 revealed that neither stimuli affected _par_ mRNA expression. With _Aspergillu_s, despite the ability to inhibit trypsin signaling,
culture supernatant did not affect either PAR expression while, similar to other protease allergens,26 Aspf18 downregulated the expression of _par1_ (Supplementary Figure S1 online). Thus,
_Candida_ activates PAR1 and PAR2 by a TLR2-dependent mechanism, an effect that is not mediated by fungal proteases, while _Aspergillus_ proteases may contribute to the downregulation of
trypsin-induced activation of PAR2, which occurs by a TLR4-dependent mechanism. PAR SIGNALING DEPENDS ON THE PRESENCE OF TLRS To investigate the contribution of TLRs to PAR signaling and
_vice versa_, we first evaluated the activation of extracellular signal-regulated kinase 1/2 (ERK1/2) and p38 mitogen-activated kinases (MAPKs) as well as NF-κB in WT PMNs exposed to PAR-APs
or PAR-ANTs, as these pathways are known to be involved in PAR5, 6 and TLR27, 28 signaling and to mediate PMN antimicrobial effector functions.29 We also studied _tlr2_−/− or _tlr4_−/− PMNs
and PMNs lacking myeloid differentiation primary-response protein 88 (MyD88−/−), since TLRs couple ligand binding to cell activation through members of the MyD88 family.27, 28 Figure 4a
shows that PAR1-AP and PAR2-AP-stimulated, and the corresponding antagonists prevented ERK1/2 and p38 phosphorylation and NF-κB activation in WT PMNs. PAR1-AP and PAR2-AP did not
phosphorylate ERK1/2 or activate NF-κB in _tlr2_−/− or _myd88_−/− PMNs, whereas _tlr4_−/− PMNs, despite a strong basal level of phosphorylation, responded normally (Figure 4b). In contrast,
p38 phosphorylation by both PAR agonists was totally abolished in _tlr4_−/− PMNs and retained in _tlr2_−/− or _myd88_−/− PMNs (Figure 4b). Because phorbol esters stimulated phosphorylation
of ERK1/2 and p38 similarly in TLR-deficient PMNs (Figure 4c), this indicates that these signaling pathways are not intrinsically defective in TLR- or MyD88-deficient cells. Therefore, the
downstream pathways activated by PARs in PMNs depend upon distinct TLR signaling. Agonists of PAR1 and PAR2 activate either ERK1/2 and NF-κB in a TLR2-/MyD88-dependent manner or p38 in a
TLR4-dependent but MyD88-independent manner. As a matter of fact, the basal level of p38 phosphorylation was undetectable in _tlr4_−/− PMNs. This finding is consistent with the existence of
divergent signaling pathways, originating upon TLR4 activation,30, 31 which control MAPK activity.32 TLR SIGNALING DEPENDS ON THE PRESENCE OF PARS The above results showed how the activation
of PARs depends upon TLR signaling. To assess instead how PARs contribute to TLR signaling, we silenced the expression of _par1_ and _par2_ using short interfering RNA (siRNA) in TLR2- or
TLR4-transfected HEK293 cells, respectively, and assessed the effects of the TLR2 ligand, zymosan, or the TLR4 ligand, lipopolysaccharide, on activation of NF-κB. While the levels of the p65
and p50 NF-κB proteins were unmodified by siRNA treatments (Figure 4d), zymosan promoted NF-κB activation in TLR2-transfected cells and _par1_ silencing inhibited this activation (Figure
4d). In contrast, lipopolysaccharide also activated NF-κB in TLR4-transfected cells, but _par2_ silencing further increased this stimulation (Figure 4d). These data show that downstream
pathways activated by TLRs depend on the expression of _par1_ and _par2_. Altogether, the data indicate that PAR signaling depends on the expression of TLRs and _vice versa._ SIGNALING
PATHWAYS ACTIVATED BY PARS ARE SUBVERTED BY FUNGI Given that fungi modulate PAR expression in infection _in vitro_ and _in vivo_, we assessed whether signaling pathways activated by either
PAR are also subverted by fungi. For this purpose, we sequentially exposed WT PMNs to fungi and PAR-APs. Figure 4e shows that stimulation with _Candida_ and either PAR-AP markedly enhanced
ERK1/2 phosphorylation and NF-κB activation. However, disparate results were obtained by concomitant stimulation with _Aspergillus_. ERK1/2 phosphorylation and NF-κB activation were still
promoted by PAR1-AP but inhibited by PAR2-AP. Interestingly, PAR2-AP markedly enhanced p38 phosphorylation. As activation of p38 has been associated with inhibition of NF-κB activation,33 we
assessed whether SB202190, a highly specific p38 MAPK inhibitor,33 could restore ERK1/2 and NF-κB activation by PAR2-AP. The results showed that p38 inhibition was associated with restored
ERK1/2 phosphorylation and NF-κB activation (Figure 4e), a finding implicating p38 in the inhibitory action of PAR2 in the presence of the fungus. Experiments in _myd88_−/− PMNs confirmed
that ERK1/2 phosphorylation and NF-κB activation occurred in an MyD88-dependent pathway and p38 phosphorylation in an MyD88-independent pathway (Figure 4e). Therefore, PAR1 and PAR2 activate
distinct pathways in response to fungi. Consistent with the effects of the fungus on PAR1 expression, PAR1-AP promoted _Candida_-induced ERK1/2 and NF-κB activation, which occurs by a TLR2-
and MyD88-dependent mechanism. In contrast, PAR2 had divergent effects depending on the fungus. PAR2-AP enhances _Candida_-induced activation of ERK1/2 and NF-κB but suppresses these
pathways in response to _Aspergillus_ through a p38-dependent mechanism. DIVERGENT ROLE OF PAR1 AND PAR2 IN PMNS' INFLAMMATORY RESPONSE Further studies showed that the agonistic and
antagonistic effects of PAR1 and PAR2 on fungal-induced TLR signaling elicited distinct inflammatory responses in PMNs. Consistent with the pattern of ERK1/2 activation, PAR1-AP increased
(by 25–30%) and PAR1-ANT decreased the respiratory burst of WT PMNs in response to both fungi, but particularly to _Candida_ (80% inhibition compared to 30% to _Aspergillus_) (Figure 4f).
PAR2-AP greatly inhibited the respiratory burst (by ∼60%) in response to _Aspergillus_ but not to _Candida,_ and the PAR2-ANT did the opposite (Figure 4f). In terms of degranulation, PAR1-AP
enhanced the production of matrix metalloproteinase 9 (MMP9) in response to _Candida_ and _Aspergillus_, and PAR2-AP dampened it in response to _Aspergillus_ (Figure 4f). PAR1 agonistic
activity was lost in _tlr2_−/− and _myd88_−/− PMNs and retained in _tlr4_−/− PMNs, whereas the PAR2 inhibitory activity was retained in _tlr2_−/− and _myd88_−/− PMNs and lost in _tlr4_−/−
PMNs (Figure 4g). Considered together, these results suggest the existence of functional interactions between TLRs and PARs that modulate inflammatory signaling. The coengagement by TLRs and
PARs of downstream adaptor modules may explain the proinflammatory role of PAR1 and the anti-inflammatory role of PAR2. If extrapolated to host responses in infections, this will predict
that (i) PAR1 promotes the inflammatory response to _Candida_, which is contingent upon TLR2 activation; (ii) PAR2 dampens inflammation to _Aspergillus_ in a TLR4-dependent fashion; and
(iii) PAR2 shows disparate effects in inflammation and immunity to each fungus. These predictions were all confirmed by the following _in vivo_ studies. PAR1 PROMOTES INFLAMMATION AND
IMMUNITY TO _CANDIDA_ THROUGH TLR2 Wild type and _tlr2_−/− mice were infected with _C. albicans_ and treated with selective agonists or antagonists of PAR1, which have been shown to modulate
PAR1 function in mice with colitis.34 PAR1-AP exacerbated and PAR1-ANT attenuated the inflammatory pathology to the fungus, as revealed by signs of acanthosis, parakeratosis, recruitment of
inflammatory cells, and local production of oxidants and MMP9 in the stomach of infected WT mice (Figure 5a). Treatment with PAR1-AP was also associated with a significant increase of
fungal growth (from 2.1 × 104 to 3.9 × 104 CFU,*_P_ < 0.05, untreated vs. treated mice). Consistent with the low level of _par1_ expression (Figure 2), local inflammatory and secretory
responses were not increased in the stomach of infected _tlr2_−/− mice as compared to WT mice and were not modified upon treatments (Figure 5b). Interestingly, both treatments oppositely
affected parameters of adaptive immunity to the fungus, such as the interleukin (IL)-12/IL-10 production by Peyer's patch dendritic cells, known to be regulated by PAR signaling,8 and
the pattern of _ifnγ_ or _il10_ gene expression in CD4+ T cells from mesenteric lymph nodes of WT mice. PAR1-AP promoted the IL-12/Th1 response and PAR1-ANT promoted the IL-10+ T regulatory
cell response35 to the fungus (Figure 5c). Thus, the upregulated expression of PAR1 in candidiasis correlates with the occurrence of local and adaptive inflammatory responses. Similar
treatments of mice with aspergillosis revealed that PAR1-AP exacerbated the lung inflammatory pathology and the local oxidant/MMP9 production, which were largely unaffected by treatment with
PAR1-ANT (Supplementary Figure S3 online). PAR2 ATTENUATES INFLAMMATION AND IMMUNITY TO _ASPERGILLUS_ THROUGH TLR4 Wild type and _tlr4_−/− mice were infected with _A. fumigatus_ and treated
with agonists and antagonists of PAR2 _in vivo_.12 PAR2-ANT exacerbated and PAR2-AP slightly decreased the inflammatory response to _Aspergillus_ in WT mice. The number of abscesses
consisting of inflammatory cells associated with signs of parenchymal destruction and the oxidant/MMP9 production were both higher in the lungs after treatment with PAR2-ANT (Figure 6a).
Although the high level expression of _par2_ (Figure 2) would predict a low inflammatory and secretory response in the lungs of infected _tlr4_−/− mice, foci of inflammation (inset of Figure
6a) could be detected, a finding confirming the contribution of TLR2 to inflammation in aspergillosis.22, 23 However, both inflammatory and secretory responses were augmented by PAR2-ANT
and left unaffected by PAR2-AP (Figure 6b), a finding suggesting that PAR2 negatively regulates inflammation in pulmonary aspergillosis. Since _par2_ expression was downregulated by
_Aspergillus_ through TLR4, our findings suggest that inhibition of PAR2 is a mechanism through which host anti-inflammatory pathways could be subverted to promote inflammation in
aspergillosis. DIVERGENT ROLE OF PAR2 IN INFLAMMATION AND IMMUNITY TO FUNGI _IN VIVO_ Studies in mice with genetic deficiency of (_par2_−/−) or overexpressing (_par2_-Tg) _par2_ confirmed
that PAR2 perturbations mainly affected inflammation and immunity to _Aspergillus_ but not to _Candida_. PAR2 deficiency greatly exacerbated fungal growth (from 2.5 × 104 CFU of WT to 2.2 ×
106 CFU of _par2_−/−) and the inflammatory pathology in the lungs of mice with aspergillosis but not in the stomach of mice with candidiasis. Numerous abscesses and fungal elements as well
as severe signs of parenchymal destruction were present in the lungs of mice with aspergillosis but not in the stomach of mice with candidiasis (Figure 7a). In contrast, the inflammatory
pathology and fungal growth were both significantly reduced, compared to WT mice, in lungs (from 2.5 × 104 to 1.0 × 104 CFU, *_P_<0.05, _par2_-Tg vs. WT) and stomach (from 2.9 × 104 to
1.8 × 104 CFU, _P_<0.05, _par2_-Tg vs. WT) of _par2_-Tg mice with aspergillosis or candidiasis, respectively (Figure 7a). Concomitantly, the local generation of oxidants and MMP9 was
significantly increased in _par2_−/− mice with aspergillosis and impaired in _par2_-Tg mice with either infection (Figure 7b). Interestingly, oxidant generation by _par2_−/− PMNs was greatly
decreased upon exposure to PAR1-ANT (inset), a finding indicating a possible reciprocal PAR regulation. PAR2 also affected the production of cytokines known to reflect the innate immune
response to _Aspergillus in vivo_.1 Tumor necrosis factor-α production was increased and IL-10 production decreased in lung homogenates from infected _par2_−/− mice; the opposite pattern was
observed in _par2_-Tg mice (Figure 7c). As a whole, these data suggest that PAR1 and PAR2 serve an opposite role in governing the inflammatory and immune responses to each fungus and that
the pattern of PAR1 and PAR2 expression at sites of infection may contribute to the occurrence of local inflammation. DISCUSSION Our results reveal novel interactions between TLRs and PARs
that contribute to signal diversity in inflammation and host antimicrobial responses to fungal infections. PAR1 has a dominant role in determining inflammation and Th1 immunity to fungi upon
which PAR2 may exert an inhibitory control. These effects may depend on the functional interactions between PARs and TLRs, which control inflammatory signaling of PMNs. Because PAR1 or PAR2
has disparate effects on the fungicidal activity of PMNs (unpublished data), a combined action of PARs on inflammation and fungicidal activity may account for disparate inflammatory
pathology and fungal burdens in conditions of either PAR deficiency or overexpression. The PAR–TLR interactions exhibit distinct features. First, fungi differentially regulate PAR expression
through TLR2 and TLR4, both in PMNs _in vitro_ and in the stomach and lungs of infected mice. Second, proteases released from PMNs, in a TLR-dependent manner, and fungal proteases can
cleave PARs and alter their capacity to signal. Third, PMN TLRs are required for PAR activation of downstream signaling pathways, and _vice versa_. Finally, the inflammatory responses of
infected animals are dramatically altered by pharmacological (agonism and antagonism) or genetic (deletion and overexpression) manipulation of PARs. We have thus characterized a novel level
of cooperation between innate immune receptors in infections, which may establish a new paradigm of recognition at the fungus–host interface. After microbial recognition by TLRs, PARs may
become activated to sense tissue injury, mediate inflammatory responses, and modulate the activity of TLRs. Thus, fungi recognition by TLRs may be licensed by damage-associated molecular
pattern recognition of the host, as has been suggested.36 Therefore, proteolytic events associated with PARs may be the missing “activator” of mammalian TLRs, for which no extracellular
proteolytic events have been demonstrated upstream of the receptor.30, 31 These events identify an indirect mode of non-self recognition that has been recently described in the mechanics of
the perception system of plants.37, 38 Indeed, a dual-sensor system to detect fungal infection seems to work throughout evolution,39 because _Drosophila_ senses fungi by sensing both the
fungal cell wall and the activity of proteolytic virulence factors.40 Several possible mechanisms have been described that may underlie the potential for receptor transactivation within the
PAR or TLR family.3, 6 Our results suggest that TLR2 implicates PAR1 and MAPK to activate the PMN's inflammatory pathway to _Candida_. Conversely, PAR2 is apparently deactivated on PMNs
by _Aspergillus_ through TLR4. There is a good precedent of TLR4-dependent desensitization of G-protein-coupled receptors on PMNs.41 PMN proteases triggered by fungi could be responsible
for PAR1 activation as well as PAR2 deactivation, a finding consistent with the ability of proteases to contribute to fungal septic shock through vascular damage and plasma leakage
associated with tissue destruction in the lungs.42 TLR4 and TLR2 induced distinct patterns of degranulation against either fungus,22 and degranulated PMNs from TLR2- or TLR4-deficient PMNs
have disparate activity on PAR functioning. It is therefore conceivable that an action on the protease/antiprotease balance may contribute to the ability of TLRs to condition heterologous or
homologous PAR activation/desensitization at the infectious site. Interestingly, PAR activity was differently modulated by PMN degranulated in response to fungal hyphae more than conidia or
yeasts (data not shown), a finding linking fungal morphogenesis to virulence through host inflammatory responses.1 The subversion of the host p38-dependent anti-inflammatory pathway by
_Aspergillus_ implies that the host anti-inflammatory pathways could be exploited therapeutically to attenuate signs of inflammatory pathology in fungal infections and sepsis. Not only is
p38 a critically important mediator in the activation of Interferon regulatory factor 3,30, 31 a transcription factor of the MyD88-independent pathway associated with TLR4 signaling, but
also there is mounting evidence for a negative cross-signaling between this and other MAPK pathways in inflammation.33 p38MAPK has a central role in the activation of homeostatic
cyclooxygenase-2 in the airways11 and in the inhibition of NF-κB activation by salicylates.43 Thus, PAR2 agonists will share with salicylates a common mechanism of anti-inflammatory action
and, at the same time, will rescue the host from toxicity associated with glucocorticoids, which are known to antagonize both p38MAPK44 and PAR2.19 The ability of PARs and TLRs to have
_cis-_ and _trans-_interactions with other receptors as well as the redundancy in their signaling pathways6 precludes a definite mechanistic view of events regulating the inflammatory
response at the sites of infection. However, our study is consistent with a model in which the inflammatory response is regulated by positive or negative signals that originated from the
TLR2/PAR1- or the TLR4/PAR2-dependent pathway, the relative contribution of each receptor pair being dependent on the fungus (Figure 8). Inflammation is promoted through a
PAR1/ERK/NF-κB-dependent pathway and inhibited through a PAR2/p38-dependent pathway. This occurs by delegation from TLRs, since PAR1 activity is promoted by TLR2 through the MyD88-dependent
pathway and PAR2 activity is promoted by TLR4 through an MyD88-independent pathway. The TLR2/PAR1-dependent pathway was promoted by _Candida_ but the TLR4/PAR2-dependent pathway was
subverted by _Aspergillus_, a finding indicating divergence in pathway exploitation by fungi in the promotion of host inflammatory response. METHODS MICE. Female C57BL6 and BALB/c mice (8–10
weeks old) were from Charles River (Calco, Italy). Breeding pairs of homozygous _tlr2_−/−_, tlr4_−/−,23 and _par2_−/− mice, raised on a C57BL6 background and breeding pairs of mice
overexpressing PAR2 (_par2_-Tg) raised on a BALB/c background,45 were bred under specific pathogen-free conditions at the breeding facilities of the University of Perugia, Italy. Experiments
were performed according to the Italian Approved Animal Welfare Assurance A-3143-01. PAR AGONISTS AND ANTAGONISTS. Since proteases can activate multiple PARs and exert effects by many other
mechanisms, synthetic peptides mimicking the tethered ligand domains of PARs (PAR-activating peptides, PAR-APs) are commonly used to selectively activate these receptors.5, 6 The tethered
ligand of PAR2 (SLIGRL-NH2) and an analog of the tethered ligand of PAR1 (Tfllr-NH2), both of which selectively activate these receptors, were used as PAR-APs. Scrambled sequences, which do
not activate these receptors (PAR-sAP), were used as controls.12, 34 PAR1-ANT and PAR2-ANT (ENMD-1068) were as described.46, 47 MICROORGANISMS, INFECTIONS, AND TREATMENT. The strains of _C.
albicans_ and _A. fumigatus_ have been described.23 For protease production _in vitro_, 106 yeasts or conidia/ml were grown in YEPD broth (1% yeast extract, 2% peptone, 2% dextrose) or
minimum essential medium with 10% fetal bovine serum, respectively, for 60 h, the time at which the protease activity (QuantiCleave Protease Assay Kit, Pierce, Milan, Italy) of supernatant
was 142 ng ml−1 (_Candida_) or 264 ng ml−1 (_Aspergillus_), respectively. The lyophilized supernatant was resuspended in RPMI before use. For _Aspergillus_ infection, conidia were given
intratracheally (1 × 108 per 20 μl saline) to mice immunosuppressed with cyclophosphamide (150 mg kg−1 intraperitoneally) 6 h before and anesthetized by intraperitoneally injection of 2.5%
avertin (Sigma). For the gastrointestinal candidiasis, 108 _Candida_ yeasts were administered by gavage.23 PAR2-AP and PAR2-ANT (1.5 mg kg−1 20 μl−1)12, 34 were administered intratracheally
the day of the _Aspergillus_ infection and intranasally twice daily for 2 days. In candidiasis, PAR1-AP and PAR1-ANT (1.5 mg kg−1 200 μl−1) were administered intraperitoneally the day of the
infection and twice daily for 5 days. PAR agonists and antagonists were dissolved as described.12, 34 Neither PAR agonist nor antagonist modified the inflammatory status when injected into
uninfected mice (data not shown). Vehicles alone had no effects on infections (data not shown). Fungal growth was quantified and tissues were stained for Periodic Acid Schiff as described.22
PMN STIMULATION, RESPIRATORY BURST, AND ZYMOGRAPHY. Purified Gr-1+ PMNs (>98% pure on flow cytometric analysis) were obtained by positive selection from the peritoneal cavity of
thioglycolate-injected mice.22, 23 PMNs were stimulated at 37 °C with 3 × 10−5 M PAR agonists and/or antagonists and/or live fungi (at a PMN:fungus ratio of 10:1) for 30 min for each single
stimulation or for a total of 60 min in sequential stimulation for MAPK phosphorylation and NF-κB activation.34, 46 Production of oxidants was performed by quantifying the release of
superoxide anion (O2−) through the measure of the superoxide dismutase-inhibitable reduction of cytochrome _c_.22 Experiments were performed in triplicate and the results were expressed as
nanomoles O2−/106 cells. Gelatinolytic activity of MMP9 was assessed by gelatin zymography and determined by scanning the lysis band in the 72-kD area using a BioRad Gel DOC 1000 imaging
densitometer (BioRad, Milan, Italy). The protease activity (200 or 140 ng ml−1 in _Candida_- or _Aspergillus_-stimulated PMNs, respectively) was quantified by the QuantiCleave Protease Assay
Kit (Pierce). FLOW CYTOMETRIC ANALYSIS. For surface staining, PMNs were incubated with fungi for 30 min and HEK293 cells with human thrombin (3 U ml−1) or trypsin (100 nM) (Sigma) for 10
min. HEK293 cells were also pre-exposed to supernatants (containing 20 ng ml−1 of protease) from fungus-exposed WT PMNs for 30 min, at the time at which peak activity was observed. Cells
were stained with goat polyclonal C-18, recognizing thrombin receptor of both human and mouse origins, followed by rabbit anti-goat IgG-PE or with the anti-murine PAR2 SAM11-PE antibody (all
from Santa Cruz Biotechnology) raised against amino acids 37–50 of human PAR2, and evaluated by the FACScan flow cytofluorometer (Becton Dickinson, Mountain View, CA) equipped with Lysis II
software. Before surface staining, cells were incubated at room temperature with 5 μg of anti-FcγR mAb (2.4G2; PharMingen, Palo Alto, CA). ERK/P38 MAPK PHOSPHORYLATION AND ELECTROPHORETIC
MOBILITY SHIFT ASSAYS. Extracellular signal-regulated kinase and p38 phosphorylation and NF-κB activation were assessed on 20 × 106 PMNs stimulated as above. Blots of cell lysates were
incubated with rabbit polyclonal Abs recognizing the unphosphorylated form of ERK and p38 or the phospho-p38 MAPK (Thr180/Tyr182) and phospho-p44/42 MAPK (Thr202/Tyr294) antibodies (Cell
Signaling Technology, Milan, Italy) followed by horseradish peroxidase-conjugated goat anti-rabbit IgG (Cell Signaling Technology), as per the manufacturer's instructions. Blots were
developed with the Enhanced Chemiluminescence detection kit (Amersham Pharmacia Biotech, Milan, Italy). Cells were pre-exposed to the p38 inhibitor SB202190 (Calbiochem, San Diego, CA) at 10
μM in 0.1% dimethylsulphoxide for 60 min. For electrophoretic mobility shift assay (EMSA), the double-stranded probe containing an NF-κB consensus site 5-agttgaggggactttcccaggc-3 was
terminally labeled with T4 PNK. The EMSA experimental reaction contains 10 μg of nuclear extracts, 2 μg of nonspecific competitor poly (dI-dC), 200 ng of single-stranded oligonucleotide. The
binding reaction mixture was made with 50,000 c.p.m. (40 fmol) of radiolabeled probe for 20 min in 20 mM HEPES (pH 7.6), 100 mM NaCl, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl
fluoride, 1 × complete protease inhibitor mixture (Roche Molecular Biochemicals, Indianapolis, IN), and 5% glycerol. Complexes were resolved on a 6% native polyacrylamide gel for 120 min at
170 V in TBE (0.5 ×). After electrophoresis, the gel was dried and processed for autoradiography. For band specificity, the nuclear extracts were incubated with the antibodies to p65 and p50
(Santa Cruz Biotechnology, Santa Cruz, CA) for 45 min at 4 °C, before the probe was added. SHORT INTERFERING RNA. The human HEK293 embryonic kidney cell lines stably transfected with human
CD14/TLR2 or CD14/TLR4 were maintained as described.1 Synthetic RNA sequences of PAR1 and PAR2 were designed, synthesized, and purified by Dharmacon Research (siGENOME SMART pool: human
_par1_ cat. no. M005094-01 and human _par2_ cat. no. M-061445-00). Transfections of siRNA (at 13 nM) were performed using the Transit TKO Transfection reagent, as per the manufacturer's
instructions (Mirus, Madison, WI). siRNA direct against human β-actin was used as control (siGENOME SMART pool: human ACTB cat. no. M003451-01). The relative amounts of _par1_ and _paR2_
mRNA in cells transfected with _par1_ or _par2_ siRNA, but not scrambled siRNA, were found to be reduced by 80% compared to WT cells 48 h post-transfection, as assessed by quantitative
reverse transcriptase-PCR. At this time, CD14/TLR2/HEK293 cells transfected with siRNA PAR1 were stimulated with 10 μg ml−1 zymosan and CD14/TLR4/HEK293 cells transfected with siRNA PAR2
were stimulated with 10 μg ml−1 lipopolysaccharide for 2 h before EMSA. Western blotting with specific polyclonal antibodies (Santa Cruz Biotechnology) was performed to assess the level of
p50 and p65 upon siRNA in cells lysed in buffer containing Tris-Hcl 1 M (pH 6.8), 1% Triton X-100, 2 mM phenylmethylsulfonyl fluoride, 1 mM sodium orthovanadate, 10 μg ml−1 leupeptin, and
phosphatase inhibitors cocktail 1 (Sigma). CALCIUM MOBILIZATION. Confluent HEK293 cells were nonenzymatically collected and suspended at 2 × 106 cells/ml in phosphate-buffered saline. Cells
were loaded with 1 μM Fura -2-AM (Molecular Probes, Eugene, OR) and shaken for 30 min at room temperature. A total of 2 × 106 cells/ml loaded cells were transferred to stirred quartz
cuvettes in an LS-50B spectrofluorimeter (Perkin Elmer, Padova, Italy), pretreated with elastase E+catepsin G (HE/CatG, each at 600 nM) or supernatants from degranulated WT (20 ng ml−1 of
protease), _tlr2_−/− or _tlr4_−/− PMNs, and stimulated with human thrombin (3 U ml−1) or trypsin (100 nM) (Sigma) 4 min later. Fura 2 fluorescence was measured at 340 and 380 nm excitation
and 510 nm emission, and the ratio of the fluorescence at the two excitation wavelengths, which is proportional to [Ca2+]i, was calculated. For inhibitory studies, culture supernatants were
preincubated with 4 mM of the serine protease inhibitor, phenylmethylsulfonyl fluoride, dissolved in ethanol, and 10 μg ml−1 of the cathepsin B inhibitor, leupeptin, dissolved in dimethyl
sulfoxide (both from Sigma, St Louis, MO) for 15 min at 7 °C. The addition of each diluent alone did not modify calcium mobilization in HEK293. CYTOKINE ASSAY. The levels of cytokines in
tissue homogenates and culture supernatants from dendritic cells purified from Payer's patches by magnetic-activated sorting using CD11c MicroBeads and MidiMacs (Miltenyi Biotec,
Bergisch Gladbach, Germany) were determined by Kit ELISA (R&D Systems, Milan, Italy). The detection limits (pg ml−1) of the assays were <32 for tumor necrosis factor-α, <10 for
IL-12p70, and <3 for IL-10. REVERSE TRANSCRIPTASE-PCR AND REAL-TIME PCR. Total RNA from organs or purified CD4+ T cells35 was extracted with TRIZOL (Invitrogen SRL Life Technologies,
Milano, Italy). Synthesis and PCR of cDNA were performed as described.12, 34 The forward and reverse PCR primers used for murine and human _par_ and _gapdh_ and cycles were as described.12,
34, 35 Semiquantitative PCR was performed using the “primer-dropping” method, in which _gapdh_ was coamplified as an internal control in all reactions. Band intensity was quantified using
laser scanning densitometry and ratios of _par1_ or _par2_ to _gapdh_ were plotted for each autoradiogram. Results are representative of three experiments. STATISTICAL ANALYSIS.
Student's _t_-test was used to determine differences between the experimental groups (significance was defined as _P_<0.05). _In vivo_ groups consisted of four to six animals. The
data reported were pooled from three experiments. Blots are from one representative experiment out of three. CONFLICT OF INTEREST The authors declared no conflict of interest. REFERENCES *
Romani, L. Immunity to fungal infections. _Nat. Rev. Immunol_. 4, 1–23 (2004). Article Google Scholar * Gantner, B.N., Simmons, R.M., Canavera, S.J., Akira, S. & Underhill, D.M.
Collaborative induction of inflammatory responses by dectin-1 and Toll-like receptor 2. _J. Exp. Med._ 197, 1107–1117 (2003). Article CAS Google Scholar * Mukhopadhyay, S., Herre, J.,
Brown, G.D. & Gordon, S. The potential for Toll-like receptors to collaborate with other innate immune receptors. _Immunology_ 112, 521–530 (2004). Article CAS Google Scholar * Brown,
G.D. Dectin-1: a signalling non-TLR pattern-recognition receptor. _Nat. Rev. Immunol_. 6, 33–43 (2006). Article CAS Google Scholar * Coughlin, S.R. Thrombin signalling and
protease-activated receptors. _Nature_ 407, 258–264 (2000). Article CAS Google Scholar * Ossovskaya, V.S. & Bunnett, N.W. Protease-activated receptors: contribution to physiology and
disease. _Physiol. Rev_. 84, 579–621 (2004). Article CAS Google Scholar * Monod, M., Capoccia, S., Lechenne, B., Zaugg, C., Holdom, M. & Jousson, O. Secreted proteases from pathogenic
fungi. _Int. J. Med. Microbiol_. 292, 405–419 (2002). Article CAS Google Scholar * Shpacovitch, V., Feld, M., Bunnett, N.W. & Steinhoff, M. Protease-activated receptors: novel
PARtners in innate immunity. _Trends Immunol_. 28, 535–544 (2007). Article Google Scholar * Kauffman, H.F. & van der Heide, S. Exposure, sensitization, and mechanisms of fungus-induced
asthma. _Curr. Allergy Asthma Rep_. 3, 430–437 (2003). Article Google Scholar * Cirino, G. & Vergnolle, N. Proteinase-activated receptors (PARs): crossroads between innate immunity
and coagulation. _Curr. Opin. Pharmacol_. 6, 428–434 (2006). Article CAS Google Scholar * Cocks, T.M. _et al_. A protective role for protease-activated receptors in the airways. _Nature_
398, 156–160 (1999). Article CAS Google Scholar * Fiorucci, S. _et al_. Proteinase-activated receptor 2 is an anti-inflammatory signal for colonic lamina propria lymphocytes in a mouse
model of colitis. _Proc. Natl. Acad. Sci. USA_ 98, 13936–13941 (2001). Article CAS Google Scholar * Vergnolle, N., Wallace, J.L., Bunnett, N.W. & Hollenberg, M.D. Protease-activated
receptors in inflammation* neuronal signaling and pain. _Trends Pharmacol. Sci_. 22, 146–152 (2001). Article CAS Google Scholar * Fiorucci, S. & Distrutti, E. Role of PAR2 in pain and
inflammation. _Trends Pharmacol. Sci._ 23, 153–155 (2002). Article CAS Google Scholar * Coughlin, S.R. & Camerer, E. PARticipation in inflammation. _J. Clin. Invest._ 111, 25–27
(2003). Article CAS Google Scholar * Noorbakhsh, F. _et al_. Proteinase-activated receptor 2 modulates neuroinflammation in experimental autoimmune encephalomyelitis and multiple
sclerosis. _J. Exp. Med._ 203, 425–435 (2006). Article Google Scholar * Turner, M.W. The role of mannose-binding lectin in health and disease. _Mol. Immunol._ 40, 423–429 (2003). Article
CAS Google Scholar * Kauffman, H.F., Tomee, J.F., van de Riet, M.A., Timmerman, A.J. & Borger, P. Protease-dependent activation of epithelial cells by fungal allergens leads to
morphologic changes and cytokine production. _J. Allergy Clin. Immunol._ 105 (6 Part 1), 1185–1193 (2000). Article CAS Google Scholar * Reed, C.E. & Kita, H. The role of protease
activation of inflammation in allergic respiratory diseases. _J. Allergy Clin. Immunol._ 114, 997–1008 (2004)quiz 1009. Article CAS Google Scholar * Lopes Bezerra, L.M. & Filler, S.G.
Interactions of _Aspergillus fumigatus_ with endothelial cells: internalization, injury, and stimulation of tissue factor activity. _Blood_ 103, 2143–2149 (2004). Article Google Scholar *
Howells, G.L. _et al_. Proteinase-activated receptor-2: expression by human neutrophils. _J. Cell Sci._ 110 (Part 7), 881–887 (1997). CAS PubMed Google Scholar * Bellocchio, S. _et al_.
TLRs govern neutrophil activity in aspergillosis. _J. Immunol._ 173, 7406–7415 (2004). Article CAS Google Scholar * Bellocchio, S. _et al_. The contribution of the Toll-Like/IL-1 receptor
superfamily to innate and adaptive immunity to fungal pathogens _in vivo_. _J. Immunol._ 172, 3059–3069 (2004). Article CAS Google Scholar * Dulon, S., Cande, C., Bunnett, N.W.,
Hollenberg, M.D., Chignard, M. & Pidard, D. Proteinase-activated receptor-2 and human lung epithelial cells: disarming by neutrophil serine proteinases. _Am. J. Respir. Cell Mol. Biol._
28, 339–346 (2003). Article CAS Google Scholar * Kawabata, A., Saifeddine, M., al-Ani, B. & Hollenberg, M.D. Protease-activated receptors: development of agonists selective for
receptors triggered by either thrombin (PAR1) or trypsin (PAR2). _Proc. West Pharmacol. Soc._ 40, 49–51 (1997). CAS PubMed Google Scholar * Asokananthan, N. _et al_. House dust mite
allergens induce proinflammatory cytokines from respiratory epithelial cells: the cysteine protease allergen, Der p 1, activates protease-activated receptor (PAR)-2 and inactivates PAR-1.
_J. Immunol._ 169, 4572–4578 (2002). Article CAS Google Scholar * Barton, G.M. & Medzhitov, R. Toll-like receptor signaling pathways. _Science_ 300, 1524–1525 (2003). Article CAS
Google Scholar * Akira, S. & Takeda, K. Toll-like receptor signalling. _Nat. Rev. Immunol._ 4, 499–511 (2004). Article CAS Google Scholar * Zychlinsky, A., Weinrauch, Y. & Weiss,
J. Introduction: Forum in immunology on neutrophils. _Microbes Infect._ 5, 1289–1291 (2003). Article Google Scholar * Palsson-McDermott, E.M. & O'Neill, L.A. Signal transduction
by the lipopolysaccharide receptor, Toll-like receptor-4. _Immunology_ 113, 153–162 (2004). Article CAS Google Scholar * Beutler, B. Inferences, questions and possibilities in Toll-like
receptor signalling. _Nature_ 430, 257–263 (2004). Article CAS Google Scholar * LA, O.N. Therapeutic targeting of Toll-like receptors for inflammatory and infectious diseases. _Curr.
Opin. Pharmacol._ 3, 396–403 (2003). Article Google Scholar * Martin-Blanco, E. p38 MAPK signalling cascades: ancient roles and new functions. _Bioessays_ 22, 637–645 (2000). Article CAS
Google Scholar * Vergnolle, N. _et al_. A role for proteinase-activated receptor-1 in inflammatory bowel diseases. _J. Clin. Invest._ 114, 1444–1456 (2004). Article CAS Google Scholar
* De Luca, A. _et al_. Functional yet balanced reactivity to _Candida albicans_ requires TRIF, MyD88, and IDO-dependent inhibition of Rorc. _J. Immunol._ 179, 5999–6008 (2007). Article CAS
Google Scholar * Matzinger, P. The danger model: a renewed sense of self. _Science_ 296, 301–305 (2002). Article CAS Google Scholar * Coaker, G., Falick, A. & Staskawicz, B.
Activation of a phytopathogenic bacterial effector protein by a eukaryotic cyclophilin. _Science_ 308, 548–550 (2005). Article CAS Google Scholar * Rooney, H.C., Van't Klooster,
J.W., Van der Hoorn, R.A., Joosten, M.H., Jones, J.D. & De Wit, P.J. Cladosporium Avr2 inhibits tomato Rcr3 protease required for Cf-2-dependent disease resistance. _Science_ 308,
1783–1786 (2005). Article CAS Google Scholar * Sexton, A.C. & Howlett, B.J. Parallels in fungal pathogenesis on plant and animal hosts. _Eukaryot. Cell_ 5, 1941–1949 (2006). Article
CAS Google Scholar * Gottar, M. _et al_. Dual detection of fungal infections in _Drosophila_ via recognition of glucans and sensing of virulence factors. _Cell_ 127, 1425–1437 (2006).
Article CAS Google Scholar * Fan, J. & Malik, A.B. Toll-like receptor-4 (TLR4) signaling augments chemokine-induced neutrophil migration by modulating cell surface expression of
chemokine receptors. _Nat. Med._ 9, 315–321 (2003). Article CAS Google Scholar * Tkalcevic, J., Novelli, M., Phylactides, M., Iredale, J.P., Segal, A.W. & Roes, J. Impaired immunity
and enhanced resistance to endotoxin in the absence of neutrophil elastase and cathepsin G. _Immunity_ 12, 201–210 (2000). Article CAS Google Scholar * Kopp, E. & Ghosh, S. Inhibition
of NF-kappa B by sodium salicylate and aspirin. _Science_ 265, 956–959 (1994). Article CAS Google Scholar * Shuto, T. _et al_. Glucocorticoids synergistically enhance nontypeable
_Haemophilus influenzae_-induced Toll-like receptor 2 expression via a negative cross-talk with p38 MAP kinase. _J. Biol. Chem._ 277, 17263–17270 (2002). Article CAS Google Scholar *
Schmidlin, F. _et al_. Protease-activated receptor 2 mediates eosinophil infiltration and hyperreactivity in allergic inflammation of the airway. _J. Immunol._ 169, 5315–5321 (2002). Article
Google Scholar * Fiorucci, S. _et al_. PAR1 antagonism protects against experimental liver fibrosis. Role of proteinase receptors in stellate cell activation. _Hepatology_ 39, 365–375
(2004). Article CAS Google Scholar * Kelso, E.B. _et al_. Therapeutic promise of proteinase-activated receptor-2 antagonism in joint inflammation. _J. Pharmacol. Exp. Ther._ 316,
1017–1024 (2006). Article CAS Google Scholar Download references ACKNOWLEDGEMENTS We thank Flavia De Bernardis (Istituto Superiore di Sanità, Rome, Italy) and Sabine Flückinger, (Swiss
Institute of Allergy and Asthma Research, Davos, Switzerland) for the generous gift of the secreted aspartyl proteinase of _C. albicans_ (SAP2) and recombinant serine protease from _A_.
_fumigatus_ Aspf18; G. Lungarella (University of Siena, Siena, Italy) and P. Geppetti (University of Florence, Florence, Italy) for the supply of _par2_−/− and _par2_-Tg mice, obtained
through the courtesy of Professor S. Fiorucci (University of Perugia, Perugia, Italy), and Dr Hembrough (ENTREMED Rockville, MD, USA) for the generous gift of the PAR2-ANT, obtained through
the courtesy of Professor G. Cirino (University of Naples-Federico II, Naples, Italy). We thank Professor Nigel W. Bunnett (University of California, San Francisco), S. Fiorucci, and G.
Cirino for invaluable intellectual support. _Sources of support in the form of grants_: This study was supported by the Specific Targeted Research Project “EURAPS” (LSHM-CT-2005), contract
number 005223 (FP6) and “MANASP” (LSHE-CT-2006), contract number 037899 (FP6). AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Experimental Medicine and Biochemical Science,
University of Perugia, Via del Giochetto, Perugia, Italy S Moretti, S Bellocchio, P Bonifazi, S Bozza, T Zelante, F Bistoni & L Romani Authors * S Moretti View author publications You
can also search for this author inPubMed Google Scholar * S Bellocchio View author publications You can also search for this author inPubMed Google Scholar * P Bonifazi View author
publications You can also search for this author inPubMed Google Scholar * S Bozza View author publications You can also search for this author inPubMed Google Scholar * T Zelante View
author publications You can also search for this author inPubMed Google Scholar * F Bistoni View author publications You can also search for this author inPubMed Google Scholar * L Romani
View author publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to L Romani. ADDITIONAL INFORMATION SUPPLEMENTARY MATERIAL is linked
to the online version of the paper at http://www.nature.com/mi SUPPLEMENTARY INFORMATION SUPPLEMENTARY FIGURE 1 (JPG 327 KB) SUPPLEMENTARY FIGURE 2 (JPG 364 KB) SUPPLEMENTARY FIGURE 3 (JPG
447 KB) POWERPOINT SLIDES POWERPOINT SLIDE FOR FIG. 1 POWERPOINT SLIDE FOR FIG. 2 POWERPOINT SLIDE FOR FIG. 3 POWERPOINT SLIDE FOR FIG. 4 POWERPOINT SLIDE FOR FIG. 5 POWERPOINT SLIDE FOR
FIG. 6 POWERPOINT SLIDE FOR FIG. 7 POWERPOINT SLIDE FOR FIG. 8 RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Moretti, S., Bellocchio, S., Bonifazi, P.
_et al._ The contribution of PARs to inflammation and immunity to fungi. _Mucosal Immunol_ 1, 156–168 (2008). https://doi.org/10.1038/mi.2007.13 Download citation * Received: 03 August 2007
* Accepted: 22 November 2007 * Published: 09 January 2008 * Issue Date: March 2008 * DOI: https://doi.org/10.1038/mi.2007.13 SHARE THIS ARTICLE Anyone you share the following link with will
be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt
content-sharing initiative