Play all audios:
ABSTRACT Comparative analysis of multiple genomes in a phylogenetic framework dramatically improves the precision and sensitivity of evolutionary inference, producing more robust results
than single-genome analyses can provide. The genomes of 12 _Drosophila_ species, ten of which are presented here for the first time (_sechellia_, _simulans_, _yakuba_, _erecta_, _ananassae_,
_persimilis_, _willistoni_, _mojavensis_, _virilis_ and _grimshawi_), illustrate how rates and patterns of sequence divergence across taxa can illuminate evolutionary processes on a genomic
scale. These genome sequences augment the formidable genetic tools that have made _Drosophila melanogaster_ a pre-eminent model for animal genetics, and will further catalyse fundamental
research on mechanisms of development, cell biology, genetics, disease, neurobiology, behaviour, physiology and evolution. Despite remarkable similarities among these _Drosophila_ species,
we identified many putatively non-neutral changes in protein-coding genes, non-coding RNA genes, and _cis_-regulatory regions. These may prove to underlie differences in the ecology and
behaviour of these diverse species. You have full access to this article via your institution. Download PDF SIMILAR CONTENT BEING VIEWED BY OTHERS STRUCTURAL AND FUNCTIONAL CHARACTERIZATION
OF A PUTATIVE DE NOVO GENE IN _DROSOPHILA_ Article Open access 12 March 2021 EVOLUTIONARY DYNAMICS OF GENOME SIZE AND CONTENT DURING THE ADAPTIVE RADIATION OF HELICONIINI BUTTERFLIES Article
Open access 12 September 2023 POPULATION-SCALE LONG-READ SEQUENCING UNCOVERS TRANSPOSABLE ELEMENTS ASSOCIATED WITH GENE EXPRESSION VARIATION AND ADAPTIVE SIGNATURES IN _DROSOPHILA_ Article
Open access 12 April 2022 MAIN As one might expect from a genus with species living in deserts, in the tropics, on chains of volcanic islands and, often, commensally with humans,
_Drosophila_ species vary considerably in their morphology, ecology and behaviour1. Species in this genus span a wide range of global distributions: the 12 sequenced species originate from
Africa, Asia, the Americas and the Pacific Islands, and also include cosmopolitan species that have colonized the planet (_D. melanogaster_ and _D. simulans_) as well as closely related
species that live on single islands (_D. sechellia_)2. A variety of behavioural strategies is also encompassed by the sequenced species, ranging in feeding habit from generalist, such as _D.
ananassae_, to specialist, such as _D. sechellia,_ which feeds on the fruit of a single plant species. Despite this wealth of phenotypic diversity, _Drosophila_ species share a distinctive
body plan and life cycle. Although only _D. melanogaster_ has been extensively characterized, it seems that the most important aspects of the cellular, molecular and developmental biology of
these species are well conserved. Thus, in addition to providing an extensive resource for the study of the relationship between sequence and phenotypic diversity, the genomes of these
species provide an excellent model for studying how conserved functions are maintained in the face of sequence divergence. These genome sequences provide an unprecedented dataset to contrast
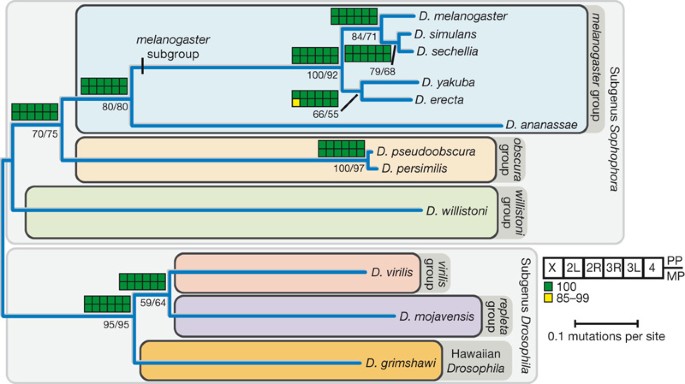
genome structure, genome content, and evolutionary dynamics across the well-defined phylogeny of the sequenced species (Fig. 1). GENOME ASSEMBLY, ANNOTATION AND ALIGNMENT GENOME SEQUENCING
AND ASSEMBLY We used the previously published sequence and updated assemblies for two _Drosophila_ species, _D. melanogaster_3,4 (release 4) and _D. pseudoobscura_5 (release 2), and
generated DNA sequence data for 10 additional _Drosophila_ genomes by whole-genome shotgun sequencing6,7. These species were chosen to span a wide variety of evolutionary distances, from
closely related pairs such as _D. sechellia_/_D. simulans_ and _D. persimilis_/_D. pseudoobscura_ to the distantly related species of the _Drosophila_ and _Sophophora_ subgenera. Whereas the
time to the most recent common ancestor of the sequenced species may seem small on an evolutionary timescale, the evolutionary divergence spanned by the genus _Drosophila_ exceeds that of
the entire mammalian radiation when generation time is taken into account, as discussed further in ref. 8. We sequenced seven of the new species (_D. yakuba_, _D. erecta_, _D. ananassae_,
_D. willistoni_, _D. virilis_, _D. mojavensis_ and _D. grimshawi_) to deep coverage (8.4× to 11.0×) to produce high quality draft sequences. We sequenced two species, _D. sechellia_ and _D.
persimilis_, to intermediate coverage (4.9× and 4.1×, respectively) under the assumption that the availability of a sister species sequenced to high coverage would obviate the need for deep
sequencing without sacrificing draft genome quality. Finally, seven inbred strains of _D. simulans_ were sequenced to low coverage (2.9× coverage from _w__501_ and ∼1× coverage of six other
strains) to provide population variation data9. Further details of the sequencing strategy can be found in Table 1, Supplementary Table 1 and section 1 in Supplementary Information. We
generated an initial draft assembly for each species using one of three different whole-genome shotgun assembly programs (Table 1). For _D. ananassae_, _D. erecta_, _D. grimshawi_, _D.
mojavensis_, _D. virilis_ and _D. willistoni,_ we also generated secondary assemblies; reconciliation of these with the primary assemblies resulted in a 7–30% decrease in the estimated
number of misassembled regions and a 12–23% increase in the N50 contig size10 (Supplementary Table 2). For _D. yakuba_, we generated 52,000 targeted reads across low-quality regions and gaps
to improve the assembly. This doubled the mean contig and scaffold sizes and increased the total fraction of high quality bases (quality score (Q) > 40) from 96.5% to 98.5%. We improved
the initial 2.9× _D. simulans w__501_ whole-genome shotgun assembly by filling assembly gaps with contigs and unplaced reads from the ∼1× assemblies of the six other _D. simulans_ strains,
generating a ‘mosaic’ assembly (Supplementary Table 3). This integration markedly improved the _D. simulans_ assembly: the N50 contig size of the mosaic assembly, for instance, is more than
twice that of the initial _w__501_ assembly (17 kb versus 7 kb). Finally, one advantage of sequencing genomes of multiple closely related species is that these evolutionary relationships can
be exploited to dramatically improve assemblies. _D. yakuba_ and _D. simulans_ contigs and scaffolds were ordered and oriented using pairwise alignment to the well-validated _D.
melanogaster_ genome sequence (Supplementary Information section 2). Likewise, the 4–5× _D. persimilis_ and _D. sechellia_ assemblies were improved by assisted assembly using the sister
species (_D. pseudoobscura_ and _D. simulans_, respectively) to validate both alignments between reads and linkage information. For the remaining species, comparative syntenic information,
and in some cases linkage information, were also used to pinpoint locations of probable genome mis-assembly, to assign assembly scaffolds to chromosome arms and to infer their order and
orientation along euchromatic chromosome arms, supplementing experimental analysis based on known markers (A. Bhutkar, S. Russo, S. Schaeffer, T. F. Smith and W. M. Gelbart, personal
communication) (Supplementary Information section 2). The mitochondrial (mt)DNA of _D. melanogaster_, _D. sechellia_, _D. simulans_ (siII), _D. mauritiana_ (maII) and _D. yakuba_ have been
previously sequenced11,12. For the remaining species (except _D. pseudoobscura_, the DNA from which was prepared from embryonic nuclei), we were able to assemble full mitochondrial genomes,
excluding the A+T-rich control region (Supplementary Information section 2)13. In addition, the genome sequences of three _Wolbachia_ endosymbionts (_Wolbachia wSim, Wolbachia wAna_ and
_Wolbachia wWil)_ were assembled from trace archives, in _D. simulans_, _D. ananassae_ and _D. willistoni_, respectively14. All of the genome sequences described here are available in
FlyBase (www.flybase.org) and GenBank (www.ncbi.nlm.nih.gov) (Supplementary Tables 4 and 5). REPEAT AND TRANSPOSABLE ELEMENT ANNOTATION Repetitive DNA sequences such as transposable elements
pose challenges for whole-genome shotgun assembly and annotation. Because the best approach to transposable element discovery and identification is still an active and unresolved research
question, we used several repeat libraries and computational strategies to estimate the transposable element/repeat content of the 12 _Drosophila_ genome assemblies (Supplementary
Information section 3). Previously curated transposable element libraries in _D. melanogaster_ provided the starting point for our analysis; to limit the effects of ascertainment bias, we
also developed _de novo_ repeat libraries using PILER-DF15,16 and ReAS17. We used four transposable element/repeat detection methods (RepeatMasker, BLASTER-TX, RepeatRunner and CompTE) in
conjunction with these transposable element libraries to identify repetitive elements in non-_melanogaster_ species. We assessed the accuracy of each method by calibration with the estimated
5.5% transposable element content in the _D. melanogaster_ genome, which is based on a high-resolution transposable element annotation18 (Supplementary Fig. 1). On the basis of our results,
we suggest a hybrid strategy for new genome sequences, employing translated BLAST with general transposable element libraries and RepeatMasker with species-specific ReAS libraries to
estimate the upper and lower bound on transposable element content. PROTEIN-CODING GENE ANNOTATION We annotated protein-coding sequences in the 11 non-_melanogaster_ genomes, using four
different _de novo_ gene predictors (GeneID19, SNAP20, N-SCAN21 and CONTRAST22); three homology-based predictors that transfer annotations from _D. melanogaster_ (GeneWise23, Exonerate24,
GeneMapper25); and one predictor that combined _de novo_ and homology-based evidence (Gnomon26). These gene prediction sets were combined using GLEAN, a gene model combiner that chooses the
most probable combination of start, stop, donor and acceptor sites from the input predictions27,28. All analyses reported here, unless otherwise noted, relied on a reconciled consensus set
of predicted gene models—the GLEAN-R set (Table 2, and Supplementary Information section 4.1). QUALITY OF GENE MODELS As the first step in assessing the quality of the GLEAN-R gene models,
we used expression data from microarray experiments on adult flies, with arrays custom-designed for _D. simulans_, _D. yakuba_, _D. ananassae_, _D. pseudoobscura_, _D. virilis_ and _D.
mojavensis_29 (GEO series GSE6640; Supplementary Information section 4.2). We detected expression significantly above negative controls (false-discovery-rate-corrected Mann–Whitney U (MWU)
_P_ < 0.001) for 77–93% of assayed GLEAN-R models, representing 50–68% of the total GLEAN-R predictions in each species (Supplementary Table 6). Evolutionarily conserved gene models are
much more likely to be expressed than lineage-specific ones (Fig. 2). Although these data cannot confirm the detailed structure of gene models, they do suggest that the majority of GLEAN-R
models contain sequence that is part of a poly-adenylated transcript. Approximately 20% of transcription in _D. melanogaster_ seems to be unassociated with protein-coding genes30, and our
microarray experiments fail to detect conditionally expressed genes. Thus, transcript abundance cannot conclusively establish the presence or absence of a protein-coding gene. Nonetheless,
we believe these expression data increase our confidence in the reliability of the GLEAN-R models, particularly those supported by homology evidence (Fig. 2). Because the GLEAN-R gene models
were built using assemblies that were not repeat masked, it is likely that some proportion of gene models are false positives corresponding to coding sequences of transposable elements. We
used RepeatMasker with _de novo_ ReAS libraries and PFAM structural annotations of the GLEAN-R gene set to flag potentially transposable element-contaminated gene models (Supplementary
Information section 4.2). These procedures suggest that 5.6–32.3% of gene models in non-_melanogaster_ species correspond to protein-coding content derived from transposable elements
(Supplementary Table 7); these transposable element-contaminated gene models are almost exclusively confined to gene predictions without strong homology support (Fig. 2). Transposable
element-contaminated gene models are excluded from the final gene prediction set used for subsequent analysis, unless otherwise noted. HOMOLOGY ASSIGNMENT Two independent approaches were
used to assign orthology and paralogy relationships among euchromatic _D. melanogaster_ gene models and GLEAN-R predictions. The first approach was a fuzzy reciprocal BLAST (FRB) algorithm,
which is an extension of the reciprocal BLAST method31 applicable to multiple species simultaneously (Supplementary Information section 5.1). Because the FRB algorithm does not integrate
syntenic information, we also used a second approach based on Synpipe (Supplementary Information section 5.2), a tool for synteny-aided orthology assignment32. To generate a reconciled set
of homology calls, pairwise Synpipe calls (between each species and _D. melanogaster_) were mapped to GLEAN-R models, filtered to retain only 1:1 relationships, and added to the FRB calls
when they did not conflict and were non-redundant. This reconciled FRB + Synpipe set of homology calls forms the basis of our subsequent analyses. There were 8,563 genes with single-copy
orthologues in the _melanogaster_ group and 6,698 genes with single-copy orthologues in all 12 species; similar numbers of genes were also obtained with an independent approach33. Most
single-copy orthologues are expressed and are free from potential transposable element contamination, suggesting that the reconciled orthologue set contains robust and high-quality gene
models (Fig. 2). VALIDATION OF HOMOLOGY CALLS Because both the FRB algorithm and Synpipe rely on BLAST-based methods to infer similarities, rapidly evolving genes may be overlooked.
Moreover, assembly gaps and poor-quality sequence may lead to erroneous inferences of gene loss. To validate putative gene absences, we used a synteny-based GeneWise pipeline to find
potentially missed homologues of _D. melanogaster_ proteins (Supplementary Information section 5.4). Of the 21,928 cases in which a _D. melanogaster_ gene was absent from another species in
the initial homology call set, we identified plausible homologues for 13,265 (60.5%), confirmed 4,546 (20.7%) as genuine absences, and were unable to resolve 4,117 (18.8%). Because this
approach is conservative and only confirms strongly supported absences, we are probably underestimating the number of genuine absences. CODING GENE ALIGNMENT AND FILTERING Investigating the
molecular evolution of orthologous and paralogous genes requires accurate multi-species alignments. Initial amino acid alignments were generated using TCOFFEE34 and converted to nucleotide
alignments (Supplementary Table 8). To reduce biases in downstream analyses, a simple computational screen was developed to identify and mask problematic regions of each alignment
(Supplementary Information section 6). Overall, 2.8% of bases were masked in the _melanogaster_ group alignments, and 3.0% of bases were masked in the full 12 species alignments,
representing 8.5% and 13.8% of alignment columns, respectively. The vast majority of masked bases are masked in no more than one species (Supplementary Fig. 3), suggesting that the masking
procedure is not simply eliminating rapidly evolving regions of the genome. We find an appreciably higher frequency of masked bases in lower-quality _D. simulans_ and _D. sechellia_
assemblies, compared to the more divergent (from _D. melanogaster_) but higher-quality _D. erecta_ and _D. yakuba_ assemblies, suggesting a higher error rate in accurately predicting and
aligning gene models in lower-quality assemblies (Supplementary Information section 6 and Supplementary Fig. 3). We used masked versions of the alignments, including only the longest _D.
melanogaster_ transcripts for all subsequent analysis unless otherwise noted. ANNOTATION OF NON-CODING (NC)RNA GENES Using _de novo_ and homology-based approaches we annotated over 9,000
ncRNA genes from recognized ncRNA classes (Table 2, and Supplementary Information section 7). In contrast to the large number of predictions observed for many ncRNA families in vertebrates
(due in part to large numbers of ncRNA pseudogenes35,36), the number of ncRNA genes per family predicted by RFAM and tRNAscan in _Drosophila_ is relatively low (Table 2). This suggests that
ncRNA pseudogenes are largely absent from _Drosophila_ genomes, which is consistent with the low number of protein-coding pseudogenes in _Drosophila_37. The relatively low numbers of some
classes of ncRNA genes (for example, small nucleolar (sno)RNAs) in the _Drosophila_ subgenus are likely to be an artefact of rapid rates of evolution in these types of genes and the
limitation of the homology-based methods used to annotate distantly related species. EVOLUTION OF GENOME STRUCTURE COARSE-LEVEL SIMILARITIES AMONG DROSOPHILIDS At a coarse level, genome
structure is well conserved across the 12 sequenced species. Total genome size estimated by flow cytometry varies less than threefold across the phylogeny, ranging from 130 Mb (_D.
mojavensis)_ to 364 Mb (_D. virilis_)38 (Table 2), in contrast to the order of magnitude difference between _Drosophila_ and mammals. Total protein-coding sequence ranges from 38.9 Mb in _D.
melanogaster_ to 65.4 Mb in _D. willistoni._ Intronic DNA content is also largely conserved, ranging from 19.6 Mb in _D. simulans_ to 24.0 Mb in _D. pseudoobscura_ (Table 2). This contrasts
dramatically with transposable element-derived genomic DNA content, which varies considerably across genomes (Table 2) and correlates significantly with euchromatic genome size (estimated
as the summed length of contigs > 200 kb) (Kendall's τ = 0.70, _P_ = 0.0016). To investigate overall conservation of genome architecture at an intermediate scale, we analysed synteny
relationships across species using Synpipe32 (Supplementary Information section 9.1). Synteny block size and average number of genes per block varies across the phylogeny as expected, with
the number of blocks increasing and the average size of blocks decreasing with increasing evolutionary distance from _D. melanogaster_ (A. Bhutkar, S. Russo, T. F. Smith and W. M. Gelbart,
personal communication) (Supplementary Fig. 4). We inferred 112 syntenic blocks between _D. melanogaster_ and _D. sechellia_ (with an average of 122 genes per block), compared to 1,406
syntenic blocks between _D. melanogaster_ and _D. grimshawi_ (with an average of 8 genes per block). On average, 66% of each genome assembly was covered by syntenic blocks, ranging from 68%
in _D. sechellia_ to 58% in _D. grimshawi_. Similarity across genomes is largely recapitulated at the level of individual genes, with roughly comparable numbers of predicted protein-coding
genes across the 12 species (Table 2). The majority of predicted genes in each species have homologues in _D. melanogaster_ (Table 2, Supplementary Table 9). Moreover, most of the 13,733
protein-coding genes in _D. melanogaster_ are conserved across the entire phylogeny: 77% have identifiable homologues in all 12 genomes, 62% can be identified as single-copy orthologues in
the six genomes of the _melanogaster_ group and 49% can be identified as single-copy orthologues in all 12 genomes. The number of functional non-coding RNA genes predicted in each
_Drosophila_ genome is also largely conserved, ranging from 584 in _D. mojavensis_ to 908 in _D. ananassae_ (Table 2). There are several possible explanations for the observed interspecific
variation in gene content. First, approximately 700 _D. melanogaster_ gene models have been newly annotated since the FlyBase Release 4.3 annotations used in the current study, reducing the
discrepancy between _D. melanogaster_ and the other sequenced genomes in this study. Second, because low-coverage genomes tend to have more predicted gene models, we suspect that artefactual
duplication of genomic segments due to assembly errors inflates the number of predicted genes in some species. Finally, the non_-melanogaster_ species have many more predicted
lineage-specific genes than _D. melanogaster_, and it is possible that some of these are artefactual. In the absence of experimental evidence, it is difficult to distinguish genuine
lineage-specific genes from putative artefacts. Future experimental work will be required to fully disentangle the causes of interspecific variation in gene number. ABUNDANT GENOME
REARRANGEMENTS DURING _DROSOPHILA_ EVOLUTION To study the structural relationships among genomes on a finer scale, we analysed gene-level synteny between species pairs. These synteny maps
allowed us to infer the history and locations of fixed genomic rearrangements between species. Although _Drosophila_ species vary in their number of chromosomes, there are six fundamental
chromosome arms common to all species. For ease of denoting chromosomal homology, these six arms are referred to as ‘Muller elements’ after Hermann J. Muller, and are denoted A–F. Although
most pairs of orthologous genes are found on the same Muller element, there is extensive gene shuffling within Muller elements between even moderately diverged genomes (Fig. 3, and
Supplementary Information section 9.1). Previous analysis has revealed heterogeneity in rearrangement rates among close relatives: careful inspection of 29 inversions that differentiate the
chromosomes of _D. melanogaster_ and _D. yakuba_ revealed that 28 were fixed in the lineage leading to _D. yakuba_, and only one was fixed on the lineage leading to _D. melanogaster_39.
Rearrangement rates are also heterogeneous across the genome among the 12 species: simulations reject a random-breakage model, which assumes that all sites are free to break in inversion
events, but fail to reject a model of coldspots and hotspots for breakpoints (S. Schaeffer, personal communication). Furthermore, inversions seem to have played important roles in the
process of speciation in at least some of these taxa40. One particularly striking example of the dynamic nature of genome micro-structure in _Drosophila_ is the homeotic _homeobox (Hox)_
gene cluster(s)41. _Hox_ genes typically occur in genomic clusters, and this clustering is conserved across many vertebrate and invertebrate taxa, suggesting a functional role for the
precise and collinear arrangement of these genes. However, several cluster splits have been previously identified in _Drosophila_42,43, and the 12 _Drosophila_ genome sequences provide
additional evidence against the functional importance of _Hox_ gene clustering in _Drosophila_. There are seven different gene arrangements found across 13 _Drosophila_ species (the 12
sequenced genomes and _D. buzzatii_), with no species retaining the inferred ancestral gene order44. It thus seems that, in _Drosophila,_ _Hox_ genes do not require clustering to maintain
proper function, and are a powerful illustration of the dynamism of genome structure across the sequenced genomes. TRANSPOSABLE ELEMENT EVOLUTION Mobile, repetitive transposable element
sequences are a particularly dynamic component of eukaryotic genomes. Transposable element/repeat content (in scaffolds >200 kb) varies by over an order of magnitude across the genus,
ranging from ∼2.7% in _D. simulans_ and _D. grimshawi_ to ∼25% in _D. ananassae_ (Table 2, and Supplementary Fig. 1). These data support the lower euchromatic transposable element content in
_D. simulans_ relative to _D. melanogaster_45, and reveal that euchromatic transposable element/repeat content is generally similar within the _melanogaster_ subgroup. Within the
_Drosophila_ subgenus, _D. grimshawi_ has the lowest transposable element/repeat content, possibly relating to its ecological status as an island endemic, which may minimize the chance for
horizontal transfer of transposable element families. Finally, the highest levels of transposable element/repeat content are found in _D. ananassae_ and _D. willistoni_. These species also
have the highest numbers of pseudo-transfer (t)RNA genes (Table 2), indicating a potential relationship between pseudo-tRNA genesis and repetitive DNA, as has been established in the mouse
genome36. Different classes of transposable elements can vary in abundance owing to a variety of host factors, motivating an analysis of the intragenomic ecology of transposable elements in
the 12 genomes. In _D. melanogaster_, long terminal repeat (LTR) retrotransposons have the highest abundance, followed by LINE (long interspersed nuclear element)-like retrotransposons and
terminal inverted repeat (TIR) DNA-based transposons18. An unbiased, conservative approach (Supplementary Information section 3) for estimating the rank order abundance of major transposable
element classes suggests that these abundance trends are conserved across the entire genus (Supplementary Fig. 5). Two exceptions are an increased abundance of TIR elements in _D. erecta_
and a decreased abundance of LTR elements in _D. pseudoobscura_; the latter observation may represent an assembly artefact because the sister species _D. persimilis_ shows typical LTR
abundance. Given that individual instances of transposable element repeats and transposable element families themselves are not conserved across the genus, the stability of abundance trends
for different classes of transposable elements is striking and suggests common mechanisms for host–transposable element co-evolution in _Drosophila_. Although comprehensive analysis of the
structural and evolutionary relationships among families of transposable elements in the 12 genomes remains a major challenge for _Drosophila_ genomics, some initial insights can be gleaned
from analysis of particularly well-characterized transposable element families. Previous analysis has shown variable dynamics for the most abundant transposable element family _(DINE-1)_46
in the _D. melanogaster_ genome18,47: although inactive in _D. melanogaster_48, _DINE-1_ has experienced a recent transpositional burst in _D. yakuba_49. Our analysis confirms that this
element is highly abundant in all of the other sequenced genomes of _Drosophila_, but is not found outside of Diptera50,51. Moreover, the inferred phylogenetic relationship of _DINE-1_
paralogues from several _Drosophila_ species suggests vertical transmission as the major mechanism for _DINE-1_ propagation. Likewise, analysis of the _Galileo_ and _1360_ transposons
reveals a widespread but discontinuous phylogenetic distribution for both families, notably with both families absent in the geographically isolated Hawaiian species, _D. grimshawi_52. These
results are consistent with an ancient origin of the _Galileo_ and _1360_ families in the genus and subsequent horizontal transfer and/or loss in some lineages. The use of these 12 genomes
also facilitated the discovery of transposable element lineages not yet documented in _Drosophila_, specifically the P instability factor (_PIF_) superfamily of DNA transposons. Our analysis
indicates that there are four distinct lineages of this transposon in _Drosophila_, and that this element has indeed colonized many of the sequenced genomes53. This superfamily is
particularly intriguing given that _PIF_-transposase-like genes have been implicated in the origin of at least seven different genes during the _Drosophila_ radiation53, suggesting that not
only do transposable elements affect the evolution of genome structure, but that their domestication can play a part in the emergence of novel genes. _D. melanogaster_ maintains its
telomeres by occasional targeted transposition of three telomere-specific non-LTR retrotransposons (_HeT-A_, _TART_ and _TAHRE_) to chromosome ends54,55 and not by the more common mechanism
of telomerase-generated G-rich repeats56. Multiple telomeric retrotransposons have originated within the genus, where they now maintain telomeres, and recurrent loss of most of the ORF2 from
telomeric retrotransposons (for example, _TAHRE_) has given rise to half-telomeric-retrotransposons (for example, _HeT-A_) during _Drosophila_ evolution57. The phylogenetic relationship
among these telomeric elements is congruent with the species phylogeny, suggesting that they have been vertically transmitted from a common ancestor57. NCRNA GENE FAMILY EVOLUTION Using
ncRNA gene annotations across the 12-species phylogeny, we inferred patterns of gene copy number evolution in several ncRNA families. Transfer RNA genes are the most abundant family of ncRNA
genes in all 12 genomes, with 297 tRNAs in _D. melanogaster_ and 261–484 tRNA genes in the other species (Table 2). Each genome encodes a single selenocysteine tRNA, with the exception of
_D. willistoni_, which seems to lack this gene (R. Guigo, personal communication). Elevated tRNA gene counts in _D. ananassae_ and _D. willistoni_ are explained almost entirely by
pseudo-tRNA gene predictions. We infer from the lack of pseudo-tRNAs in most _Drosophila_ species, and from similar numbers of tRNAs obtained from an analysis of the chicken genome (_n_ =
280)58, that the minimal metazoan tRNA set is encoded by ∼300 genes, in contrast to previous estimates of 497 in human and 659 in _Caenorhabditis elegans_59,60. Similar numbers of snoRNAs
are predicted in the _D. melanogaster_ subgroup (_n_ = 242–255), in which sequence similarity is high enough for annotation by homology, with fewer snoRNAs (_n_ = 194–216) annotated in more
distant members of the _Sophophora_ subgenus, and even fewer snoRNAs (_n_ = 139–165) predicted in the _Drosophila_ subgenus, in which annotation by homology becomes much more difficult. Of
78 previously reported micro (mi)RNA genes, 71 (91%) are highly conserved across the entire genus, with the remaining seven genes (_mir-2b-1_, -_289_, -_303_, -_310_, -_311_, -_312_ and
-_313_) restricted to the subgenus _Sophophora_ (Supplementary Information section 7.2). All the species contain similar numbers of spliceosomal snRNA genes (Table 2), including at least one
copy each of the four U12-dependent (minor) spliceosomal RNAs, despite evidence for birth and death of these genes and the absence of stable subtypes61. The unusual, lineage-specific
expansion in size of U11 snRNA, previously described in _Drosophila_61,62, is even more extreme in _D. willistoni_. We annotated 99 copies of the 5S ribosomal (r)RNA gene in a cluster in _D.
melanogaster_, and between 13 and 73 partial 5S rRNA genes in clusters in the other genomes. Finally, we identified members of several other classes of ncRNA genes, including the RNA
components of the RNase P (1 per genome) and the signal recognition particle (SRP) RNA complexes (1–3 per genome), suggesting that these functional RNAs are involved in similar biological
processes throughout the genus. We were only able to locate the _roX_ (RNA on X)63,64 genes involved in dosage compensation using nucleotide homology in the _melanogaster_ subgroup, although
analyses incorporating structural information have identified _roX_ genes in other members of the genus65. We investigated the evolution of rRNA genes in the 12 sequenced genomes, using
trace archives to locate sequence variants within the transcribed portions of these genes. This analysis revealed moderate levels of variation that are not distributed evenly across the rRNA
genes, with fewest variants in conserved core coding regions, more variants in coding expansion regions, and higher still variant abundances in non-coding regions. The level and
distribution of sequence variation in rRNA genes are suggestive of concerted evolution, in which recombination events uniformly distribute variants throughout the rDNA loci, and selection
dictates the frequency to which variants can expand66. PROTEIN-CODING GENE FAMILY EVOLUTION For a general perspective on how the protein-coding composition of these 12 genomes has changed,
we examined gene family expansions and contractions in the 11,434 gene families (including those of size one in each species) predicted to be present in the most recent common ancestor of
the two subgenera. We applied a maximum likelihood model of gene gain and loss67 to estimate rates of gene turnover. This analysis suggests that gene families expand or contract at a rate of
0.0012 gains and losses per gene per million years, or roughly one fixed gene gain/loss across the genome every 60,000 yr68. Many gene families (4,692 or 41.0%) changed in size in at least
one species, and 342 families showed significantly elevated (_P_ < 0.0001) rates of gene gain and loss compared to the genomic average, indicating that non-neutral processes may play a
part in gene family evolution. Twenty-two families exhibit rapid copy number evolution along the branch leading to _D. melanogaster_ (eighteen contractions and four expansions; Supplementary
Table 10). The most common Gene Ontology (GO) terms among families with elevated rates of gain/loss include ‘defence response’, ‘protein binding’, ‘zinc ion binding’, ‘proteolysis’, and
‘trypsin activity’. Interestingly, genes involved in ‘defence response’ and ‘proteolysis’ also show high rates of protein evolution (see below). We also found heterogeneity in overall rates
of gene gain and loss across lineages, although much of this variation could result from interspecific differences in assembly quality68. LINEAGE-SPECIFIC GENES The vast majority of _D.
melanogaster_ proteins that can be unambiguously assigned a homology pattern (Supplementary Information section 5) are inferred to be ancestrally present at the genus root (11,348/11,644, or
97.5%). Of the 296 non-ancestrally present genes, 252 are either _Sophophora_-specific, or have a complicated pattern of homology requiring more than one gain and/or loss on the phylogeny,
and are not discussed further. The remaining 44 proteins include 14 present in the _melanogaster_ group, 23 present only in the _melanogaster_ subgroup, 3 unique to the _melanogaster_
species complex, and 4 found in _D. melanogaster_ only. Because we restricted this analysis to unambiguous homologues of high-confidence protein-coding genes in _D. melanogaster_8, we are
probably undercounting the number of genes that have arisen _de novo_ in any particular lineage. However, ancestrally heterochromatic genes that are currently euchromatic in _D.
melanogaster_ may spuriously seem to be lineage-specific. The 44 lineage-specific genes (Supplementary Table 11) differ from ancestrally present genes in several ways. They have a shorter
median predicted protein length (lineage-specific median 177 amino acids, other median 421 amino acids, MWU, _P_ = 3.6 × 10-13), are more likely to be intronless (Fisher's exact test
(FET), _P_ = 6.2 × 10-6), and are more likely to be located in the intron of another gene on the opposite strand (FET, _P_ = 3.5 × 10-4). In addition, 18 of these 44 genes are testis- or
accessory-gland-specific in _D. melanogaster_, a significantly greater fraction than is found in the ancestral set (FET, _P_ = 1.25 × 10-4). This is consistent with previous observations
that novel genes are often testis-specific in _Drosophila_69,70,71,72,73 and expression studies on seven of the species show that species-restricted genes are more likely to exhibit
male-biased expression29. Further, these genes are significantly more tissue-specific in expression (as measured by τ; ref. 74) (MWU, _P_ = 9.6 × 10-6), and this pattern is not solely driven
by genes with testis-specific expression patterns. PROTEIN-CODING GENE EVOLUTION POSITIVE SELECTION AND SELECTIVE CONSTRAINTS IN _DROSOPHILA_ GENOMES To study the molecular evolution of
protein-coding genes, we estimated rates of synonymous and non-synonymous substitution in 8,510 single-copy orthologues within the six _melanogaster_ group species using PAML75
(Supplementary Information section 11.1); synonymous site saturation prevents analysis of more divergent comparisons. We investigate only single-copy orthologues because when paralogues are
included, alignments become increasingly problematic. Rates of amino acid divergence for single-copy orthologues in all 12 species were also calculated; these results are largely consistent
with the analysis of non-synonymous divergence in the _melanogaster_ group, and are not discussed further. To understand global patterns of divergence and constraint across functional
classes of genes, we examined the distributions of ω ( = _d_N/_d_S, the ratio of non-synonymous to synonymous divergence) across Gene Ontology categories (GO)76, excluding GO annotations
based solely on electronic support (Supplementary Information section 11.2). Most functional categories of genes are strongly constrained, with median estimates of ω much less than one. In
general, functionally similar genes are similarly constrained: 31.8% of GO categories have significantly lower variance in ω than expected (_q_-value true-positive test77). Only 11% of GO
categories had statistically significantly elevated ω (relative to the median of all genes with GO annotations) at a 5% false-discovery rate (FDR), suggesting either positive selection or a
reduction in selective constraint. The GO categories with elevated ω include the biological process terms ‘defence response’, ‘proteolysis’, ‘DNA metabolic process’ and ‘response to biotic
stimulus’; the molecular function terms ‘transcription factor activity’, ‘peptidase activity’, ‘receptor binding’, ‘odorant binding’, ‘DNA binding’, ‘receptor activity’ and
‘G-protein-coupled receptor activity’; and the cellular location term ‘extracellular’ (Fig. 4, and Supplementary Table 12). Similar results are obtained when _d_N is compared across GO
categories, suggesting that in most cases differences in ω among GO categories is driven by amino acid rather than synonymous site substitutions. The two exceptions are the molecular
function terms ‘transcription factor activity’ and ‘DNA binding activity’, for which we observe significantly decelerated _d_S (FDR = 7.2 × 10-4 for both; Supplementary Information section
11.2) and no significant differences in _d_N. To distinguish possible positive selection from relaxed constraint, we tested explicitly for genes that have a subset of codons with signatures
of positive selection, using codon-based likelihood models of molecular evolution, implemented in PAML78,79 (Supplementary Information section 11.1). Although this test is typically regarded
as a conservative test for positive selection, it may be confounded by selection at synonymous sites. However, selection at synonymous sites (that is, codon bias, see below) is quite weak.
Moreover, variability in ω presented here tends to reflect variability in _d_N. We therefore believe that it is appropriate to treat synonymous sites as nearly neutral and sites with ω >
1 as consistent with positive selection. Despite a number of functional categories with evidence for elevated ω, ‘helicase activity’ is the only functional category significantly more likely
to be positively selected (permutation test, _P_ = 2 × 10-4, FDR = 0.007; Supplementary Table 12); the biological significance of this finding merits further investigation. Furthermore,
within each GO class, there is greater dispersion among genes in their probability of positive selection than in their estimate of ω (MWU one-tailed, _P_ = 0.011; Supplementary Information
section 11.1), suggesting that although functionally similar genes share patterns of constraint, they do not necessarily show similar patterns of positive selection (Fig. 4). Interestingly,
protein-coding genes with no annotated (‘unknown’) function in the GO database seem to be less constrained (permutation test, _P_ < 1 × 10-4, FDR = 0.006)80 and to have on average lower
_P_-values for the test of positive selection than genes with annotated functions (permutation test, _P_ = 0.001, FDR = 0.058). It is unlikely that this observation results entirely from an
over-representation of mis-annotated or non-protein-coding genes in the ‘unknown’ functional class, because this finding is robust to the removal of all _D. melanogaster_ genes predicted to
be non-protein-coding in ref. 8. The bias in the way biological function is ascribed to genes (to laboratory-induced, easily scorable functions) leaves open the possibility that unannotated
biological functions may have an important role in evolution. Indeed, genes with characterized mutant alleles in FlyBase evolve significantly more slowly than other genes (median ωwith
alleles = 0.0525 and ωwithout alleles = 0.0701; MWU, _P_ < 1 × 10-16). Previous work has suggested that a substantial fraction of non-synonymous substitutions in _Drosophila_ were fixed
through positive selection81,82,83,84,85. We estimate that 33.1% of single-copy orthologues in the _melanogaster_ group have experienced positive selection on at least a subset of codons
(_q_-value true-positive tests77) (Supplementary Information section 11.1). This may be an underestimate, because we have only examined single-copy orthologues, owing to difficulties in
producing accurate alignments of paralogues by automated methods. On the basis of the 878 genes inferred to have experienced positive selection with high confidence (FDR < 10%), we
estimated that an average of 2% of codons in positively selected genes have ω > 1. Thus, several lines of evidence, based on different methodologies, suggest that patterns of amino acid
fixation in _Drosophila_ genomes have been shaped extensively by positive selection. The presence of functional domains within a protein may lead to heterogeneity in patterns of constraint
and adaptation along its length. Among genes inferred to be evolving by positive selection at a 10% FDR, 63.7% (_q_-value true-positive tests77) show evidence for spatial clustering of
positively selected codons (Supplementary Information section 11.2). Spatial heterogeneity in constraint is further supported by contrasting ω for codons inside versus outside defined
InterPro domains (genes lacking InterPro domains are treated as ‘outside’ a defined InterPro domain). Codons within InterPro domains were significantly more conserved than codons outside
InterPro domains (median ω: 0.062 InterPro domains, 0.084 outside InterPro domains; MWU, _P_ < 2.2 × 10-16; Supplementary Information section 11.2). Similarly, there were significantly
more positively selected codons outside of InterPro domains than inside domains (FET _P_ < 2.2 × 10-16), suggesting that in addition to being more constrained, codons in protein domains
are less likely to be targets of positive selection (Supplementary Fig. 6). FACTORS AFFECTING THE RATE OF PROTEIN EVOLUTION IN _DROSOPHILA_ The sequenced genomes of the _melanogaster_ group
provide unprecedented statistical power to identify factors affecting rates of protein evolution. Previous analyses have suggested that although the level of gene expression consistently
seems to be a major determinant of variation in rates of evolution among proteins86,87, other factors probably play a significant, if perhaps minor, part88,89,90,91. In _Drosophila_,
although highly expressed genes do evolve more slowly, breadth of expression across tissues, gene essentiality and intron number all also independently correlate with rates of protein
evolution, suggesting that the additional complexities of multicellular organisms are important factors in modulating rates of protein evolution78. The presence of repetitive amino acid
sequences has a role as well: non-repeat regions in proteins containing repeats evolve faster and show more evidence for positive selection than genes lacking repeats92. These data also
provide a unique opportunity to examine the impact of chromosomal location on evolutionary rates. Population genetic theory predicts that for new recessive mutations, both purifying and
positive selection will be more efficient on the X chromosome given its hemizygosity in males93. In contrast, the lack of recombination on the small, mainly heterochromatic dot
chromosome94,95 is expected to reduce the efficacy of selection96. Because codon bias, or the unequal usage of synonymous codons in protein-coding sequences, reflects weak but pervasive
selection, it is a sensitive metric for evaluating the efficacy of purifying selection. Consistent with expectation, in all 12 species, we find significantly elevated levels of codon bias on
the X chromosome and significantly reduced levels of codon bias on the dot chromosome97. Furthermore, X-chromosome-linked genes are marginally over-represented within the set of positively
selected genes in the _melanogaster_ group (FET, _P_ = 0.055), which is consistent with increased rates of adaptive substitution on this chromosome. This analysis suggests that chromosomal
context also serves to modulate rates of molecular evolution in protein-coding genes. To examine further the impact of genomic location on protein evolution, we examined the subset of genes
that have moved within or between chromosome arms32,98. Genes inferred to have moved between Muller elements have a significantly higher rate of protein evolution than genes inferred to have
moved within a Muller element (MWU, _P_ = 1.32 × 10-14) and genes that have maintained their genomic position (MWU, _P_ = 0.008) (Supplementary Fig. 7). Interestingly, genes that move
within Muller elements have a significantly lower rate of protein evolution than those for which genomic locations have been maintained (MWU, _P_ = 3.85 × 10-14). It remains unclear whether
these differences reflect underlying biases in the types of genes that move inter- versus intra-chromosomally, or whether they are due to _in situ_ patterns of evolution in novel genomic
contexts. CODON BIAS Codon bias is thought to enhance the efficiency and/or accuracy of translation99,100,101 and seems to be maintained by mutation–selection–drift balance101,102,103,104.
Across the 12 _Drosophila_ genomes, there is more codon bias in the _Sophophora_ subgenus than in the _Drosophila_ subgenus, and a previously noted105,106,107,108,109 striking reduction in
codon bias in _D. willistoni_110,111 (Fig. 5). However, with only minor exceptions, codon preferences for each amino acid seem to be conserved across 11 of the 12 species. The striking
exception is _D. willistoni_, in which codon usage for 6 of 18 redundant amino acids has diverged (Fig. 5). Mutation alone is not sufficient to explain codon-usage bias in _D. willistoni_,
which is suggestive of a lineage-specific shift in codon preferences111,112. We found evidence for a lineage-specific genomic reduction in codon bias in _D. melanogaster_ (Fig. 5), as has
been suggested previously113,114,115,116,117,118,119. In addition, maximum-likelihood estimation of the strength of selection on synonymous sites in 8,510 _melanogaster_ group single-copy
orthologues revealed a marked reduction in the number of genes under selection for increased codon bias in _D. melanogaster_ relative to its sister species _D. sechellia_120. EVOLUTION OF
GENES ASSOCIATED WITH ECOLOGY AND REPRODUCTION Given the ecological and environmental diversity encompassed by the 12 _Drosophila_ species, we examined the evolution of genes and gene
families associated with ecology and reproduction. Specifically, we selected genes with roles in chemoreception, detoxification/metabolism, immunity/defence, and sex/reproduction for more
detailed study. CHEMORECEPTION _Drosophila_ species have complex olfactory and gustatory systems used to identify food sources, hazards and mates, which depend on odorant-binding proteins,
and olfactory/odorant and gustatory receptors (_Or_s and _Gr_s). The _D. melanogaster_ genome has approximately 60 _Or_s, 60 _Gr_s and 50 odorant-binding protein genes. Despite overall
conservation of gene number across the 12 species and widespread evidence for purifying selection within the _melanogaster_ group, there is evidence that a subset of _Or_ and _Gr_ genes
experiences positive selection121,122,123. Furthermore, clear lineage-specific differences are detectable between generalist and specialist species within the _melanogaster_ subgroup. First,
the two independently evolved specialists (_D. sechellia_ and _D. erecta_) are losing _Gr_ genes approximately five times more rapidly than the generalist species121,124. We believe this
result is robust to sequence quality, because all pseudogenes and deletions were verified by direct re-sequencing and synteny-based orthologue searches, respectively. Generalists are
expected to encounter the most diverse set of tastants and seem to have maintained the greatest diversity of gustatory receptors. Second, _Or_ and _Gr_ genes that remain intact in _D.
sechellia_ and _D. erecta_ evolve significantly more rapidly along these two lineages (ω = 0.1556 for _Or_s and 0.1874 for _Gr_s) than along the generalist lineages (ω = 0.1049 for _Or_s and
0.1658 for _Gr_s; paired Wilcoxon, _P_ = 0.0003 and 0.003, respectively124). There is some evidence that odorant-binding protein genes also evolve significantly faster in specialists
compared to generalists122. This elevated ω reflects a trend observed throughout the genomes of the two specialists and is likely to result, at least in part, from demographic phenomena.
However, the difference between specialist and generalist ω for _Or_/_Gr_ genes (0.0292) is significantly greater than the difference for genes across the genome (0.0091; MWU, _P_ =
0.0052)121, suggesting a change in selective regime. Moreover, the observation that elevated ω as well as accelerated gene loss disproportionately affect groups of _Or_ and _Gr_ genes that
respond to specific chemical ligands and/or are expressed during specific life stages suggests that rapid evolution at _Or_/_Gr_ loci in specialists is related to the ecological shifts these
species have sustained121. DETOXIFICATION/METABOLISM The larval food sources for many _Drosophila_ species contain a cocktail of toxic compounds, and consequently _Drosophila_ genomes
encode a wide variety of detoxification proteins. These include members of the cytochrome P450 (P450), carboxyl/choline-esterase (CCE) and glutathione _S_-transferase (GST) multigene
families, all of which also have critical roles in resistance to insecticides125,126,127. Among the P450s, the five enzymes associated with insecticide resistance are highly dynamic across
the phylogeny, with 24 duplication events and 4 loss events since the last common ancestor of the genus, which is in striking contrast to genes with known developmental roles, eight of which
are present as a single copy in all 12 species (C. Robin, personal communication). As with chemoreceptors, specialists seem to lose detoxification genes at a faster rate than generalists.
For instance, _D. sechellia_ has lost the most P450 genes; these 14 losses comprise almost one-third of all P450 loss events (Supplementary Table 13) (C. Robin, personal communication).
Positive selection has been implicated in detoxification-gene evolution as well, because a search for positive selection among GSTs identified the parallel evolution of a radical glycine to
lysine amino acid change in GSTD1, an enzyme known to degrade DDT128. Finally, although metabolic enzymes in general are highly constrained (median ω = 0.045 for enzymes, 0.066 for
non-enzymes; MWU, _P_ = 5.7 × 10-24), enzymes involved in xenobiotic metabolism evolve significantly faster than other enzymes (median ω = 0.05 for the xenobiotic group versus ω = 0.045
overall, two-tailed permutation test, _P_ = 0.0110; A. J. Greenberg, personal communication). Metazoans deal with excess selenium in the diet by sequestration in selenoproteins, which
incorporate the rare amino acid selenocysteine (Sec) at sites specified by the TGA codon. The recoding of the normally terminating signal TGA as a Sec codon is mediated by the selenocystein
insertion sequence (SECIS), a secondary structure in the 3′ UTR of selenoprotein messenger RNAs. All animals examined so far have selenoproteins; three have been identified in _D.
melanogaster_ (SELG, SELM and SPS2129,130). Interestingly, although the three known _melanogaster_ selenoproteins are all present in the genomes of the other _Drosophila_ species, in _D.
willistoni_ the TGA Sec codons have been substituted by cysteine codons (TGT/TGC). Consistent with this finding, analysis of the seven genes implicated to date in selenoprotein synthesis
including the Sec-specific tRNA suggests that most of these genes are absent in _D. willistoni_ (R. Guigo, personal communication). _D. willistoni_ thus seems to be the first animal known to
lack selenoproteins. If correct, this observation is all the more remarkable given the ubiquity of selenoproteins and the selenoprotein biosynthesis machinery in metazoans, the toxicity of
excess selenium, and the protection from oxidative stress mediated by selenoproteins. However, it remains possible that this species encodes selenoproteins in a different way, and this
represents an exciting avenue of future research. IMMUNITY/DEFENCE _Drosophila_, like all insects, possesses an innate immune system with many components analogous to the innate immune
pathways of mammals, although it lacks an antibody-mediated adaptive immune system131. Immune system genes often evolve rapidly and adaptively, driven by selection pressures from pathogens
and parasites132,133,134. The genus _Drosophila_ is no exception: immune system genes evolve more rapidly than non-immune genes, showing both high total divergence rates and specific signs
of positive selection135. In particular, 29% of receptor genes involved in phagocytosis seem to evolve under positive selection, suggesting that molecular co-evolution between _Drosophila_
pattern recognition receptors and pathogen antigens is driving adaptation in the immune system135. Somewhat surprisingly, genes encoding effector proteins such as antimicrobial peptides are
far less likely to exhibit adaptive sequence evolution. Only 5% of effector genes (and no antimicrobial peptides) show evidence of adaptive evolution, compared to 10% of genes genome-wide.
Instead, effector genes seem to evolve by rapid duplication and deletion. Whereas 49% of genes genome-wide, 63% of genes involved in pathogen recognition and 81% of genes implicated in
immune-related signal transduction can be found as single-copy orthologues in all 12 species, only 40% of effector genes exist as single-copy orthologues across the genus (χ2 = 41.13, _P_ =
2.53 × 10-8), suggesting rapid radiation of effector protein classes along particular lineages135. Thus, much of the _Drosophila_ immune system seems to evolve rapidly, although the mode of
evolution varies across immune-gene functional classes. SEX/REPRODUCTION Genes encoding sex- and reproduction-related proteins are subject to a wide array of selective forces, including
sexual conflict, sperm competition and cryptic female choice, and to the extent that these selective forces are of evolutionary consequence, this should lead to rapid evolution in these
genes136 (for an overview see refs 137, 138). The analysis of 2,505 sex- and reproduction-related genes within the _melanogaster_ group indicated that male sex- and reproduction-related
genes evolve more rapidly at the protein level than genes not involved in sex or reproduction or than female sex- and reproduction-related genes (Supplementary Fig. 8). Positive selection
seems to be at least partially responsible for these patterns, because genes involved in spermatogenesis have significantly stronger evidence for positive selection than do
non-spermatogenesis genes (permutation test, _P_ = 0.0053). Similarly, genes that encode components of seminal fluid have significantly stronger evidence for positive selection than
‘non-sex’ genes139. Moreover, protein-coding genes involved in male reproduction, especially seminal fluid and testis genes, are particularly likely to be lost or gained across _Drosophila_
species29,139. EVOLUTIONARY FORCES IN THE MITOCHONDRIAL GENOME Functional elements in mtDNA are strongly conserved, as expected: tRNAs are relatively more conserved than the mtDNA overall
(average pairwise nucleotide distance = 0.055 substitutions per site for tRNAs versus 0.125 substitutions per site overall). We observe a deficit of substitutions occurring in the stem
regions of the stem-loop structure in tRNAs, consistent with strong selective pressure to maintain RNA secondary structure, and there is a strong signature of purifying selection in
protein-coding genes13. However, despite their shared role in aerobic respiration, there is marked heterogeneity in the rates of amino acid divergence between the oxidative phosphorylation
enzyme complexes across the 12 species (NADH dehydrogenase, 0.059 > ATPase, 0.042 > CytB, 0.037 > cytochrome oxidase, 0.020; mean pairwise _d__N_ ), which contrasts with the
relative homogeneity in synonymous substitution rates. A model with distinct substitution rates for each enzyme complex rather than a single rate provides a significantly better fit to the
data (_P_ < 0.0001), suggesting complex-specific selective effects of mitochondrial mutations13. NON-CODING SEQUENCE EVOLUTION NCRNA SEQUENCE EVOLUTION The availability of complete
sequence from 12 _Drosophila_ genomes, combined with the tractability of RNA structure predictions, offers the exciting opportunity to connect patterns of sequence evolution directly with
structural and functional constraints at the molecular level. We tested models of RNA evolution focusing on specific ncRNA gene classes in addition to inferring patterns of sequence
evolution using more general datasets that are based on predicted intronic RNA structures. The exquisite simplicity of miRNAs and their shared stem-loop structure makes these ncRNAs
particularly amenable to evolutionary analysis. Most miRNAs are highly conserved within the _Drosophila_ genus: for the 71 previously described miRNA genes inferred to be present in the
common ancestor of these 12 species, mature miRNA sequences are nearly invariant. However, we do find a small number of substitutions and a single deletion in mature miRNA sequences
(Supplementary Table 14), which may have functional consequences for miRNA–target interactions and may ultimately help identify targets through sequence covariation. Pre-miRNA sequences are
also highly conserved, evolving at about 10% of the rate of synonymous sites. To link patterns of evolution with structural constraints, we inferred ancestral pre-miRNA sequences and deduced
secondary structures at each ancestral node on the phylogeny (Supplementary Information section 12.1). Although conserved miRNA genes show little structural change (little change in free
energy), the five _melanogaster_ group-specific miRNA genes (_miR-303_ and the _mir-310/311/312/313_ cluster) have undergone numerous changes across the entire pre-miRNA sequence, including
the ordinarily invariant mature miRNA. Patterns of polymorphism and divergence in these lineage-specific miRNA genes, including a high frequency of derived mutations, are suggestive of
positive selection140. Although lineage-specific miRNAs may evolve under less constraint because they have fewer target transcripts in the genome, it is also possible that recent integration
into regulatory networks causes accelerated rates of miRNA evolution. We further investigated patterns of sequence evolution for the subset of 38 conserved pre-miRNAs with mature miRNA
sequences at their 3′ end by calculating evolutionary rates in distinct site classes (Fig. 6, and Supplementary Information section 12.2). Outside the mature miRNA and its complementary
sequence, loops had the highest rate of evolution, followed by unpaired sites, with paired sites having the lowest rate of evolution. Inside the mature miRNA, unpaired sites evolve more
slowly than paired sites, whereas the opposite is true for the sequence complementary to the mature miRNA. Surprisingly, a large fraction of unpaired bulges or internal loops in the mature
miRNA seem to be conserved—a pattern which may have implications for models of miRNA biogenesis and the degree of mismatch allowed in miRNA–target prediction methods. Overall these results
support the qualitative model proposed in ref. 141 for the canonical progression of miRNA evolution, and show that functional constraints on the miRNA itself supersede structural constraints
imposed by maintenance of the hairpin-loop. To assess constraint on stem regions of RNA structures more generally, we compared substitution rates in stems (_S_) to those in nominally
unconstrained loop regions (_L_) in a wide variety of ncRNAs (Supplementary Information section 12.3). We estimated substitution rates using a maximum likelihood framework, and compared the
observed _L_/_S_ ratio with the average _L_/_S_ ratio estimated from published secondary structures in RFAM, which we normalized to 1.0. _L_/_S_ ratios for _Drosophila_ ncRNA families range
from a highly constrained 2.57 for the nuclear RNase P family to 0.56 for the 5S ribosomal RNA (Supplementary Table 15). Finally, we predicted a set of conserved intronic RNA structures and
analysed patterns of compensatory nucleotide substitution in _D. melanogaster_, _D. yakuba_, _D. ananassae_, _D. pseudoobscura_, _D. virilis_ and _D. mojavensis_ (Supplementary Information
section 13). Signatures of compensatory evolution in RNA helices are detected as covarying nucleotide sites or ‘covariations’ (that is, two Watson–Crick bases that interact in species A
replaced by a different Watson–Crick pair in species B). The number of covariations (per base pair of a helix) depends on the physical distance between the interacting nucleotides
(Supplementary Fig. 9), as has been observed for the RNA helices in the _Drosophila bicoid_ 3′ UTR region142. Short-range pairings exhibit a higher average number of covariations with a
larger variance among helices than longer-range pairings. The decrease in rate of covariation with increasing distance may be explained by physical properties of a helix, which may impose
selective constraints on the evolution of covarying nucleotides within a helix. Alternatively, if individual mutations at each locus are deleterious but compensated by mutations at a second
locus, given sufficiently strong selection against the first deleterious mutation these epistatic fitness interactions could generate the observed distance effect143. EVOLUTION OF
_CIS_-REGULATORY DNAS Comparative analyses of _cis_-regulatory sequences may provide insights into the evolutionary forces acting on regulatory components of genes, shed light on the
constraints of the _cis_-regulatory code and aid in annotation of new regulatory sequences. Here we rely on two recently compiled databases, and present results comparing _cis_-regulatory
modules144 and transcription factor binding sites (derived from DNase I footprints)145 between _D. melanogaster_ and _D. simulans_ (Supplementary Information section 8). We estimated mean
selective constraint (_C_, the fraction of mutations removed by natural selection) relative to the ‘fastest evolving intron’ sites at the 5′ end of short introns, which represent putatively
unconstrained neutral standards (Supplementary Information section 8.2)146. Note that this approach ignores the contribution of positively selected sites, potentially underestimating the
fraction of functionally relevant sites147. Consistent with previous findings, _Drosophila_ _cis_-regulatory sequences are highly constrained148,149. Mean constraint within _cis_-regulatory
modules is 0.643 (95% bootstrap confidence interval = 0.621–0.662) and within footprints is 0.692 (0.655–0.723), both of which are significantly higher than mean constraint in non-coding DNA
overall (0.555 (0.546–0.563)) and significantly lower than constraint at non-degenerate coding sites (0.862 (0.856–0.868)) and ncRNA genes (0.864 (0.846–0.880)) (Supplementary Fig. 10). The
high level of constraint in _cis_-regulatory sequences also extends into flanking sequences, only declining to constraint levels typical of non-coding DNA 40 bp away. This is consistent
with previous findings that transcription factor binding sites tend to be found in larger blocks of constraint that cluster to form _cis_-regulatory modules150. To understand selective
constraints on nucleotides within _cis_-regulatory sequences that have direct contact with transcription factors, we estimated the selective constraint for the best match to position weight
matrices within each footprint151; core motifs in transcription-factor-binding sites have a mean constraint of 0.773 (0.729–0.814), significantly greater than the mean for the footprints as
a whole, and approaching the level of constraint found at non-degenerate coding sites and in ncRNA genes (Supplementary Fig. 10). We next examined the variation in selective constraint
across _cis_-regulatory sequences. Surprisingly, we find no evidence that selective constraint is correlated with predicted transcription-factor-binding strength (estimated as the position
weight matrix score _P_-value) (Spearman’s _r_ = 0.0681, _P_ = 0.0609). We observe significant variation in constraint both among target genes (Kruskal–Wallis tests, footprints, _P_ <
0.0001; and position weight matrix matches within footprints, _P_ = 0.0023) and among chromosomes (_cis_-regulatory modules, _P_ = 0.0186; footprints, _P_ = 0.0388; and position weight
matrix matches within footprints, _P_ = 0.0108; Supplementary Table 16). DISCUSSION AND CONCLUSION Each new genome sequence affords novel opportunities for comparative genomic inference.
What makes the analysis of these 12 _Drosophila_ genomes special is the ability to place every one of these genomic comparisons on a phylogeny with a taxon separation that is ideal for
asking a wealth of questions about evolutionary patterns and processes. It is without question that this phylogenomic approach places additional burdens on bioinformatics efforts,
multiplying the amount of data many-fold, requiring extra care in generating multi-species alignments, and accommodating the reality that not all genome sequences have the same degree of
sequencing or assembly accuracy. These difficulties notwithstanding, phylogenomics has extraordinary advantages not only for the analyses that are possible, but also for the ability to
produce high-quality assemblies and accurate annotations of functional features in a genome by using closely related genomes as guides. The use of multi-species orthology provides especially
convincing evidence in support of particular gene models, not only for protein-coding genes, but also for miRNA and other ncRNA genes. Many attributes of the genomes of _Drosophila_ are
remarkably conserved across species. Overall genome size, number of genes, distribution of transposable element classes, and patterns of codon usage are all very similar across these 12
genomes, although _D. willistoni_ is an exceptional outlier by several criteria, including its unusually skewed codon usage, increased transposable element content and potential lack of
selenoproteins. At a finer scale, the number of structural changes and rearrangements is much larger; for example, there are several different rearrangements of genes in the _Hox_ cluster
found in these _Drosophila_ species. The vast majority of multigene families are found in all 12 genomes, although gene family size seems to be highly dynamic: almost half of all gene
families change in size on at least one lineage, and a noticeable fraction shows rapid and lineage-specific expansions and contractions. Particularly notable are cases consistent with
adaptive hypotheses, such as the loss of _Gr_ genes in ecological specialists and the lineage-specific expansions of antimicrobial peptides and other immune effectors. All species were found
to have novel genes not seen in other species. Although lineage-specific genes are challenging to verify computationally, we can confirm at least 44 protein-coding genes unique to the
_melanogaster_ group, and these proteins have very different properties from ancestral proteins. Similarly, although the relative abundance of transposable element subclasses across these
genomes does not differ dramatically, total genomic transposable element content varies substantially among species, and several instances of lineage-specific transposable elements were
discovered. There is considerable variation among protein-coding genes in rates of evolution and patterns of positive selection. Functionally similar proteins tend to evolve at similar
rates, although variation in genomic features such as gene expression level, as well as chromosomal location, are also associated with variation in evolutionary rate among proteins. Whereas
broad functional classes do not seem to share patterns of positive selection, and although very few GO categories show excesses of positive selection, a number of genes involved in
interactions with the environment and in sex and reproduction do show signatures of adaptive evolution. It thus seems likely that adaptation to changing environments, as well as sexual
selection, shape the evolution of protein-coding genes. Annotation of ncRNA genes across all 12 species allows comprehensive analysis of the evolutionary divergence of these genes. MicroRNA
genes in particular are more conserved than protein-coding genes with respect to their primary DNA sequence, and the substitutions that do occur often have compensatory changes such that the
average estimated free energy of the folding structures remains remarkably constant across the phylogeny. Surprisingly, mismatches in miRNAs seem to be highly conserved, which may impact
models of miRNA biogenesis and target recognition. Lineage-restricted miRNAs, however, have considerably elevated rates of change, suggesting either reduced constraint due to novel miRNAs
having fewer targets, or adaptive evolution of evolutionarily young miRNAs. Virtually any question about the function of genome features in _Drosophila_ is now empowered by being embedded in
the context of this 12 species phylogeny, allowing an analysis of the ways by which evolution has tuned myriad biological processes across the hundreds of millions of years spanned in total
by this phylogeny. The analyses presented herein have generated more questions than they have answered, and these results represent a small fraction of that which is possible. Because much
of this rich and extraordinary comparative genomic dataset remains to be explored, we believe that these 12 _Drosophila_ genome sequences will serve as a powerful tool for gleaning further
insight into genetic, developmental, regulatory and evolutionary processes. METHODS The full methods for this paper are described in Supplementary Information. Here, we describe the datasets
generated by this project and their availability. GENOMIC SEQUENCE Scaffolds and assemblies for all genomic sequence generated by this project are available from GenBank (Supplementary
Tables 4 and 5), and FlyBase (ftp://ftp.flybase.net/12_species_analysis/). Genome browsers are available from UCSC (http://genome.ucsc.edu/cgi-bin/hgGateway?hgsid =98180333&clade =
insect&org = 0&db = 0) and Flybase (http://flybase.org/cgi-bin/gbrowse/dmel/). BLAST search of these genomes is available at FlyBase (http://flybase.org/blast). PREDICTED GENE MODELS
Consensus gene predictions for the 11 non-_melanogaster_ species, produced by combining several different GLEAN runs that weight homology evidence more or less strongly, are available from
FlyBase as GFF files for each species (ftp://ftp.flybase.net/12_species_analysis/). These gene models can also be accessed from the Genome Browser in FlyBase (Gbrowse;
http://flybase.org/cgi-bin/gbrowse/dmel/). Predictions of non-protein-coding genes are also available in GFF format for each species, from FlyBase
(ftp://ftp.flybase.net/12_species_analysis/). HOMOLOGY Multiway homology assignments are available from FlyBase (ftp://ftp.flybase.net/12_species_analysis/), and also in the Genome Browser
(Gbrowse). ALIGNMENTS All alignment sets produced are available in FASTA format from FlyBase (ftp://ftp.flybase.net/12_species_analysis/). PAML PARAMETERS Output from PAML models for the
alignments of single copy orthologues in the _melanogaster_ group, including the _q_-value for the test for positive selection, are available from FlyBase
(ftp://ftp.flybase.net/12_species_analysis/). REFERENCES * Markow, T. A. & O'Grady, P. M. Drosophila biology in the genomic age. _Genetics_ doi: 10.1534/genetics.107.074112 (in the
press) * Powell, J. R. _Progress and Prospects in Evolutionary Biology: The Drosophila Model_ (Oxford Univ. Press, Oxford, 1997) Google Scholar * Adams, M. D. et al. The genome sequence of
_Drosophila melanogaster_ . _Science_ 287, 2185–2195 (2000) PubMed Google Scholar * Celniker, S. E. et al. Finishing a whole-genome shotgun: release 3 of the _Drosophila melanogaster_
euchromatic genome sequence. _Genome Biol._ 3, research0079.1–0079.14 (2002) Google Scholar * Richards, S. et al. Comparative genome sequencing of _Drosophila pseudoobscura_: chromosomal,
gene, and _cis_-element evolution. _Genome Res._ 15, 1–18 (2005) CAS PubMed PubMed Central Google Scholar * Myers, E. W. et al. A whole-genome assembly of _Drosophila_ . _Science_ 287,
2196–2204 (2000) ADS CAS PubMed Google Scholar * Fleischmann, R. D. et al. Whole-genome random sequencing and assembly of _Haemophilus influenzae_ Rd. _Science_ 269, 496–512 (1995) ADS
CAS PubMed Google Scholar * Stark et al Discovery of functional elements in 12 _Drosophila_ genomes using evolutionary signatures. _Nature_ doi: 10.1038/nature06340 (this issue) * Begun,
D. J. et al. Population genomics: whole-genome analysis of polymorphism and divergence in _Drosophila simulans_ . _PLoS Biol._ 5 e310 doi: 10.1371/journal.pbio.0050310 (2007) Article CAS
PubMed PubMed Central Google Scholar * Zimin, A. V., Smith, D. R., Sutton, G. & Yorke, J. A. Assembly reconciliation. _Bioinformatics_ (in the press) * Clary, D. O. &
Wolstenholme, D. R. The mitochondrial DNA molecule of _Drosophila_ _yakuba_: nucleotide sequence, gene organization, and genetic code. _J. Mol. Evol._ 22, 252–271 (1985) ADS CAS PubMed
Google Scholar * Ballard, J. W. When one is not enough: introgression of mitochondrial DNA in _Drosophila_ . _Mol. Biol. Evol._ 17, 1126–1130 (2000) CAS PubMed Google Scholar * Montooth,
K. L., Abt, D. N., Hoffman, J. & Rand, D. M. Evolution of the mitochondrial DNA across twelve species of _Drosophila_ . _Mol. Biol. Evol._ (submitted) * Salzberg, S. et al.
Serendipitous discovery of _Wolbachia_ genomes in multiple _Drosophila_ species. _Genome Biol._ 6, R23 (2005) PubMed PubMed Central Google Scholar * Edgar, R. C. & Myers, E. W. PILER:
identification and classification of genomic repeats. _Bioinformatics_ 21, i152–i158 (2005) CAS PubMed Google Scholar * Smith, C. D. et al. Improved repeat identification and masking in
Dipterans. _Gene_ 389, 1–9 (2007) CAS PubMed Google Scholar * Li, Q. et al. ReAS: Recovery of ancestral sequences for transposable elements from the unassembled reads of a whole shotgun.
_PloS Comput. Biol._ 1, e43 (2005) ADS PubMed PubMed Central Google Scholar * Bergman, C. M., Quesneville, H., Anxolabehere, D. & Ashburner, M. Recurrent insertion and duplication
generate networks of transposable element sequences in the _Drosophila melanogaster_ genome. _Genome Biol._ 7, R112 (2006) PubMed PubMed Central Google Scholar * Guigo, R., Knudsen, S.,
Drake, N. & Smith, T. Prediction of gene structure. _J. Mol. Biol._ 226, 141–157 (1992) CAS PubMed Google Scholar * Korf, I. Gene finding in novel genomes. _BMC Bioinformatics_ 5, 59
(2004) PubMed PubMed Central Google Scholar * Gross, S. S. & Brent, M. R. Using multiple alignments to improve gene prediction. _J. Comput. Biol._ 13, 379–393 (2006) MathSciNet CAS
PubMed MATH Google Scholar * Gross, S. S., Do, C. B. & Batzoglou, S. in _BCATS 2005 Symposium Proc._ 82. (2005) * Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise.
_Genome Res._ 14, 988–995 (2004) CAS PubMed PubMed Central Google Scholar * Slater, G. & Birney, E. Automated generation of heuristics for biological sequence comparison. _BMC
Bioinformatics_ 6, 31 (2005) PubMed PubMed Central Google Scholar * Chatterji, S. & Pachter, L. Reference based annotation with GeneMapper. _Genome Biol._ 7, R29 (2006) PubMed PubMed
Central Google Scholar * Souvorov, A. et al. in _NCBI News Fall/Winter, NIH Publication No. 04-3272_ (eds Benson, D & Wheeler, D). (2006) Google Scholar * Honeybee Genome Sequencing
Consortium. Insights into social insects from the genome of the honeybee _Apis mellifera_ . _Nature_ 443, 931–949 (2006) * Elsik, C. G. et al. Creating a honey bee consensus gene set.
_Genome Biol._ 8, R13 (2007) PubMed PubMed Central Google Scholar * Zhang, Y., Sturgill, D., Parisi, M., Kumar, S. & Oliver, B. Constraint and turnover in sex-biased gene expression
in the genus _Drosophila_ . _Nature_ doi: 10.1038/nature06323 (this issue). * Manak, J. R. et al. Biological function of unannotated transcription during the early development of _Drosophila
melanogaster_ . _Nature Genet._ 38, 1151–1158 (2006) CAS PubMed Google Scholar * Tatusov, R. L., Koonin, E. V. & Lipman, D. J. A genomic perspective on protein families. _Science_
278, 631–637 (1997) ADS CAS PubMed Google Scholar * Bhutkar, A., Russo, S., Smith, T. F. & Gelbart, W. M. Techniques for multi-genome synteny analysis to overcome assembly
limitations. _Genome Informatics_ 17, 152–161 (2006) CAS PubMed Google Scholar * Heger, A. & Ponting, C. Evolutionary rate analyses of orthologues and paralogues from twelve
_Drosophila_ genomes. doi: 10.1101/gr6249707 _Genome Res_. (in the press) * Notredame, C., Higgins, D. G. & Heringa, J. T-Coffee: A novel method for fast and accurate multiple sequence
alignment. _J. Mol. Biol._ 302, 205–217 (2000) CAS PubMed Google Scholar * Rat Genome Sequencing Project Consortium. Genome sequence of the Brown Norway rat yields insights into mammalian
evolution. _Nature_ 428, 493–521 (2004) * Waterston, R. H. et al. Initial sequencing and comparative analysis of the mouse genome. _Nature_ 420, 520–562 (2002) ADS CAS PubMed Google
Scholar * Harrison, P. M., Milburn, D., Zhang, Z., Bertone, P. & Gerstein, M. Identification of pseudogenes in the _Drosophila melanogaster_ genome. _Nucleic Acids Res._ 31, 1033–1037
(2003) CAS PubMed PubMed Central Google Scholar * Bosco, G., Campbell, P., Leiva-Neto, J. & Markow, T. Analysis of _Drosophila_ species genome size and satellite DNA content reveals
significant differences among strains as well as between species. _Genetics_ doi: 10.1534/Genetics107.075069. (in the press) * Ranz, J. et al. Principles of genome evolution in the
_Drosophila melanogaster_ species group. _PLoS Biol._ 5 e152 doi: 10.1371/journal.pbio.0050152 (2007) Article CAS PubMed PubMed Central Google Scholar * Noor, M. A. F., Garfield, D. A.,
Schaeffer, S. W. & Machado, C. A. Divergence between the _Drosophila pseudoobscura_ and _D. _ _persimilis_ genome sequences in relation to chromosomal inversions. _Genetics_ doi:
10.1534/genetics.107.070672. (in the press) * Lewis, E. B. A gene complex controlling segmentation in _Drosophila_ . _Nature_ 276, 565–570 (1978) ADS CAS PubMed Google Scholar * Negre,
B., Ranz, J. M., Casals, F., Caceres, M. & Ruiz, A. A new split of the _Hox_ gene complex in _Drosophila_: relocation and evolution of the gene labial. _Mol. Biol. Evol._ 20, 2042–2054
(2003) CAS PubMed Google Scholar * Von Allmen, G. et al. Splits in fruitfly _Hox_ gene complexes. _Nature_ 380, 116 (1996) ADS CAS PubMed Google Scholar * Negre, B. & Ruiz, A.
HOM-C evolution in _Drosophila_: is there a need for _Hox_ gene clustering? _Trends Genet._ 23, 55–59 (2007) CAS PubMed Google Scholar * Dowsett, A. P. & Young, M. W. Differing levels
of dispersed repetitive DNA among closely related species of _Drosophila_ . _Proc. Natl Acad. Sci._ 79, 4570–4574 (1982) ADS CAS PubMed PubMed Central Google Scholar * Kapitonov, V. V.
& Jurka, J. DNAREP1_DM. (Repbase Update Release 3.4, 1999) * Kapitonov, V. V. & Jurka, J. Molecular paleontology of transposable elements in the _Drosophila melanogaster_ genome.
_Proc. Natl Acad. Sci. USA_ 100, 6569–6574 (2003) ADS CAS PubMed PubMed Central Google Scholar * Singh, N. D., Arndt, P. F. & Petrov, D. A. Genomic heterogeneity of background
substitutional patterns in _Drosophila melanogaster_ . _Genetics_ 169, 709–722 (2004) PubMed Google Scholar * Yang, H.-P., Hung, T.-L., You, T.-L. & Yang, T.-H. Genomewide comparative
analysis of the highly abundant transposable element _DINE-1_ suggests a recent transpositional burst in _Drosophila_ _yakuba_ . _Genetics_ 173, 189–196 (2006) CAS PubMed PubMed Central
Google Scholar * Yang, H.-P. & Barbash, D. Abundant and species-specific miniature inverted-repeat transposable elements in 12 _Drosophila_ genomes. _Genome Biol._ (submitted) * Wilder,
J. & Hollocher, H. Mobile elements and the genesis of microsatellites in dipterans. _Mol. Biol. Evol._ 18, 384–392 (2001) CAS PubMed Google Scholar * Marzo, M., Puig, M. & Ruiz,
A. The foldback-like element _Galileo_ belongs to the P superfamily of DNA transposons and is widespread within the genus _Drosophila_ . _Proc. Natl Acad. Sci. USA_ (submitted) * Casola, C.,
Lawing, A., Betran, E. & Feschotte, C. PIF-like transposons are common in _Drosophila_ and have been repeatedly domesticated to generate new host genes. _Mol. Biol. Evol._ 24, 1872–1888
(2007) CAS PubMed Google Scholar * Abad, J. P. et al. Genomic analysis of _Drosophila melanogaster_ telomeres: full-length copies of _HeT-A_ and _TART_ elements at telomeres. _Mol. Biol.
Evol._ 21, 1613–1619 (2004) CAS PubMed Google Scholar * Abad, J. P. et al. _TAHRE_, a novel telomeric retrotransposon from _Drosophila melanogaster_, reveals the origin of _Drosophila_
telomeres. _Mol. Biol. Evol._ 21, 1620–1624 (2004) CAS PubMed Google Scholar * Blackburn, E. H. Telomerases. _Annu. Rev. Biochem._ 61, 113–129 (1992) CAS PubMed Google Scholar *
Villasante, A. et al. _Drosophila_ telomeric retrotransposons derived from an ancestral element that as recruited to replace telomerase. _Genome Res._ (in the press) * International Chicken
Genome Sequencing Consortium. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. _Nature_ 432, 695–716 (2004) * Lander, E. S. et al.
Initial sequencing and analysis of the human genome. _Nature_ 409, 860–921 (2001) ADS CAS PubMed Google Scholar * _C. elegans_ Sequencing Consortium. Genome sequence of the nematode _C.
_ _elegans_: a platform for investigating biology. _Science_ 282, 2012–2018 (1998) * Mount, S. M., Gotea, V., Lin, C. F., Hernandez, K. & Makalowski, W. Spliceosomal small nuclear RNA
genes in 11 insect genomes. _RNA_ 13, 5–14 (2007) CAS PubMed PubMed Central Google Scholar * Schneider, C., Will, C. L., Brosius, J., Frilander, M. J. & Luhrmann, R. Identification
of an evolutionarily divergent U11 small nuclear ribonucleoprotein particle in _Drosophila_ . _Proc. Natl Acad. Sci. USA_ 101, 9584–9589 (2004) ADS CAS PubMed PubMed Central Google
Scholar * Deng, X. & Meller, V. H. Non-coding RNA in fly dosage compensation. _Trends Biochem. Sci._ 31, 526–532 (2006) CAS PubMed Google Scholar * Amrien, H. & Axel, R. Genes
expressed in neurons of adult male _Drosophila_ . _Cell_ 88, 459–469 (1997) Google Scholar * Park, S.-W. et al. An evolutionarily conserved domain of roX2 RNA is sufficient for induction of
H4-Lys16 acetylation on the _Drosophila_ X chromosome. _Genetics_ (in the press) * Stage, D. E. & Eickbush, T. H. Sequence variation within the rRNA gene loci of 12 _Drosophila_
species. _Genome Res._ (in the press) * Hahn, M. W., De Bie, T., Stajich, J. E., Nguyen, C. & Cristianini, N. Estimating the tempo and mode of gene family evolution from comparative
genomic data. _Genome Res._ 15, 1153–1160 (2005) CAS PubMed PubMed Central Google Scholar * Hahn, M. W., Han, M. V. & Han, S.-G. Gene family evolution across 12 _Drosophila_ genomes.
_PLoS Biol._ 3, e197 (2007) Google Scholar * Levine, M. T., Jones, C. D., Kern, A. D., Lindfors, H. A. & Begun, D. J. Novel genes derived from noncoding DNA in _Drosophila
melanogaster_ are frequently X-linked and exhibit testis-biased expression. _Proc. Natl Acad. Sci. USA_ 103, 9935–9939 (2006) ADS CAS PubMed PubMed Central Google Scholar * Ponce, R.
& Hartl, D. L. The evolution of the novel _Sdic_ gene cluster in _Drosophila melanogaster_ . _Gene_ 376, 174–183 (2006) CAS PubMed Google Scholar * Arguello, J. R., Chen, Y., Tang,
S., Wang, W. & Long, M. Originiation of an X-linked testes chimeric gene by illegitimate recombination in _Drosophila_ . _PLoS Genet._ 2, e77 (2006) PubMed PubMed Central Google
Scholar * Begun, D. J., Lindfore, H. A., Thompson, M. E. & Holloway, A. K. Recently evolved genes identified from _Drosophila_ _yakuba_ and _D. _ _erecta_ accessory gland expressed
sequence tags. _Genetics_ 172, 1675–1681 (2006) CAS PubMed PubMed Central Google Scholar * Betran, E., Thornton, K. & Long, M. Retroposed new genes out of the X in _Drosophila_ .
_Genome Res._ 12, 1854–1859 (2002) CAS PubMed PubMed Central Google Scholar * Yanai, I. et al. Genome-wide midrange transcription profiles reveal expression level relationships in human
tissue specification. _Bioinformatics_ 21, 650–659 (2005) CAS PubMed Google Scholar * Yang, Z. PAML: a program package for phylogenetic analysis by maximum likelihood. _Comput. Appl.
Biosci._ 13, 555–556 (1997) CAS PubMed Google Scholar * The Gene Ontology Consortium. Gene Ontology: tool for the unification of biology. _Nature Genet._ 25, 25–29 (2000) * Storey, J. D.
A direct approach to false discovery rates. _J. R. Stat. Soc. B_ 64, 479–498 (2002) MathSciNet MATH Google Scholar * Larracuente, A. M. et al. Evolution of protein-coding genes in
_Drosophila_ . _Trends Genet._ (submitted) * Yang, Z., Nielsen, R., Goldman, N. & Pedersen, A. M. Codon-substitution models for heterogeneous selection pressure at amino acid sites.
_Genetics_ 155, 431–449 (2000) CAS PubMed PubMed Central Google Scholar * Bergman, C. M. et al. Assessing the impact of comparative genomic sequence data on the functional annotation of
the _Drosophila_ genome. _Genome Biol._ 3, research0086.1–0086.20 (2002) Google Scholar * Bierne, N. & Eyre Walker, A. C. The genomic rate of adaptive amino acid substitution in
_Drosophila_ . _Mol. Biol. Evol._ 21, 1350–1360 (2004) CAS PubMed Google Scholar * Sawyer, S. A., Kulathinal, R. J., Bustamante, C. D. & Hartl, D. L. Bayesian analysis suggests that
most amino acid replacements in _Drosophila_ are driven by positive selection. _J. Mol. Evol._ 57 (suppl. 1). S154–S164 (2003) ADS CAS PubMed Google Scholar * Sawyer, S. A., Parsch, J.,
Zhang, Z. & Hartl, D. L. Prevalence of positive selection among nearly neutral amino acid replacements in _Drosophila_ . _Proc. Natl Acad. Sci. USA_ 104, 6504–6510 (2007) ADS CAS
PubMed PubMed Central Google Scholar * Smith, N. G. & Eyre-Walker, A. Adaptive protein evolution in _Drosophila_ . _Nature_ 415, 1022–1024 (2002) ADS CAS PubMed Google Scholar *
Welch, J. J. Estimating the genomewide rate of adaptive protein evolution in _Drosophila_ . _Genetics_ 173, 821–837 (2006) CAS PubMed PubMed Central Google Scholar * Drummond, D. A.,
Bloom, J. D., Adami, C., Wilke, C. O. & Arnold, F. H. Why highly expressed proteins evolve slowly. _Proc. Natl Acad. Sci. USA_ 102, 14338–14343 (2005) ADS CAS PubMed PubMed Central
Google Scholar * Drummond, D. A., Raval, A. & Wilke, C. O. A single determinant dominates the rate of yeast protein evolution. _Mol. Biol. Evol._ 23, 327–337 (2006) CAS PubMed Google
Scholar * Pal, C., Papp, B. & Hurst, L. D. Highly expressed genes in yeast evolve slowly. _Genetics_ 158, 927–931 (2001) CAS PubMed PubMed Central Google Scholar * Pal, C., Papp, B.
& Lercher, M. J. An integrated view of protein evolution. _Nature Rev. Genet._ 7, 337–348 (2006) CAS PubMed Google Scholar * Wall, D. P. et al. Functional genomic analysis of the
rates of protein evolution. _Proc. Natl Acad. Sci. USA_ 102, 5483–5488 (2005) ADS CAS PubMed PubMed Central Google Scholar * Rocha, E. P. The quest for the universals of protein
evolution. _Trends Genet._ 22, 412–416 (2006) CAS PubMed Google Scholar * Huntley, M. A. & Clark, A. G. Evolutionary analysis of amino acid repeats across the genomes of 12
_Drosophila_ species. _Mol. Biol. Evol._ (in the press) * Charlesworth, B., Coyne, J. A. & Barton, N. H. The relative rates of evolution of sex chromosomes and autosomes. _Am. Nat._ 130,
113–146 (1987) Google Scholar * Larsson, J. & Meller, V. H. Dosage compensation, the origin and the afterlife of sex chromosomes. _Chromosome Res._ 14, 417–431 (2006) CAS PubMed
Google Scholar * Riddle, N. C. & Elgin, S. C. The dot chromosome of _Drosophila_: insights into chromatin states and their change over evolutionary time. _Chromosome Res._ 14, 405–416
(2006) CAS PubMed Google Scholar * Gordo, I. & Charlesworth, B. Genetic linkage and molecular evolution. _Curr. Biol._ 11, R684–R686 (2001) CAS PubMed Google Scholar * Singh, N.
D., Larracuente, A. M. & Clark, A. G. Contrasting the efficacy of selection on the X and autosomes in _Drosophila_ . _Mol. Biol. Evol._ (submitted) * Bhutkar, A., Russo, S. M., Smith, T.
F. & Gelbart, W. M. Genome scale analysis of positionally relocated genes. _Genome Res._ (in the press) * Akashi, H. & Eyre-Walker, A. Translational selection and molecular
evolution. _Curr. Opin. Genet. Dev._ 8, 688–693 (1998) CAS PubMed Google Scholar * Akashi, H., Kliman, R. M. & Eyre-Walker, A. Mutation pressure, natural selection, and the evolution
of base composition in _Drosophila_ . _Genetica (Dordrecht)_ 102–103, 49–60 (1998) Google Scholar * Bulmer, M. The selection–mutation–drift theory of synonymous codon usage. _Genetics_ 129,
897–908 (1991) CAS PubMed PubMed Central Google Scholar * McVean, G. A. T. & Charlesworth, B. A population genetic model for the evolution of synonymous codon usage: Patterns and
predictions. _Genet. Res._ 74, 145–158 (1999) Google Scholar * Sharp, P. M. & Li, W. H. An evolutionary perspective on synonymous codon usage in unicellular organisms. _J. Mol. Evol._
24, 28–38 (1986) ADS CAS PubMed Google Scholar * Akashi, H. & Schaeffer, S. W. Natural selection and the frequency distributions of “silent” DNA polymorphism in _Drosophila_ .
_Genetics_ 146, 295–307 (1997) CAS PubMed PubMed Central Google Scholar * Powell, J. R., Sezzi, E., Moriyama, E. N., Gleason, J. M. & Caccone, A. Analysis of a shift in codon usage
in _Drosophila_ . _J. Mol. Evol._ 57, S214–S225 (2003) ADS CAS PubMed Google Scholar * Anderson, C. L., Carew, E. A. & Powell, J. R. Evolution of the _Adh_ locus in the _Drosophila
willistoni_ group: The loss of an intron, and shift in codon usage. _Mol. Biol. Evol._ 10, 605–618 (1993) CAS PubMed Google Scholar * Rodriguez-Trelles, F., Tarrio, R. & Ayala, F. J.
Switch in codon bias and increased rates of amino acid substitution in the _Drosophila saltans_ species group. _Genetics_ 153, 339–350 (1999) CAS PubMed PubMed Central Google Scholar *
Rodriguez-Trelles, F., Tarrio, R. & Ayala, F. J. Evidence for a high ancestral GC content in _Drosophila_ . _Mol. Biol. Evol._ 17, 1710–1717 (2000) CAS PubMed Google Scholar *
Rodriguez-Trelles, F., Tarrio, R. & Ayala, F. J. Fluctuating mutation bias and the evolution of base composition in _Drosophila_ . _J. Mol. Evol._ 50, 1–10 (2000) ADS CAS PubMed
Google Scholar * Heger, A. & Ponting, C. Variable strength of translational selection among twelve _Drosophila_ species. _Genetics_ (in the press) * Vicario, S., Moriyama, E. N. &
Powell, J. R. Codon Usage in Twelve Species of _Drosophila_ . _BMC Evol. Biol._ (submitted) * Singh, N. D., Arndt, P. F. & Petrov, D. A. Minor shift in background substitutional patterns
in the _Drosophila saltans_ and _willistoni_ lineages is insufficient to explain GC content of coding sequences. _BMC Biol._ 4, 10.1186/1741–7007–4-37. (2006) * Akashi, H. Inferring weak
selection from patterns of polymorphism and divergence at “silent” sites in _Drosophila_ DNA. _Genetics_ 139, 1067–1076 (1995) CAS PubMed PubMed Central Google Scholar * Akashi, H.
Molecular evolution between _Drosophila melanogaster_ and _D. _ _simulans_: reduced codon bias, faster rates of amino acid substitution, and larger proteins in _D._ _ melanogaster_ .
_Genetics_ 144, 1297–1307 (1996) CAS PubMed PubMed Central Google Scholar * Akashi, H. et al. Molecular evolution in the _Drosophila melanogaster_ species subgroup: Frequent parameter
fluctuations on the timescale of molecular divergence. _Genetics_ 172, 1711–1726 (2006) CAS PubMed PubMed Central Google Scholar * Bauer DuMont, V., Fay, J. C., Calabrese, P. P. &
Aquadro, C. F. DNA variability and divergence at the _Notch_ locus in _Drosophila melanogaster_ and _D. _ _simulans_: a case of accelerated synonymous site divergence. _Genetics_ 167,
171–185 (2004) Google Scholar * McVean, G. A. & Vieira, J. The evolution of codon preferences in _Drosophila_: a maximum-likelihood approach to parameter estimation and hypothesis
testing. _J. Mol. Evol._ 49, 63–75 (1999) ADS CAS PubMed Google Scholar * Nielsen, R., Bauer DuMont, V., Hubisz, M. J. & Aquadro, C. F. Maximum likelihood estimation of ancestral
codon usage bias parameters in _Drosophila_ . _Mol. Biol. Evol._ 24, 228–235 (2007) CAS PubMed Google Scholar * Begun, D. J. The frequency distribution of nucleotide variation in
_Drosophila_ simulans. _Mol. Biol. Evol._ 18, 1343–1352 (2001) CAS PubMed Google Scholar * Singh, N. S., Bauer DuMont, V. L., Hubisz, M. J., Nielsen, R. & Aquadro, C. F. Patterns of
mutation and selection at synonymous sites in _Drosophila_ . _Mol. Biol. Evol._ doi: 10.1093/mobev.msm196. (in the press) * McBride, C. S. & Arguello, J. R. Five _Drosophila_ genomes
reveal non-neutral evolution and the signature of host specialization in the chemoreceptor superfamily. _Genetics_ (in the press) * Vieira, F. G., Sanchez-Gracia, A. & Rozas, J.
Comparative genomic analysis of the odorant-binding protein family in 12 _Drosophila_ genomes: Purifying selection and birth-and-death evolution. _Genome Biol._ 8, R235 (2007) PubMed PubMed
Central Google Scholar * Gardiner, A., Barker, D., Butilne, R. K., Jordan, W. C. & Ritchie, M. G. _Drosophila_ chemoreceptor evolution: Selection, specialisation and genome size.
_Genome Biol._ (submitted) * McBride, C. S. Rapid evolution of smell and taste receptor genes during host specialization in _Drosophila_ _sechellia_ . _Proc. Natl Acad. Sci. USA_ 104,
4996–5001 (2007) ADS CAS PubMed PubMed Central Google Scholar * Ranson, H. et al. Evolution of supergene families associated with insecticide resistance. _Science_ 298, 179–181 (2002)
ADS CAS PubMed Google Scholar * Tijet, N., Helvig, C. & Feyereisen, R. The cytochrome P450 gene superfamily in _Drosophila melanogaster._ . _Gene_ 262, 189–198 (2001) CAS PubMed
Google Scholar * Claudianos, C. et al. A deficit of detoxification enzymes: pesticide sensitivity and environmental response in the honeybee. _Insect Mol. Biol._ 15, 615–636 (2006) CAS
PubMed PubMed Central Google Scholar * Low, W. L. et al. Molecular evolution of glutathione _S_-transferases in the genus _Drosophila_ . _Genetics_ (in the press) * Castellano, S. et al.
_In silico_ identification of novel selenoproteins in the _Drosophila melanogaster_ genome. _EMBO Rep._ 2, 697–702 (2001) CAS PubMed PubMed Central Google Scholar * Martin-Romero, F. J.
et al. Selenium metabolism in _Drosophila_: selenoproteins, selenoprotein mRNA expression, fertility, and mortality. _J. Biol. Chem._ 276, 29798–29804 (2001) CAS PubMed Google Scholar *
Lemaitre, B. & Hoffmann, J. The host defense of _Drosophila melanogaster_ . _Annu. Rev. Immunol._ 25, 697–743 (2007) CAS PubMed Google Scholar * Hughes, A. L. & Nei, M. Pattern of
nucleotide substitution at major histocompatibility complex class I loci reveals overdominant selection. _Nature_ 335, 167–170 (1988) ADS CAS PubMed Google Scholar * Murphy, P. M.
Molecular mimicry and the generation of host defense protein diversity. _Cell_ 72, 823–826 (1993) CAS PubMed Google Scholar * Schlenke, T. A. & Begun, D. J. Natural selection drives
_Drosophila_ immune system evolution. _Genetics_ 164, 1471–1480 (2003) CAS PubMed PubMed Central Google Scholar * Sackton, T. B. et al. The evolution of the innate immune system across
_Drosophila_ . _Nature Genet._ (submitted) * Civetta, A. & Singh, R. S. High divergence of reproductive tract proteins and their association with postzygotic reproductive isolation in
_Drosophila melanogaster_ and _Drosophila_ _virilis_ group species. _J. Mol. Evol._ 41, 1085–1095 (1995) ADS CAS PubMed Google Scholar * Civetta, A. Shall we dance or shall we fight?
Using DNA sequence data to untangel controversies surrounding sexual selection. _Genome_ 46, 925–929 (2003) CAS PubMed Google Scholar * Clark, N. L., Aagard, J. E. & Swanson, W. J.
Evolution of reproductive proteins from animals and plants. _Reproduction_ 131, 11–22 (2006) CAS PubMed Google Scholar * Haerty, W. et al. Evolution in the fast lane: rapidly evolving
sex- and reproduction-related genes in _Drosophila_ species. _Genetics_ (in the press) * Lu, J. et al. Adaptive evolution of newly-emerged microRNA genes in _Drosophila_ . _Mol. Biol. Evol._
(submitted) * Lai, E. C., Tomancak, P., Williams, R. W. & Rubin, G. M. Computational identification of _Drosophila_ microRNA genes. _Genome Biol._ 4, R42 (2003) PubMed PubMed Central
Google Scholar * Parsch, J., Braverman, J. M. & Stephan, W. Comparative sequence analysis and patterns of covariation in RNA secondary structures. _Genetics_ 154, 909–921 (2000) CAS
PubMed PubMed Central Google Scholar * Stephan, W. The rate of compensatory evolution. _Genetics_ 144, 419–426 (1996) CAS PubMed PubMed Central Google Scholar * Gallo, S. M., Li, L.,
Hu, Z. & Halfon, M. S. REDfly: a Regulatory Element Database for _Drosophila_ . _Bioinformatics_ 22, 381–383 (2006) CAS PubMed Google Scholar * Bergman, C. M., Carlson, J. W. &
Celniker, S. E. _Drosophila_ DNase I footprint database: a systematic genome annotation of transcription factor binding sites in the fruitfly, _Drosophila melanogaster_ . _Bioinformatics_
21, 1747–1749 (2005) CAS PubMed Google Scholar * Halligan, D. L. & Keightley, P. D. Ubiquitous selective constraints in the _Drosophila_ genome revealed by a genome-wide interspecies
comparison. _Genome Res._ 16, 875–884 (2006) CAS PubMed PubMed Central Google Scholar * Andolfatto, P. Adaptive evolution of non-coding DNA in _Drosophila_ . _Nature_ 437, 1149–1152
(2005) ADS CAS PubMed Google Scholar * Bird, C. P., Stranger, B. E. & Dermitzakis, E. T. Functional variation and evolution of non-coding DNA. _Curr. Opin. Genet. Dev._ 16, 559–564
(2006) CAS PubMed Google Scholar * Wittkopp, P. J. Evolution of _cis_-regulatory sequence and function in Diptera. _Heredity_ 97, 139–147 (2006) CAS PubMed Google Scholar * Ludwig, M.
Z., Patel, N. H. & Kreitman, M. Functional analysis of _eve stripe 2_ enhancer evolution in _Drosophila_ . _Development_ 125, 949–958 (1998) CAS PubMed Google Scholar * Down, A. T.
A., Bergman, C. M., Su, J. & Hubbard, T. J. P. Large scale discovery of promoter motifs in _Drosophila melanogaster_ . _PloS Comput. Biol._ 3, e7 (2007) ADS PubMed PubMed Central
Google Scholar * Tamura, K., Subramanian, S. & Kumar, S. Temporal patterns of fruit fly (_Drosophila_) evolution revealed by mutation clocks. _Mol. Biol. Evol._ 21, 36–44 (2004) CAS
PubMed Google Scholar * Kumar, S., Tamura, K. & Nei, M. MEGA3: Integrated software for molecular evolutionary genetics analysis and sequence alignment. _Brief. Bioinform._ 5, 150–163
(2004) CAS PubMed Google Scholar * Pollard, D. A., Iyer, V. N., Moses, A. M. & Eisen, M. B. Widespread discordance of gene trees with species tree in _Drosophila_: evidence for
incomplete lineage sorting. _PLoS Genet._ 2, e173 (2006) PubMed PubMed Central Google Scholar * Bhutkar, A., Gelbart, W. M. & Smith, T. F. Inferring genome-scale rearrangement
phylogeny and ancestral gene order: A _Drosophila_ case study. _Genome Biol._ (in the press) Download references ACKNOWLEDGEMENTS Agencourt Bioscience Corporation, The Broad Institute of MIT
and Harvard and the Washington University Genome Sequencing Center were supported by grants and contracts from the National Human Genome Research Insititute (NHGRI). T.C. Kaufman
acknowledges support from the Indian Genomics Initiative. AUTHOR CONTRIBUTIONS The laboratory groups of A. G. Clark (including A. M. Larracuente, T. B. Sackton, and N. D. Singh) and Michael
B. Eisen (including V. N. Iyer and D. A. Pollard) played the part of coordinating the primary writing and editing of the manuscript with the considerable help of D. R. Smith, C. M. Bergman,
W. M. Gelbart, B. Oliver, T. A. Markow, T. C. Kaufman and M. Kellis. D. R. Smith served as primary coordinator for the assemblies. The remaining authors contributed either through their
efforts in sequence production, assembly and annotation, or in the analysis of specific topics that served as the focus of more than 40 companion papers. AUTHOR INFORMATION AUTHORS AND
AFFILIATIONS * Department of Molecular Biology and Genetics, Cornell University, Ithaca, New York 14853, USA., Andrew G. Clark, Timothy B. Sackton, Amanda M. Larracuente, Nadia D. Singh,
Charles F. Aquadro, Daniel A. Barbash, Anthony J. Greenberg, Melanie A. Huntley, Kristipati Ravi Ram, Laura Sirot, Mariana F. Wolfner, Alex Wong & Hsiao-Pei Yang * Life Sciences
Division, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA., Michael B. Eisen, Susan E. Celniker & Roger A. Hoskins * Department of Molecular and Cell Biology,
Center for Integrative Genomics, University of California at Berkeley, Berkeley, California 94720, USA., Michael B. Eisen * Agencourt Bioscience Corporation, Beverly, Massachusetts 01915,
USA., Douglas R. Smith, Stephanie A. Bosak, Adrianne D. Brand, Robert G. David, Heather Ebling, Barry E. Garvin, Erik A. Gustafson, Brendan McKernan, Kevin McKernan, Matthew J. Parisi, Marc
J. Rubenfield, David J. Saranga, Wei Tao & Jason M. Tsolas * Faculty of Life Sciences, University of Manchester, Manchester M13 9PT, UK., Casey M. Bergman & Sam Griffiths-Jones *
Laboratory of Cellular and Developmental Biology, National Institutes of Health, Bethesda, Maryland 20892, USA., Brian Oliver, Michael Parisi, David Sturgill & Yu Zhang * Department of
Ecology and Evolutionary Biology, University of Arizona, Tucson, Arizona 85721, USA., Therese A. Markow, Carlos A. Machado, Luciano Matzkin & Thomas Watts * Department of Biology,
Indiana University, Bloomington, Indiana 47405, USA., Thomas C. Kaufman, Don Gilbert, Matthew W. Hahn & Roberta Kwok * Computer Science and Artificial Intelligence Laboratory, Cambridge,
Massachusetts 02139, USA., Manolis Kellis, Pouya Kheradpour, Michael F. Lin, Leopold Parts, Matthew D. Rasmussen & Alexander Stark * Broad Institute of MIT and Harvard, 7 Cambridge
Center, Cambridge, Massachusetts 02142, USA., Manolis Kellis, Jean L. Chang, Sante Gnerre, David B. Jaffe, Eric Lander, Michael F. Lin, Kerstin Lindblad-Toh, Alexander Stark, Jennifer
Baldwin, Amr Abdouelleil, Jamal Abdulkadir, Adal Abebe, Brikti Abera, Justin Abreu, St Christophe Acer, Lynne Aftuck, Allen Alexander, Peter An, Erica Anderson, Scott Anderson, Harindra
Arachi, Marc Azer, Pasang Bachantsang, Andrew Barry, Tashi Bayul, Aaron Berlin, Daniel Bessette, Toby Bloom, Jason Blye, Leonid Boguslavskiy, Claude Bonnet, Boris Boukhgalter, Imane
Bourzgui, Adam Brown, Patrick Cahill, Sheridon Channer, Yama Cheshatsang, Lisa Chuda, Mieke Citroen, Alville Collymore, Patrick Cooke, Maura Costello, Katie D'Aco, Riza Daza, Georgius
De Haan, Stuart DeGray, Christina DeMaso, Norbu Dhargay, Kimberly Dooley, Erin Dooley, Missole Doricent, Passang Dorje, Kunsang Dorjee, Alan Dupes, Richard Elong, Jill Falk, Abderrahim
Farina, Susan Faro, Diallo Ferguson, Sheila Fisher, Chelsea D. Foley, Alicia Franke, Dennis Friedrich, Loryn Gadbois, Gary Gearin, Christina R. Gearin, Georgia Giannoukos, Tina Goode, Joseph
Graham, Edward Grandbois, Sharleen Grewal, Kunsang Gyaltsen, Nabil Hafez, Birhane Hagos, Jennifer Hall, Charlotte Henson, Andrew Hollinger, Tracey Honan, Monika D. Huard, Leanne Hughes,
Brian Hurhula, M Erii Husby, Asha Kamat, Ben Kanga, Seva Kashin, Dmitry Khazanovich, Peter Kisner, Krista Lance, Marcia Lara, William Lee, Niall Lennon, Frances Letendre, Rosie LeVine, Alex
Lipovsky, Xiaohong Liu, Jinlei Liu, Shangtao Liu, Tashi Lokyitsang, Yeshi Lokyitsang, Rakela Lubonja, Annie Lui, Pen MacDonald, Vasilia Magnisalis, Kebede Maru, Charles Matthews, William
McCusker, Susan McDonough, Teena Mehta, James Meldrim, Louis Meneus, Oana Mihai, Atanas Mihalev, Tanya Mihova, Rachel Mittelman, Valentine Mlenga, Anna Montmayeur, Leonidas Mulrain, Adam
Navidi, Jerome Naylor, Tamrat Negash, Thu Nguyen, Nga Nguyen, Robert Nicol, Choe Norbu, Nyima Norbu, Nathaniel Novod, Barry O'Neill, Sahal Osman, Eva Markiewicz, Otero L. Oyono,
Christopher Patti, Pema Phunkhang, Fritz Pierre, Margaret Priest, Sujaa Raghuraman, Filip Rege, Rebecca Reyes, Cecil Rise, Peter Rogov, Keenan Ross, Elizabeth Ryan, Sampath Settipalli, Terry
Shea, Ngawang Sherpa, Lu Shi, Diana Shih, Todd Sparrow, Jessica Spaulding, John Stalker, Nicole Stange-Thomann, Sharon Stavropoulos, Catherine Stone, Christopher Strader, Senait Tesfaye,
Talene Thomson, Yama Thoulutsang, Dawa Thoulutsang, Kerri Topham, Ira Topping, Tsamla Tsamla, Helen Vassiliev, Andy Vo, Tsering Wangchuk, Tsering Wangdi, Michael Weiand, Jane Wilkinson, Adam
Wilson, Shailendra Yadav, Geneva Young, Qing Yu, Lisa Zembek, Danni Zhong, Andrew Zimmer, Zac Zwirko, David B. Jaffe, Pablo Alvarez, Will Brockman, Jonathan Butler, CheeWhye Chin, Sante
Gnerre, Manfred Grabherr, Michael Kleber, Evan Mauceli & Iain MacCallum * Department of Molecular and Cellular Biology, Harvard University, Cambridge, Massachusetts 02138, USA., William
Gelbart, Arjun Bhutkar, Susan Russo & Peili Zhang * FlyBase, The Biological Laboratories, Harvard University, Cambridge, Massachusetts 02138, USA., William Gelbart, Susan Russo &
Peili Zhang * Department of Molecular and Cell Biology, University of California at Berkeley, Berkeley, California 94720, USA., Venky N. Iyer, Angela N. Brooks & Gary H. Karpen *
Biophysics Graduate Group, University of California at Berkeley, Berkeley, California 94720, USA., Daniel A. Pollard * Field of Ecology and Evolutionary Biology, Cornell University, Ithaca,
New York 14853, USA., Timothy B. Sackton * Centro de Biología Molecular Severo Ochoa, Universidad Autónoma de Madrid, Madrid 28049, Spain., Jose P. Abad, Maria Mendez-Lago & Alfredo
Villasante * Department of Ecology and Evolutionary Biology, Brown University, Providence, Rhode Island 02912, USA., Dawn N. Abt, Kristi Montooth & David Rand * Structural Studies
Division, MRC Laboratory of Molecular Biology, Cambridge CB2 2QH, UK., Boris Adryan, Sarah Teichmann & Derek Wilson * Departament de Genètica, Universitat de Barcelona, Barcelona 08071,
Spain., Montserrat Aguade, Montserrat Papaceit, Julio Rozas, Alejandro Sanchez-Gracia, Carmen Segarra, Filipe G. Vieira & Albert J. Vilella * Department of Biology, Pennsylvania State
University, University Park, Pennsylvania 16802, USA., Hiroshi Akashi, Valer Gotea, Gillian M. Halter, Chiao-Feng Lin, Wojciech Makalowski, Michael U. Mollenhauer, Xu Mu & Stephen W.
Schaeffer * Department of Genetics, University of Georgia, Athens, Georgia 30602, USA., Wyatt W. Anderson * Linnaeus Centre for Bioinformatics, Uppsala Universitet, Uppsala, SE-75124,
Sweden., David H. Ardell & Eva Freyhult * Department of Ecology and Evolution, University of Chicago, Chicago, Illinois 60637, USA., Roman Arguello, Jerry A. Coyne, Manyuan Long, Jian Lu
& Chung-I Wu * Department of Biology, McMaster University, Hamilton, Ontario, L8S 4K1, Canada., Carlo G. Artieri, Wilfried Haerty, Santosh Jagadeeshan & Rama S. Singh * School of
Biology, University of St. Andrews, Fife KY16 9TH, UK., Daniel Barker, Anastasia Gardiner & Michael G. Ritchie * Dipartimento di Genetica e Microbiologia dell'Università di Bari,
Bari, 70126, Italy., Paolo Barsanti, Corrado Caggese & Damiano Porcelli * Department of Genetics, University of Melbourne, Melbourne 3010, Australia., Phil Batterham, Robert Good, Lloyd
Low & Charles Robin * Computer Science Department, Stanford University, Stanford, California 94305, USA., Serafim Batzoglou, Chuong B. Do & Samuel Gross * Section of Evolution and
Ecology and Center for Population Biology, University of California at Davis, Davis, California 95616, USA., Dave Begun, Charles H. Langley & Carolyn S. McBride * BioMolecular
Engineering Research Center, Boston University, Boston, Massachusetts 02215, USA., Arjun Bhutkar & Temple F. Smith * Research Group in Biomedical Informatics, Institut Municipal
d'Investigacio Medica, Universitat Pompeu Fabra, Barcelona 08003, Catalonia, Spain., Enrico Blanco, Charles Chapple & Roderic Guigo * Department of Bioengineering, University of
California at Berkeley, Berkeley, California 94720, USA., Robert K. Bradley, Anat Caspi, Ian Holmes & Gabriel Wu * Laboratory for Computational Genomics, Washington University, St Louis,
Missouri 63108, USA., Michael R. Brent & Randall H. Brown * Animal and Plant Sciences, The University of Sheffield, Sheffield S10 2TN, UK., Roger K. Butlin * Department of Biology,
Syracuse University, Syracuse, New York 13244, USA., Brian R. Calvi * Departamento de Genética, Universidade Federal do Rio de Janeiro, Rio de Janeiro 21944-970, Brazil., A. Bernardo de
Carvalho & Leonardo B. Koerich * Tucson Stock Center, Tucson, Arizona 85721, USA., Sergio Castrezana * Department of Mathematics, University of California at Berkeley, Berkeley,
California 94720, USA., Sourav Chatterji & Lior Pachter * Genome Center, University of California at Davis, Davis, California 95616, USA., Sourav Chatterji * Genome Sequencing Center,
Washington University School of Medicine, St Louis, Missouri 63108, USA., Asif Chinwalla, Sandra W. Clifton, Kim Delehaunty, Lucinda Fulton, Robert Fulton, Jennifer Godfrey, LaDeana Hillier,
Patrick Minx, Amy Reily, John Spieth, Richard K. Wilson & Shiaw-Pyng Yang * Department of Biology, University of Winnipeg, Winnipeg, Manitoba R3B 2E9, Canada., Alberto Civetta &
Lisa Levesque * Department of Biological Sciences, University of Iowa, Iowa City, Iowa 52242, USA., Josep M. Comeron, Ana Llopart & Bryant McAllister * School of Informatics, Indiana
University, Bloomington, Indiana 47405, USA., James C. Costello, Matthew W. Hahn & Mira V. Han * Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge CB10
1SA, UK., Jennifer Daub & Sam Griffiths-Jones * Center for Bioinformatics and Computational Biology, University of Maryland, College Park, Maryland 20742, USA., Arthur L. Delcher,
Stephen M. Mount, Adam M. Phillippy, Mihai Pop, Steven L. Salzberg & Michael C. Schatz * Department of Biological Sciences, Illinois State University, Normal, Illinois 61790, USA., Kevin
Edwards * Department of Biology, University of Rochester, Rochester, New York 14627, USA., Thomas Eickbush & Deborah E. Stage * Bee Research Lab, USDA-ARS, Beltsville, Maryland 20705,
USA., Jay D. Evans * Center for Evolutionary Functional Genomics, Biodesign Institute, Arizona State University, Tempe, Arizona 85287, USA., Alan Filipski, Sven Findeiß, Sudhir Kumar &
Sonja Prohaska * Department of Computer Science, University of Leipzig, Leipzig 04107, Germany., Sven Findeiß * Departamento de Genética, Universidade Federal do Rio Grande do Sul, Porto
Alegre/RS 68011, Brazil., Ana C. L. Garcia & Vera L. S. Valente * Department of Biology, Duke University, Durham, New Carolina 27708, USA., David A. Garfield & Mohamed A. F. Noor *
Department of Genetics, North Carolina State University, Raleigh, North Caroline 27695, USA., Greg Gibson & Laura K. Reed * Health Center, University of Connecticut, Farmington,
Connecticut 06030, USA., Brenton Gravely * Center of Genomic Regulation, Barcelona 8003, Catalonia, Spain., Roderic Guigo * Institute of Evolutionary Biology, University of Edinburgh,
Edinburgh EH9 3JT, UK., Daniel L. Halligan & Peter D. Keightley * J. Craig Venter Institute, Rockville, Maryland 20850, USA., Aaron L. Halpern, Justin Johnson, Ewen F. Kirkness, Karin A.
Remington, Yu-Hui Rogers, Robert L. Strausberg, Granger Sutton, Granger G. Sutton, Eli Venter, J. Craig Venter & Brian Walenz * MRC Functional Genetics Unit, University of Oxford,
Oxford OX1 3QX, UK., Andreas Heger & Chris P. Ponting * Department of Physiology, Anatomy and Genetics, University of Oxford, Oxford OX1 3QX, UK., Andreas Heger & Chris P. Ponting *
Center for Biomolecular Science and Engineering, University of California at Santa Cruz, Santa Cruz, California 95064, USA., Angie S. Hinrichs & Jakob S. Pedersen * Department of Human
Genetics, University of Chicago, Chicago, Illinois 60637, USA., Melissa J. Hubisz * Umeå Center for Molecular Pathogenesis, Umeå University, Umeå SE-90187, Sweden., Dan Hultmark * Department
of Biology and Carolina Center for Genome Sciences, The University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, USA., William R. Jeck & Corbin D. Jones *
Institute of Zoology, Regent's Park, London NW1 4RY, UK., William C. Jordan * Department of Genome and Computational Biology, Drosophila Heterochromatin Genome Project, Lawrence
Berkeley National Laboratory, Berkeley, California 94720, USA., Gary H. Karpen & Chris D. Smith * Kyorin University, School of Medicine, Mitaka, Tokyo 181-8611, Japan., Eiko Kataoka,
Muneo Matsuda & Hajime Sato * Department of Biochemistry and Molecular Biology, University of Southern Denmark, Odense M DK-5230, Denmark, Karsten Kristiansen, Ruiqiang Li & Jun Wang
* Department of Plant Sciences and BIO5, Arizona Genomics Institute, University of Arizona, Tucson, Arizona 85721, USA., Dave Kudrna, So-Jeong Lee, Meizhong Luo, Nicholas B. Sisneros &
Rod A. Wing * Department of Organismic and Evolutionary Biology, Harvard University, Cambridge, Massachusetts 02138, USA., Rob J. Kulathinal & Elena Lozovsky * School of Life Sciences,
Arizona State University, Tempe, Arizona 85287, USA., Sudhir Kumar & Stuart Newfeld * Department of Environmental Science, Policy and Management, University of California at Berkeley,
Berkeley, California 94720, USA., Richard Lapoint & Patrick O'Grady * Department of Entomology, Cornell University, Ithaca, New York 14853, USA., Brian P. Lazzaro * Beijing Genomics
Institute at ShenZhen, ShenZhen 518083, China., Ruiqiang Li, Jun Wang & Gane Ka-Shu Wong * Departament Genètica i Microbiologia, Universitat Autònoma de Barcelona, Bellaterra 08193,
Spain., Mar Marzo, Marta Puig & Alfredo Ruiz * Department of Cell Biology and Molecular Genetics, University of Maryland, College Park, Maryland 20742, USA., Stephen M. Mount * Janelia
Farm Research Campus, Howard Hughes Medical Institue, Ashburn, Viginia 20147-2408, USA., Eugene Myers * Department of Zoology, University of Cambridge, Cambridge, CB2 3EJ, UK., Barbara Negre
* Institue of Biology, University of Copenhagen, DK-2100 Copenhagen Ø, Denmark., Rasmus Nielsen * Department of Molecular Biology, Bioinformatics Centre, University of Copenhagen, DK-2200
Copenhagen N, Denmark., Jakob S. Pedersen * Dipartimento di Biochimica e Biologia Molecolare, Università di Bari and Istituto Tecnologie Biomediche del Consiglio Nazionale delle Ricerche,
Bari 70126, Italy., Graziano Pesole * Department of Ecology and Evolutionary Biology, Yale University, New Haven, Connecticut 06520, USA., Jeffrey R. Powell & Saverio Vicario *
Department of Biomedical Informatics, Arizona State University, Tempe, Arizona 85287, USA., Sonja Prohaska * National Center for Biotechnology Information, National Institutes of Health,
Bethesda, Maryland 20894, USA., Kim Pruitt * Bioinformatics and Genomics Laboratory, Institut Jacques Monod, Paris, 75251, France., Hadi Quesneville * Department of Molecular Biology, Cell
Biology and Biochemistry, Brown University, Providence, Rhode Island 02912, USA., Robert Reenan * Departamento de Genética, Centro de Ciências Biológicas, Universidade Federal de Pernambuco,
Recife/PE 68011, Brazil., Tania T. Rieger * Centro Acadêmico de Vitória, Universidade Federal de Pernambuco, Vitória de Santo Antão/PE, Brazil., Claudia Rohde * Cajal Institute, CSIC,
Madrid 28002, Spain., Alejandro Sanchez-Gracia * Department of Biology, Emory University, Atlanta, Georgia 30322, USA., Todd Schlenke * Department of Biological Sciences, Carnegie Mellon
University, Pittsburgh, Pennsylvania 15213, USA., Russell Schwartz * Biomedical Informatics, Stanford University, Stanford, California 94305, USA., Marina Sirota * Department of Biology, San
Francisco State University, San Francisco, California 94132, USA., Chris D. Smith * Department of Biology, University of Munich, 82152 Planegg-Martinsried, Germany., Wolfgang Stephan &
Sebastian Strempel * Institute of Evolutionary Biology, Setagaya-ku, Tokyo 158-0098, Japan., Yoshiko N. Tobari * Shiba Gakuen, Minato-ku, Tokyo 105-0011, Japan., Yoshihiko Tomimura *
European Bioinformatics Institute, Hinxton, CB10 1SD, UK., Albert J. Vilella * Department of Biology, City University of New York at Queens, Flushing, New York 11367, USA., Marvin Wasserman
* Department of Biological Sciences and Department of Medicine, University of Alberta, Edmonton, Alberta T6G 2E9, Canada., Gane Ka-Shu Wong * Department of Developmental Biology and
Neurosciences, Tohoku University, Sendai 980-8578, Japan., Daisuke Yamamoto * Institute for Physical Science and Technology, University of Maryland, College Park, Maryland 20742, USA., James
A. Yorke & Aleksey V. Zimin * Hokkaido University, EESBIO, Sapporo, Hokkaido 060-0810, Japan., Kiyohito Yoshida * Faculty of Medecine, Universite de Geneve, Geneva CH-1211,
Switzerland., Evgeny Zdobnov CONSORTIA DROSOPHILA 12 GENOMES CONSORTIUM * PROJECT LEADERS * Andrew G. Clark * , Michael B. Eisen * , Douglas R. Smith * , Casey M. Bergman * , Brian Oliver *
, Therese A. Markow * , Thomas C. Kaufman * , Manolis Kellis * & William Gelbart * ANNOTATION COORDINATION * Venky N. Iyer * & Daniel A. Pollard * ANALYSIS/WRITING COORDINATION *
Timothy B. Sackton * , Amanda M. Larracuente * & Nadia D. Singh * SEQUENCING, ASSEMBLY, ANNOTATION AND ANALYSIS CONTRIBUTORS * Jose P. Abad * , Dawn N. Abt * , Boris Adryan * ,
Montserrat Aguade * , Hiroshi Akashi * , Wyatt W. Anderson * , Charles F. Aquadro * , David H. Ardell * , Roman Arguello * , Carlo G. Artieri * , Daniel A. Barbash * , Daniel Barker * ,
Paolo Barsanti * , Phil Batterham * , Serafim Batzoglou * , Dave Begun * , Arjun Bhutkar * , Enrico Blanco * , Stephanie A. Bosak * , Robert K. Bradley * , Adrianne D. Brand * , Michael R.
Brent * , Angela N. Brooks * , Randall H. Brown * , Roger K. Butlin * , Corrado Caggese * , Brian R. Calvi * , A. Bernardo de Carvalho * , Anat Caspi * , Sergio Castrezana * , Susan E.
Celniker * , Jean L. Chang * , Charles Chapple * , Sourav Chatterji * , Asif Chinwalla * , Alberto Civetta * , Sandra W. Clifton * , Josep M. Comeron * , James C. Costello * , Jerry A. Coyne
* , Jennifer Daub * , Robert G. David * , Arthur L. Delcher * , Kim Delehaunty * , Chuong B. Do * , Heather Ebling * , Kevin Edwards * , Thomas Eickbush * , Jay D. Evans * , Alan Filipski *
, Sven Findeiß * , Eva Freyhult * , Lucinda Fulton * , Robert Fulton * , Ana C. L. Garcia * , Anastasia Gardiner * , David A. Garfield * , Barry E. Garvin * , Greg Gibson * , Don Gilbert *
, Sante Gnerre * , Jennifer Godfrey * , Robert Good * , Valer Gotea * , Brenton Gravely * , Anthony J. Greenberg * , Sam Griffiths-Jones * , Samuel Gross * , Roderic Guigo * , Erik A.
Gustafson * , Wilfried Haerty * , Matthew W. Hahn * , Daniel L. Halligan * , Aaron L. Halpern * , Gillian M. Halter * , Mira V. Han * , Andreas Heger * , LaDeana Hillier * , Angie S.
Hinrichs * , Ian Holmes * , Roger A. Hoskins * , Melissa J. Hubisz * , Dan Hultmark * , Melanie A. Huntley * , David B. Jaffe * , Santosh Jagadeeshan * , William R. Jeck * , Justin Johnson *
, Corbin D. Jones * , William C. Jordan * , Gary H. Karpen * , Eiko Kataoka * , Peter D. Keightley * , Pouya Kheradpour * , Ewen F. Kirkness * , Leonardo B. Koerich * , Karsten Kristiansen
* , Dave Kudrna * , Rob J. Kulathinal * , Sudhir Kumar * , Roberta Kwok * , Eric Lander * , Charles H. Langley * , Richard Lapoint * , Brian P. Lazzaro * , So-Jeong Lee * , Lisa Levesque * ,
Ruiqiang Li * , Chiao-Feng Lin * , Michael F. Lin * , Kerstin Lindblad-Toh * , Ana Llopart * , Manyuan Long * , Lloyd Low * , Elena Lozovsky * , Jian Lu * , Meizhong Luo * , Carlos A.
Machado * , Wojciech Makalowski * , Mar Marzo * , Muneo Matsuda * , Luciano Matzkin * , Bryant McAllister * , Carolyn S. McBride * , Brendan McKernan * , Kevin McKernan * , Maria Mendez-Lago
* , Patrick Minx * , Michael U. Mollenhauer * , Kristi Montooth * , Stephen M. Mount * , Xu Mu * , Eugene Myers * , Barbara Negre * , Stuart Newfeld * , Rasmus Nielsen * , Mohamed A. F.
Noor * , Patrick O'Grady * , Lior Pachter * , Montserrat Papaceit * , Matthew J. Parisi * , Michael Parisi * , Leopold Parts * , Jakob S. Pedersen * , Graziano Pesole * , Adam M.
Phillippy * , Chris P. Ponting * , Mihai Pop * , Damiano Porcelli * , Jeffrey R. Powell * , Sonja Prohaska * , Kim Pruitt * , Marta Puig * , Hadi Quesneville * , Kristipati Ravi Ram * ,
David Rand * , Matthew D. Rasmussen * , Laura K. Reed * , Robert Reenan * , Amy Reily * , Karin A. Remington * , Tania T. Rieger * , Michael G. Ritchie * , Charles Robin * , Yu-Hui Rogers *
, Claudia Rohde * , Julio Rozas * , Marc J. Rubenfield * , Alfredo Ruiz * , Susan Russo * , Steven L. Salzberg * , Alejandro Sanchez-Gracia * , David J. Saranga * , Hajime Sato * , Stephen
W. Schaeffer * , Michael C. Schatz * , Todd Schlenke * , Russell Schwartz * , Carmen Segarra * , Rama S. Singh * , Laura Sirot * , Marina Sirota * , Nicholas B. Sisneros * , Chris D. Smith *
, Temple F. Smith * , John Spieth * , Deborah E. Stage * , Alexander Stark * , Wolfgang Stephan * , Robert L. Strausberg * , Sebastian Strempel * , David Sturgill * , Granger Sutton * ,
Granger G. Sutton * , Wei Tao * , Sarah Teichmann * , Yoshiko N. Tobari * , Yoshihiko Tomimura * , Jason M. Tsolas * , Vera L. S. Valente * , Eli Venter * , J. Craig Venter * , Saverio
Vicario * , Filipe G. Vieira * , Albert J. Vilella * , Alfredo Villasante * , Brian Walenz * , Jun Wang * , Marvin Wasserman * , Thomas Watts * , Derek Wilson * , Richard K. Wilson * , Rod
A. Wing * , Mariana F. Wolfner * , Alex Wong * , Gane Ka-Shu Wong * , Chung-I Wu * , Gabriel Wu * , Daisuke Yamamoto * , Hsiao-Pei Yang * , Shiaw-Pyng Yang * , James A. Yorke * , Kiyohito
Yoshida * , Evgeny Zdobnov * , Peili Zhang * , Yu Zhang * & Aleksey V. Zimin * *BROAD INSTITUTE GENOME SEQUENCING PLATFORM * Jennifer Baldwin * , Amr Abdouelleil * , Jamal Abdulkadir *
, Adal Abebe * , Brikti Abera * , Justin Abreu * , St Christophe Acer * , Lynne Aftuck * , Allen Alexander * , Peter An * , Erica Anderson * , Scott Anderson * , Harindra Arachi * , Marc
Azer * , Pasang Bachantsang * , Andrew Barry * , Tashi Bayul * , Aaron Berlin * , Daniel Bessette * , Toby Bloom * , Jason Blye * , Leonid Boguslavskiy * , Claude Bonnet * , Boris
Boukhgalter * , Imane Bourzgui * , Adam Brown * , Patrick Cahill * , Sheridon Channer * , Yama Cheshatsang * , Lisa Chuda * , Mieke Citroen * , Alville Collymore * , Patrick Cooke * , Maura
Costello * , Katie D'Aco * , Riza Daza * , Georgius De Haan * , Stuart DeGray * , Christina DeMaso * , Norbu Dhargay * , Kimberly Dooley * , Erin Dooley * , Missole Doricent * , Passang
Dorje * , Kunsang Dorjee * , Alan Dupes * , Richard Elong * , Jill Falk * , Abderrahim Farina * , Susan Faro * , Diallo Ferguson * , Sheila Fisher * , Chelsea D. Foley * , Alicia Franke * ,
Dennis Friedrich * , Loryn Gadbois * , Gary Gearin * , Christina R. Gearin * , Georgia Giannoukos * , Tina Goode * , Joseph Graham * , Edward Grandbois * , Sharleen Grewal * , Kunsang
Gyaltsen * , Nabil Hafez * , Birhane Hagos * , Jennifer Hall * , Charlotte Henson * , Andrew Hollinger * , Tracey Honan * , Monika D. Huard * , Leanne Hughes * , Brian Hurhula * , M Erii
Husby * , Asha Kamat * , Ben Kanga * , Seva Kashin * , Dmitry Khazanovich * , Peter Kisner * , Krista Lance * , Marcia Lara * , William Lee * , Niall Lennon * , Frances Letendre * , Rosie
LeVine * , Alex Lipovsky * , Xiaohong Liu * , Jinlei Liu * , Shangtao Liu * , Tashi Lokyitsang * , Yeshi Lokyitsang * , Rakela Lubonja * , Annie Lui * , Pen MacDonald * , Vasilia Magnisalis
* , Kebede Maru * , Charles Matthews * , William McCusker * , Susan McDonough * , Teena Mehta * , James Meldrim * , Louis Meneus * , Oana Mihai * , Atanas Mihalev * , Tanya Mihova * , Rachel
Mittelman * , Valentine Mlenga * , Anna Montmayeur * , Leonidas Mulrain * , Adam Navidi * , Jerome Naylor * , Tamrat Negash * , Thu Nguyen * , Nga Nguyen * , Robert Nicol * , Choe Norbu * ,
Nyima Norbu * , Nathaniel Novod * , Barry O'Neill * , Sahal Osman * , Eva Markiewicz * , Otero L. Oyono * , Christopher Patti * , Pema Phunkhang * , Fritz Pierre * , Margaret Priest *
, Sujaa Raghuraman * , Filip Rege * , Rebecca Reyes * , Cecil Rise * , Peter Rogov * , Keenan Ross * , Elizabeth Ryan * , Sampath Settipalli * , Terry Shea * , Ngawang Sherpa * , Lu Shi * ,
Diana Shih * , Todd Sparrow * , Jessica Spaulding * , John Stalker * , Nicole Stange-Thomann * , Sharon Stavropoulos * , Catherine Stone * , Christopher Strader * , Senait Tesfaye * , Talene
Thomson * , Yama Thoulutsang * , Dawa Thoulutsang * , Kerri Topham * , Ira Topping * , Tsamla Tsamla * , Helen Vassiliev * , Andy Vo * , Tsering Wangchuk * , Tsering Wangdi * , Michael
Weiand * , Jane Wilkinson * , Adam Wilson * , Shailendra Yadav * , Geneva Young * , Qing Yu * , Lisa Zembek * , Danni Zhong * , Andrew Zimmer * & Zac Zwirko * BROAD INSTITUTE WHOLE
GENOME ASSEMBLY TEAM * David B. Jaffe * , Pablo Alvarez * , Will Brockman * , Jonathan Butler * , CheeWhye Chin * , Sante Gnerre * , Manfred Grabherr * , Michael Kleber * , Evan Mauceli *
& Iain MacCallum CORRESPONDING AUTHORS Correspondence to Andrew G. Clark, Michael B. Eisen, Douglas R. Smith, Casey M. Bergman, Brian Oliver, Therese A. Markow, Thomas C. Kaufman,
Manolis Kellis, William Gelbart, Venky N. Iyer, Daniel A. Pollard, Timothy B. Sackton, Amanda M. Larracuente, Nadia D. Singh or Michael Kleber. ETHICS DECLARATIONS COMPETING INTERESTS The
author declare no competing financial interests. ADDITIONAL INFORMATION A list of participants and affiliations appears at the end of the paper. SUPPLEMENTARY INFORMATION SUPPLEMENTARY
INFORMATION The file contains all methods for the project as well as further detail on certain sections. Also contains Supplementary Tables 1-10 and 13-20, and Supplementary Figures 1-10
with Legends. This document has a detailed table of contents. (PDF 1381 kb) SUPPLEMENTARY TABLE 11 The file contains Supplementary Table 11 which is a list of 44 lineage-specific genes
arising in the melanogaster group or some subset of the melanogaster group phylogeny. (XLS 22 kb) SUPPLEMENTARY TABLE 12 The file contains Supplementary Table 12 which shows median value of
ω, the negative log of the P-value from the test of positive selection and dN for each of the 115 GO (XLS 58 kb) RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS
ARTICLE Drosophila 12 Genomes Consortium. Evolution of genes and genomes on the _Drosophila_ phylogeny. _Nature_ 450, 203–218 (2007). https://doi.org/10.1038/nature06341 Download citation *
Received: 19 July 2007 * Accepted: 05 October 2007 * Issue Date: 08 November 2007 * DOI: https://doi.org/10.1038/nature06341 SHARE THIS ARTICLE Anyone you share the following link with will
be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt
content-sharing initiative