Play all audios:
ABSTRACT One-fourth of _Plasmodium falciparum_ proteins have asparagine repeats that increase the propensity for aggregation, especially at elevated temperatures that occur routinely in
malaria-infected patients. Here we report that a _Plasmodium_ Asn repeat-containing protein (PFI1155w) formed aggregates in mammalian cells at febrile temperatures, as did a yeast
Asn/Gln-rich protein (Sup35). Co-expression of the cytoplasmic _P. falciparum_ heat shock protein 110 (_Pf_Hsp110c) prevented aggregation. Human or yeast orthologs were much less effective.
All-Asn and all-Gln versions of Sup35 were protected from aggregation by PfHsp110c, suggesting that this chaperone is not limited to handling runs of asparagine. _PfHsp110c_ gene-knockout
parasites were not viable and conditional knockdown parasites died slowly in the absence of protein-stabilizing ligand. When exposed to brief heat shock, these knockdowns were unable to
prevent aggregation of PFI1155w or Sup35 and died rapidly. We conclude that _Pf_Hsp110c protects the parasite from harmful effects of its asparagine repeat-rich proteome during febrile
episodes. You have full access to this article via your institution. Download PDF SIMILAR CONTENT BEING VIEWED BY OTHERS A HEAT-SHOCK RESPONSE REGULATED BY THE PFAP2-HS TRANSCRIPTION FACTOR
PROTECTS HUMAN MALARIA PARASITES FROM FEBRILE TEMPERATURES Article 16 August 2021 PLASMEPSIN X ACTIVATES THE PCRCR COMPLEX OF _PLASMODIUM FALCIPARUM_ BY PROCESSING PFRH5 FOR ERYTHROCYTE
INVASION Article Open access 19 April 2023 THE STRUCTURE OF A _PLASMODIUM VIVAX_ TRYPTOPHAN RICH ANTIGEN DOMAIN SUGGESTS A LIPID BINDING FUNCTION FOR A PAN-_PLASMODIUM_ MULTI-GENE FAMILY
Article Open access 14 September 2023 INTRODUCTION The deadly malaria parasite, _P. falciparum_, has a proteome that is replete with amino-acid repeats; the majority of these repeats consist
of asparagine (Asn) residues1,2. One in four proteins in the _P. falciparum_ proteome have Asn repeat-rich sequences, comprising up to 83 Asn residues and an average size of 37 residues3.
There can be multiple repeats in a given protein. The presence of Asn-rich sequences in proteins is known to increase their propensity for aggregate formation4,5,6. Formation of aggregates
is enhanced under heat shock stress conditions owing to an increase in protein unfolding7,8. In a recent survey of prion proteins in yeast, numerous Asn-rich sequences were found to form
aggregates4. Prion-like aggregates have been shown to be responsible for the inheritance of several phenotypes in yeast9,10, to have a functional biological role in bacteria11, to be
important for persistence of synaptic facilitation12 and to be vital for antiviral innate immunity13. While such regulated aggregation of proteins is benign or beneficial, unregulated
aggregation of proteins can lead to cell death6,14. The malaria parasite is able to thrive with an Asn repeat-rich proteome in face of the periodic heat shock stress that is a hallmark of
clinical malaria. Patients suffer recurrent bouts of fever, often exceeding 40 °C and lasting several hours at a time. The ability of _P. falciparum_ to survive this insult is likely owing
to processes that mask Asn repeat-rich protein aggregation. Certain chaperones, particularly heat shock proteins (Hsps), have been shown to have a vital role in controlling aggregate
formation15,16. _In vitro_, these chaperones act in concert to refold proteins and unfold preformed aggregates17. The Hsp70–Hsp110–Hsp40 network refolds proteins via repeated cycles of
binding and release of unfolded proteins by Hsp70 that is governed by its ADP or ATP bound state, respectively15,16. Hsp110 acts as a nucleotide exchange factor for Hsp70, exchanging ADP for
ATP and thereby completing the refolding cycle18,19,20,21. Hsp110 can also bind unfolded proteins, via its substrate-binding domain, but the role of this binding event in the refolding
cycle is unclear22,23,24 and its biological function remains undefined. Recently, mammalian Hsp110 was found to localize with aggregates in human embryonic kidney (HEK) 293T cells but the
significance of the association is not understood14. _Drosophila_ Hsp110 was identified in a genome-wide RNA interference screen as a mitigating factor for aggregation of Huntingtin
protein25. This chaperone system clearly has a role in unfolded protein handling, but its capacity is easily overwhelmed by the expression of aggregation-prone proteins, especially under
heat shock conditions. Heat shock stress increases the propensity of proteins to unfold, enhancing the formation of aggregates7,8. We show here that the cytoplasmic Hsp110 of _P. falciparum_
(_Pf_Hsp110c) is able to prevent aggregation of Asn repeat-rich proteins in cultured malaria parasites and in mammalian cells. The _P. falciparum_ chaperone is much better at this than
orthologs from yeast or humans. We conclude that _Pf_Hsp110c, in particular its substrate-binding activity, is vital for proteostasis of the Asn repeat-rich proteome of _P. falciparum_ and
propose that its presence allows the propagation of these repeats within the parasite proteome. RESULTS A _P. FALCIPARUM_ ASN-RICH PROTEIN AGGREGATES IN HUMAN CELLS Aggregation of Asn
repeat-rich proteins has not been observed within _P. falciparum_ and we have reported that the presence of these repeats within a protein does not seem to affect its cellular function or
location, even under heat shock26. This suggested that the parasite chaperone network has adapted to mask the aggregation propensity of its Asn repeat-rich proteome or that the parasite Asn
repeat-rich proteins are not capable of aggregating even during febrile episodes. To distinguish between these possibilities, we transiently transfected HEK293T cells with a _P. falciparum_
protein that contains a stretch of 83 Asn residues (PlasmoDB ID: PF3D7_0923500 or PFI1155w). For comparison, we transfected HEK293T cells with the N-terminal Asn/glutamine (Gln)-rich
prion-forming domain (PrD, amino acids 1–125) of the yeast protein Sup35 (Sup35PrD)9,27. Each was fused to a monomeric derivative of red fluorescent protein (tagRFP-T)28 The transfected
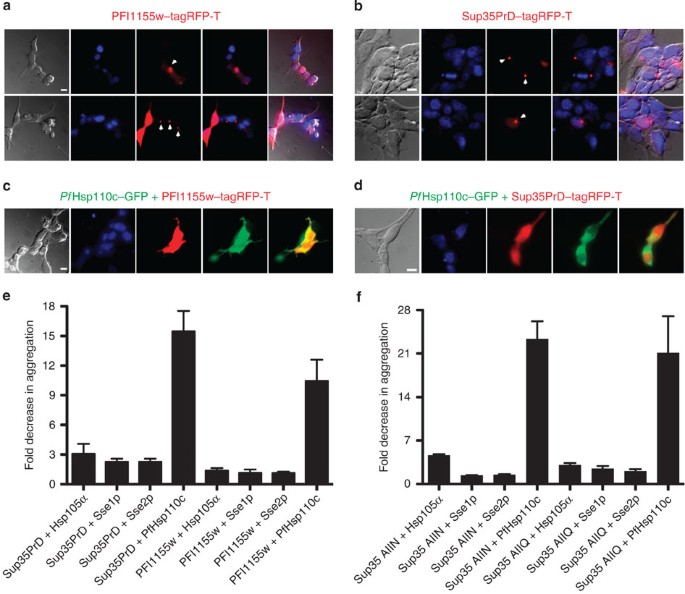
HEK293T cells were incubated at 40 °C for 6 h and then assessed by fluorescence microscopy (Fig. 1a). Protein aggregation, observed as distinct fluorescent foci, was seen in a large
proportion of the HEK293T cells expressing either Asn-rich protein (Fig. 1a and Supplementary Fig. S1a). The fluorescent fusion protein, tagRFP-T, expressed alone in HEK293T cells did not
form aggregates (Supplementary Fig. S2). _PF_HSP110C PREVENTS PROTEIN AGGREGATION IN HUMAN CELLS We tested the ability of Hsp110 proteins from yeast, human and _P. falciparum_ to prevent
aggregation. Two Hsp110 proteins in _P. falciparum_ were identified via sequence homology to human and yeast Hsp110 proteins (Supplementary Fig. S3). One of the _P. falciparum_ Hsp110
proteins (PF3D7_134200 or MAL13P1.540) has a predicted signal sequence as well as an endoplasmic reticulum retention signal (Supplementary Fig. S3). We focused our efforts on the cytoplasmic
Hsp110 (PF3D7_0708800 or PF07_0033, referred to as _Pf_Hsp110c). Co-expression of a _Pf_Hsp110c–green fluorescent protein (GFP) fusion with PFI1155w or Sup35PrD in heat-shocked HEK293T
cells gave uniform cytoplasmic distribution of the Asn-rich proteins (Fig. 1c). In contrast, substituting orthologs from human (Hsp105α) or yeast (Sse1p or Sse2p) for _Pf_Hsp110c allowed
substantial formation of fluorescent foci in co-transfected cells (Supplementary Fig. S1b–g). All fusion proteins were expressed at comparable levels in HEK293T cells (Supplementary Fig.
S1h,i). The formation of protein aggregates was quantified (Fig. 1e and Supplementary Fig. S1a). While Hsp105α, Sse1p and Sse2p co-expression gave 2–3-fold reductions in the appearance of
Sup35PrD or PFI1155w protein aggregates after heat shock, co-expression of _Pf_Hsp110c reduced the formation of protein aggregates by 10–15-fold compared with Asn-rich protein fusion
construct alone (Fig. 1e). We conclude that Hsp110 homologues can reduce aggregation of Asn-rich proteins, and that _Pf_Hsp110c is substantially better at doing so than its yeast and human
counterparts. To determine if PfHsp110c is selective for Asn-rich proteins, all Asn or all Gln versions of Sup35PrD5, in which every Gln residue in Sup35PrD was changed to Asn or every Asn
residue in Sup35PrD was changed to Gln, were substituted for the wild-type Sup35PrD in the HEK293T transfection experiment. PfHsp110c protected against aggregation of both versions and to a
greater extent than its homologues in all cases (Fig. 1f). PfHsp110c appears to have a general aggregation protection function. _PF_HSP110C IS AN ESSENTIAL GENE To investigate the role of
_Pf_Hsp110c in the intraerythrocytic life cycle of the malaria parasite, we attempted to knock out the _PfHsp110c_ gene using a double-crossover homologous recombination approach29 (Fig.
2a). Successful double-crossover integration of the hDHFR drug resistance cassette into the _PfHsp110c_ gene was observed in all clones isolated from independent transfections (Supplementary
Fig. S4). However, in the Southern blot probed with the 5′-homologous region utilized for generating the knockout, we observed a second band corresponding to an uninterrupted endogenous
gene (Fig. 2b). The data suggest that the _PfHsp110c_ gene underwent a duplication event and the hDHFR selection cassette integrated into one copy. Locus mapping showed that duplication was
limited to the _PfHsp110c_ gene and did not extend to neighbouring genes (Supplementary Fig. S4c). The inability to obtain disruptants of the _PfHsp110c_ gene without gene duplication
suggests an essential function for this gene30. The study of essential genes in the haploid malaria parasite has been enhanced by degradation-domain-based conditional expression
systems31,32,33. We recently reported the use of a regulated fluorescent affinity (RFA) tag based on the _Escherichia coli_ DHFR degradation domain (DDD)26. In the absence of the folate
analogue trimethoprim (TMP), the fusion protein is unstable and thus targeted for degradation by the proteasome. In the presence of TMP, the fusion protein is stabilized and able to carry
out its normal biological function. The _PfHsp110c_ gene was RFA-tagged via a single-crossover homologous recombination strategy (Fig. 2c). Clones isolated from independent transfections
were used for further analysis (1F9 and 2D8). Growth of _PfHsp110c–RFA_ parasite lines in the absence or presence of 10 μM TMP was monitored over several days by flow cytometry (Fig. 3a).
_PfHsp110c–RFA_ parasites did not grow in the absence of TMP and by 48 h no evidence of live parasites was seen by microscopy. Growth of _PfHsp110c–RFA_ parasite lines was TMP concentration
dependant (Fig. 3b). We conclude that _Pf_Hsp110c provides to blood-stage parasites an essential function that is disrupted upon TMP removal owing to destabilization of the RFA tag.
_PF_HSP110C IS REQUIRED FOR SURVIVING HEAT SHOCK High fever, which occurs periodically in malaria patients, is a cellular stress that results in a global tendency towards unfolding of
proteins7,8. Asynchronous _PfHsp110c–RFA_ parasites were incubated at 40 °C for 4–6 h with or without TMP (Fig. 3c). After the heat shock, the parasites were transferred back into medium
containing TMP and incubated at the normal growth temperature, 37 °C (Fig. 3c). The recovery of _PfHsp110c–RFA_ parasites was monitored over several days. In the absence of TMP, even a brief
4-h incubation at 40 °C severely inhibited parasite viability (Fig. 3c). In contrast, parasites incubated at 37 °C for 6 h without TMP were fully viable (Supplementary Fig. S5). Indeed,
cultures kept at 37 °C without TMP took 2 days to lose viability (Fig. 3a). The results demonstrate that _Pf_Hsp110c is vital for the parasite to survive even brief febrile temperatures,
suggesting a role for _Pf_Hsp110c in the proteostasis of the Asn repeat-rich parasite proteome. TMP CONTROLS _PF_HSP110C–RFA FUNCTION We assessed protein levels in _PfHsp110c–RFA_ parasites.
Asynchronous _PfHsp110c–RFA_ parasites were incubated at 37 or 40 °C in varying amounts of TMP for 24 h. Protein levels were monitored by western blots immunoprobed for GFP, which is a
component of the RFA-tag (Supplementary Fig. S6a). Surprisingly, there was no TMP dose-dependent variation in _Pf_Hsp110c–RFA levels at either temperature (Supplementary Fig. S6a). In
addition, parasites incubated in the absence of TMP showed no time-dependent degradation at 37 or 40 °C (Supplementary Fig. S6b). In contrast, we have seen robust degradation of other
RFA-tagged proteins26. As Hsp110 proteins act in concert with Hsp70 and their nucleotide exchange factor activity is required for Hsp70 function18,19,20,24, we hypothesized that in the
absence of TMP the RFA-tag interferes with essential _Pf_Hsp110c interactions resulting in parasite death. We tested this model using co-immunoprecipitation experiments. _Pf_Hsp110c–RFA
parasites were incubated at 37 or 40 °C with or without TMP for 6 h as in Fig. 2c. Total _Pf_Hsp110c–RFA and _Pf_Hsp70 levels in lysates were stable and no degradation of _Pf_Hsp110c–RFA was
observed in the absence of TMP (Fig. 4a). When _Pf_Hsp70 was immunoprecipitated from the lysates, higher amounts of _Pf_Hsp110c–RFA co-precipitated at 40 °C compared with 37 °C (Fig. 4b)
indicating that more Hsp70–Hsp110 complex exists at the higher temperature. However, there was no TMP-dependent difference in the amount of co-precipitated _Pf_Hsp110c–RFA at either
temperature. We assessed the reciprocal interaction by immunoprecipitating _Pf_Hsp110c–RFA using an anti-GFP monoclonal antibody, 3E6 (Fig. 4c), and western blotting with a second anti-GFP
monoclonal antibody, JL8. The amounts of _Pf_Hsp110c–RFA and of co-precipitated _Pf_Hsp70 did not vary at 37 °C with or without TMP. However, at 40 °C, less _Pf_Hsp110c–RFA was
immunoprecipitated in the absence of TMP and correspondingly lower amounts of _Pf_Hsp70 were seen (Fig. 4c). The data show that at 40 °C without TMP, the 3E6 monoclonal anti-GFP antibody
used for immunoprecipitation of _Pf_Hsp110c–RFA is unable to recognize the GFP epitope within the RFA-tag, even though the protein is clearly in the lysates (Fig. 4a). Similar results were
obtained using a second approach, semi-denaturing detergent agarose gel electrophoresis (SDD-AGE)34,35, and another anti-GFP antibody (Supplementary Fig. S7). The co-immunoprecipitation and
SDD-AGE data suggest that the unfolded DDD within the _Pf_Hsp110c–RFA is occluding the GFP within the RFA-tag by binding intramolecularly to the substrate-binding domain of _Pf_Hsp110c.
_PF_HSP110C AND THIOFLAVIN T RESCUE THE KNOCKDOWN PHENOTYPE _PfHsp110c–RFA_ parasites die upon TMP removal even though _Pf_Hsp110c–RFA protein levels do not decrease. To test if the death of
_PfHsp110c–RFA_ parasites is owing to the absence of chaperone function or owing to fusion protein toxicity, we episomally expressed _Pf_Hsp110c fused to tagRFP-T, under the control of the
native _PfHsp110c_ promoter in _PfHsp110c–RFA_ parasites. The complemented _PfHsp110c–RFA_ parasites were subjected to heat shock as in Fig. 2c. Parasites complemented with
_Pf_Hsp110c–tagRFP-T but not with tagRFP-T alone were able to recover after the heat shock in the absence of TMP (Fig. 5a). The recovery is partial because plasmid maintenance is incomplete
(data not shown). In contrast, _PfHsp110–RFA_ parasites expressing either the human Hsp105α or the yeast Hsp110 homologues (Sse1p and Sse2p) under the control of the _PfHsp110c_ promoter
were unable to recover after heat shock in the absence of TMP (Fig. 5b). These findings support our model that the death of _PfHsp110c–RFA_ parasites in the absence of TMP is specifically
owing to the loss of _Pf_Hsp110c function. Thioflavin T (ThT) has been utilized to monitor protein aggregation _in vitro_36. Recently, ThT was successfully used to maintain proteostasis and
extend the life span of _Caenorhabditis elegans_ expressing a polyglutamine protein37. _PfHsp110c–RFA_ parasites were incubated at 40 °C for 6 h in the absence of TMP and in the presence or
absence of 100 nM ThT and then allowed to recover. There was substantial and reproducible rescue of parasite growth (Fig. 5c). Higher ThT concentrations had toxic effects independent of TMP.
This result suggests that in the absence of a functional _Pf_Hsp110c, parasites die owing to disruption of proteostasis in its Asn repeat-rich proteome. SUP35PRD AND PFI1155W EXPRESSION IN
_PFHSP110C–RFA_ PARASITES To independently test this idea, we episomally expressed PFI1155w–tagRFP-T and Sup35PrD–tagRFP-T in _PfHsp110c–RFA_ parasite clones. Transfected parasites were
incubated at 40 °C for 6 h with or without TMP (Fig. 6). Those subjected to heat shock in the presence of TMP showed uniform cytoplasmic distribution of PFI1155w–tagRFP-T (Fig. 6a) and
Sup35PrD–tagRFP-T (Fig. 6c), as well as _Pf_Hsp110c–RFA (in green). However, parasites that were subjected to heat shock without TMP showed foci of PFI1155w–tagRFP-T (Fig. 6b) and
Sup35PrD–tagRFP-T (Fig. 6d) fluorescence indicative of protein aggregation9,27. The distribution of _Pf_Hsp110c–RFA also was more focal upon heat shock in the absence of TMP (Fig. 6b) and
there was minimal overlap between either PFI1155w–tagRFP-T or Sup35PrD–tagRFP-T and _Pf_Hsp110c–RFA foci (Fig. 6b). Expression of tagRFP-T alone in _PfHsp110c–RFA_ lines gave diffuse
cytoplasmic fluorescence in all conditions (Supplementary Fig. S8), showing that aggregation was dependent on the Asn-rich sequence. These observations support our model that _Pf_Hsp110c has
an essential role in preventing parasite Asn-rich protein aggregation. DISCUSSION The reason for the widespread presence of Asn repeat-rich proteins in the proteome of the deadly malaria
parasite, _P. falciparum_, remains a mystery. Indeed, proteins with large Asn-rich regions are prone to forming aggregates and prion-like fibrils4,5,6 and thus might be expected to reduce
the fitness of malaria parasites. Furthermore, the propensity of Asn-rich proteins to aggregate is enhanced by elevated temperatures7,8, a scenario that _P. falciparum_ parasites routinely
encounter during febrile episodes that are a hallmark clinical manifestation of malaria. In this study, we have uncovered a central role of _Pf_Hsp110c in maintaining proteostasis and
preventing aggregation of the Asn repeat-rich proteome of _P. falciparum_ (Fig. 7). This chaperone is essential for parasite growth within the red blood cell (Figs 2 and 3) and its
substrate-binding activity is necessary for surviving even a brief exposure to febrile temperatures (Figs 3 and 4 and Supplementary Fig. S7). The sequence homology of _Pf_Hsp110c to human
and yeast Hsp110 proteins is only about 24%, with most of the homology in the nucleotide-binding region (Supplementary Fig. S3). The divergent susbtrate-binding region of _Pf_Hsp110c could
be responsible for the better aggregation prevention properties of the _P. falciparum_ homologue. The malaria parasite has to deal with regular exposure to temperatures of 40 °C (during
febrile episodes). We therefore tested the ability of _PfHsp110c–RFA_ parasites to survive heat shock in the absence of the stabilizing ligand (Fig. 3c). The _PfHsp110c–RFA_ parasites were
killed within a few hours, faster than killing achieved by most antimalarial drugs. However, when we assessed cellular levels, we found that unlike other RFA-tagged parasite proteins26,
_Pf_Hsp110c–RFA is not degraded in the absence of TMP (Fig. 4a and Supplementary Fig. S6). _PfHsp110c–RFA_ parasite lines were able to recover from heat shock when they were complemented
with _Pf_Hsp110c but not with the human or yeast Hsp110 homologues, underscoring the specificity of the phenotype (Fig. 5a). By using monoclonal antibodies against the GFP and HA modules of
the RFA tag under more or less denaturing conditions, we were able to show that the unfolded degradation domain within the RFA-tag was occluding the epitopes of two anti-GFP monoclonal
antibodies when _PfHsp110c–RFA_ parasites were heat shocked (Fig. 4b and Supplementary Fig. S7). Presumably, _Pf_Hsp110c–RFA was busy binding its own tag, preventing it from carrying out its
usual role in binding other aggregation-prone, heat-shocked proteins and thus allowing the use of TMP to modulate chaperone function (Fig. 7). Utilizing degradation domains whose unfolding
is controlled by small molecules could be a general technique to manipulate chaperone function _in vivo_. Our experiments failed to detect any TMP-rescued inactivation of _Pf_Hsp110c–RFA
function at 37 °C (Fig. 4b and Supplementary Fig. S6). We believe that because death of _PfHsp110c–RFA_ parasites upon removal of TMP at 37 °C takes much longer (2 days) than at 40 °C (4–6
h) (Fig. 3a), the imbalance in proteostasis is more subtle at 37 °C. Does _Pf_Hsp110c affect proteostasis of the Asn repeat-rich parasite proteome? We investigated this question in two ways:
by testing the ability of the aggregate-binding small molecule, ThT, to rescue the growth of _PfHsp110c–RFA_ parasites heat shocked in the absence of TMP (Fig. 5c) and by expressing Asn
repeat-rich proteins (PFI1155w and Sup35PrD), in _PfHsp110c–RFA_ parasites (Fig. 6). In _PfHsp110c–RFA_ clones, ThT was able to substantially rescue parasite viability (Fig. 5c). In
addition, expression of PFI1155w or Sup35PrD in _P. falciparum_ did not lead to formation of aggregates (Fig. 6a), in contrast to what happens in yeast9,27 and in mammalian cells (Fig. 1a).
Only in the absence of TMP (functional knockdown of _Pf_Hsp110c) and after heat shock did aggregates of PFI1155w and Sup35PrD form (Fig. 6b). In HEK293T cells, Hsp110 orthologs from multiple
organisms were able to modulate aggregation of the Asn-rich proteins Sup35PrD and PFI1155w but _Pf_Hsp110c did so to a much greater extent (Fig. 1). _Pf_Hsp110c prevented aggregation of not
only Asn-rich proteins but also of a Gln-only version of the Sup35PrD (Fig. 1f), suggesting that its ability to handle aggregates does not depend on the sequence of the aggregating protein.
These findings show that _P. falciparum_ has evolved an Hsp110-dependent aggregation-resistance mechanism (Fig. 7). We propose that _Pf_Hsp110c may function as a cellular capacitor38,
allowing the rampant expansion of Asn repeats in surface loops, where tolerated1. These repeats could then evolve over time into new functional domains of advantage to the organism. We have
demonstrated that _Pf_Hsp110c is essential for parasite survival within the host red blood cell. We have also shown that _Pf_Hsp110c is vital for maintaining proteostasis in _P. falciparum_
(Fig. 7). In fact, _Pf_Hsp110c is able to prevent aggregation even in HEK293T cells, suggesting that its interacting partners in the chaperone network can be interchanged with distant
orthologs. The identification of other chaperones that act in concert with _Pf_Hsp110c and their roles in maintaining the proteostasis of the Asn repeat-rich parasite proteome are active
areas of future research. The ability to hamper the proteostasis of the imbalanced parasite proteome by inhibiting _Pf_Hsp110c function should make it an attractive target for drug
development. Our findings also raise the intriguing possibility of utilizing _Pf_Hsp110c to prevent protein-misfolding diseases. METHODS DNA SEQUENCES AND CLONING Genomic DNAs were isolated
from _P. falciparum_ using the Qiagen Blood and Cell Culture kit. Primers and restriction sites (New England Biolabs) used in this study are listed in Supplementary Table S1. All constructs
utilized in this study were confirmed by sequencing. All PCR products were inserted into the respective plasmids using the In-Fusion cloning system (Clonetech). The p110-KO-TK vector was
derived from pHHT-TK vector29. A 1-kb homologous sequence from the 5′-end of the _Pf_Hsp110c gene and a 1-kb homologous sequence from the 3′-end of the _Pf_Hsp110c gene were amplified by PCR
using primers in Supplementary Table S1. The 5′-homologous region was introduced using SacII and BglII (New England Biolabs) restriction sites and the 3′-homologous region was introduced
using ClaI and NcoI (New England Biolabs) restriction sites into the pHHT-TK vector, flanking the hDHFR cassette, to make p110-KO-TK vector. Episomal vectors for parasites were constructed
from the plasmid containing Plasmepsin II in-frame with GFP (pPM2GT)39 by inserting a PCR product (Supplementary Table S1) comprising the _hsp86_ or the _hsp110_ promoter using AatII and
XhoI (New England Biolabs) restriction sites. These vectors were further modified by replacing the hDHFR drug selection cassette with the yeast dihydroorotate dehydrogenase selection
cassette40 using the BamH1 (New England Biolabs) restriction site (plasmid kindly provided by Eva Istvan). The GFP was replaced with the PCR product containing the fluorescent protein,
tagRFP-T (Supplementary Table S1), that was introduced into these vectors using AvrII and EagI (New England Biolabs) restriction sites. For expressing tagRFP-T alone, the PCR product
comprising tagRFP-T (Supplementary Table S1) was introduced into the vector using XhoI and EagI (New England Biolabs) restriction sites. The PCR products comprising Sup35PrD or PFI1155w
(Supplementary Table S1) were introduced, in-frame with tagRFP-T, into the episomal vector with the _hsp86_ promoter using XhoI and AvrII restriction sites. The _Pf_Hsp110c, human Hsp105α
and the two yeast Hsp110s (Sse1p and Sse2p) open reading frame (ORF) PCR products (Supplementary Table S1) were introduced, in-frame with tagRFP-T, into the episomal vector containing the
_hsp110_ promoter using XhoI and AvrII restriction sites. The PCR products comprising Sup35PrD or PFI1155w (Supplementary Table S1) were inserted into pcDNA3.1 in-frame with tagRFP-T using
HindIII and BamHI (New England Biolabs) restriction sites. The PCR product _Pf_Hsp110c ORF in-frame with GFP (Supplementary Table S1) was introduced into pLexm vector using NotI and XhoI
(New England Biolabs) restriction sites. The _Pf_Hsp110c ORF from this modified pLexm vector was replaced, in-frame with GFP, with PCR products comprising human Hsp105α (Origene), Sse1p and
Sse2p (Supplementary Table S1) using NotI and AvrII restriction sites. CELL CULTURE AND TRANSFECTIONS 3D7 parasites were cultured in RPMI medium supplemented with Albumax and transfected as
described earlier41,42. Parasites transfected with p110-KO-TK underwent positive selection at 48 h with 10 nM WR99210. After parasites appeared from WR99210 selection, they were cycled off
drug for 3 weeks. Double-crossover integrants were then selected by applying 10 nM WR99210 and 20 μM ganciclovir. Plasmepsin I-knockout parasites26,43 transfected with p110-GDB underwent
positive selection 48 h after transfection with 2.5 μg ml−1 BSD (Calbiochem) and 10 μM TMP (Sigma). Integration was detected after two rounds of BSD cycling. Parasites were always cultured
with 10 μM TMP after it was initially introduced into the medium. In all cases, clones were isolated via limiting dilution. _PfHsp110c–RFA_ parasites were also transfected with episomal
vectors that contained a yeast dihydroorotate dehydrogenase selection marker40 allowing selection for parasites maintaining the episome using 2 μM DSM-1. For growth curves, parasites were
washed twice and incubated in the required medium and temperature. Medium was changed everyday and parasites were subcultured with fresh red blood cells every 3 days. Human kidney cell line
293T (HEK293T) was maintained as recommended by the ATCC. HEK293T cells were transfected using the X-tremeGENE 9 DNA Transfection Reagent (Roche) as per the manufacturer’s instructions.
After 40-h incubation with transfection reagent and plasmid DNA, cells were heat shocked for 6 h and fixed with 4% paraformaldehyde (Electron Microscopy Sciences) in PBS to be analysed by
microscopy. SOUTHERN BLOT Southern blots were performed with genomic DNA isolated using the Qiagen Blood and Cell Culture kit. For _Pf_Hsp110c-knockout parasites, 1 μg of DNA was digested
overnight with EcoRV (New England Biolabs) and integrants were screened using probes against the positive selection marker, hDHFR, and the 5′-homologous region used for integration. For
_PfHsp110c–RFA_ parasites, 1 μg of DNA was digested overnight with BbsI (New England Biolabs) and integrants were screened using probes against the 3′-end of the _Pf_Hsp110c ORF. All
Southern blots were performed as described earlier39. CO-IMMUNOPRECIPITATION Immunoprecipitation was carried out as described earlier26. The soluble fraction of the parasite lysates were
incubated for 1 h with 0.2 μg mouse monoclonal anti-GFP, 3E6 (Invitrogen), to immunoprecipitate _Pf_Hsp110c–RFA or 0.5 μg rabbit polyclonal anti-Hsp70 (Agrisera) to immunoprecipitate
_Pf_Hsp70 and 50 μl of Protein G-linked Dynabeads (Invitrogen). The beads were then washed four times with PBS containing protease inhibitor cocktail (Roche). The washed beads were
solubilized in SDS–PAGE loading buffer (LICOR Biosciences) and fractionated by 10% SDS–PAGE to be analysed by western blot. WESTERN BLOT AND SDD-AGE Western blots were performed as described
previously26. For SDD-AGE, parasites were collected and host red blood cells were permeabilized selectively by treatment with ice-cold 0.04% saponin in PBS for 10 min, followed by a wash in
ice-cold PBS. Lysates from 5 × 107 cells were loaded per lane. SDD-AGE was performed as described34. The antibodies used in this study were mouse monoclonal anti-GFP, JL8 (1:4000)
(Clonetech), mouse monoclonal anti-HA, 3F10 (1:3000) (Roche), monoclonal anti-cyclinB1, GNS1 (1: 2500) (Santa Cruz), rabbit polyclonal anti-Hsp70 (1:3000) (Agrisera) and rabbit polyclonal
anti-EF1α (1: 3000)44. The primary antibodies were detected using IRDye 680CW (1: 15,000) conjugated goat anti-rabbit IgG (LICOR Biosciences and IRDye 800CW (1:15,000) conjugated goat
anti-mouse IgG (LICOR Biosciences). The western blot images were processed and analysed using the Odyssey infrared imaging system software (LICOR Biosciences). FLOW CYTOMETRY Aliquots of
parasite cultures (5 μl) were stained with 1.5 μg ml−1 Acridine Orange (Molecular Probes) in PBS. The fluorescence profiles of infected erythrocytes were analysed by flow cytometry on a BD
FACSCanto (BD Biosystems) or MACSQuant Analyser (Miltenyi Biotec). The parasitemia data were fit to standard growth curve or dose–response equations (nonlinear least-squares analysis) in the
software package GraphPad Prism v.5.0a. MICROSCOPY Live parasites were stained with 2 μM Hoechst 33342 (Molecular Probes) as described previously39. HEK293T cells were grown on coverslips
pretreated with 0.01% poly-L-lysine (Sigma), fixed and mounted on ProLong Gold with DAPI (Invitrogen), before microscopy. Cells were observed on an Axioscope Microscope (Carl Ziess
Microimaging)42. Images were analysed and processed using ImageJ (National Institutes of Health) and merged images were generated using Adobe Photoshop. ADDITIONAL INFORMATION HOW TO CITE
THIS ARTICLE: Muralidharan, V. _et al_. _Plasmodium falciparum_ heat shock protein 110 stabilizes the asparagine repeat-rich parasite proteome during malarial fevers. _Nat. Commun._ 3:1310
doi: 10.1038/ncomms2306 (2012). REFERENCES * Aravind L., Iyer L. M., Wellems T. E. & Miller L. H. Plasmodium biology: genomic gleanings. _Cell_ 115, 771–785 (2003). Article CAS Google
Scholar * Singh G. P. et al. Hyper-expansion of asparagines correlates with an abundance of proteins with prion-like domains in _Plasmodium falciparum_. _Mol. Biochem. Parasitol._ 137,
307–319 (2004). Article CAS Google Scholar * Zilversmit M. M. et al. Low-complexity regions in _Plasmodium falciparum_: missing links in the evolution of an extreme genome. _Mol. Biol.
Evol._ 27, 2198–2209 (2010). Article CAS Google Scholar * Alberti S., Halfmann R., King O., Kapila A. & Lindquist S. A systematic survey identifies prions and illuminates sequence
features of prionogenic proteins. _Cell_ 137, 146–158 (2009). Article CAS Google Scholar * Halfmann R. et al. Opposing effects of glutamine and asparagine govern prion formation by
intrinsically disordered proteins. _Mol. Cell._ 43, 72–84 (2011). Article ADS CAS Google Scholar * Peters T. W. & Huang M. Protein aggregation and polyasparagine-mediated cellular
toxicity in _Saccharomyces cerevisiae_. _Prion_ 1, 144–153 (2007). Article Google Scholar * Liberek K., Lewandowska A. & Zietkiewicz S. Chaperones in control of protein disaggregation.
_EMBO J._ 27, 328–335 (2008). Article CAS Google Scholar * Dill K. A., Ghosh K. & Schmit J. D. Physical limits of cells and proteomes. _Proc. Natl Acad. Sci. USA_ 108, 17876–17882
(2011). Article ADS CAS Google Scholar * Patino M. M., Liu J. J., Glover J. R. & Lindquist S. Support for the prion hypothesis for inheritance of a phenotypic trait in yeast.
_Science_ 273, 622–626 (1996). Article ADS CAS Google Scholar * Wickner R. B. [Ure3] as an altered Ure2 protein—evidence for a prion analog in _Saccharomyces-cerevisiae_. _Science_ 264,
566–569 (1994). Article ADS CAS Google Scholar * Sabate R., de Groot N. S. & Ventura S. Protein folding and aggregation in bacteria. _Cell. Mol. Life Sci._ 67, 2695–2715 (2010).
Article CAS Google Scholar * Si K., Choi Y. B., White-Grindley E., Majumdar A. & Kandel E. R. Aplysia CPEB can form prion-like multimers in sensory neurons that contribute to
long-term facilitation. _Cell_ 140, 421–435 (2010). Article CAS Google Scholar * Hou F. et al. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate
immune response. _Cell_ 146, 448–461 (2011). Article CAS Google Scholar * Olzscha H. et al. Amyloid-like aggregates sequester numerous metastable proteins with essential cellular
functions. _Cell_ 144, 67–78 (2011). Article CAS Google Scholar * Hartl F. U., Bracher A. & Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. _Nature_ 475,
324–332 (2011). Article CAS Google Scholar * Young J. C. Mechanisms of the Hsp70 chaperone system. _Biochem. Cell. Biol._ 88, 291–300 (2010). Article CAS Google Scholar * Sharma S. K.,
De los Rios P., Christen P., Lustig A. & Goloubinoff P. The kinetic parameters and energy cost of the Hsp70 chaperone as a polypeptide unfoldase. _Nat. Chem. Biol._ 6, 914–920 (2010).
Article CAS Google Scholar * Dragovic Z., Broadley S. A., Shomura Y., Bracher A. & Hartl F. U. Molecular chaperones of the Hsp110 family act as nucleotide exchange factors of Hsp70s.
_EMBO J._ 25, 2519–2528 (2006). Article CAS Google Scholar * Raviol H., Sadlish H., Rodriguez F., Mayer M. P. & Bukau B. Chaperone network in the yeast cytosol: Hsp110 is revealed as
an Hsp70 nucleotide exchange factor. _EMBO J._ 25, 2510–2518 (2006). Article CAS Google Scholar * Polier S., Dragovic Z., Hartl F. U. & Bracher A. Structural basis for the cooperation
of Hsp70 and Hsp110 chaperones in protein folding. _Cell_ 133, 1068–1079 (2008). Article CAS Google Scholar * Schuermann J. P. et al. Structure of the Hsp110:Hsc70 nucleotide exchange
machine. _Mol. Cell_ 31, 232–243 (2008). Article CAS Google Scholar * Goeckeler J. L. et al. The yeast Hsp110, Sse1p, exhibits high-affinity peptide binding. _FEBS Lett._ 582, 2393–2396
(2008). Article CAS Google Scholar * Liu Q. & Hendrickson W. A. Insights into Hsp70 chaperone activity from a crystal structure of the yeast Hsp110 Sse1. _Cell_ 131, 106–120 (2007).
Article CAS Google Scholar * Polier S., Hartl F. U. & Bracher A. Interaction of the Hsp110 molecular chaperones from _S. cerevisiae_ with substrate protein. _J. Mol. Biol._ 401,
696–707 (2010). Article CAS Google Scholar * Zhang S., Binari R., Zhou R. & Perrimon N. A genomewide RNA interference screen for modifiers of aggregates formation by mutant Huntingtin
in _Drosophila_. _Genetics_ 184, 1165–1179 (2010). Article CAS Google Scholar * Muralidharan V., Oksman A., Iwamoto M., Wandless T. J. & Goldberg D. E. Asparagine repeat function in
a _Plasmodium falciparum_ protein assessed via a regulatable fluorescent affinity tag. _Proc. Natl Acad. Sci. USA_ 108, 4411–4416 (2011). Article ADS CAS Google Scholar * DePace A. H.,
Santoso A., Hillner P. & Weissman J. S. A critical role for amino-terminal glutamine/asparagine repeats in the formation and propagation of a yeast prion. _Cell_ 93, 1241–1252 (1998).
Article CAS Google Scholar * Shaner N. C. et al. Improving the photostability of bright monomeric orange and red fluorescent proteins. _Nat. Methods_ 5, 545–551 (2008). Article CAS
Google Scholar * Duraisingh M. T., Triglia T. & Cowman A. F. Negative selection of _Plasmodium falciparum_ reveals targeted gene deletion by double crossover recombination. _Int. J.
Parasitol._ 32, 81–89 (2002). Article CAS Google Scholar * Cruz A. K., Titus R. & Beverley S. M. Plasticity in chromosome number and testing of essential genes in Leishmania by
targeting. _Proc. Natl Acad. Sci. USA_ 90, 1599–1603 (1993). Article ADS CAS Google Scholar * Armstrong C. M. & Goldberg D. E. An FKBP destabilization domain modulates protein levels
in _Plasmodium falciparum_. _Nat. Methods_ 4, 1007–1009 (2007). Article CAS Google Scholar * Dvorin J. D. et al. A plant-like kinase in _Plasmodium falciparum_ regulates parasite egress
from erythrocytes. _Science_ 328, 910–912 (2010). Article ADS CAS Google Scholar * Russo I., Oksman A., Vaupel B. & Goldberg D. E. A calpain unique to alveolates is essential in
_Plasmodium falciparum_ and its knockdown reveals an involvement in pre-S-phase development. _Proc. Natl Acad. Sci. USA_ 106, 1554–1559 (2009). Article ADS CAS Google Scholar * Halfmann
R., Lindquist S. Screening for amyloid aggregation by Semi-Denaturing Detergent-Agarose Gel Electrophoresis. _J Vis Exp_ 17, e838 (2008). Google Scholar * Kryndushkin D. S., Alexandrov I.
M., Ter-Avanesyan M. D. & Kushnirov V. V. Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104. _J. Biol. Chem._ 278, 49636–49643 (2003). Article CAS
Google Scholar * Groenning M. Binding mode of Thioflavin T and other molecular probes in the context of amyloid fibrils-current status. _J. Chem. Biol._ 3, 1–18 (2009). Article Google
Scholar * Alavez S., Vantipalli M. C., Zucker D. J. S., Klang I. M. & Lithgow G. J. Amyloid-binding compounds maintain protein homeostasis during ageing and extend lifespan. _Nature_
472, 226–229 (2011). Article ADS CAS Google Scholar * Rutherford S. L. & Lindquist S. Hsp90 as a capacitor for morphological evolution. _Nature_ 396, 336–342 (1998). Article ADS
CAS Google Scholar * Klemba M., Beatty W., Gluzman I. & Goldberg D. E. Trafficking of plasmepsin II to the food vacuole of the malaria parasite _Plasmodium falciparum_. _J. Cell.
Biol._ 164, 47–56 (2004). Article CAS Google Scholar * Ganesan S. M. et al. Yeast dihydroorotate dehydrogenase as a new selectable marker for _Plasmodium falciparum_ transfection. _Mol.
Biochem. Parasitol._ 177, 29–34 (2011). Article CAS Google Scholar * Drew M. E. et al. Plasmodium food vacuole plasmepsins are activated by falcipains. _J. Biol. Chem._ 283, 12870–12876
(2008). Article CAS Google Scholar * Russo I., Oksman A. & Goldberg D. E. Fatty acid acylation regulates trafficking of the unusual _Plasmodium falciparum_ calpain to the nucleolus.
_Mol. Microbiol._ 72, 229–245 (2009). Article CAS Google Scholar * Liu J., Gluzman I. Y., Drew M. E. & Goldberg D. E. The role of _Plasmodium falciparum_ food vacuole plasmepsins. _J.
Biol. Chem._ 280, 1432–1437 (2005). Article CAS Google Scholar * Mamoun C. B. & Goldberg D. E. Plasmodium protein phosphatase 2C dephosphorylates translation elongation factor 1beta
and inhibits its PKC-mediated nucleotide exchange activity _in vitro_. _Mol. Microbiol._ 39, 973–981 (2001). Article CAS Google Scholar Download references ACKNOWLEDGEMENTS We thank
Lauren Pepper for providing Sse1p and Sse2p and for helpful discussions, Heather True for providing Sup35 and anti-Sup35 antibody, Akhil Vaidya for providing DSM-1 and DHODH plasmid, Mike
Diamond and Hyelim Cho for HEK293T cells, Roger Tsien for tagRFP-T, Niraj Tolia for pLexm, Barb Vaupel for technical assistance, Paul Sigala for comments on the manuscript, Andrey Shaw for
helpful suggestions and the US National Institutes of Health (grant 1K99AI099156-01 to V.M.) for funding. AUTHOR INFORMATION Author notes * Vasant Muralidharan Present address: Present
address: Center for Tropical and Emerging Global Diseases and Department of Cellular Biology, University of Georgia, Athens, Georgia 30602, USA, AUTHORS AND AFFILIATIONS * Howard Hughes
Medical Institute, Washington University School of Medicine in St. Louis, St. Louis, 63110, Missouri, USA Vasant Muralidharan, Anna Oksman, Priya Pal, Susan Lindquist & Daniel E.
Goldberg * Departments of Medicine and Molecular Microbiology, Washington University School of Medicine in St. Louis, St. Louis, 63110, Missouri, USA Vasant Muralidharan, Anna Oksman, Priya
Pal & Daniel E. Goldberg * Whitehead Institute for Biomedical Research, Cambridge, 02139, Massachusetts, USA Susan Lindquist * Department of Biology, Massachusetts Institute of
Technology, Cambridge, 02142, Massachusetts, USA Susan Lindquist Authors * Vasant Muralidharan View author publications You can also search for this author inPubMed Google Scholar * Anna
Oksman View author publications You can also search for this author inPubMed Google Scholar * Priya Pal View author publications You can also search for this author inPubMed Google Scholar *
Susan Lindquist View author publications You can also search for this author inPubMed Google Scholar * Daniel E. Goldberg View author publications You can also search for this author
inPubMed Google Scholar CONTRIBUTIONS V.M. and D.E.G. designed the study, interpreted the results and wrote the manuscript. V.M., A.O. and P.P. performed the experiments and interpreted the
results. S.L. contributed novel reagents before publication and helped interpret the results. CORRESPONDING AUTHOR Correspondence to Daniel E. Goldberg. ETHICS DECLARATIONS COMPETING
INTERESTS The authors declare no competing financial interests. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION Supplementary Figures S1-S8, Supplementary Table S1 (PDF 9923 kb) RIGHTS
AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Muralidharan, V., Oksman, A., Pal, P. _et al._ _Plasmodium falciparum_ heat shock protein 110 stabilizes the
asparagine repeat-rich parasite proteome during malarial fevers. _Nat Commun_ 3, 1310 (2012). https://doi.org/10.1038/ncomms2306 Download citation * Received: 12 June 2012 * Accepted: 15
November 2012 * Published: 18 December 2012 * DOI: https://doi.org/10.1038/ncomms2306 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get
shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative