Play all audios:
ABSTRACT There is increasing evidence from genome-wide association studies for a strong inherited genetic basis of susceptibility to acute lymphoblastic leukaemia (ALL) in children, yet the
effects of protein-coding variants on ALL risk have not been systematically evaluated. Here we show a missense variant in _CDKN2A_ associated with the development of ALL at genome-wide
significance (rs3731249, _P_=9.4 × 10−23, odds ratio=2.23). Functional studies indicate that this hypomorphic variant results in reduced tumour suppressor function of p16INK4A, increases the
susceptibility to leukaemic transformation of haematopoietic progenitor cells, and is preferentially retained in ALL tumour cells. Resequencing the _CDKN2A_–_CDKN2B_ locus in 2,407
childhood ALL cases reveals 19 additional putative functional germline variants. These results provide direct functional evidence for the influence of inherited genetic variation on ALL
risk, highlighting the important and complex roles of _CDKN2A_–_CDKN2B_ tumour suppressors in leukaemogenesis. SIMILAR CONTENT BEING VIEWED BY OTHERS GENOME-WIDE ANALYSES OF 200,453
INDIVIDUALS YIELD NEW INSIGHTS INTO THE CAUSES AND CONSEQUENCES OF CLONAL HEMATOPOIESIS Article Open access 14 July 2022 FUNCTIONAL DISSECTION OF INHERITED NON-CODING VARIATION INFLUENCING
MULTIPLE MYELOMA RISK Article Open access 10 January 2022 ADVANCES IN GERMLINE PREDISPOSITION TO ACUTE LEUKAEMIAS AND MYELOID NEOPLASMS Article 16 December 2020 INTRODUCTION The risk of
developing acute lymphoblastic leukaemia (ALL) is highest between 2 and 5 years after birth1,2, with initiating sentinel somatic genomic lesions (for example, chromosomal translocations)
detectable at the time of birth in many cases3,4. This early disease onset suggests a strong inherited genetic basis for ALL susceptibility, and recent genome-wide association studies (GWAS)
have discovered at least six risk loci: _ARID5B_, _IKZF1_, _CEBPE_, _PIP4K2A-BMI1_, _GATA3_ and _CDKN2A_–_CDKN2B_5,6,7,8,9,10. These ALL risk genes are directly involved in haematopoietic
stem cell function, lymphocyte differentiation and development, and cell cycle regulation11,12,13,14,15, several of which are also commonly targeted by somatic genomic lesions. In
particular, the _CDKN2A_–_CDKN2B_ locus is one of the most frequently deleted genomic regions in childhood ALL with focal copy number loss in both B- and T-cell ALL14,16. The vast majority
of variants examined in previous ALL GWAS are intronic or intergenic. Although it is now evident that non-coding variants related to disease traits are significantly over-represented in
regulatory DNA and often function as modulators of local or distal gene transcription17,18, questions also arise whether coding variants within ALL susceptibility genes might confer even
greater effects on disease development. Moreover, a large number of low-frequency and rare-coding germline variants have been discovered by exome-sequencing efforts19, but their
contributions to ALL pathogenesis have yet to be examined systematically. In the present study, we perform an exome-focused GWAS to systematically examine the impact of germline-coding
variants on the development of ALL in children of European descent, experimentally explore the functional consequences of the genome-wide significant variant in the _CDKN2A_ gene, and
comprehensively characterize coding variation at this locus by targeted resequencing. RESULTS EXOME-FOCUSED GWAS OF ALL SUSCEPTIBILITY In the discovery GWAS, we genotyped 1,773 children with
B-ALL and 10,448 non-ALL controls of European descent20,21 for 247,505 variants using the Illumina Infinium HumanExome array. Three loci with genome-wide significant association signals
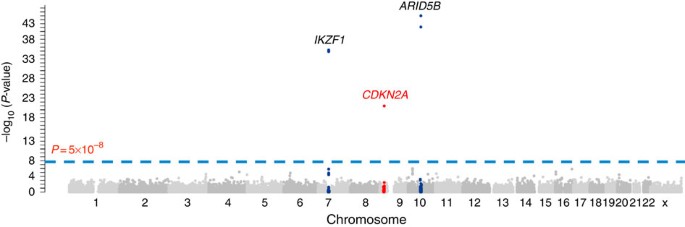
were observed: _ARID5B_ (10q21.2), _IKZF1_ (7q12.2) and _CDKN2A_ (9p21.3) (Fig. 1). Non-coding variants rs10821936 in _ARID5B_ and rs4132601 in _IKZF1_ showed the strongest association
(_P_=9.9 × 10−46 and 4.3 × 10−37, the logistic regression test, respectively; Fig. 1 and Supplementary Table 1), confirming previous GWAS findings from our group and others5,6. No coding
variants in _ARID5B_ and _IKZF1_ were significantly associated with ALL susceptibility. The third genome-wide significant hit was a missense SNP at the _CDKN2A_ locus (rs3731249, _P_=9.4 ×
10−23, the logistic regression test, Fig. 1, Table 1). The T allele at rs3731249 was over-represented in ALL compared with controls (6.8% versus 3.0%, Table 1), with every copy of the allele
conferring 2.23-fold increase in disease risk (95% confidence interval 1.90–2.61). The C-to-T nucleotide substitution at rs3731249 (c.C442T) resulted in an alanine-to-threonine change in
amino-acid sequence (p.A148T) for tumour suppressor p16INK4A. This variant also locates in the 3′ untranslated region (3′-UTR) of the p14ARF transcript, an alternative open reading frame at
this locus encoding a different tumour suppressor. Interestingly, previous GWAS had identified an intronic variant in _CDKN2A_ (rs3731217) to be strongly associated with susceptibility to
ALL in populations of European descent9. Genotype correlation between the coding variant rs3731249 and the intronic rs3731217 is exceedingly low (_r_2<0.01 in Europeans, Supplementary
Fig. 1), and multivariate analyses including both SNPs indicated their independent contribution to ALL risk (Supplementary Table 2). In the replication cohort of 409 childhood ALL cases and
1,599 non-ALL controls of European descent in Denmark, the association signal at rs3731249 was validated (_P_=5.2 × 10−4, odds ratio=1.73 (1.27–2.36), the logistic regression test, Table 1)
and this variant also remained significant after adjusting for rs3731217. FUNCTIONAL CHARACTERIZATION OF THE RS3731249 VARIANT To experimentally evaluate the effects of rs3731249 on ALL
leukaemogenesis, we directly compared the effect of wildtype versus variant allele p16INK4A (p.148A versus p.148 T) on _BCR_–_ABL1_-mediated leukaemic transformation _in vitro_. We chose
mouse haematopoietic progenitor Ba/f3 cell line because it is inherently p16Ink4a-defective due to methylation at the _Ink4a-Arf_ locus22, and ectopic expression of _BCR-ABL1_ in Ba/f3 cells
efficiently induces exogenous cytokine (interleukin 3 (IL3))-independent proliferation. Over-expression of wild-type p16INK4A(p.148A) significantly inhibited leukaemic transformation by
_BCR_–_ABL1_ (Fig. 2a, Supplementary Fig. 2), consistent with its role as a critical tumour suppressor in ALL. In contrast, Ba/f3 cells overexpressing variant p16INK4A(p.148 T) were
significantly more susceptible to _BCR_–_ABL1_ transformation measured by IL3-independent growth, suggesting that the p.148 T variant is likely hypomorphic with reduced tumour suppressor
function. In Ba/f3 cells transfected with both variant and wild-type _p16__INK4A_, the relative ratio of the p.148 T (variant) to p.148A (wildtype) transcript increased substantially upon
_BCR_–_ABL1_-mediated transformation (Supplementary Fig. 3), consistent with the increased leukaemia risk conferred by the variant allele at rs3731249. To further examine the potential
susceptibility to ALL conferred by the rs3731249 in patients, we compared the genotype distribution in RNA and DNA from primary leukaemic blasts and matched germline samples from children
with ALL (Fig. 2b). Of 15 cases with the heterozygous germline genotype at this SNP, six exhibited somatic deletion of one copy of _CDKN2A_, all of which retained the risk allele in tumour
cells. Even in cases not affected by somatic copy number loss at this locus, the variant _p16__INK4A_(c.442 T) was preferentially transcribed relative to wildtype (c.442C), with
allele-biased expression ranging from 61 to 100%, Fig. 2b). Altogether, these results pointed to the possibility that cells carrying the hypomorphic risk allele at rs3731249 might have been
enriched during leukaemogenesis. TARGETED RESEQUENCING OF _CDKN2A_ AND _CDKN2B_ IN CHILDHOOD ALL To comprehensively identify putative functional ALL susceptibility variants at this locus, we
resequenced the coding region of the _CDKN2A_ and _CDKN2B_ genes in germline DNA from 2,407 childhood ALL cases (1,450 of which were also included in the discovery GWAS). In addition to
rs3731249, we observed another 13 germline exonic variants in tumour suppressors p16INK4A and p14ARF encoded by the _CDKN2A_ gene, 12 of which result in amino-acid sequence changes (Fig. 3,
Supplementary Table 3). These missense variants were all singletons, except for the p.D125H variant in p16INK4A and the p.A121T variant in p14ARF observed in two and five cases,
respectively. Five variants were predicted to be damaging based on combined annotation dependent depletion23 (CADD score>13, Supplementary Table 3), and we did not observe germline
insertions or deletions in _CDKN2A_ in our ALL cohort. Comparing with 4,300 European American individuals from the NHLBI GO Exome Sequencing Project (ESP), there was a trend for a higher
burden of rare missense variants in relative to controls the _CDKN2A_ gene (p16INK4A and p14ARF) in children with ALL (0.71% versus 0.23%, _P_=0.0045, Fisher’s exact test, Fig. 3). In
addition, we identified six germline-coding variants in the adjacent _CDKN2B_ gene in this cohort of children with ALL, although there was no significant over-representation compared with
European controls in the ESP cohort (0.83% versus 0.79%, Fig. 3). DISCUSSION Encoding three tumour suppressor proteins (p16INK4A, p14ARF and p15INK4B), the _CDKN2A_–_CDKN2B_ locus at 9p21 is
promiscuously associated with tumorigenesis and commonly targeted by somatic mutation, deletion and/or hypermethylation in various cancers. p16INK4A and p15INK4B are highly homologous
inhibitors of cyclin-dependent kinase and function mainly as master regulators of cell cycle entry via the Rb-E2F signalling axis24. Although also encoded by the _CDKN2A_ gene, p14ARF
utilizes a completely different reading frame with distinct tumour suppression functions by inhibiting MDM2 and activating p5325. Suppressed during normal haematopoiesis, p16INK4A and p14ARF
expression is activated on oncogenic stimuli (for example, constitutive expression of _BCR-ABL1_ fusion) to trigger cell cycle exit (senescence) or apoptosis as a means of eliminating
oncogene-stressed cells26. In fact, the _CDKN2A_–_CDKN2B_ locus is either bi- or monoallelicly deleted in 64% of _BCR_–_ABL1_-positive ALL cases and in 32–72% of T- or B-ALL cases without
the _BCR-ABL1_ translocation, suggesting positive selection for cells with defective p16INK4A, p14ARF and p15INK4B (or some combinations thereof) during leukaemogenesis. The previously
reported ALL susceptibility variant rs3731217 is located in a non-coding region downstream of exon 1β (specific for p14ARF), but distal to exon 1α (specific for p16INK4A) of the _CDKN2A_
gene. The germline genotype at this SNP was not associated with overall _CDKN2A_ expression in lymphoblastoid cell lines9 but transcript-specific analyses may be needed to definitively
determine the effects of this variant on p14ARF versus p16INK4A expression. In contrast, the genome-wide significant variant rs3731249 in our current GWAS localizes to exon 2 of _CDKN2A_.
While this exon is shared by both p16INK4A and p14ARF, the C-to-T nucleotide transition causes a missense change for the p16INK4A open reading frame but is in the UTR of the p14ARF,
therefore, likely to have a more direct effect on the former. This hypothesis is supported by the fact that haematopoietic progenitor cells (Ba/f3) expressing variant p16INK4A were
substantially more susceptible to _BCR_–_ABL1_-mediated leukaemic transformation compared with cells with the wild-type protein (Fig. 2a), pointing to rs3731249 as a possible functional
variant directly contributing to the association with ALL risk. The structural basis of the hypomorphic effects of the p.A148T variant is unclear, since this residue is not directly involved
in binding to CDK4 or CDK627. However, there was evidence that the variant p16INK4A (p.148 T) is preferentially retained in the nucleus compared with the wild-type p16INK4A (p.148A),
compromising its ability to inhibit CDKs in the cytoplasm28,29. The relative contribution of p16INK4A versus p14ARF to ALL pathogenesis is not unequivocal because somatic deletions at this
locus almost always lead to the loss of both genes. Although the rs3731249 variant also results in sequence changes of the 3′-UTR of the _p14__ARF_ transcript, bioinformatic prediction did
not identify any potential effects on mRNA stability or microRNA binding and no difference was observed in reporter gene transcription under the influence of 3′-UTR containing either the
wildtype or variant allele at rs3731249 (Supplementary Fig. 4), suggesting minimal effects of this variant on _p14__ARF_ transcription. Finally, rs3731249 is also observed in non-European
populations, for example, there was a trend for a higher frequency of the risk allele in African American children with ALL than that in individuals from this racial background in the NHLBI
ESP cohort (0.58% in 260 ALL cases versus 0.38% in 2,203 controls), although a much larger sample size is needed to rigorously examine the statistical significance of such differences. It
should be noted that we and others previously showed that the non-coding ALL risk variants (rs17756311 and rs3731217) at this locus had much stronger effects in European Americans than in
other race/ethnic groups7,30, suggesting potential racial differences in genetic susceptibility to ALL. We subsequently identified additional coding variants in p16INK4A, p14ARF and p15INK4B
by resequencing, most of which were low frequency or rare. While there was a modest over-representation of potentially damaging coding variants in ALL cases compared with controls (Fig. 3),
our data do not suggest that rare variants contribute substantially to the associations with ALL susceptibility observed at this locus. It should also be noted that the vast majority of
coding variants within the _CDKN2A_ gene affects only one of the two tumour suppressors (either p16INK4A or p14ARF). Interestingly, rs199888003 is the only variant that is located in the
coding region of both p16INK4A and p14ARF, resulting in an alanine-to-threonine change in p14ARF (p.A121T) with synonymous effect on p16INK4A. This is also the most frequent germline
missense variant in p14ARF in our cohort and was over-represented in ALL compared with non-ALL controls (0.21% versus 0.046%, respectively, Fig. 3). This substitution of threonine in p14ARF
adds a possible glycosylation and phosphorylation site and also introduces a phosphoprotein-binding FHA domain implicated in DNA damage response and cell cycling31. Future studies are
warranted to determine the exact consequences of this variant on p14ARF functions. To systemically evaluate the contribution of low frequency and rare-coding variants to ALL risk, we also
performed genome-wide gene-level burden test but did not observe any genome-wide significant associations (Supplementary Table 4). Of the six known ALL risk loci, we noted two coding
variants in _CEBPE_ (rs141903485 and rs146580935, Supplementary Table 5) nominally associated with ALL susceptibility. In conclusion, we comprehensively evaluated exonic genetic variations
for association with ALL susceptibility and identified novel coding risk variants at the _CDKN2A_–_CDKN2B_ locus that may directly affect tumour suppressor functions and potentiate leukaemic
transformation. These results provided functional evidence for the influence of inherited genetic variants on ALL leukaemogenesis, further indicating that a continuum of genetic variations
in both host and tumour genomes contribute to malignant transformation and cancer risk. METHODS SUBJECTS AND SAMPLES The discovery GWAS consisted of 1,773 childhood B-ALL cases and 10,448
non-ALL controls of European descent (>90% European genetic ancestry as estimated using STRUCTURE32,33). ALL cases were from the Children’s Oncology Group (COG) AALL0232 study
(_N_=1,277)8, the COG P9906 protocol (_N_=115)34 and St Jude Total Therapy XIIIB and XV protocols (_N_=381)5. Unrelated individuals of European descent from the Atherosclerosis Risk in
Communities (ARIC) study20,21 were used as non-ALL controls because the prevalence of adult survivors of childhood ALL is less than 1 in 10,000 in the US. The replication series included 409
children with ALL from NOPHO ALL92, ALL2000 and ALL2008 protocols35 and 1,599 unrelated non-ALL controls from Danish Childhood Obesity Biobank study (clinicaltrials.gov: NCT00928473) in
Holbæk and at random schools in Zealand, Denmark. ALL cases were selected only on the basis of sample availability, and we did not observe any statistically significant differences in
demographic or clinical features of children included versus not included in this genetic study. We elected to focus on individuals of European descent to minimize population
stratification36. Germline DNA for cases was extracted from peripheral blood or bone marrow samples obtained during clinical remission (<5% ALL blasts by morphology). This study was
approved by the Institutional Review Board at St Jude Children’s Research Hospital and COG member institutions and the Ethics Committee at the Danish Data Protection Agency, Region Zealand
and the University Hospital Rigshospitalet, Denmark. Informed consent was obtained from parents, guardians, or patients, as appropriate. GENOTYPING AND QUALITY CONTROL SNP genotyping was
performed in germline DNA using the Illumina Infinium HumanExome Array v1.0 in the discovery GWAS, and using Illumina HumanCoreExome chip for the replication series. Genotype calls (coded as
0, 1, and 2 for AA, AB and BB genotypes) were determined using the Illumina GenomeStudio Software. For the ALL cases, samples for which genotype was ascertained at <98% of SNPs on the
array were deemed to have failed and were excluded from the analyses. Quality control procedures were performed for both samples and SNPs on the basis of call rate, minor allele frequency
(MAF), and Hardy Weinberg equilibrium (Supplementary Fig. 5). Detailed quality control for the non-ALL controls from the ARIC study was performed at the University of Texas Health Science
Center following established protocols21. We performed principal component analysis of cases and controls in the discovery GWAS to characterize population substructure (Supplementary Fig.
6). GENOME-WIDE ANALYSES In the discovery GWAS, the association of each SNP individually with ALL susceptibility was tested by comparing the genotype frequency between ALL cases and non-ALL
controls in logistic regression models, after adjusting for top 10 principal components to control for population stratification. A quantile–quantile (Q–Q) plot was constructed and there was
only minimal inflation at the upper tail of the distribution (_λ_=1.08, Supplementary Fig. 7). In the replication studies, we evaluated the novel genome-wide significant variant rs3731249,
using the same logistic regression models. Multivariate logistic regression model including both rs3731217 and rs3731249 were tested to determine independent association signals at the
_CDKN2A_ locus in both discovery and replication series. We also performed gene-level analyses to evaluate the aggregated effects of low-frequency variants on ALL susceptibility, using the
SKAT test37. Missense, stop codon-altering and splice-site variants with MAF<5% were included. In total, 12,687 genes with at least two variants were tested. R (version 3.0) statistical
software was used for all analyses unless indicated otherwise. _CDKN2A_–_CDKN2B_ RESEQUENCING AND RARE VARIANT ANALYSES Germline DNA from 2,407 children with ALL was used to create
individual Illumina dual-indexed libraries. These libraries were pooled in sets of 96 and hybridized with a custom version of the Roche NimbleGen SeqCap EZ custom probes to capture the
_CDKN2A_–_CDKN2B_ region on 9p21. Quantitative PCR was used to determine the appropriate capture product titre necessary to efficiently populate an Illumina HiSeq 2000 flowcell for
paired-end 2 × 101 bp sequencing. Each sequence pool of 96 samples was demultiplexed, with coverages of >20 × depth across >90% of the targeted regions for nearly all samples. Sequence
reads in FASTQ format were mapped and aligned using the Burrows–Wheeler Aligner, and genetic variants were called using the GATK pipeline version 3.1 (ref. 38). We compared the proportion
of rare variant-carriers in ALL subjects (either homozygous or heterozygous) versus that in individuals of European descent in the ESP cohort (non-ALL controls), focusing on variants with
MAF<1%. Statistical significance of the difference was estimated using Fisher’s exact test. _CDKN2A_ sequencing was also performed in matched germline and diagnostic ALL tumour DNA by
Complete Genomics for all cases with available materials, and in tumour RNA by RNA-seq. Details regarding sequencing, data analysis and coverage are available at
ftp://caftpd.nci.nih.gov/pub/dcc_target/ALL/Phase_II/sequence/WGS/CGI_TARGET_Pipeline_README.pdf, or as previously described39 (European Genome Phenome archive: EGAS00001000654). LEUKAEMIC
TRANSFORMATION ASSAY IN BA/F3 CELLS The full-length _CDKN2A_ was purchased from GE Healthcare. The p.A148T variant (rs3731249) was introduced by site-directed mutagenesis (forward primer:
5′-TGCCCGCATAGATGCCACGGAAGGTCCCTCAGA-3′, reverse primer: 5′-TCTGAGGGACCTTCCGTGGCATCTATGCGGGCA-3′) and cloned into the cL20c-IRES–GFP lentiviral vector, and lentiviral supernatants containing
cL20c–p16INK4Ap.148A–IRES–GFP or cL20c–p16INK4Ap.148 T–IRES–GFP were produced by transient transfection of 293 T cells (American Type Culture Collection) using calcium phosphate. The MSCV
(Babe MCS)–_BCR_–_ABL1_–Luc2 construct was a gift from Dr Charles Sherr at St Jude Children’s Research Hospital22 and retroviral particles were produced using 293 T cells. Ba/f3 cells (gift
from Dr Omar Abdel-Wahab at the Memorial Sloan Kettering Cancer Center) were maintained in medium supplemented with 10 ng ml−1 recombinant mouse IL3. Ba/f3 cells were transduced with
lentiviral supernatants with wild-type or variant p16INK4A(Supplementary Fig. 8). GFP-positive cells were sorted 48 h after transduction and maintained in IL3 medium for another 24 h before
transfected by _BCR_–_ABL1_ retroviral supernatants. Forty-eight hours later, cells were washed three times and grown in the absence of cytokine. Cell growth and viability were monitored
daily by Trypan blue using a TC10 automated cell counter (BIO-RAD). Each experiment was performed three times. For immunoblotting assays, Ba/f3 cells were washed and resuspended in lysis
buffer (10 × PBS with 0.5 M EDTA, 10% NP-40 and 50% glycerol) with protease inhibitors and phosphatase inhibitors. Lysates were sonicated six times and centrifuged at 13,000 g for 10 min at
4 °C. Supernatants were quantified for protein concentration by BCA kit, electrophoresed, and transferred to nitrocellulose membranes. Membranes were probed with 1: 1,000 anti-p16INK4A
antibody (Abcam, ab81278), with α-tubulin as a loading control (1: 1,000 anti-tubulin antibody, Sigma-Aldrich, T5618). For quantitative reverse transcription-PCR (qRT–PCR), total RNA was
extracted using the RNeasy Micro kit (Qiagen) according to the manufacturer’s protocol. Total RNA (500 ng) was reverse transcribed into cDNA using oligoT primers and the SuperScript III
reverse transcriptase kit (Invitrogen). Quantitative real-time PCR was performed by using ABI Prism 7900HT detection system (Applied Biosystems) with Faststart SYBR Green master mix (Roche).
Relative expression was calculated as a ratio of _BCR-ABL1_ to _Hprt._ Primer sequences of _BCR_–_ABL1_ and _Hprt_ were as follows: _BCR-ABL1_ (forward: 5′-CTGGCCCAACGATGGCGA-3′; reverse:
5′-CACTCAGACCCTGAGGCTCAA-3′); _Hprt_ (forward: 5′-GAGCAATGATCTTGATCTTC-3′; reverse: 5′-TTCCTTCTTGGGTATGGAAT-3′). To co-express rs3731249 variant and wild-type p16INK4A, Ba/f3 cells were
transduced with equal molar cL20c–p16INK4Ap.148A–IRES–GFP and cL20c–p16INK4Ap.148 T–IRES–iYFP lentivirus and cells successfully transfected with both were selected by flow cytometry sorting
for GFP/YFP double positivity. _BCR-ABL1_-mediated transformation was performed as described above. Genomic DNA and RNA samples were collected at day 0, 2, 4 and 5 after IL3 removal. p.148A
and p.148 T transcript in RNA was quantified using allele-specific Taqman genotyping assay and normalized to allele ratio in matched DNA samples at respective time points. Each experiment
was performed three times and each sample was assayed in triplicate. LUCIFERASE REPORTER ASSAYS The _p14__INK4A_–3′-UTR vector (3′-UTR for Human NM_058195.2 was placed downstream of
luciferase reporter gene on the pEZX-MT01 backbone) was purchased from GeneCopoeia and the T variant at rs3731249 was introduced by site-directed mutagenesis (forward primer:
5′-CCATGCCCGCATAGATGCCGTGGAAGGTCCCTCAGACATCC-3′; reverse primer: 5′-GGATGTCTGAGGGACCTTCCACGGCATCTATGCGGGCATGG-3′). For reporter gene assay, 2.5 × 104 293 T cells cultured in 96-well plate
were transiently transfected with 100 ng empty vector, variant, or wild-type _p14__INK4A_ 3′UTR constructs using Lipofectamine 2000 (Invitrogen). Firefly luciferase activities were measured
24 h later using the Dual Luciferase Assay (Promega). The results were normalized against Renilla luciferase. Each reporter construct transfection was replicated at least three times, and
each sample was assayed in triplicate. ADDITIONAL INFORMATION ACCESSION CODES. The RNA-seq data have been deposited in European Genome Phenome archive under the accession codes
EGAS00001000654. HOW TO CITE THIS ARTICLE: Xu, H. _et al._ Inherited coding variants at the CDKN2A locus influence susceptibility to acute lymphoblastic leukaemia in children. _Nat. Commun._
6:7553 doi: 10.1038/ncomms8553 (2015). REFERENCES * Greaves, M. Infection, immune responses and the aetiology of childhood leukaemia. _Nat. Rev. Cancer_ 6, 193–203 (2006). Article CAS
PubMed Google Scholar * Hjalgrim, L. L. et al. Age- and sex-specific incidence of childhood leukemia by immunophenotype in the Nordic countries. _J. Natl Cancer Inst._ 95, 1539–1544
(2003). Article PubMed Google Scholar * Greaves, M. F. & Wiemels, J. Origins of chromosome translocations in childhood leukaemia. _Nat. Rev. Cancer_ 3, 639–649 (2003). Article CAS
PubMed Google Scholar * Greaves, M. F., Maia, A. T., Wiemels, J. L. & Ford, A. M. Leukemia in twins: lessons in natural history. _Blood_ 102, 2321–2333 (2003). Article CAS PubMed
Google Scholar * Trevino, L. R. et al. Germline genomic variants associated with childhood acute lymphoblastic leukemia. _Nat. Genet._ 41, 1001–1005 (2009). Article CAS PubMed PubMed
Central Google Scholar * Papaemmanuil, E. et al. Loci on 7p12.2, 10q21.2 and 14q11.2 are associated with risk of childhood acute lymphoblastic leukemia. _Nat. Genet._ 41, 1006–1010 (2009).
Article CAS PubMed PubMed Central Google Scholar * Xu, H. et al. Novel susceptibility variants at 10p12.31-12.2 for childhood acute lymphoblastic leukemia in ethnically diverse
populations. _J. Natl Cancer Inst._ 105, 733–742 (2013). Article CAS PubMed PubMed Central Google Scholar * Perez-Andreu, V. et al. Inherited GATA3 variants are associated with Ph-like
childhood acute lymphoblastic leukemia and risk of relapse. _Nat. Genet._ 45, 1494–1498 (2013). Article CAS PubMed PubMed Central Google Scholar * Sherborne, A. L. et al. Variation in
CDKN2A at 9p21.3 influences childhood acute lymphoblastic leukemia risk. _Nat. Genet._ 42, 492–494 (2010). Article CAS PubMed PubMed Central Google Scholar * Migliorini, G. et al.
Variation at 10p12.2 and 10p14 influences risk of childhood B-cell acute lymphoblastic leukemia and phenotype. _Blood_ 122, 3298–3307 (2013). Article CAS PubMed Google Scholar * Akasaka,
T. et al. Five members of the CEBP transcription factor family are targeted by recurrent IGH translocations in B-cell precursor acute lymphoblastic leukemia (BCP-ALL). _Blood_ 109,
3451–3461 (2007). Article CAS PubMed Google Scholar * Novershtern, N. et al. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. _Cell_ 144,
296–309 (2011). Article CAS PubMed PubMed Central Google Scholar * Lahoud, M. H. et al. Gene targeting of Desrt, a novel ARID class DNA-binding protein, causes growth retardation and
abnormal development of reproductive organs. _Genome Res._ 11, 1327–1334 (2001). Article CAS PubMed Google Scholar * Mullighan, C. G. et al. Deletion of IKZF1 and prognosis in acute
lymphoblastic leukemia. _N. Engl. J. Med._ 360, 470–480 (2009). Article CAS PubMed PubMed Central Google Scholar * Yagi, R., Zhu, J. & Paul, W. E. An updated view on transcription
factor GATA3-mediated regulation of Th1 and Th2 cell differentiation. _Int. Immunol._ 23, 415–420 (2011). Article CAS PubMed PubMed Central Google Scholar * Mullighan, C. G. et al.
Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. _Nature_ 446, 758–764 (2007). Article ADS CAS PubMed Google Scholar * ENCODE Project Consortium. An
integrated encyclopedia of DNA elements in the human genome. _Nature_ 489, 57–74 (2012). * Maurano, M. T. et al. Systematic localization of common disease-associated variation in regulatory
DNA. _Science_ 337, 1190–1195 (2012). Article ADS CAS PubMed PubMed Central Google Scholar * Tennessen, J. A. et al. Evolution and functional impact of rare coding variation from deep
sequencing of human exomes. _Science_ 337, 64–69 (2012). Article ADS CAS PubMed Google Scholar * The Atherosclerosis Risk in Communities (ARIC) Study: design and objectives. The ARIC
investigators. _Am. J. Epidemiol._ 129, 687–702 (1989). * Grove, M. L. et al. Best practices and joint calling of the HumanExome BeadChip: the CHARGE Consortium. _PLoS ONE_ 8, e68095 (2013).
Article ADS CAS PubMed PubMed Central Google Scholar * Williams, R. T., Roussel, M. F. & Sherr, C. J. Arf gene loss enhances oncogenicity and limits imatinib response in mouse
models of Bcr-Abl-induced acute lymphoblastic leukemia. _Proc. Natl Acad. Sci. USA_ 103, 6688–6693 (2006). Article ADS CAS PubMed PubMed Central Google Scholar * Kircher, M. et al. A
general framework for estimating the relative pathogenicity of human genetic variants. _Nat. Genet._ 46, 310–315 (2014). Article CAS PubMed PubMed Central Google Scholar * Krug, U.,
Ganser, A. & Koeffler, H. P. Tumor suppressor genes in normal and malignant hematopoiesis. _Oncogene_ 21, 3475–3495 (2002). Article CAS PubMed Google Scholar * Sherr, C. J. et al.
p53-Dependent and -independent functions of the Arf tumor suppressor. _Cold Spring Harb. Symp. Quant. Biol._ 70, 129–137 (2005). Article CAS PubMed Google Scholar * Williams, R. T. &
Sherr, C. J. The INK4-ARF (CDKN2A/B) locus in hematopoiesis and BCR-ABL-induced leukemias. _Cold Spring Harb. Symp. Quant. Biol._ 73, 461–467 (2008). Article CAS PubMed Google Scholar *
Russo, A. A., Tong, L., Lee, J. O., Jeffrey, P. D. & Pavletich, N. P. Structural basis for inhibition of the cyclin-dependent kinase Cdk6 by the tumour suppressor p16INK4a. _Nature_
395, 237–243 (1998). Article ADS CAS PubMed Google Scholar * Walker, G. J., Gabrielli, B. G., Castellano, M. & Hayward, N. K. Functional reassessment of P16 variants using a
transfection-based assay. _Int. J. Cancer_ 82, 305–312 (1999). Article CAS PubMed Google Scholar * Lilischkis, R., Sarcevic, B., Kennedy, C., Warlters, A. & Sutherland, R. L.
Cancer-associated mis-sense and deletion mutations impair p16INK4 CDK inhibitory activity. _Int. J. Cancer_ 66, 249–254 (1996). Article CAS PubMed Google Scholar * Chokkalingam, A. P. et
al. Genetic variants in ARID5B and CEBPE are childhood ALL susceptibility loci in Hispanics. _Cancer Causes Control_ 24, 1789–1795 (2013). Article PubMed PubMed Central Google Scholar *
Durocher, D., Smerdon, S. J., Yaffe, M. B. & Jackson, S. P. The FHA domain in DNA repair and checkpoint signaling. _Cold Spring Harb. Symp. Quant. Biol._ 65, 423–431 (2000). Article
CAS PubMed Google Scholar * Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. _Genetics_ 155, 945–959 (2000). CAS PubMed
PubMed Central Google Scholar * Yang, J. J. et al. Ancestry and pharmacogenomics of relapse in acute lymphoblastic leukemia. _Nat. Genet._ 43, 237–241 (2011). Article CAS PubMed
PubMed Central Google Scholar * Harvey, R. C. et al. Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome
in pediatric B-progenitor acute lymphoblastic leukemia. _Blood_ 115, 5312–5321 (2010). Article CAS PubMed PubMed Central Google Scholar * Schmiegelow, K. et al. Long-term results of
NOPHO ALL-92 and ALL-2000 studies of childhood acute lymphoblastic leukemia. _Leukemia_ 24, 345–354 (2010). Article CAS PubMed Google Scholar * Mathieson, I. & McVean, G.
Differential confounding of rare and common variants in spatially structured populations. _Nat. Genet._ 44, 243–246 (2012). Article CAS PubMed PubMed Central Google Scholar * Wu, M. C.
et al. Rare-variant association testing for sequencing data with the sequence kernel association test. _Am. J. Hum. Genet._ 89, 82–93 (2011). Article CAS PubMed PubMed Central Google
Scholar * DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. _Nat. Genet._ 43, 491–498 (2011). Article CAS PubMed PubMed
Central Google Scholar * Roberts, K. G. et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. _N. Engl. J. Med._ 371, 1005–1015 (2014). Article PubMed
PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS We thank the patients and parents who participated in the clinical protocols included in this study and the clinicians
and research staff at participating institutions. J.J.Y. is supported by the American Society of Hematology Scholar Award and by the Order of St. Francis Foundation. H.Z. is a St Baldrick’s
International Scholar. V.P.A is supported by the Spanish Ministry of Education Fellowship Grant and by the St Jude Children’s Research Hospital Academic Programs Special Fellowship. C.G.M.
is a Pew Scholar in the Biomedical Sciences and a St Baldrick’s Scholar. We thank M. Shriver (Pennsylvania State University) for sharing SNP genotype data of the Native American references
and K. Nielsen (The Technical University of Denmark) for assistance with analysing the Danish dataset. This work was supported by the National Institutes of Health (grant numbers CA156449,
CA21765, CA36401, CA98543, CA114766, CA98413, CA140729, CA176063, GM097119 and GM92666, HHSN261200800001E), the American Lebanese Syrian Associated Charities (ALSAC), the Danish Council for
Strategic Research (TARGET (0603-00484B), BIOCHILD (0603-00457B)), the Region Zealand Health Scientific Research Foundation, Danish National Research Foundation, Danish Childhood Cancer
Foundation, and Swedish Childhood Cancer Foundation. The Atherosclerosis Risk in Communities (ARIC) study is carried out as a collaborative study supported by the National Heart, Lung, and
Blood Institute contracts (HHSN268201100005C, HHSN268201100006C, HHSN268201100007C, HHSN268201100008C, HHSN268201100009C, HHSN268201100010C, HHSN268201100011C and HHSN268201100012C). Funding
support for ‘Building on GWAS for NHLBI-diseases: the U.S. CHARGE consortium’ was provided by the NIH through the American Recovery and Reinvestment Act of 2009 (ARRA) (5RC2HL102419). The
authors also thank the staff and participants of the ARIC study for their important contributions. AUTHOR INFORMATION Author notes * Heng Xu and Hui Zhang: These authors contributed equally
to this work. AUTHORS AND AFFILIATIONS * Department of Pharmaceutical Sciences, St. Jude Children’s Research Hospital, Memphis, 38105, Tennessee, USA Heng Xu, Hui Zhang, Wenjian Yang,
Maoxiang Qian, Virginia Perez-Andreu, Xujie Zhao, William E. Evans, Mary V. Relling & Jun J. Yang * Department of Laboratory Medicine, National Key Laboratory of Biotherapy/Collaborative
Innovation Center of Biotherapy, and Cancer Center, West China Hospital, Sichuan University, Chengdu, 610041, Sichuan, China Heng Xu * Department of Pediatrics, The first affiliated
hospital of Guangzhou Medical University, Guangzhou, 510120, Guangdong, China Hui Zhang * Centre for Biological Sequence Analysis, The Technical University of Denmark, Kgs, Lyngby, DK-2800,
Denmark Rachita Yadav & Ramneek Gupta * Department of Epidemiology, Human Genetics, and Environmental Sciences, School of Public Health, University of Texas Health Science Center,
Houston, 77030, Texas, USA Alanna C. Morrison * Department of Biostatistics, Epidemiology and Health Policy Research, College of Medicine, University of Florida, Gainesville, 32610, Florida,
USA Meenakshi Devidas * Department of Computational Biology, St. Jude Children’s Research Hospital, Memphis, 38105, Tennessee, USA Yu Liu & Jinghui Zhang * Department of Pathology and
Laboratory Medicine, and Departments of Pathology and Pediatrics, Nationwide Children’s Hospital, Ohio State University College of Medicine, Columbus, 43205, Ohio, USA Julie M.
Gastier-Foster * Department of Pediatrics, Texas Children's Cancer Center, Baylor College of Medicine, Houston, 77030, Texas, USA Philip J. Lupo * Hartwell Center for Bioinformatics
& Biotechnology, St. Jude Children’s Research Hospital, Memphis, 38105, Tennessee, USA Geoff Neale * Huntsman Cancer Institute, The University of Utah, Salt Lake City, 84112, Utah, USA
Elizabeth Raetz * Maine Children’s Cancer Program, Scarborough, 04074, Maine, USA Eric Larsen * Cook Children's Medical Center, Ft. Worth, 38754, Texas, USA W. Paul Bowman * Pediatric
Oncology, Cancer Institute New York University, New York City, 10016, New York, USA William L. Carroll * Pediatric Hematology/Oncology, University of Texas Southwestern Medical Center,
Dallas, 75235, Texas, USA Naomi Winick * Puma Biotechnology Inc., Los Angeles, 90024, California, USA Richard Williams * The Novo Nordisk Foundation Center for Basic Metabolic Research,
Faculty of Health and Medical Sciences, University of Copenhagen, Copenhagen, DK-2200, Denmark Torben Hansen * Department of Pediatrics, The Children’s Obesity Clinic, Copenhagen University
Hospital Holbaek, Holbaek, DK-4300, Denmark Jens-Christian Holm * McDonnell Genome Institute, Washington University School of Medicine, St Louis, 63108, Missouri, USA Elaine Mardis &
Robert Fulton * Hematological Malignancies Program, Comprehensive Cancer Center, St. Jude Children's Research Hospital, Memphis, 38105, Tennessee, USA Ching-Hon Pui, Charles G.
Mullighan, William E. Evans, Mary V. Relling & Jun J. Yang * Department of Oncology, St. Jude Children’s Research Hospital, Memphis, 38105, Tennessee, USA Ching-Hon Pui * Department of
Pathology, St. Jude Children’s Research Hospital, Memphis, 38105, Tennessee, USA Charles G. Mullighan * Division of Oncology and Center for Childhood Cancer Research, Children’s Hospital of
Philadelphia, Philadelphia, 19104, Pennsylvania, USA Stephen P. Hunger * Department of Paediatrics and Adolescent Medicine, The Juliane Marie Centre, The University Hospital Rigshospitalet,
and the Institute of Clinical Medicine, Faculty of Health, University of Copenhagen, Copenhagen, DK-2100, Denmark Kjeld Schmiegelow * Department of Pediatrics, Benioff Children’s Hospital
and the Helen Diller Family Comprehensive Cancer Center, University of California at San Francisco, San Francisco, 94115, California, USA Mignon L. Loh Authors * Heng Xu View author
publications You can also search for this author inPubMed Google Scholar * Hui Zhang View author publications You can also search for this author inPubMed Google Scholar * Wenjian Yang View
author publications You can also search for this author inPubMed Google Scholar * Rachita Yadav View author publications You can also search for this author inPubMed Google Scholar * Alanna
C. Morrison View author publications You can also search for this author inPubMed Google Scholar * Maoxiang Qian View author publications You can also search for this author inPubMed Google
Scholar * Meenakshi Devidas View author publications You can also search for this author inPubMed Google Scholar * Yu Liu View author publications You can also search for this author
inPubMed Google Scholar * Virginia Perez-Andreu View author publications You can also search for this author inPubMed Google Scholar * Xujie Zhao View author publications You can also search
for this author inPubMed Google Scholar * Julie M. Gastier-Foster View author publications You can also search for this author inPubMed Google Scholar * Philip J. Lupo View author
publications You can also search for this author inPubMed Google Scholar * Geoff Neale View author publications You can also search for this author inPubMed Google Scholar * Elizabeth Raetz
View author publications You can also search for this author inPubMed Google Scholar * Eric Larsen View author publications You can also search for this author inPubMed Google Scholar * W.
Paul Bowman View author publications You can also search for this author inPubMed Google Scholar * William L. Carroll View author publications You can also search for this author inPubMed
Google Scholar * Naomi Winick View author publications You can also search for this author inPubMed Google Scholar * Richard Williams View author publications You can also search for this
author inPubMed Google Scholar * Torben Hansen View author publications You can also search for this author inPubMed Google Scholar * Jens-Christian Holm View author publications You can
also search for this author inPubMed Google Scholar * Elaine Mardis View author publications You can also search for this author inPubMed Google Scholar * Robert Fulton View author
publications You can also search for this author inPubMed Google Scholar * Ching-Hon Pui View author publications You can also search for this author inPubMed Google Scholar * Jinghui Zhang
View author publications You can also search for this author inPubMed Google Scholar * Charles G. Mullighan View author publications You can also search for this author inPubMed Google
Scholar * William E. Evans View author publications You can also search for this author inPubMed Google Scholar * Stephen P. Hunger View author publications You can also search for this
author inPubMed Google Scholar * Ramneek Gupta View author publications You can also search for this author inPubMed Google Scholar * Kjeld Schmiegelow View author publications You can also
search for this author inPubMed Google Scholar * Mignon L. Loh View author publications You can also search for this author inPubMed Google Scholar * Mary V. Relling View author publications
You can also search for this author inPubMed Google Scholar * Jun J. Yang View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS J.J.Y. is the
principal investigator of this study and has full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. H.X.,
W.Y., M.Q., R.Y., R.G. and V.P.A. performed data analysis, H.Z., H.X. and X.Z. performed the experiments. J.J.Y., H.X, H.Z. and M.Q. wrote the manuscript. R.Y., A.C.M., M.D., J.M.G-F.,
P.J.L., G.N., Y.L., E.R., E.L., W.P-B., W.L.C., N.W., R.W., T.H., J.H., E.M., R.F., C.P., J.Z., C.G.M., W.E.E, S.P.H., R.G., K.S., M.L.L. and M.V.R. contributed reagents, materials and/or
data. J.J.Y., H.X., H.Z., W.Y., M.Q., V.P.A., R.Y., R.G., R. W. and K. S. interpreted the data and the research findings. All of the co-authors reviewed the manuscript. CORRESPONDING AUTHOR
Correspondence to Jun J. Yang. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing financial interests. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION Supplementary
Figures 1-8 and Supplementary Tables 1-5 (PDF 1183 kb) RIGHTS AND PERMISSIONS This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third
party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative
Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Xu, H., Zhang, H., Yang, W. _et al._ Inherited coding variants at the _CDKN2A_ locus influence susceptibility to acute
lymphoblastic leukaemia in children. _Nat Commun_ 6, 7553 (2015). https://doi.org/10.1038/ncomms8553 Download citation * Received: 06 January 2015 * Accepted: 20 May 2015 * Published: 24
June 2015 * DOI: https://doi.org/10.1038/ncomms8553 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link
is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative