Play all audios:
ABSTRACT Abdominal aortic aneurysm (AAA) is a permanent expansion of the abdominal aorta that has a high mortality but limited treatment options. Phosphodiesterase (PDE) 4 family members are
cAMP-specific hydrolyzing enzymes and have four isoforms (PDE4A-PDE4D). Several pan-PDE4 inhibitors are used clinically. However, the regulation and function of PDE4 in AAA remain largely
unknown. Herein, we showed that PDE4D expression is upregulated in human and angiotensin II-induced mouse AAA tissues using RT-PCR, western blotting, and immunohistochemical staining.
Furthermore, smooth muscle cell (SMC)-specific _Pde4d_ knockout mice showed significantly reduced vascular destabilization and AAA development in an experimental AAA model. The PDE4
inhibitor rolipram also suppressed vascular pathogenesis and AAA formation in mice. In addition, PDE4D deficiency inhibited caspase 3 cleavage and SMC apoptosis in vivo and in vitro, as
shown by bulk RNA-seq, western blotting, flow cytometry and TUNEL staining. Mechanistic studies revealed that PDE4D promotes apoptosis by suppressing the activation of cAMP-activated protein
kinase A (PKA) instead of the exchange protein directly activated by cAMP (Epac). Additionally, the phosphorylation of BCL2-antagonist of cell death (Bad) was reversed by PDE4D siRNA in
vitro, which indicates that PDE4D regulates SMC apoptosis via the cAMP-PKA-pBad axis. Overall, these findings indicate that PDE4D upregulation in SMCs plays a causative role in AAA
development and suggest that pharmacological inhibition of PDE4 may represent a potential therapeutic strategy. SIMILAR CONTENT BEING VIEWED BY OTHERS MIR-126-5P PROMOTES CONTRACTILE
SWITCHING OF AORTIC SMOOTH MUSCLE CELLS BY TARGETING VEPH1 AND ALLEVIATES ANG II-INDUCED ABDOMINAL AORTIC ANEURYSM IN MICE Article 01 July 2020 LILRB4 KNOCKDOWN INHIBITS AORTIC DISSECTION
DEVELOPMENT BY REGULATING PYROPTOSIS AND THE JAK2/STAT3 SIGNALING PATHWAY Article Open access 06 July 2024 NCOR1 MAINTAINS THE HOMEOSTASIS OF VASCULAR SMOOTH MUSCLE CELLS AND PROTECTS
AGAINST AORTIC ANEURYSM Article 23 September 2022 INTRODUCTION Abdominal aortic aneurysm (AAA) is a chronic vessel wall degenerative disease characterized by the irreversible progressive
dilatation of the abdominal aorta over 3.0 cm (1.5-fold of normal abdominal aorta)1. AAA is a multifactorial disease that predominantly affects elderly males 65 years or older2. AAA rupture
is a life-threatening medical emergency, with mortality rates > 81%3. At present, there are no effective clinical medicines to prevent, delay, or reverse the growth or rupture of AAA,
except surgical repair4. Therefore, understanding the molecular mechanisms and identifying regulators underlying the pathogenesis of AAA are essential for developing potential therapeutic
strategies. Smooth muscle cells (SMCs) are the major cell type in the medial layer of large arteries. SMCs regulate vascular contractility in response to pulsatile blood flow and pressure in
aortas. SMCs are the major contributors to elastin fibers that maintain the elasticity of aortas. Increasing evidence has indicated that the loss of vascular SMCs contributes to aortic wall
weakening, dilation, and aneurysm formation5,6. The second messenger cyclic nucleotides 3′,5′-cyclic adenosine monophosphate (3′,5′-cAMP) and 3′,5′-cyclic guanosine monophosphate
(3′,5′-cGMP) are important in regulating smooth muscle contractile function and vessel wall structure integrity. Abnormal cyclic nucleotide homeostasis contributes to a variety of
cardiovascular diseases7,8,9. Cyclic nucleotide phosphodiesterases (PDEs) are a superfamily of enzymes responsible for hydrolyzing cyclic nucleotides and thus play crucial roles in
regulating the duration, magnitude, and compartmentalization of cyclic nucleotide responses10. There are 11 families within the PDE superfamily (PDE1 to PDE11), and each family contains
multiple subtypes. PDE dysregulation has been associated with numerous diseases11. Over the past decades, PDEs have been proven to be ideal and feasible drug targets. Several family-specific
PDE inhibitors are currently used or are currently under clinical trials for the treatment of a variety of diseases12,13,14. A few reports have suggested potential links between cAMP
signaling and AAA. For example, ablation of SMC-specific _Gsα_, a G-protein responsible for cAMP production, exaggerated AAA formation in mice15. Cilostazol, an inhibitor of cAMP-hydrolyzing
PDE3 and adenosine uptake16, suppressed AAA in rats and mice17. The mechanistic action of cilostazol in AAA remains to be clarified: cAMP versus adenosine. Recently, we reported that PDE1C,
a Ca2+/calmodulin-stimulated PDE, contributed to AAA by promoting SMC senescence by suppressing cAMP-mediated activation of SIRT118. These previous studies showed a protective effect of
cAMP on AAA prevention. However, the roles of different cAMP signaling pathways regulated by distinct cAMP-PDEs in AAA remain unclear. PDE4 family members are cAMP-specific hydrolyzing
enzymes encoded by four isoforms: PDE4A, 4B, 4C, and 4D. Several PDE4 family-selective inhibitors—such as rolipram, roflumilast, apremilast, and crisaborole—have been used to clinically
treat inflammatory diseases, including inflammatory airway diseases, psoriatic arthritis, and atopic dermatitis19. Understanding the roles of PDE4 isozymes and the effects of PDE4 inhibitors
in AAA is critical for developing potential therapeutic strategies. In this study, we aimed to explore the regulation, function, and mechanistic action of the PDE4 isozyme in AAA
pathogenesis and development. Because our pilot study showed that PDE4D is most prominently upregulated in SMCs of human and mouse AAA tissues, we focused on SMC PDE4D in the current study.
Furthermore, PDE4D deficiency improved vascular pathogenesis and AAA development in SMC-specific _Pde4d_ knockout mice. The PDE4 inhibitor rolipram protected against AAA in mice. In
addition, we demonstrated that PDE4D promotes SMC apoptosis via the cAMP-PKA-pBad axis. MATERIALS AND METHODS ANIMAL MODELS All animal procedures were performed in accordance with Peking
Union Medical College guidelines (Animal Care and Use Committee of Peking Union Medical College accreditation number: ACUC-A01-2017-007). _Pde4d_-floxed mice _(Pde4d__flox/flox_) were
generated in and obtained from the Shanghai Model Organisms Center (Shanghai, China) through homologous recombination using CRISPR/Cas9 technology. _Pde4d_-floxed mice carry floxed exon 10
of the _Pde4d_ gene. _Pde4d__flox/flox_ mice were crossed with _Apoe__−/−_ mice. For generation of SMC-specific knockout mice on the _Apoe__−/−_ background (_Apoe__−/−__Pde4d__SMC−/-_),
_Apoe__−/−_ mice carrying the floxed _Pde4d_ allele were crossed with _Tagln_-Cre mice (Shanghai Model Organisms Center, China). For genotyping, a One Step Mouse Genotyping Kit (Vazyme
Biotech Co., Ltd., PD101-01, Nanjing, China) was used to test tail biopsies of mice by PCR. Primers targeting the wild-type allele, the floxed allele, and cre recombinase are listed in
Supplementary Table 2. After genotyping analysis, _Apoe__−/−__Pde4d__flox/flox_ and _Apoe__−/−__Pde4d__SMC−/−_ littermates were used for study. Only male mice were used, as AAA primarily
affects elderly males in humans2. For induction of AAA, eight-week-old male mice were infused with angiotensin II (Ang II, 1000 ng kg−1 min−1; Sigma, Cat#: A9525-50MG, US) subcutaneously
implanted with osmotic pumps (Alzet MODEL 2004; DURECT, US) for twenty-eight days and fed a high-fat diet (HFD; Research Diets, Cat#: D12108C, US), as described previously20. For a model
with the important features of human AAA, the main method used in animal models is subcutaneous infusion of Ang IIto induce AAA in mice20,21,22. Based on previous experience and literature
reports20,23, four groups of mice were randomly treated as follows: _Apoe__−/−__Pde4d__flox/flox_ mice treated with saline (_n_ = 6), _Apoe__−/−__Pde4d__SMC−/−_ mice treated with saline (_n_
= 6), _Apoe__−/−__Pde4d__flox/flox_ mice treated with Ang II (_n_ = 17), and _Apoe__−/−__Pde4d__SMC−/−_ mice treated with Ang II (_n_ = 16). For determination of the effect of rolipram on
AAA, _Apoe__−/−_ male mice (C57BL/6, N11, The Jackson Laboratory, US) at eight weeks old were treated as mentioned above. A total of 0.375 mg mL−1 rolipram (PDE4 inhibitor, 8 mL kg−1 d−1,
dissolved in ethyl alcohol; Sigma-Aldrich, R6520) was administered orally via gavage daily for twenty-eight days. The mice were randomly divided into four groups: Ang II (−) mice treated
with 7% ethyl alcohol (3 mg kg−1 d−1; _n_ = 5), Ang II (−) mice treated with rolipram (_n_ = 6), Ang II (+) mice treated with 7% ethyl alcohol (3 mg kg−1 d−1; _n_ = 23), and Ang II (+) mice
treated with rolipram (_n_ = 16). AAA was defined as 50% dilation in the diameter of the external abdominal aorta compared with the normal abdominal aorta of the control group. The aortas
were excised, and the maximal external diameter of the abdominal aorta was measured by two different investigators using a stereoscope at the time of necropsy. The animals were raised in a
specific pathogen-free (SPF) facility. HUMAN AORTIC TISSUES Human specimens were obtained under protocols approved by the Ethics Committee of Beijing Anzhen Hospital, Capital Medical
University. Human samples from the anterior region of the aneurysmal aortic wall were obtained from nine AAA patients who had undergone open aortic aneurysm surgery repair (Project Number:
2017-051X). The patients were diagnosed with AAA by repeated ultrasonography or CT angiography. Specimens from patients with collagen vascular disease or severe chronic kidney disease
(estimated glomerular filtration rate < 30 mL [min·1.73 m2]−1) were excluded. Samples were snap-frozen in liquid nitrogen directly after surgery. Corresponding adjacent human aortic
non-AAA tissue specimens used for controls were collected from the bodies of six deceased donors with no detectable vascular disease (Volunteer Corpse Donation Reception Station). Samples in
the freezers were snap-frozen in liquid nitrogen after dissection. Informed consent was obtained from all study participants. The clinical information associated with the samples is shown
in Supplementary Table 1. RNA SEQUENCING ANALYSIS The filtered and trimmed reads of each sample were aligned to the rat reference genome (Rnor_6.0) using HISAT2 software with default
parameters24,25. The read counts of genes were calculated using the R packages Genomic Features and Genomic Alignments26. The FPKM (fragments per kilobase million) of genes was determined
using the DESeq2 package27. Differentially expressed genes were identified using a paired _t_ test with _p_ value < 0.1. Pathway enrichment analysis was performed using the MetaCore
online tool (https://portal.genego.com/). An FDR cutoff of < 0.05 was set to select for significant pathways. The relationship between pathways and genes was visualized using the R
package GOplot (v1.0.2)28. HISTOLOGICAL, IMMUNOHISTOCHEMICAL, AND IMMUNOFLUORESCENT ANALYSIS Human aortic tissue samples were vertically embedded in optimal cutting temperature compound
(O.C.T, Thermo Fisher, US) and stored at −80 °C. Mouse AAA aortas were dissected at the maximal suprarenal outer aortic diameter and embedded vertically in O.C.T compound for histological
evaluation. Corresponding adjacent normal aortic vascular tissues were collected from non-Ang II-infused control mice. At least ten-fifteen serial frozen sections, six μm thick, were
sectioned from the AAA portion, covering the maximal dilated aorta and control tissues using a freezing microtome (Leica CM1860, Germany). Immunohistochemical staining was performed as
previously described29,30. Tissue sections were incubated with rabbit anti-PDE4D (1:100, Abcam, ab14613, Britain) antibody overnight at 4 °C, followed by incubation with anti-rabbit
IgG-peroxidase conjugate (Beijing Zsbio Biotechnology, China) for thirty mins at room temperature and developed by adding the substrate 3-amino-9-ethylcarbazole (AEC). Images were taken with
a microscope (Nikon, digital sight DS—Fi2, Germany). For quantitative analysis, the positive staining intensity was semiquantified with Image-Pro Plus Software (Media Cybernetics, Bethesda,
MD). For each animal, a total of five fields were randomly selected, quantified, and averaged. For negative controls, sections were incubated with goat anti-rabbit secondary antibody using
the primary antibody omitted (Supplementary Fig. 1a–c). In addition, mouse aortic sections were stained for elastin staining (Modified Verhoeff Van Gieson Elastic Stain Kit, Sigma, Cat#:
HT25A, US). Elastin staining is indicated by the darkest color, and elastin degradation was rated according to the key provided by the manufacturer31. Mouse heart tissues were stained with
hematoxylin and eosin (H&E) (Solarbio, Cat#: G1120, China). Immunofluorescence analysis of PDE4D (1:50, Abcam, ab14613, Britain), α-SMA (1:250, Invitrogen, 14-9760-82, Britain), and
terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labeling (TUNEL; Beyotime Biotechnology, China, Cat#: C1088) was performed. The sections were coincubated overnight at 4 °C
with primary antibodies against PDE4D and α-SMA. The antibodies were detected using fluorescein-labeled secondary antibodies. The nuclei were stained with 4’,6-diamidino-2-phenylindole
(DAPI) (Abcam, Cat#: ab228549, Britain). Images were photographed using a fluorescence microscope (Nikon, digital sight DS—Fi2, Japan). For negative controls, adjacent sections were
incubated with fluorescein-labeled secondary antibodies using the primary antibody omitted (Supplementary Fig. 1e, f). Apoptotic cells were examined by TUNEL staining and quantified as the
ratio of apoptotic cells to total cells in 5 fields per randomly selected mouse using Image-Pro Plus Software. For negative controls, adjacent sections were incubated with dUTP and without
TdT (Supplementary Fig. 15). MEASUREMENT OF BLOOD PRESSURE BY TAIL-CUFF PLETHYSMOGRAPHY Systolic and diastolic blood pressure were measured via the noninvasive tail-cuff method (CODA, Kent
Scientific, US) before osmotic pump implantation and later sacrifice as described32. Briefly, mice were warmed to ~37 °C in restraint tubes on the heating pad with their tails restrained in
the occlusion tail cuff. Mice were trained for ~3–15 min until a stable blood pressure was recorded. The average of at least five successful measurements was considered the blood pressure of
the mice on that recording day. CELL CULTURE AND SMALL INTERFERING RNA (SIRNA) Rat aortic smooth muscle cells (SMCs) were purchased from ScienCell (San Diego, California, US, Cat# R6110)
and cultured in smooth muscle cell medium (SMCM, ScienCell, US, Cat#: 1101) containing 5% fetal bovine serum (FBS), 100 U mL−1 penicillin, and 100 g mL−1 streptomycin. SMCs have a stable
phenotype, are not prone to phenotypic transition, and can be passaged for a long time. SMCs between passages five and twelve were used in all experiments. Cells were maintained at 37 °C in
a humidified incubator with an atmosphere of 5% CO2. For in vitro experiments, cells were cultured in 6-well plates (6 × 105 cells/well; NEST, Cat#: 703001) and stimulated with 100 nM Ang II
(Sigma, Cat#: A9525-50MG) or 300 μM H2O2 (Sigma-Aldrich, H1009, US) for 24 h. For inhibition of PDE4D, 500 nM rolipram (PDE4 inhibitor, Sigma-Aldrich, R6520, US) was added 0.5 h before Ang
II or H2O2 treatment. For knockdown of PDE4D, PDE4D siRNA (RiboBio, siB180730051733) and control scrambled siRNA (RiboBio, siN0000001-1-5) were purchased from RiboBio. siRNA transfections
were performed according to the manufacturer’s protocol. Briefly, SMCs at 80% confluence were transfected with 200 nM PDE4D siRNA duplexes in 5 μL of Oligofectamine for 48 h (Invitrogen,
Carlsbad, CA, US, Cat#: 12252011). siRNA efficiency was determined by RT-PCR and western blot. REAL-TIME POLYMERASE CHAIN REACTION (RT-PCR) Total RNA was extracted from aortic tissues or
SMCs using TRIzol reagent (Invitrogen, Carlsbad, CA, Cat#: 15596018) according to the manufacturer’s protocol. Equal amounts of RNA (1000 ng) were reverse transcribed into cDNA (TianGen,
KR116-02), and quantitative real-time PCR was performed in triplicate using a single-color RT-PCR detection system (Bio-Rad, Hercules, CA, US). _Pde4a_, _Pde4b_, _Pde4c_, _Pde4d_, _Il-1α_,
_Il-1β_, _Il-6_, _Mrc-1_, _Arg-1_, _Cd163_, _Mmp2_, _Mmp9_, _Sm22α_, _Cnn_, _Opn_, _Col1a1_, _Col3a1_, _Fn_, _Tnf-α_, and _Ccl2_ levels were normalized to the level of the housekeeping gene
glyceraldehyde-3-phosphate dehydrogenase (_Gapdh_). The mRNA levels were recorded as the expression fold change compared with the control group or a single control mouse and were calculated
using the 2-ΔΔCt method. The RT-PCR primers are shown in Supplementary Table 2. WESTERN BLOT ANALYSIS Protein was extracted from aortic tissues or SMCs in RIPA lysis buffer with protease
inhibitor cocktail (Roche, NJ, US) for thirty mins and centrifuged at 12,000 rpm for 10 min. The protein concentration was determined using a BCA Protein Assay Kit (Pierce, Holmdel, NJ, US,
Cat#: 23225). Equal amounts of protein (25–30 μg) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE; 8%, 10%, or 12% gel) and transferred onto a
polyvinylidene difluoride (PVDF) membrane. The PVDF membranes were blocked with 5% (m/v) fat-free milk powder/Tris buffered saline-Tween (TBST) for 1 h and incubated overnight at 4 °C with
primary antibodies against PDE4D (1:1000, Abcam, Britain, Cat#: ab171750), cleaved caspase-3 (1:1000, Cell Signaling Technology, US, Cat#: 9662S), caspase-3 (1:1000, Cell Signaling
Technology, US, Cat#: 9662S), phospho-Bad (pBad, 1:1000, Thermo Fisher, US, Cat#: PAS-105022) and Bad (1:1000, Cell Signaling Technology, US, Cat#: 9292S). Immunoblotting of the housekeeping
protein GAPDH (1:8000, Proteintech, US, Cat#: 60004-1-Ig) was performed to ensure equal protein loading. After three washes with TBST, the membranes were incubated with horseradish
peroxidase (HRP)-labeled rabbit (1:5000, Invitrogen, Britain, Cat#: A24531) or mouse (1:5000, Invitrogen, Britain, Prod#: 04-6020) secondary antibodies for 1 h at room temperature.
Immunoreactive bands were visualized with SuperSignal™ West Pico PLUS Chemiluminescent Substrate (Pierce, Cat#: 34577). Protein expression was measured by analyzing the intensities of the
protein bands with Image-Pro Plus 6.0 software. FLOW CYTOMETRY SMC apoptosis was investigated via flow cytometry. Briefly, the digested SMC suspension was passed through a 40 μm Cell
Strainer (Biologix Group, Ltd., Jiangsu, China) and stained according to the protocol of the Annexin V-FITC/PI Apoptosis Detection Kit (Dojindo, Japan, AD10). All groups of cells in the
mixture were used as the gating control samples stained with only Annexin V-FITC or PI or without any dye. The data were acquired and analyzed by BD Accuri C6 flow cytometry (BD Biosciences,
US). The gating strategy was to divide positive and negative events in negative, Annexin V-FITC, and PI gating control samples (Supplementary Fig. 12a–d). MEASUREMENT OF CAMP A cAMP Direct
Fluorometric Immunoassays Kit (Abcam, ab138880) was used to measure the cAMP level according to the manufacturer’s instructions. For in vitro cAMP quantification, cells plated in sterile
96-well plates (1 × 105 cells/well) were lysed with 100 μL/well of Cell Lysis Buffer. For in vivo cAMP quantification, aortic tissues were lysed in 20 μL/mg of Cell Lysis Buffer. Briefly,
all samples and standards (75 μL each) mixed with 25 μL of 1x HRP-cAMP were incubated in plates at room temperature for 2 h on a plate shaker. After four washes, the plate was incubated with
100 μL/well of AbRed Working Solution for 45 min. The fluorescence was measured at Ex/Em = 540/590 nm using a Biotek Synergy™ H1 microplate reader. The standard curve was determined by
regression analysis using a logistic curve-fit and prepared for every experiment independently. CELL PROLIFERATION ASSAY SMC proliferation was assayed using BeyoClick™ EdU-488 Cell
Proliferation Assay Kit (Beyotime, Beijing, China) following the manufacturer’s instructions. Images were taken by a fluorescence microscope (Nikon Eclipse Ti2, Japan), and the signals were
counted in three random visual fields for each sample. STATISTICAL ANALYSIS Data were statistically analyzed using GraphPad Prism 9 (GraphPad Software, LLC, San Diego, CA). Data are
expressed as the mean ± SEM. The Shapiro-Wilk test was used to test for normal distribution. The Brown-Forsythe test or F test was used to test for equality of variances. Normally
distributed datasets with equal variance were analyzed with the parametric unpaired Student’s _t_ test for 2 independent groups and the parametric one-way ANOVA or two-way ANOVA followed by
the Holm-Sidak’s post hoc test for ≥3 groups. Normally distributed datasets without equal variance were analyzed with a parametric Welch’s t test for 2 independent groups and the parametric
Welch ANOVA with Dunnett’s T3 post hoc test for ≥3 groups. Where a normal distribution could not be confirmed, the nonparametric Mann-Whitney test was used for 2 independent groups. All
tests were two sided. A _p_ value < 0.05 was considered statistically significant. Detailed statistical analyses and plotting methods are listed in Supplementary Table 3. RESULTS PDE4D
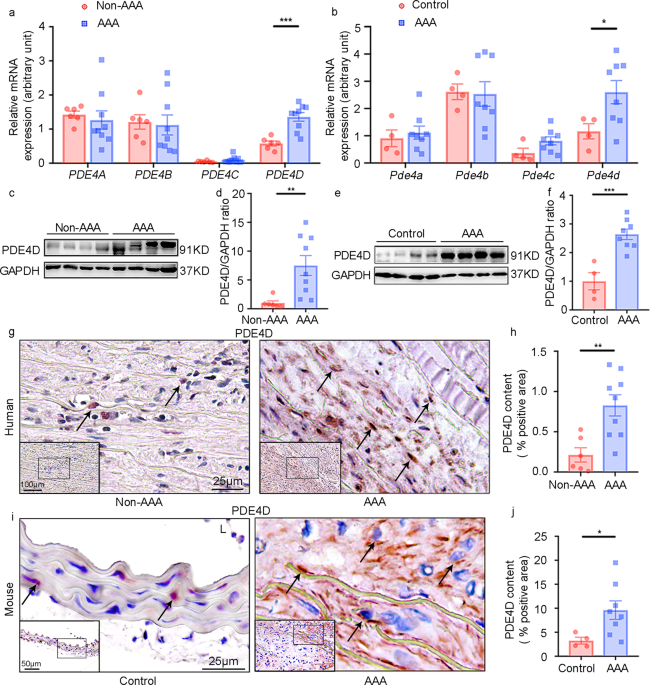
EXPRESSION IS UPREGULATED IN HUMAN AND MOUSE AAA TISSUES To explore PDE4 expression during AAA formation, we assessed the mRNA levels of individual PDE4 family isoforms (PDE4A-D) in both
human and mouse AAA tissues. Human AAA tissues were collected from AAA patients via surgery, and control non-AAA tissues were corresponding tissues collected from the bodies of deceased
donors with no detectable vascular disease (Supplementary Table 1). Mouse AAA was induced by a chronic infusion of Ang II (1000 ng kg−1 min−1) for twenty-eight days via osmotic minipumps in
_Apoe__−/_− male mice fed a high-fat diet (HFD): a well-established murine AAA model20. Control mice received vehicle infusion. Mouse AAA tissues and corresponding normal aortic tissues were
dissected from the suprarenal aortic regions of the mice with AAA and the control mice, respectively. Among the four PDE4 isozymes, only _PDE4D_ expression levels were significantly
increased in human AAA compared to non-AAA tissues (Fig. 1a). Similar observations were obtained for mouse AAA tissues (Fig. 1b). Henceforth, we focused on PDE4D in this study. Consistently,
we found that the PDE4D protein levels were significantly higher in human (Fig. 1c, d) and mouse AAA tissues (Fig. 1e, f). Immunohistochemical staining further confirmed the PDE4D protein
increase in human (Fig. 1g, h) and mouse AAA lesions (Fig. 1i, j). PDE4D antibody specificity was supported by negative controls performed in mouse and human tissues (Supplementary Fig.
1a–g). These results demonstrated an upregulation of PDE4D expression in AAA. PDE4D expression was largely observed in the medial areas where SMCs resided (Fig. 1g–j), suggesting the
expression PDE4D in SMCs. To further confirm the expression of PDE4D in SMCs, we performed double immunofluorescence staining of PDE4D and α-smooth muscle actin (α-SMA), a marker for
differentiated contractile and dedifferentiated synthetic SMCs (or myofibroblasts). We observed prominent PDE4D staining in α-SMA-positive cells in human (Fig. 2a and Supplementary Fig. 2a)
and mouse AAA lesions (Fig. 2b and Supplementary Fig. 2b). Consistently, in cultured rat aorta SMCs, Ang II also increased PDE4D mRNA and protein levels (Fig. 2c–e). Therefore, in this
study, we focused on the role of SMC PDE4D in SMC pathogenesis and AAA development. SMC-SPECIFIC _PDE4D_ DEFICIENCY DECREASES ANG II-INDUCED AAA FORMATION To explore the role of SMC PDE4D in
AAA development, we used SMC-specific _Pde4d_ knockout mice on an _Apoe__−/−_ background (_Apoe__−/−__Pde4d__SMC−/−_). We generated _Pde4d_-floxed mice (_Pde4d__flox/flox_) carrying floxed
exon 10 of the _Pde4d_ gene through homologous recombination via CRISPR/Cas9 technology. We then crossed _Pde4d__flox/flox_ mice with _Apoe__−/−_ mice and subsequently crossed
_Apoe__−/−__Pde4d__flox/flox_ mice with _Tagln_-Cre mice (Supplementary Fig. 3a). The resultant _Apoe__−/−__Pde4d__flox/flox_ and _Apoe__−/−__Pde4d__SMC−/−_ littermates served as
experimental mouse groups. RT-PCR (Supplementary Fig. 3b), immunoblotting (Supplementary Fig. 3c), and immunostaining (Supplementary Fig. 1d–g) confirmed PDE4D depletion in mouse aortic
SMCs. We also observed no significant alterations in other PDE4 isozymes between _Apoe__−/−__Pde4d__flox/flox_ and _Apoe__−/−__Pde4d__SMC−/_− aortic tissues (Supplementary Fig. 3b). AAA was
induced by Ang II infusion (1000 ng kg−1 min−1) for twenty-eight days along with an HFD, while the controls were infused with saline (Fig. 3a). The aortas were excised, and the maximal
external diameter of the abdominal aorta was measured by two different investigators via stereoscopy at the time of necropsy. AAA was defined as a 50% dilation in the diameter of the
external abdominal aorta compared with the normal mouse abdominal aorta. Under Ang II infusion, AAA development was significantly attenuated in the _Apoe__−/−__Pde4d__SMC−/−_ mice compared
to the _Apoe__−/−__Pde4d__flox/flox_ mice, as illustrated by the aortic morphology (Fig. 3b). The average external diameter of the abdominal aortas was significantly smaller in the
_Apoe__−/−__Pde4d__SMC−/−_ Ang II group than in the _Apoe__−/−__Pde4d__flox/flox_ Ang II group (1.936 ± 0.203 mm vs. 3.073 ± 0.327 mm; Fig. 3c). Supplementary Fig. 4a includes images of all
AAA samples shown in Fig. 3b, c. In addition, the fragmentation and degradation of the aortic elastic lamina—a characteristic feature of AAA—was evaluated. Verhoeff Van Gieson staining for
elastin and semiquantitative analysis revealed more profound elastin degradation in the _Apoe__−/−__Pde4d__flox/flox_ Ang II mice than in the _Apoe__−/−__Pde4d__SMC−/−_ Ang II mice (Fig. 3d,
e). _Mmp2_ and _Mmp9_ mRNA levels were both upregulated in the _Apoe__−/−__Pde4d__flox/flox_ Ang II group, but only _Mmp2_ expression was rescued in _Apoe__−/−__Pde4d__SMC−/−_ mouse aortas
(Supplementary Fig. 5a, b). In addition, aortic cAMP levels were decreased in the Ang II-induced AAA mice compared with the saline-treated mice, and SMC-specific knockout of _Pde4d_ reversed
the cAMP levels in AAA tissues (Supplementary Fig. 6a). These results suggest that PDE4D deficiency in SMCs enhances the stability of the aortic wall and decreases Ang II-induced AAA
formation. Additionally, Ang II infusion increased systolic and diastolic blood pressure (BP) in the _Apoe__−/−__Pde4d__flox/flox_ mice, both of which were significantly lower in the
_Apoe__−/−__Pde4d__SMC−/−_ Ang II mice (Supplementary Fig. 4b, c). Based on a previous report by Daugherty et al., AAA occurred in the _Apoe__−/−_ mice infused with Ang II (1000 ng kg−1
min−1) for twenty-eight days, accompanied by increased BP. Hydralazine administration (an antihypertensive drug) lowered systolic BP in the Ang II-infused _Apoe__−/−_ mice, while hydralazine
did not prevent AAA formation33. Therefore, it is generally believed that Ang II infusion-induced AAA formation is independent of BP elevation. Thus, the protective effect of
_Pde4d__SMC−/−_ against AAA development is unlikely to result from a BP reduction. THE PDE4 INHIBITOR ROLIPRAM ATTENUATES ANG II-INDUCED AAA FORMATION Next, we sought to determine the
pharmacological effect of a PDE4 inhibitor on AAA. We selected the pan-PDE4 inhibitor rolipram, which was used clinically for neuroinflammation in the early 1990s19. Male C57BL/6J
_Apoe__−/−_ mice at the age of eight weeks were infused with Ang II or saline and fed an HFD for four weeks. Rolipram (3 mg kg−1 d−1) or vehicle (7% ethyl alcohol) was given daily via gavage
for four weeks (Fig. 4a). Rolipram (+) treatment significantly reduced the AAA size compared with rolipram (−) vehicle treatment (Fig. 4b). The average external diameter of the abdominal
aorta was smaller in the rolipram (+) Ang II group (1.425 ± 0.192 mm) than in the rolipram (−) Ang II group (2.279 ± 0.279 mm; Fig. 4c). Supplementary Fig. 7a includes images of all AAA
samples shown in Fig. 4b, c. Moreover, elastin fragmentation in the aortic wall was significantly reduced in the rolipram (+) Ang II mice compared with the rolipram (−) Ang II mice (Fig. 4d,
e). Consistently, rolipram also attenuated _Mmp2_, but not _Mmp9_, in aortic tissues induced by Ang II (Supplementary Fig. 5c, d). As expected, cAMP levels in the AAA mice were lower than
those in the saline-treated mice, and rolipram reversed cAMP levels in AAA tissues (Supplementary Fig. 6b). These results demonstrate a significant pharmacological impact of rolipram in
preventing AAA development in mice. Similar to the results in _Pde4d__SMC−/−_ mice, rolipram also reduced systolic and diastolic BP in AAA mice (Supplementary Fig. 7b, c). PDE4D PROMOTES SMC
APOPTOSIS IN VITRO AND IN VIVO To determine the underlying mechanism by which PDE4D participates in AAA formation, we performed bulk RNA-seq to identify PDE4D-regulated genes in rat aortic
SMCs treated with PDE4D-specific or scramble control siRNA. RNA-seq revealed 1,848 genes upregulated in SMCs stimulated with Ang II (100 nM, 24 h; Supplementary Fig. 8a, c, d) and 1,551
genes downregulated in SMCs by PDE4D siRNA (Supplementary Fig. 8b, c) (for the full list, refer to Supplementary Dataset 1). Among these genes, 235 were upregulated by Ang II, and the
upregulation was reversed by si-PDE4D (Supplementary Fig. 8c). We next performed MetaCore pathway enrichment analysis of these 235 genes (for the full list, refer to Supplementary Dataset
1). We focused on the top 15 pathways shared by the two groups (control vs. Ang II and si-scramble vs. si-PDE4D) based on their minimum false discovery rate (FDR). Among these pathways,
apoptosis-related pathways were predominant, which is consistent with the important role of SMC apoptosis in AAA pathogenesis (Fig. 5a). We next determined the role of PDE4D in regulating
SMC apoptosis using PDE4D siRNA to knockdown PDE4D expression or rolipram to inhibit PDE4 activity in SMCs (Supplementary Fig. 9a–c). We found that Ang II increased caspase-3 cleavage, a key
event in apoptosis34. PDE4D siRNA substantially attenuated Ang II-induced caspase-3 cleavage (Fig. 5b, c). Similarly, PDE4D siRNA also reduced caspase-3 cleavage induced by H2O2 as a
reactive oxidative stress (ROS) mediator known to be important in AAA (Supplementary Fig. 10a, b). We also assessed SMC apoptosis via Annexin V/propidium iodide (PI) staining and flow
cytometry (Fig. 5d and Supplementary Fig. 10c). We found that SMC apoptosis induced by Ang II or H2O2 was significantly suppressed by PDE4D siRNA (Fig. 5e and Supplementary Fig. 10d). As
with PDE4D siRNA, rolipram also reduced cleaved caspase-3 levels in SMCs treated with Ang II (Fig. 5f, g) or H2O2 (Supplementary Fig. 11a, b) and decreased SMC apoptosis induced by Ang II
(Fig. 5h, i) or H2O2 (Supplementary Fig. 11c, d). The gating strategy of flow cytometry was to divide positive and negative events in negative, Annexin V-FITC, and PI gating control samples
(Supplementary Fig. 12a–d). These results support a proapoptotic function of PDE4D in SMCs in vitro. Consistent with the in vitro findings, cleaved caspase-3 in Ang II-induced AAA tissues
was significantly reduced in the _Apoe__−/−__Pde4d__SMC−/−_ mice (Fig. 6a, b). Apoptotic cell numbers detected by TUNEL staining were also decreased in the _Apoe__−/−__Pde4d__SMC−/−_ Ang II
mouse tissues compared to the _Apoe__−/−__Pde4d__flox/flox_ Ang II group (Fig. 6c and Supplementary Fig. 13). Accordingly, rolipram decreased cleaved caspase-3 levels and TUNEL-positive
cells in AAA tissues (Fig. 6d–f and Supplementary Fig. 14). Supplementary Fig. 15 shows the negative staining of TUNEL. These results suggest that PDE4D promotes SMC apoptosis in AAA. PDE4D
ANTAGONIZES PKA-MEDIATED PHOSPHORYLATION OF BAD AND INDUCES CLEAVED CASPASE-3 AND APOPTOSIS IN SMCS PDE4 family members are cAMP-hydrolyzing enzymes. We examined the levels of cAMP in vitro
using a cAMP kit. As expected, Ang II reduced SMC cAMP and PDE4D siRNA, and rolipram upregulated cAMP levels in vitro (Supplementary Fig. 6c, d). cAMP activates cAMP-dependent protein kinase
A (PKA) or the exchange protein directly activated by cAMP (Epac)35. To identify whether PDE4D regulates apoptosis by relying on the PKA and/or Epac pathways, we used PKA- or Epac-selective
inhibitors, including PKI (PKA inhibitor) and ESI-09 (Epac inhibitor). We found that PDE4D siRNA attenuated the apoptosis marker cleaved caspase-3 in SMCs induced by Ang II; however, the
inhibitory effect of PDE4D siRNA on cleaved caspase-3 was blocked by PKI (Fig. 7a, b) but not ESI-09 (Fig. 7c, d). These data suggest that PDE4D regulates SMC apoptosis in a cAMP-PKA
signaling-dependent manner. Based on our transcriptome sequencing results, the BCL2 antagonist of cell death (Bad) was highlighted in several apoptosis-related pathways (Supplementary Fig.
16). Bad is an important protein involved in apoptosis. Bad can be phosphorylated by PKA at Ser15536. This Bad phosphorylation (pBad) causes Bad inactivation and thus inhibits apoptosis37.
We therefore investigated the role of PDE4D in Bad phosphorylation. We found that Ang II reduced Bad phosphorylation, which was reversed by PDE4D siRNA (Fig. 7e–h). Interestingly, the effect
of PDE4D siRNA on increasing Bad phosphorylation was largely blocked in the presence of PKI (Fig. 7e–h), suggesting that PDE4D negatively regulates the PKA phosphorylation of Bad.
DISCUSSION In this study, we reported the role and regulatory mechanism of PDE4D in SMC apoptosis and AAA formation (Fig. 7i). We showed that PDE4D played an essential role in SMC apoptosis,
at least partially by attenuating PKA-mediated phosphorylation and inactivation of the proapoptotic molecule Bad. We demonstrated PDE4D upregulation in SMCs from human and mouse AAA lesions
through multiple approaches, including RT-PCR, western blotting, and immunostaining. To perform in vivo evaluation of the role of SMC PDE4D in AAA formation, we generated SMC-specific
_Pde4d_ knockout mice on an _Apoe__−/−_ genetic background and tested them in a well-established AAA mouse model. We demonstrated a causative role of SMC PDE4D in aortic wall degeneration
and AAA development. Moreover, we identified a protective effect of the PDE4 inhibitor rolipram against vascular degeneration and AAA development. Rolipram is a pan-PDE4 inhibitor that
targets all four PDE4 members38. While possessing a history of clinical use, it has been shown to induce nausea and emesis in humans39, likely due to its blood-brain barrier penetration and
inhibition of PDE4D in the brain chemoreceptor trigger zone of vomiting19,40,41. Thus, developing potential PDE4 inhibitors that are peripherally restricted will be beneficial for treating
peripheral diseases. In addition to PDE4 in SMCs, a number of different cell types in AAA tissues may also express PDE4, including endothelial cells, adventitial fibroblasts, and
macrophages. However, these different cell types may primarily express distinct PDE4 isozyme(s), among 4A, 4B, 4C, and 4D. It has been shown previously that PDE4B is highly expressed in
inflammatory cells and contributes significantly to inflammation42. The role of PDE4B in inflammation leads to the development of the PDE4 inhibitor roflumilast for the treatment of COPD.
Previous studies have also shown that PDE4D is the dominant PDE4 isozyme expressed in rodent or human arterial VSMCs43,44. Because we found that PDE4D is most significantly upregulated in
AAA tissues, particularly in SMCs, we thus focused on the role of PDE4D in SMC and AAA development in the current study using SMC-specific PDE4D deficiency animal models. Although our data
demonstrated a critical role of SMC-derived PDE4D in AAA, we cannot exclude the possible contributions of PDE4D in other cell types to AAA. We also cannot exclude the possible contribution
of PDE4B in immune cells to AAA, particularly in the rolipram treatment model, given an important role of inflammation in Ang II-induced AAA. We indeed found that the pan-PDE4 inhibitor
rolipram elicited more profound effects on suppressing AAA than SMC-specific PDE4D deficiency (aortic diameters, 1.936 ± 0.203 mm vs. 1.425 ± 0.192 mm). These results suggest that inhibiting
other PDE4 isozymes and/or PDE4D in other cell types may contribute to the protective effects of rolipram. It will be of great future interest to elucidate the contributions of distinct
PDE4 isozymes in differing cell types to AAA development, given that AAA is a multifactorial disease involving numerous cell types. In our current study, we focused on the contribution of
SMCs in AAA using SMC (Tagln-Cre)-specific _Pde4d_ KO mice. Although Tagln (also called smooth muscle 22α) is expressed on SMCs in cardiac, smooth, and skeletal muscle, Tagln mediates Cre
expression at a high level in vascular SMCs. To exclude the effect of Tagln in cardiac myocytes, we indicated that the structure of heart tissues did not change between the
_Apoe__−/−__Pde4d__flox/flox_ and _Apoe__−/−__Pde4d__SMC−/−_ mice (Supplementary Fig. 3d). Furthermore, comparing the _Pde4d__SMC−/−_ mice and the mice treated with the pan-PDE4 inhibitor
rolipram, we found that rolipram elicited more profound effects on suppressing AAA than that in the _Pde4d__SMC−/−_ mice (aortic diameters, 1.425 ± 0.192 mm vs. 1.936 ± 0.203 mm). These
results suggest that inhibiting other PDE4 isozymes and/or PDE4D in other cell types may contribute to the protective effects of rolipram. It will be of great future interest to elucidate
the contributions of distinct PDE4 isozymes in differing cell types to AAA development, given that AAA is a multifactorial disease involving numerous cell types. The roles of cAMP signaling
in vascular SMC apoptosis are controversial. Some studies have shown the antiapoptotic effect of cAMP in vascular SMCs. For example, it has been shown that rat aortic SMC apoptosis is
attenuated by the activation of cAMP/PKA signaling through the beta-adrenergic receptor (β-AR) agonist adenosine or the PDE4 inhibitor Ro-20172445. Some other studies reported a proapoptotic
effect of cAMP in vascular SMCs. For example, human vascular SMC apoptosis was promoted by the nonselective elevation of intracellular cAMP levels through 8-bromo-cAMP and forskolin or
increasing PDE3-regulated cAMP through cilostazol46. In the current study, we provided solid evidence and demonstrated that rat aortic SMC apoptosis is attenuated by stimulating cAMP/PKA
signaling through the PDE4 inhibitor rolipram or specific depletion of PDE4D isozymes. These lines of experimental evidence from ours and others suggest that SMC viability may be
differentially, even oppositely, regulated by different cAMP/PKA signalosomes that are coupled to distinct G-protein coupled receptors (GPCRs) and/or PDEs. Although we demonstrated the roles
of PDE4D in SMC apoptosis in this study, there are eleven different PDE4D variants reported to date, and the expression of different PDE4D variants in SMCs has been reported previously. The
contractile/quiescent vascular SMCs primarily express PDE4D3, while the synthetic/activated SMCs express PDE4D1 and PDE4D247. PDE4D8 is enriched in the pseudopodia of SMCs and regulates SMC
migration48. These findings suggest that different PDE4D variants may be involved in distinct SMC functions. The specific PDE4D variant(s) in SMC apoptosis remain to be elucidated in the
future. We also found that Ang II-induced elevation of systolic and diastolic BP, measured by tail cuff, was significantly reduced in the SMC-specific _Pde4d_ knockout mice and in the mice
treated with rolipram. This raised the question of whether BP reduction contributes to the effect of PDE4D deficiency/inhibition in AAA. Daugherty et al. found that both NE and Ang II raised
arterial pressure. However, Ang II but not NE induced AAA. Moreover, blocking this BP elevation using the antihypertensive drug hydroxyzine did not prevent Ang II-induced AAA occurrence and
development33. Although previous experimental evidence indicates that Ang II induces AAA formation in mice independent of its effect on BP elevation33, BP may affect the outcomes of AAA
progression and rupture49. Thus, we do not exclude the potential impact of BP reduction by PDE4D deficiency or inhibition in AAA progression or rupture. In addition to apoptosis, we also
determined whether PDE4D was associated with other phenotypes of SMCs, including proliferation, dedifferentiation, deposition of extracellular matrix (ECM), and inflammation, in AAA
development. We found that Ang II significantly increased the proliferation of SMCs; however, PDE4D siRNA could not suppress the proliferation induced by Ang II (Supplementary Fig. 17a, b).
SMCs can dedifferentiate into macrophage-like SMCs or osteoblast-like SMCs by changing from the contractile type to the synthetic type50. Our results showed that Ang II-treated SMCs
expressed higher levels of the SMC contractile marker proteins smooth muscle 22α (SM22α) and SM-calponin (CNN) as well as lower levels of the synthetic marker osteopontin (OPN), but PDE4D
siRNA did not reverse the dedifferentiation phenotype (Supplementary Fig. 18a–c). In addition, we found that _Mmp2_ and _Mmp9_ expression was upregulated by Ang II, but PDE4D deficiency only
rescued _Mmp2_ expression in vitro (Supplementary Fig. 19a, b). Moreover, expression of the ECM genes Collagen 1A1 (_Col1a1_), Collagen 3A1 (_Col3a1_) and fibronectin 1 (_Fn_)51 was
elevated by Ang II stimulation, but only _Col1a1_ expression was reversed by PDE4D siRNA (Supplementary Fig. 20a–c). Regarding inflammation, Sadhan et al. reported that Ang II promoted CCL2,
IL-6 and TNF-α expression in SMCs52. Therefore, we found that Ang II stimulation increased all three cytokines; however, suppression of PDE4D did not rescue the upregulation of _Ccl2_,
_Il-6,_ and _Tnf-α_ expression (Supplementary Fig. 21a–c). These findings indicate that PDE4D in SMCs may affect ECM deposition by regulating Col1a1 and MMP2 expression in AAA progression.
In conclusion, our study shows that PDE4D in SMCs exacerbates Ang II-induced AAA. Furthermore, the inhibition of PDE4 alleviates vascular pathogenesis and AAA formation. We verified the
mechanism by which PDE4D influences SMC apoptosis via in vitro and in vivo experimental models and identified those results using a variety of molecular biological methods. This study
illustrates that PDE4D in SMCs plays a pivotal role in AAA and that PDE4 inhibitors may be potential targets for AAA treatment. REFERENCES * Force, U. S. P. S. T. et al. Screening for
abdominal aortic aneurysm: US preventive services task force recommendation statement. _JAMA_ 322, 2211–2218 (2019). Article Google Scholar * Li, J. et al. Decoding the genomics of
abdominal aortic aneurysm. _Cell_ 174, 1361–1372 e1310 (2018). Article CAS PubMed Google Scholar * Guirguis-Blake, J. M., Beil, T. L., Senger, C. A. & Coppola, E. L. Primary care
screening for abdominal aortic aneurysm: updated evidence report and systematic review for the US preventive services task force. _JAMA_ 322, 2219–2238 (2019). Article PubMed Google
Scholar * Kumar, S., Boon, R. A., Maegdefessel, L., Dimmeler, S. & Jo, H. Role of noncoding RNAs in the pathogenesis of abdominal aortic aneurysm. _Circ. Res._ 124, 619–630 (2019).
Article CAS PubMed PubMed Central Google Scholar * Rowe, V. L. et al. Vascular smooth muscle cell apoptosis in aneurysmal, occlusive, and normal human aortas. _J. Vasc. Surg._ 31,
567–576 (2000). Article CAS PubMed Google Scholar * Crowther, M., Goodall, S., Jones, J. L., Bell, P. R. & Thompson, M. M. Increased matrix metalloproteinase 2 expression in vascular
smooth muscle cells cultured from abdominal aortic aneurysms. _J. Vasc. Surg._ 32, 575–583 (2000). Article CAS PubMed Google Scholar * Berisha, F. & Nikolaev, V. O. Cyclic
nucleotide imaging and cardiovascular disease. _Pharmacol. Ther._ 175, 107–115 (2017). Article CAS PubMed Google Scholar * Rainer, P. P. & Kass, D. A. Old dog, new tricks: novel
cardiac targets and stress regulation by protein kinase G. _Cardiovasc. Res._ 111, 154–162 (2016). Article CAS PubMed PubMed Central Google Scholar * Guellich, A., Mehel, H. &
Fischmeister, R. Cyclic AMP synthesis and hydrolysis in the normal and failing heart. _Pflug. Arch._ 466, 1163–1175 (2014). Article CAS Google Scholar * Blokland, A. et al.
Phosphodiesterase Type 4 Inhibition in CNS Diseases. _Trends Pharmacol. Sci._ 40, 971–985 (2019). Article CAS PubMed Google Scholar * Baillie, G. S., Tejeda, G. S. & Kelly, M. P.
Therapeutic targeting of 3’,5’-cyclic nucleotide phosphodiesterases: inhibition and beyond. _Nat. Rev. Drug. Discov._ 18, 770–796 (2019). Article CAS PubMed PubMed Central Google Scholar
* Galie, N. et al. Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension. _N. Engl. J. Med._ 373, 834–844 (2015). Article CAS PubMed Google Scholar * Lebwohl, M.
G. et al. Trial of roflumilast cream for chronic plaque psoriasis. _N. Engl. J. Med._ 383, 229–239 (2020). Article CAS PubMed Google Scholar * Hatemi, G. et al. Trial of apremilast for
oral ulcers in Behcet’s syndrome. _N. Engl. J. Med._ 381, 1918–1928 (2019). Article CAS PubMed Google Scholar * Qin, X. et al. Smooth muscle-specific Gsalpha deletion exaggerates
angiotensin II-induced abdominal aortic aneurysm formation in mice in vivo. _J. Mol. Cell Cardiol._ 132, 49–59 (2019). Article CAS PubMed PubMed Central Google Scholar * Liu, Y. et al.
Inhibition of adenosine uptake and augmentation of ischemia-induced increase of interstitial adenosine by cilostazol, an agent to treat intermittent claudication. _J. Cardiovasc. Pharmacol._
36, 351–360 (2000). Article CAS PubMed Google Scholar * Umebayashi, R. et al. Cilostazol attenuates angiotensin ii-induced abdominal aortic aneurysms but not atherosclerosis in
apolipoprotein E-deficient mice. _Arterioscler. Thromb. Vasc. Biol._ 38, 903–912 (2018). Article CAS PubMed Google Scholar * Zhang, C. et al. Cyclic nucleotide phosphodiesterase 1C
contributes to abdominal aortic aneurysm. _Proc. Natl Acad. Sci. USA_ 118, 31 (2021). Google Scholar * Li, H., Zuo, J. & Tang, W. Phosphodiesterase-4 inhibitors for the treatment of
inflammatory diseases. _Front. Pharmacol._ 9, 1048 (2018). Article CAS PubMed PubMed Central Google Scholar * Daugherty, A., Manning, M. W. & Cassis, L. A. Angiotensin II promotes
atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. _J. Clin. Invest._ 105, 1605–1612 (2000). Article CAS PubMed PubMed Central Google Scholar * Wang, S. et al.
Activation of AMP-activated protein kinase alpha2 by nicotine instigates formation of abdominal aortic aneurysms in mice in vivo. _Nat. Med._ 18, 902–910 (2012). Article CAS PubMed PubMed
Central Google Scholar * Moran, C. S. et al. Resveratrol inhibits growth of experimental abdominal aortic aneurysm associated with upregulation of angiotensin-converting enzyme 2.
_Arterioscler. Thromb. Vasc. Biol._ 37, 2195–2203 (2017). Article CAS PubMed Google Scholar * Li, D. Y. et al. H19 induces abdominal aortic aneurysm development and progression.
_Circulation_ 138, 1551–1568 (2018). Article CAS PubMed PubMed Central Google Scholar * Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory
requirements. _Nat. Methods_ 12, 357–360 (2015). Article CAS PubMed PubMed Central Google Scholar * Pertea, M., Kim, D., Pertea, G. M., Leek, J. T. & Salzberg, S. L.
Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. _Nat. Protoc._ 11, 1650–1667 (2016). Article CAS PubMed PubMed Central Google Scholar *
Lawrence, M. et al. Software for computing and annotating genomic ranges. _PLoS Comput. Biol._ 9, e1003118 (2013). Article CAS PubMed PubMed Central Google Scholar * Love, M. I., Huber,
W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. _Genome Biol._ 15, 550 (2014). Article PubMed PubMed Central CAS Google Scholar *
Walter, W., Sanchez-Cabo, F. & Ricote, M. GOplot: an R package for visually combining expression data with functional analysis. _Bioinformatics_ 31, 2912–2914 (2015). Article CAS
PubMed Google Scholar * Schulte, S. et al. Cystatin C deficiency promotes inflammation in angiotensin II-induced abdominal aortic aneurisms in atherosclerotic mice. _Am. J. Pathol._ 177,
456–463 (2010). Article CAS PubMed PubMed Central Google Scholar * Zhang, J. et al. Chemokine (C-C motif) receptor 2 mediates mast cell migration to abdominal aortic aneurysm lesions in
mice. _Cardiovasc. Res._ 96, 543–551 (2012). Article CAS PubMed PubMed Central Google Scholar * Deckert, V. et al. Development of abdominal aortic aneurysm is decreased in mice with
plasma phospholipid transfer protein deficiency. _Am. J. Pathol._ 183, 975–986 (2013). Article CAS PubMed Google Scholar * Wilde, E. et al. Tail-cuff technique and its influence on
central blood pressure in the mouse. _J. Am. Heart Assoc._ 6, e005204 (2017). 6. Article PubMed PubMed Central Google Scholar * Cassis, L. A. et al. ANG II infusion promotes abdominal
aortic aneurysms independent of increased blood pressure in hypercholesterolemic mice. _Am. J. Physiol. Heart Circ. Physiol._ 296, H1660–H1665 (2009). Article CAS PubMed PubMed Central
Google Scholar * Morrow, D. et al. Cyclic strain inhibits Notch receptor signaling in vascular smooth muscle cells in vitro. _Circ. Res._ 96, 567–575 (2005). Article CAS PubMed Google
Scholar * Cheng, X., Ji, Z., Tsalkova, T. & Mei, F. Epac and PKA: a tale of two intracellular cAMP receptors. _Acta Biochim. Biophys. Sin. (Shanghai)_ 40, 651–662 (2008). Article CAS
Google Scholar * Isobe, K. et al. Systems-level identification of PKA-dependent signaling in epithelial cells. _Proc. Natl Acad. Sci. USA_ 114, E8875–E8884 (2017). Article CAS PubMed
PubMed Central Google Scholar * Ying, W. Z., Zhang, H. G. & Sanders, P. W. EGF receptor activity modulates apoptosis induced by inhibition of the proteasome of vascular smooth muscle
cells. _J. Am. Soc. Nephrol._ 18, 131–142 (2007). Article CAS PubMed Google Scholar * Gale, D. D. et al. Pharmacology of a new cyclic nucleotide phosphodiesterase type 4 inhibitor,
V11294. _Pulm. Pharmacol. Ther._ 16, 97–104 (2003). Article CAS PubMed Google Scholar * Dyke, H. J. & Montana, J. G. Update on the therapeutic potential of PDE4 inhibitors. _Expert.
Opin. Investig. Drugs_ 11, 1–13 (2002). Article CAS PubMed Google Scholar * Borison, H. L. & Wang, S. C. Physiology and pharmacology of vomiting. _Pharmacol. Rev._ 5, 193–230 (1953).
CAS PubMed Google Scholar * Carpenter, D. O., Briggs, D. B., Knox, A. P. & Strominger, N. Excitation of area postrema neurons by transmitters, peptides, and cyclic nucleotides. _J.
Neurophysiol._ 59, 358–369 (1988). Article CAS PubMed Google Scholar * Manning, C. D. et al. Suppression of human inflammatory cell function by subtype-selective PDE4 inhibitors
correlates with inhibition of PDE4A and PDE4B. _Br. J. Pharm._ 128, 1393–1398 (1999). Article CAS Google Scholar * Palmer, D. & Maurice, D. H. Dual expression and differential
regulation of phosphodiesterase 3A and phosphodiesterase 3B in human vascular smooth muscle: implications for phosphodiesterase 3 inhibition in human cardiovascular tissues. _Mol.
Pharmacol._ 58, 247–252 (2000). Article CAS PubMed Google Scholar * Liu, H. & Maurice, D. H. Phosphorylation-mediated activation and translocation of the cyclic AMP-specific
phosphodiesterase PDE4D3 by cyclic AMP-dependent protein kinase and mitogen-activated protein kinases. A potential mechanism allowing for the coordinated regulation of PDE4D activity and
targeting. _J. Biol. Chem._ 274, 10557–10565 (1999). Article CAS PubMed Google Scholar * Orlov, S. N. et al. Activation of cAMP signaling transiently inhibits apoptosis in vascular
smooth muscle cells in a site upstream of caspase-3. _Cell Death Differ._ 6, 661–672 (1999). Article CAS PubMed Google Scholar * Hayashi, S. et al. Cyclic AMP inhibited proliferation of
human aortic vascular smooth muscle cells, accompanied by induction of p53 and p21. _Hypertension_ 35, 237–243 (2000). Article CAS PubMed Google Scholar * Tilley, D. G. & Maurice, D.
H. Vascular smooth muscle cell phosphodiesterase (PDE) 3 and PDE4 activities and levels are regulated by cyclic AMP in vivo. _Mol. Pharmacol._ 62, 497–506 (2002). Article CAS PubMed
Google Scholar * Raymond, D. R., Carter, R. L., Ward, C. A. & Maurice, D. H. Distinct phosphodiesterase-4D variants integrate into protein kinase A-based signaling complexes in cardiac
and vascular myocytes. _Am. J. Physiol. Heart Circ. Physiol._ 296, H263–H271 (2009). Article CAS PubMed Google Scholar * Shiraya, S. et al. Hypertension accelerated experimental
abdominal aortic aneurysm through upregulation of nuclear factor kappaB and Ets. _Hypertension_ 48, 628–636 (2006). Article CAS PubMed Google Scholar * Cai, Y. et al. Role of
cAMP-phosphodiesterase 1C signaling in regulating growth factor receptor stability, vascular smooth muscle cell growth, migration, and neointimal hyperplasia. _Circ. Res._ 116, 1120–1132
(2015). Article CAS PubMed PubMed Central Google Scholar * Xie, Y. et al. LMO7 is a negative feedback regulator of transforming growth factor beta signaling and fibrosis. _Circulation_
139, 679–693 (2019). Article CAS PubMed PubMed Central Google Scholar * Das, S. et al. A novel angiotensin II-induced long noncoding RNA giver regulates oxidative stress, inflammation,
and proliferation in vascular smooth muscle cells. _Circ. Res._ 123, 1298–1312 (2018). Article CAS PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS This work
was supported by the Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences [grant number 2021-1-I2M-016], the National Natural Science Foundation of China [grant number
82100514], and the National Key Research and Development Program of China [grant numbers 2019YFA0801804, 2019YFA0801703]. AUTHOR INFORMATION Author notes * These authors contributed equally:
Ran Gao, Wenjun Guo, Tianfei Fan. AUTHORS AND AFFILIATIONS * State Key Laboratory of Medical Molecular Biology, Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences,
Department of Pathophysiology, Peking Union Medical College, Beijing, China Ran Gao, Wenjun Guo, Tianfei Fan, Junling Pang, Yangfeng Hou, Xiaohang Feng, Bolun Li, Weipeng Ge, Tianhui Fan,
Tiantian Zhang & Jing Wang * Department of Anesthesiology, Beijing Anzhen Hospital, Capital Medical University, Beijing Institute of Heart, Lung and Blood Vessel Diseases, Beijing, China
Jiakai Lu & He Jing * Department of Anesthesiology, Beijing Friendship Hospital, Capital Medical University, Beijing, China Mu Jin * Aab Cardiovascular Research Institute, University of
Rochester, School of Medicine and Dentistry, Rochester, NY, 14642, USA Chen Yan Authors * Ran Gao View author publications You can also search for this author inPubMed Google Scholar *
Wenjun Guo View author publications You can also search for this author inPubMed Google Scholar * Tianfei Fan View author publications You can also search for this author inPubMed Google
Scholar * Junling Pang View author publications You can also search for this author inPubMed Google Scholar * Yangfeng Hou View author publications You can also search for this author
inPubMed Google Scholar * Xiaohang Feng View author publications You can also search for this author inPubMed Google Scholar * Bolun Li View author publications You can also search for this
author inPubMed Google Scholar * Weipeng Ge View author publications You can also search for this author inPubMed Google Scholar * Tianhui Fan View author publications You can also search
for this author inPubMed Google Scholar * Tiantian Zhang View author publications You can also search for this author inPubMed Google Scholar * Jiakai Lu View author publications You can
also search for this author inPubMed Google Scholar * He Jing View author publications You can also search for this author inPubMed Google Scholar * Mu Jin View author publications You can
also search for this author inPubMed Google Scholar * Chen Yan View author publications You can also search for this author inPubMed Google Scholar * Jing Wang View author publications You
can also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to Jing Wang. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests.
ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION
SUPPLEMENTAL MATERIAL RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation,
distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and
indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to
the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will
need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE
CITE THIS ARTICLE Gao, R., Guo, W., Fan, T. _et al._ Phosphodiesterase 4D contributes to angiotensin II-induced abdominal aortic aneurysm through smooth muscle cell apoptosis. _Exp Mol Med_
54, 1201–1213 (2022). https://doi.org/10.1038/s12276-022-00815-y Download citation * Received: 31 October 2021 * Revised: 05 May 2022 * Accepted: 09 May 2022 * Published: 23 August 2022 *
Issue Date: August 2022 * DOI: https://doi.org/10.1038/s12276-022-00815-y SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link
Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative