Play all audios:
ABSTRACT CD8+ T cell immunosurveillance is crucial in solid tumors and T cell dysfunction leads to tumor progression. In contrast, the role of CD8+ T cells in the control of leukemia is less
clear. We characterized the molecular signature of leukemia stem/progenitor cells (LSPCs) and paired CD8+ T cells in patients with acute myeloid leukemia (AML). Epigenetic alterations via
histone deacetylation reduced the expression of immune-related genes in bone marrow (BM)-infiltrating CD8+ T cells. Surprisingly, a silenced gene expression pattern in CD8+ T cells
significantly correlated with an improved prognosis. To define interactions between CD8+ T cells and LSPCs, we performed comprehensive correlative network modeling. This analysis indicated
that CD8+ T cells contribute to the maintenance/expansion of LSPCs, particularly in favorable risk AML. Functionally, CD8+ T cells in favorable AML induced the expansion of LSPCs by
stimulating the autocrine production of important hematopoietic cytokines such as interleukin (IL)-3. In contrast, LSPCs in aggressive AML were characterized by a higher activation of
stemness/proliferation-related pathways and develop independent of BM CD8+ T cells. Overall, our study indicates that CD8+ T cells support and expand LSPCs in favorable risk AML whereas
intermediate and adverse risk AML possess the intrinsic molecular abnormalities to develop independently. SIMILAR CONTENT BEING VIEWED BY OTHERS COOPERATION BETWEEN _KDM6B_ OVEREXPRESSION
AND _TET2_ DEFICIENCY IN THE PATHOGENESIS OF CHRONIC MYELOMONOCYTIC LEUKEMIA Article 14 June 2022 PEDIATRIC T-CELL ACUTE LYMPHOBLASTIC LEUKEMIA BLAST SIGNATURE AND MRD ASSOCIATED IMMUNE
ENVIRONMENT CHANGES DEFINED BY SINGLE CELL TRANSCRIPTOMICS ANALYSIS Article Open access 02 August 2023 THE INTRINSIC DEFECTS OF T CELLS IMPACT THE EFFICACY OF CAR-T THERAPY IN PATIENTS WITH
DIFFUSE LARGE B-CELL LYMPHOMA Article Open access 14 December 2023 INTRODUCTION Acute myeloid leukemia (AML) is a heterogeneous group of hematologic malignant diseases, characterized by
maturation arrest and increased proliferation of myeloid blasts [1, 2]. Based on the (cyto)genetic profile of AML blasts, patients can be classified into three different risk groups:
favorable risk (40–45%), intermediate risk (25–35%), and adverse risk (25–30%) [3]. Leukemia stem cells (LSCs) represent a minor fraction of the bulk leukemia cell population that
reconstitutes and propagates the disease. They are resistant to chemotherapy and irradiation and therefore are the main cause of relapse [4,5,6,7,8]. LSCs possess stem cell properties such
as self-renewal and quiescence that are regulated by cell-intrinsic and cell-extrinsic mechanisms [9,10,11]. As cell-intrinsic drivers, LSCs display molecular abnormalities that lead to
constitutive activation of the nuclear factor kB (NF-kB) [2], Wnt [12], and Notch signaling pathways [13]. In addition, similar to hematopoietic stem cells (HSCs), LSCs interact with cells
of the bone marrow (BM) microenvironment, including endothelial cells, specialized BM stromal cells, and osteoblasts [14,15,16]. These cells constitute the HSC niche and regulate
maintenance, self-renewal, and differentiation of HSCs and LSCs [17]. Immune cells are part of the BM microenvironment and interact with HSCs as well as leukemia stem and progenitor cells
(LSPCs). For example, CD8+ T cells have the potential to support the maintenance of HSCs and facilitate their engraftment after allogeneic stem-cell transplantation [18, 19]. It is well
documented that AML responds to immune-mediated therapies such as allogeneic hematopoietic stem cell transplantation and donor lymphocyte infusions [14, 20]. Similarly, AML blasts can be
lysed by adoptively transferred gene-modified T cells in xenotransplantation experiments [21]. However, several studies indicate that LSCs escape elimination by CD8+ T cell and natural
killer (NK) cells by down-regulating important molecules/pathways for immune recognition or by the expression of immune-inhibitory molecules [22, 23]. Experimental evidence indicates that
cytokines secreted by immune cells and defined cell–cell interactions such as the CD70/CD27 interaction expand HSCs and LSCs and contribute to disease progression [24, 25]. However, the role
of the adaptive immune system in the control of human AML and especially in the regulation of LSCs is poorly understood. We performed a comprehensive transcriptomic profiling of BM-derived
LSCs and leukemia progenitor cells together with paired CD8+ T cells of AML patients from different molecular risk groups. This analysis indicated that epigenetic mechanisms silence the gene
expression of CD8+ T cells in AML. Importantly, a silenced gene expression pattern correlated with improved prognosis. Correlation network modeling revealed that CD8+ T cells regulate LSPC
in favorable risk but not in adverse risk AML. Functionally, we show that CD8+ T cells induce the autocrine production of the hematopoietic cytokines such as IL-3 in favorable risk AML that
expands LSPCs. The interaction of CD8+ T cells with LSPCs was gradually lost from favorable risk to intermediate and adverse risk AML. In contrast, LSPCs from patients with intermediate and
adverse risk AML had a higher expression of genes related to stemness and cell proliferation. This study indicates that LSPCs in favorable risk AML are regulated by extrinsic signals such as
BM-infiltrating CD8+ T cells, whereas mainly cell-intrinsic mechanisms drive LSPC expansion in aggressive AML. MATERIALS AND METHODS PATIENTS Blood and BM aspirates from patients diagnosed
with AML at the Department of Medical Oncology, University Hospital Bern were prospectively collected. Thirty patients were selected from this repository based on the FACS immune-phenotype
of the AML cells and the risk category. The clinical and molecular characteristics of the AML patients and controls are listed in Supplementary Table 1. This study was approved by the local
ethical committee (Kantonale Ethikkommission Bern, KEK122/14). MOLECULAR PROFILING AND CORRELATION NETWORK MODELING Transcriptomic analysis was performed on 111 different well-defined
FACS-purified samples of hematopoietic stem/progenitors and paired CD8+ T cells from 30 AML patients and 7 controls. Modeling of several tens of thousands of predictive correlation network
was assessed to map potential links between genes expressed in stem/progenitor cells and paired CD8+ T cells in all AML patients of different risk groups and controls. Selection of
investigated genes, biological signaling, and correlations were further functionally validated. For details, see Supplementary Methods. DATA AVAILABILITY All transcriptomic data compiled for
this study have been deposited in NCBI GEO under the accession code GSE117090. In addition, expression data from GEO public repository was assessed as a validation cohort (GSE6891).
Complete methods are included in the Supplementary Appendix. RESULTS A SILENCED GENE EXPRESSION PATTERN IN CD8+ T CELLS CORRELATES WITH IMPROVED PROGNOSIS IN AML We first characterized the
complete gene expression signature of FACS-purified CD34+CD38− AML LSCs, CD34+CD38+ AML progenitors and paired CD8+ T cells in the BM of AML patients at the time point of diagnosis
(Supplementary Fig. 1, Supplementary Table 1). As expected, patients in the favorable risk group had a better overall survival in comparison to the other AML risk groups (Supplementary Fig.
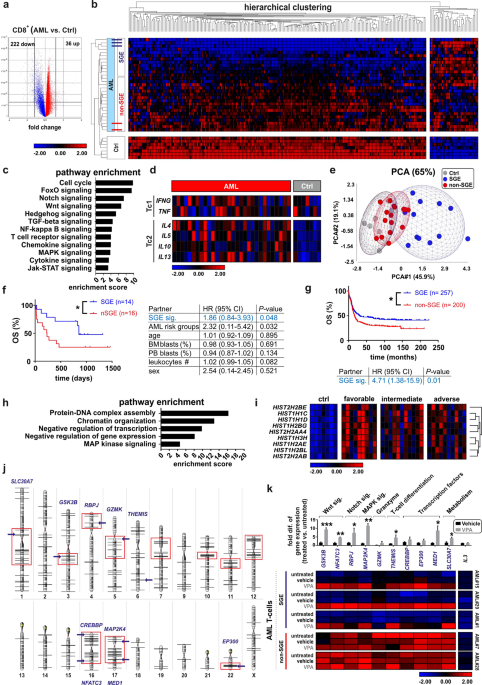
2). The expression of 258 genes was significantly changed in BM-infiltrating CD8+ T cells from AML patients compared to healthy donors (Supplemental Dataset 1). Interestingly, 222 genes were
down-regulated and only 36 genes were up-regulated (Fig. 1a, b). The down-regulated genes are involved in signaling pathways related to T cell activation, differentiation, and function,
such as NFkB, Wnt, FoxO, T cell receptor (TCR), and cytokine/chemokine signaling (Fig. 1c). The expression profile of key genes for T cell polarization in CD8+ T cells of AML patients
revealed a TC1 phenotype in controls but a skewing towards a TC2 phenotype in AML patients (Fig. 1d). Out of the 222 down-regulated genes, we defined a 40-gene panel of genes that belong to
the NFkB, Wnt, FoxO and Notch pathway, TCR and cytokine/chemokine signaling according to the results of the gene ontology enrichment and pathway analysis (Fig. 1c, Supplementary Table 2).
Based on the calculated mean gene expression level of these 40 pre-defined genes, outcome-based cut-points were defined using X-Tile software [26]. AML patients with a mean gene expression
below or above the defined cut-point were classified as patients with a “silenced gene expression (SGE) signature” or “non-silenced gene expression” (non-SGE) signature, respectively.
Principal component analysis (PCA) revealed a closer similarity between the non-SGE group and healthy controls while patients with the SGE signature exhibited more distinct gene expression
patterns (Fig. 1e). Importantly, patients with a SGE signature in BM CD8+ T cells had a significantly better overall survival compared to patients with a non-SGE signature (Fig. 1f).
Although the analysis in the different risk groups is limited by a rather small sample size, the expression of a SGE signature correlated with longer survival only in the favorable risk but
not in the intermediate or adverse risk group (Supplementary Fig. 3). Multivariate analysis for the SGE signature adjusted for AML risk group, patient age, percentage of blasts in BM and
blood, leukocyte counts, and sex, confirmed the SGE signature as an independent prognostic marker for overall survival in our analyzed AML patient cohort (Fig. 1f). To validate the
prognostic value of this 40-genes panel in a larger cohort of AML patients, we analyzed the expression of these genes in a publically available dataset comprising gene expression of
non-fractionated BM cells of AML patients [27, 28]. Our defined 40-gene panel showed significantly better overall survival for patients with a SGE signature than for patients with a non-SGE
signature. Cox-regression analysis further confirmed the SGE signature as a prognostic marker for overall survival (Fig. 1g). In our analysis, we observed a systemic down-regulation of genes
in AML CD8+ T cells compared to controls. Chromatin modification mainly via histone deacetylation is one of the key mechanisms for gene silencing [29]. Only 36 genes were up-regulated in
AML CD8+ T cells (Fig. 1b). Pathway enrichment analysis revealed that these 36 up-regulated genes are primarily involved in the control of chromatin organization or negative regulation of
transcription and gene expression (Fig. 1h). Nine of these 36 up-regulated genes are controlling chromatin organization/regulation (Fig. 1i). Chromosomal position-based gene-mapping analysis
(karyogram) showed that down-regulated genes were not randomly distributed all over the genome but significantly enriched in some particular regions. These data suggest altered histone
organization in particular chromosomal regions in leukemic CD8 T cells (Fig. 1j). Histone deacetylation catalyzed by the histone deacetylase (HDAC) is a central switch of permissive to
repressive chromatin domains leading to transcriptional silencing [30]. To functionally test the effect of histone deacetylation on the observed SGE signature, we treated FACS-purified CD8+
T cells isolated from three AML patients with SGE signature and from two AML patients with non-SGE signature with the HDAC inhibitor, valproic acid (VPA). VPA treatment significantly
reversed the SGE phenotype and increased the expression of 10 selected key genes involved in the regulation of Wnt-, Notch-, MAPK-signaling, T cell differentiation, and in metabolism. In
contrast, VPA did not change the expression of these genes in T cells with non-SGE signature (Fig. 1k). In addition, VPA treatment did not change the expression of _IL3_ in CD8+ T cells from
different AML patients, indicating that _IL3_ gene expression in T cells is not regulated by histone deacetylation (Fig. 1k). Overall, these data indicate that important genes involved in
CD8+ T cell activation, differentiation, and function are down-regulated in AML due to pathologic epigenetic alterations mainly mediated via histone deacetylation. Down-regulation of these
genes in CD8+ T cells correlates with an improved prognosis of AML patients. LSPCS FROM AML PATIENTS DISPLAY A DYSREGULATED EXPRESSION OF GENES INVOLVED IN PROLIFERATION, STEMNESS, AND
IMMUNE-RECOGNITION We next investigated the gene expression signature of CD34+CD38− AML LSCs and CD34+CD38+ AML progenitors. HSPCs from healthy donors (Ctrl) had a similar gene expression
profile and therefore clustered together in the PCA. In contrast, the gene expression of AML cells was more diverse and differed from healthy controls (Fig. 2a). In AML LSCs and progenitors,
403 and 1309 genes were differentially expressed compared to controls, respectively (Fig. 2a, Supplementary Dataset 1). A comprehensive pathway analysis of the differentially expressed
genes revealed that mainly pathways related to stemness, cell proliferation, cell cycle, or immune cell signaling were changed in AML LSCs and progenitor cells (Fig. 2b). The number of 155
genes (58 up-regulated and 97 down-regulated genes) were similarly dysregulated in AML LSCs and progenitors when compared to controls (Fig. 2c). Pathway enrichment analysis revealed that
up-regulated genes are mainly involved in cell proliferation, cell cycle, or immune-related signaling while down-regulated genes are primarily involved in signaling pathways mediating
antigen-presentation and interaction with immune cells (Fig. 2c). Taken together, AML LSCs and progenitors had a higher expression of genes involved in stemness and cell proliferation
compared to HSPCs whereas the down-regulated genes and pathways facilitate immune-escape. GENES INVOLVED IN IMMUNOSURVEILLANCE ARE DOWN-REGULATED WHEREAS STEMNESS-RELATED GENES ARE
UP-REGULATED IN ADVERSE RISK AML We next compared the gene expression profiles of AML LSCs, progenitors, and paired CD8+ T cells between the three different risk categories (Fig. 3a). AML
progenitors shared 294 dysregulated intersection genes across all AML risk categories. In contrast, only 67 intersection genes were found in AML LSCs. The gene expression profile of BM CD8+
T cells differed significantly between the different risk groups with only 39 intersection genes that were similarly altered in all three-risk categories (Fig. 3a). AML progenitors, which
comprise the highly proliferative AML cells, showed the highest expression of genes related to cell cycling. In contrast, genes involved in stemness- and leukemogenesis-related pathways were
expressed at higher levels in AML LSCs (e.g., Wnt, ErbB, and NF-kB). These stemness-related pathways were expressed at highest levels in LSCs from adverse risk AML and at lowest levels in
LSCs from the favorable risk group. This finding is in line with previous studies indicating that a LSC-related gene signature is a negative prognostic marker in AML [31, 32]. Furthermore,
genes involved in immune-related pathways such as cytokine–cytokine receptor interactions and cytokine/chemokine signaling were more expressed in LSCs from favorable risk AML but were not
expressed in adverse risk AML (Fig. 3b). In line with these findings, genes involved in pathways mediating immune cell function and target cell recognition such as TCR and chemokine
signaling as well as cytokine–cytokine receptor interaction were more expressed in paired CD8+ T cells from favorable risk AML but were down-regulated in the adverse risk group. Similarly,
signaling pathways involved in T cell effector function, such as NF-kB and Notch signaling, were significantly down-regulated in CD8+ T cells isolated from intermediate and adverse risk AML
patients [33]. The Wnt pathway, which is crucial for the differentiation into memory T cells [33, 34], was inactivated in CD8+ T cells from patients with adverse risk AML (Fig. 3b). These
results suggest that T cells in intermediate and high risk AML are less functional than in favorable risk AML and might be exhausted. However, the analysis of markers defining exhausted CD8+
T cells on gene level did not reveal differences between the AML risk groups (Supplementary Fig. 4). Importantly, the frequency of BM-infiltrating CD8+ T cells did not differ between the
AML risk groups or healthy controls (Supplementary Fig. 5). This indicates that functional but not numerical differences lead to the CD8+ T cell-mediated expansion of LSPCs in favorable risk
AML. Wnt signaling is fundamental for stemness and effector function of LSCs and CD8+ T cells, respectively [12]. Gene set enrichment analysis (GSEA) in our AML cohort revealed a
significant enrichment for genes involved in Wnt pathway activation in LSPCs from intermediate and adverse risk group AML patients, but not in LSPCs from the favorable risk group. In
contrast, Wnt-related genes were down-regulated in paired CD8+ T cells from AML patients of the intermediate and adverse risk group (Fig. 3c). These results suggest that LSPCs of adverse
risk AML patients develop and expand largely independently of CD8+ T cells, whereas CD8+ T cells are mainly active in favorable risk AML patients. CD8+ T CELLS REGULATE LSPCS IN FAVORABLE
BUT NOT ADVERSE RISK AML To define possible interactions of genes/pathways in CD8+ T cells with genes/pathways in AML LSPCs, comprehensive correlation networks were constructed. In each
network, a node was defined as a gene expressed either in CD8+ T cells or in LSPCs. A node (gene) in one cell that correlates significantly with more than 15 nodes in the other cell type was
considered as a hub (high-degree correlated node) (Fig. 4a). The assumption was that a gene expressed in CD8+ T cells communicates or coordinates other genes in LSPCs or vice versa. Three
types of correlations were detected: (1) “appear”, a correlation present in AML but not in controls, (2) “disappear”, a correlation present in the controls and absent in AML, and (3) “flip”,
where the direction of the correlation changes. The highest number of nodes was detected in AML progenitor cells and in CD8+ T cells, whereas few nodes were detected in AML LSCs (Fig. 4a,
b). Interestingly, the highest number of hubs were detected in CD8+ T cells, whereas fewer hubs were found in LSPCs (Fig. 4a, b, Supplementary Dataset 2). We next analyzed these networks in
the different AML risk categories (Fig. 4c, d). The number of nodes in the “appear network” in AML LSPCs and CD8+ T cells gradually decreased from the favorable risk to the adverse risk
group (Fig. 4d). In contrast, the number of nodes in “disappear” and “flip networks” did not change across AML risk groups (Fig. 4c, d). Importantly, the majority of hubs in CD8+ T cells
were present in the favorable risk group and their number gradually decreased in the intermediate and adverse risk groups (Fig. 4e). No CD8+ T cells hubs were detected in intermediate and
adverse risk groups that were not present in the favorable risk group (Fig. 4e, f). Only few hubs were identified in CD34+CD38+ progenitors. However, similar to CD8+ T cells, reduced number
of hubs were identified with increased risk category (Fig. 4e, Supplementary Fig. 6). The majority of nodes within AML LSPCs, which were connected to CD8+ hubs, were up-regulated compared to
controls (Supplementary Fig. 7). The number of nodes in LSPCs connected to given hubs in CD8+ T cells was gradually decreased from favorable to intermediate and adverse risk AML
(Supplementary Table 3). In order to identify the signaling pathways in AML LSPCs, which are modulated by the identified CD8+ T cell hubs, we performed a pathway analysis. The significantly
altered pathways in LSPCs included stemness, cell proliferation and survival, immune-related signaling, gene expression regulation, growth factor signaling, and metabolism which were most
activated in favorable risk AML (Fig. 4g, Supplementary Fig. 7). Taken together, our correlation network modeling indicated that CD8+ T cells regulate LSPCs in favorable risk AML. This
interaction is reduced or absent in intermediate and adverse risk AML. CD8+ T CELLS INDUCE THE EXPANSION AND MAINTENANCE OF LSPCS IN FAVORABLE RISK AML To functionally analyze the potential
influence of CD8+ T cells on LSPCs, we FACS-purified both LSCs or AML progenitor cells together with paired CD8+ T cells from BM of nine different AML patients (Fig. 5a, Supplementary Table
1). The co-culture of CD8+ T cells with LSCs resulted in an up to 2-fold increase in colony-formation compared to LSC mono-culture in the favorable risk group. In contrast, the increase in
colony formation in intermediate and adverse risk was less pronounced and only observed at higher T cell:LSC ratios (Fig. 5b). In addition, the number of cells per colonies was not
significantly changed, suggesting that differentiation was not affected by co-culture of LSPCs with CD8+ T cells (Fig. 5c). Further re-plating of cells derived from primary colony assays
showed a significant difference in the colony-formation capacity of co-cultured LSCs with a higher ratio of CD8+ T cells compared to LSC mono-culture. This indicates that LSCs with
unimpaired self-renewal capacity are expanded by CD8+ T cells (Supplementary Fig. 8). _MGAT5_ was identified as a hub in CD8+ T cells of all risk groups. However, while _MGAT5_ positively
correlated with the expression of _IL3_ and other important genes in CD34+CD38+ progenitors in favorable risk AML patients, this correlation is lost in intermediate and adverse risk AML
(Fig. 5d, e, Supplementary Fig. 9, Supplementary Dataset 2). Our results suggest that CD8+ T cells regulate LSPCs in favorable AML by inducing the production of important hematopoietic
cytokines such as IL-3. To address this possibility, we co-cultured FACS-purified CD8+ T cells with paired LSPCs derived from the same patients of favorable risk AML and analyzed the
expression of _IL3_ mRNA and protein. LSPCs of all risk groups expressed the IL-3 receptor alpha (IL-3Ra). While, CD8+ T cells did not express IL3Ra on mRNA and protein levels (Supplementary
Fig. 10). Co-culture with CD8+ T cells resulted in an up to 3-fold increase in _IL3_ mRNA expression in LSPCs and an up to 2-fold increase in IL-3 protein expression in culture supernatants
compared to LSPCs mono-culture from favorable risk AML (Fig. 5f). In healthy controls, the expression of _MGAT5_ in CD8+ T cells did not correlate with _IL3_ expression in progenitors
(Supplementary Fig. 11a). In addition, co-culture of CD8+ T cells with CD34+ HSPCs derived from healthy donors did not increase the level of _IL3_ mRNA expression (Supplementary Fig. 11b).
However, co-culture of pooled CD8+ T cells derived from three good risk AML patients significantly increased the colony formation capacity of both, AML LSCs and control HSCs (Supplementary
Fig. 11c). Interestingly, colony formation in LSCs was increased significantly more than in HSCs. This suggests that AML LSCs have a higher proliferative capacity in response to stimulation
by CD8+ T cells than normal HSCs. Therefore, favorable risk AML LSPCs respond to similar interactions with CD8+ T cells as normal HSPCs. However, only BM-infiltrating CD8+ T cells of AML
patients but not of healthy donors induce IL-3 production in LSCs and HSCs. Knockdown of _MGAT5_ gene in CD8+ T cells using a siRNA before initiation of the co-culture revealed an up to
2-fold decrease in _IL3_ mRNA expression in LSPCs (Fig. 5g). The level of _MGAT5_ gene expression after siRNA treatment of CD8 T cells significantly correlated with the _IL3_ expression in
co-cultured LSPCs (Fig. 5h). In silico pathway analysis predicted that _MGAT5_ expressed in CD8+ T cells triggers the expression of _IL3_ in LSPCs via EGF/EGFR, IL-2 or IFNγ signaling
(Supplementary Fig. 9a). Correlation analysis confirmed a significant positive correlation between _IL3_ expression in CD34+CD38+ progenitors and _EGF_/_EGFR_ or _IL2_ expression in CD8+ T
cells in favorable risk AML but not in intermediate or adverse risk AML (Supplementary Fig. 9b–c). We tested this concept experimentally by co-culturing FACS-purified CD34+ LSPCs with paired
CD8+ T cells in the presence of neutralizing antibodies to these specific cytokines and growth factor receptor. Blocking EGFR and IL-2, but not IFNγ reduced _IL3_ mRNA expression in
FACS-purified CD34+ LSPCs (Fig. 5i). This finding indicates that CD8+ T cells induce IL-3 production in LSPCs mainly by IL-2 and EGF/EGFR signaling. Neutralization of IL-3 in the co-culture
of CD8+ T cells with LSCs from favorable risk AML patients significantly decreased the colony-formation capacity (Fig. 5j). In addition, co-culture of CD34+ LSPCs with CD8+ T cells resulted
in the up-regulation of selected genes that are regulated by hubs with documented function in hematopoiesis, leukemia development, cell proliferation, or immune tolerance (_CD47_, _CD58_,
_CD63_, _CD99_, and _IL17b_) (Fig. 5k, Supplementary Table 3) [35,36,37,38,39]. Taken together, these findings indicated that BM CD8+ T cells induce pathways in favorable risk LSPCs that
promote leukemia development, such as the autocrine production of hematopoietic cytokines. In contrast, LSPCs in more aggressive forms of AML develop largely independent of CD8+ T cells
(Fig. 5l). DISCUSSION It is well documented that the immune system contributes to the control of solid tumors, a process called “tumor immunosurveillance”. However, cancer cells evade immune
recognition and elimination by cytotoxic CD8+ lymphocytes via various mechanisms, including loss of antigen and the expression of immune inhibitory molecules [40, 41]. Thus,
tumor-infiltrating lymphocytes (TILs) are often dysfunctional or “exhausted” due to the interaction with immune inhibitory ligands expressed by tumor cells [40]. The presence of
dysfunctional CD8+ T cells in TILs is a negative prognostic and predictive factor for the response to treatment with immune-checkpoint inhibitors [42, 43]. In the present study, we analyzed
the gene expression signature of BM CD8+ T cells together with paired AML LSCs and progenitor cells. The majority of differentially expressed genes were down-regulated in CD8+ T cells
derived from AML patients. The down-regulated genes are involved in key functions of T cell activation and differentiation including NFkB, Notch, Wnt, FoxO, TCR, and cytokine/chemokine
signaling. This indicates that similar to TILs in solid tumors, CD8+ T cells in the AML BM are dysfunctional. Surprisingly, patients with dysfunctional CD8+ T cells as indicated by a SGE
signature had a significantly better survival compared to patients with non-SGE signature. This difference may be explained by the fact that TILs comprise a population of activated
tumor-specific T cells whereas the BM is a secondary lymphoid organ that contains mainly memory T cells with different specificities [44]. The up-regulated genes in CD8+ T cells of AML
patients included main drivers of histone deacetylation and epigenetic regulation, which correlated with the SGE signature in our study population. HDAC enzymes regulate key functions in T
cells, such as maturation, migration, and TCR signaling [45]. Recently, it was reported that the transcription factors Tcf1 and Lef1 favor CD8+ T cell development by HDAC mediated
suppression of lineage-inappropriate genes [46]. In addition, HDAC inhibitors enhance anti-tumor activity of antigen-specific cytotoxic T cells against solid tumors and multiple myeloma
[47]. Similarly, treatment of AML and myelodysplasia patients with azacitidine and VPA induces a CD8+ T-cell response to the MAGE cancer testis antigen [48]. This suggests that a SGE
signature of T cells might be reversed by treatment with demethylating agents. To define possible interactions of CD8+ T cells with human AML LSPCs, we performed a comprehensive correlation
network analysis. The goal of this analysis was to identify genes in CD8+ T cells that regulate pathways in LSPCs and vice versa (hubs). Interestingly, most hub genes were identified in CD8+
T cells whereas only few hubs were present in LSPCs. The number of CD8+ T cell hubs was the highest in favorable risk AML and then gradually lower from intermediate to adverse risk AML. In
addition, a given CD8+ T cell hub correlated with the highest number of genes (nodes) in LSPCs from favorable risk group and this number was gradually reduced in intermediate to adverse risk
AML. In addition, AML progenitors expressed more node genes than AML LSCs, indicating that CD8+ T cells predominantly interact with progenitor cells. Our finding suggests that CD8+ T cells
regulate LSPCs in favorable risk AML while more aggressive forms of AML develop independently of BM-infiltrating CD8+ T cells. According to our comprehensive network modeling, hubs in CD8+ T
cells modulate stemness, cell-proliferation and cell-survival, immune-related signaling, and growth factor signaling mainly in favorable risk AML. CD8+ T cells’ hub genes in favorable risk
AML, correlate with an increased gene expression of cytokine/chemokines such as _IL3_ and _IL17B_, and cell surface molecules (e.g., _CD47_, _CD58_, _CD63_, and _CD99_) that have been
associated with hematopoiesis, leukemia development, cell proliferation, or immune tolerance [35,36,37,38,39, 49]. Our results indicated that CD8+ T cells induce IL3 production in AML LSPCs
via IL2 and EGF signaling leading to their expansion. This is in agreement with reports documenting that IL-3 increases proliferation and expansion of CML stem cells and AML blasts [50, 51].
Interestingly, the level of IL-3Ra expression on AML blasts is associated with increased cellularity, enhanced proliferation, and with poor prognosis [52]. CD8+ T cells regulate LSPCs
mainly in favorable risk AML, while more aggressive forms of AML develop independently of BM-infiltrating CD8+ T cells. The frequency of AML-specific T cells is rather low [53, 54].
Therefore, the majority of the analyzed CD8+ T cells in the BM may be BM-resident memory CD8+ T cells that are not leukemia-specific. We document that in adverse risk AML, the positive
correlation between _IL2_ or _EGF_ expressed in CD8+ T cells to _IL3_ in AML progenitors was lost (Supplementary Fig. 9c). We did not detect differences in the level of gene silencing in
CD8+ T cells in different risk categories (Fig. 1, Supplementary Dataset 1). In addition, we also tested the expression of markers used to characterize exhausted CD8+ T cells (_CD244_ (2B4),
_CD160_, _TIGIT_, _HAVCR2_, _LAG3_, _PDCD1_ (PD-1)). This analysis did not reveal a preferential expression of exhaustion markers in T cells of intermediate or adverse risk AML
(Supplementary Fig. 4). In addition, genes involved in immune-related pathways such as cytokine–cytokine receptor interactions and cytokine/chemokine signaling were expressed in LSCs from
favorable risk AML but were not expressed in adverse risk AML (Fig. 3b). Therefore, we suggest that the lack of interaction between CD8+ T cells and LSPCs in intermediate and adverse risk
AML is not due to alterations in T cells but due to molecular changes in LSPCs in intermediate and high risk AML that render them independent in regard of proliferation and expansion. AML
LSCs and progenitors in aggressive AML had a higher expression of genes involved in stemness and cell proliferation (as an intrinsic driver of leukemia) compared to LSPCs in favorable risk
AML, whereas the down-regulated genes and pathways facilitated immune-escape and immune-tolerance. Stemness signatures in blasts are an important negative prognostic marker [12, 24]. The
canonical Wnt pathway, which is central for HSC maintenance and development, is constitutively active in myeloid leukemia and of crucial importance for LSCs [24, 55, 56]. Fusion proteins
such as AML1-ETO, MLL-ENL, PLZF-RARα, and PML-RARα induce Wnt signaling via activation of γ-catenin [57,58,59]. In addition, activating mutations in FLT3 are associated with high β-catenin
levels and correlate with poor overall survival in AML patients [59, 60]. We document that stemness-related pathways, especially the Wnt pathway, were more active in adverse risk than in
intermediate or favorable risk AML. AML is a very heterogeneous disease. It is therefore not surprising that the interaction of CD8+ T cells with LSPCs also varies in different molecular
subtypes of AML. Interestingly, although the prognostic risk groups still comprise a molecular very heterogeneous group of diseases, we found a clear difference in the regulation of LSPCs.
Favorable risk AML have less intrinsic molecular abnormalities that drive proliferation and stemness. The disease development depends on external cues from the niche, e.g., from CD8+ T
cells. In contrast, more aggressive AML is propagated mainly by cell-intrinsic mechanisms and develop independent of immune cells. REFERENCES * Estey E, Dohner H. Acute myeloid leukaemia.
Lancet. 2006;368:1894–907. Article PubMed Google Scholar * Hoesel B, Schmid JA. The complexity of NF-κB signaling in inflammation and cancer. Mol Cancer. 2013;12:86. Article CAS PubMed
PubMed Central Google Scholar * Zeisig BB, Kulasekararaj AG, Mufti GJ, So CW. SnapShot: acute myeloid leukemia. Cancer Cell. 2012;22:698. Article CAS PubMed Google Scholar * Lapidot
T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–8. Article
CAS PubMed Google Scholar * Hope KJ, Jin L, Dick JE. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat Immunol.
2004;5:738–43. Article CAS PubMed Google Scholar * Guzman ML, Allan JN. Concise review: leukemia stem cells in personalized medicine. Stem Cells. 2014;32:844–51. Article CAS PubMed
PubMed Central Google Scholar * Murone M, Radpour R, Attinger A, Chessex AV, Huguenin AL, Schurch CM, et al. The multi-kinase inhibitor Debio 0617B reduces maintenance and self-renewal of
primary human AML CD34(+) stem/progenitor cells. Mol Cancer Ther. 2017;16:1497–510. Article CAS PubMed Google Scholar * Radpour R, Forouharkhou F. Single-cell analysis of tumors:
creating new value for molecular biomarker discovery of cancer stem cells and tumor-infiltrating immune cells. World J Stem Cells. 2018;10:160–71. Article PubMed PubMed Central Google
Scholar * Rice KN, Jamieson CH. Molecular pathways to CML stem cells. Int J Hematol. 2010;91:748–52. Article PubMed Google Scholar * Passegue E, Weisman IL. Leukemic stem cells: where do
they come from? Stem Cell Rev. 2005;1:181–8. Article CAS PubMed Google Scholar * Radpour R. Tracing and targeting cancer stem cells: new venture for personalized molecular cancer
therapy. World J Stem Cells. 2017;9:169–78. Article PubMed PubMed Central Google Scholar * Staal FJ, Famili F, Garcia Perez L, Pike-Overzet K. Aberrant Wnt signaling in leukemia.
Cancers. 2016;8:78. Article PubMed Central Google Scholar * Tohda S. NOTCH signaling roles in acute myeloid leukemia cell growth and interaction with other stemness-related signals.
Anticancer Res. 2014;34:6259–64. CAS PubMed Google Scholar * Sanchez-Aguilera A, Mendez-Ferrer S. The hematopoietic stem-cell niche in health and leukemia. Cell Mol Life Sci.
2017;74:579–90. Article CAS PubMed Google Scholar * Doron B, Handu M, Kurre P. Concise review: adaptation of the bone marrow stroma in hematopoietic malignancies: current concepts and
models. Stem Cells. 2018;36:304–12. Article PubMed Google Scholar * Riether C, Schurch CM, Ochsenbein AF. Regulation of hematopoietic and leukemic stem cells by the immune system. Cell
Death Differ. 2015;22:187–98. Article CAS PubMed Google Scholar * Shiozawa Y, Havens AM, Pienta KJ, Taichman RS. The bone marrow niche: habitat to hematopoietic and mesenchymal stem
cells, and unwitting host to molecular parasites. Leukemia. 2008;22:941–50. Article CAS PubMed PubMed Central Google Scholar * Geerman S, Brasser G, Bhushal S, Salerno F, Kragten NA,
Hoogenboezem M, et al. Memory CD8(+) T cells support the maintenance of hematopoietic stem cells in the bone marrow. Haematologica. 2018;103:e230–3. Article CAS PubMed PubMed Central
Google Scholar * Gandy KL, Domen J, Aguila H, Weissman IL. CD8+TCR+ and CD8+TCR− cells in whole bone marrow facilitate the engraftment of hematopoietic stem cells across allogeneic
barriers. Immunity. 1999;11:579–90. Article CAS PubMed Google Scholar * Hale G, Waldmann H. Control of graft-versus-host disease and graft rejection by T cell depletion of donor and
recipient with Campath-1 antibodies. Results of matched sibling transplants for malignant diseases. Bone Marrow Transplant. 1994;13:597–611. CAS PubMed Google Scholar * Tasian SK,
Kenderian SS, Shen F, Ruella M, Shestova O, Kozlowski M, et al. Optimized depletion of chimeric antigen receptor T cells in murine xenograft models of human acute myeloid leukemia. Blood.
2017;129:2395–407. Article CAS PubMed PubMed Central Google Scholar * Austin R, Smyth MJ, Lane SW. Harnessing the immune system in acute myeloid leukaemia. Crit Rev Oncol Hematol.
2016;103:62–77. Article PubMed Google Scholar * Masarova L, Kantarjian H, Garcia-Mannero G, Ravandi F, Sharma P, Daver N. Harnessing the immune system against leukemia: monoclonal
antibodies and checkpoint strategies for AML. Adv Exp Med Biol. 2017;995:73–95. Article CAS PubMed Google Scholar * Riether C, Schurch CM, Buhrer ED, Hinterbrandner M, Huguenin AL,
Hoepner S, et al. CD70/CD27 signaling promotes blast stemness and is a viable therapeutic target in acute myeloid leukemia. J Exp Med. 2017;214:359–80. Article CAS PubMed PubMed Central
Google Scholar * Riether C, Schurch CM, Flury C, Hinterbrandner M, Druck L, Huguenin AL, et al. Tyrosine kinase inhibitor-induced CD70 expression mediates drug resistance in leukemia stem
cells by activating Wnt signaling. Sci Transl Med. 2015;7:298ra119. Article PubMed Google Scholar * Camp RL, Dolled-Filhart M, Rimm DL. X-tile: a new bio-informatics tool for biomarker
assessment and outcome-based cut-point optimization. Clin Cancer Res. 2004;10:7252–9. Article CAS PubMed Google Scholar * Verhaak RG, Wouters BJ, Erpelinck CA, Abbas S, Beverloo HB,
Lugthart S, et al. Prediction of molecular subtypes in acute myeloid leukemia based on gene expression profiling. Haematologica. 2009;94:131–4. Article PubMed Google Scholar * de Jonge
HJ, Valk PJ, Veeger NJ, ter Elst A, den Boer ML, Cloos J, et al. High VEGFC expression is associated with unique gene expression profiles and predicts adverse prognosis in pediatric and
adult acute myeloid leukemia. Blood. 2010;116:1747–54. Article PubMed Google Scholar * Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. Article CAS
PubMed Google Scholar * Verdone L, Caserta M, Di Mauro E. Role of histone acetylation in the control of gene expression. Biochem Cell Biol. 2005;83:344–53. Article CAS PubMed Google
Scholar * Gentles AJ, Plevritis SK, Majeti R, Alizadeh AA. Association of a leukemic stem cell gene expression signature with clinical outcomes in acute myeloid leukemia. J Am Med Assoc.
2010;304:2706–15. Article CAS Google Scholar * Ng SW, Mitchell A, Kennedy JA, Chen WC, McLeod J, Ibrahimova N, et al. A 17-gene stemness score for rapid determination of risk in acute
leukaemia. Nature. 2016;540:433–7. Article CAS PubMed Google Scholar * Backer RA, Hombrink P, Helbig C, Amsen D. The fate choice between effector and memory T cell lineages: asymmetry,
signal integration, and feedback to create bistability. Adv Immunol. 2018;137:43–82. Article CAS PubMed Google Scholar * Sorcini D, Bruscoli S, Frammartino T, Cimino M, Mazzon E, Galuppo
M, et al. Wnt/β-catenin signaling induces integrin α4β1 in T cells and promotes a progressive neuroinflammatory disease in mice. J Immunol. 2017;199:3031–41. Article CAS PubMed Google
Scholar * Galli S, Zlobec I, Schurch C, Perren A, Ochsenbein AF, Banz Y. CD47 protein expression in acute myeloid leukemia: a tissue microarray-based analysis. Leuk Res. 2015;39:749–56.
Article CAS PubMed Google Scholar * Kong F, Gao F, Li H, Liu H, Zhang Y, Zheng R, et al. CD47: a potential immunotherapy target for eliminating cancer cells. Clin Transl Oncol.
2016;18:1051–5. Article CAS PubMed Google Scholar * McArdel SL, Terhorst C, Sharpe AH. Roles of CD48 in regulating immunity and tolerance. Clin Immunol. 2016;164:10–20. Article CAS
PubMed PubMed Central Google Scholar * Seubert B, Cui H, Simonavicius N, Honert K, Schafer S, Reuning U, et al. Tetraspanin CD63 acts as a pro-metastatic factor via beta-catenin
stabilization. Int J Cancer. 2015;136:2304–15. Article CAS PubMed Google Scholar * Yamaguchi Y, Fujio K, Shoda H, Okamoto A, Tsuno NH, Takahashi K, et al. IL-17B and IL-17C are
associated with TNF-alpha production and contribute to the exacerbation of inflammatory arthritis. J Immunol. 2007;179:7128–36. Article CAS PubMed Google Scholar * Baitsch L,
Baumgaertner P, Devevre E, Raghav SK, Legat A, Barba L, et al. Exhaustion of tumor-specific CD8(+) T cells in metastases from melanoma patients. J Clin Invest. 2011;121:2350–60. Article CAS
PubMed PubMed Central Google Scholar * Gorenshteyn D, Zaslavsky E, Fribourg M, Park CY, Wong AK, Tadych A, et al. Interactive big data resource to elucidate human immune pathways and
diseases. Immunity. 2015;43:605–14. Article CAS PubMed PubMed Central Google Scholar * Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on
clinical outcome. Nat Rev Cancer. 2012;12:298–306. Article CAS PubMed Google Scholar * Ji RR, Chasalow SD, Wang L, Hamid O, Schmidt H, Cogswell J, et al. An immune-active tumor
microenvironment favors clinical response to ipilimumab. Cancer Immunol Immunother. 2012;61:1019–31. Article CAS PubMed Google Scholar * Di Rosa F, Gebhardt T. Bone marrow T cells and
the integrated functions of recirculating and tissue-resident memory T cells. Front Immunol. 2016;7:51 PubMed PubMed Central Google Scholar * Haery L, Thompson RC, Gilmore TD. Histone
acetyltransferases and histone deacetylases in B- and T-cell development, physiology and malignancy. Genes Cancer. 2015;6:184–213. CAS PubMed PubMed Central Google Scholar * Xing S, Li
F, Zeng Z, Zhao Y, Yu S, Shan Q, et al. Tcf1 and Lef1 transcription factors establish CD8(+) T cell identity through intrinsic HDAC activity. Nat Immunol. 2016;17:695–703. Article CAS
PubMed PubMed Central Google Scholar * Bae J, Hideshima T, Tai YT, Song Y, Richardson P, Raje N, et al. Histone deacetylase (HDAC) inhibitor ACY241 enhances anti-tumor activities of
antigen-specific central memory cytotoxic T lymphocytes against multiple myeloma and solid tumors. Leukemia. 2018;32:1932–47. Article CAS PubMed PubMed Central Google Scholar * Goodyear
O, Agathanggelou A, Novitzky-Basso I, Siddique S, McSkeane T, Ryan G, et al. Induction of a CD8+ T-cell response to the MAGE cancer testis antigen by combined treatment with azacitidine and
sodium valproate in patients with acute myeloid leukemia and myelodysplasia. Blood. 2010;116:1908–18. Article CAS PubMed Google Scholar * Chung SS, Eng WS, Hu W, Khalaj M,
Garrett-Bakelman FE, Tavakkoli M, et al. CD99 is a therapeutic target on disease stem cells in myeloid malignancies. Sci Transl Med. 2017;9:eaaj2025. Article PubMed PubMed Central Google
Scholar * Nievergall E, Ramshaw HS, Yong AS, Biondo M, Busfield SJ, Vairo G, et al. Monoclonal antibody targeting of IL-3 receptor alpha with CSL362 effectively depletes CML progenitor and
stem cells. Blood. 2014;123:1218–28. Article CAS PubMed Google Scholar * Onetto-Pothier N, Aumont N, Haman A, Park L, Clark SC, De Lean A, et al. IL-3 inhibits the binding of GM-CSF to
AML blasts, but the two cytokines act synergistically in supporting blast proliferation. Leukemia. 1990;4:329–36. CAS PubMed Google Scholar * Testa U, Riccioni R, Militi S, Coccia E,
Stellacci E, Samoggia P, et al. Elevated expression of IL-3Ralpha in acute myelogenous leukemia is associated with enhanced blast proliferation, increased cellularity, and poor prognosis.
Blood. 2002;100:2980–8. Article CAS PubMed Google Scholar * Le Dieu R, Taussig DC, Ramsay AG, Mitter R, Miraki-Moud F, Fatah R, et al. Peripheral blood T cells in acute myeloid leukemia
(AML) patients at diagnosis have abnormal phenotype and genotype and form defective immune synapses with AML blasts. Blood. 2009;114:3909–16. Article PubMed PubMed Central Google Scholar
* Narita M, Takahashi M, Liu A, Nikkuni K, Furukawa T, Toba K, et al. Leukemia blast-induced T-cell anergy demonstrated by leukemia-derived dendritic cells in acute myelogenous leukemia.
Exp Hematol. 2001;29:709–19. Article CAS PubMed Google Scholar * Staal FJ, Clevers HC. WNT signalling and haematopoiesis: a WNT–WNT situation. Nat Rev Immunol. 2005;5:21–30. Article CAS
PubMed Google Scholar * Wang Y, Krivtsov AV, Sinha AU, North TE, Goessling W, Feng Z, et al. The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML.
Science. 2010;327:1650–3. Article CAS PubMed PubMed Central Google Scholar * Yeung J, Esposito MT, Gandillet A, Zeisig BB, Griessinger E, Bonnet D, et al. β-Catenin mediates the
establishment and drug resistance of MLL leukemic stem cells. Cancer Cell. 2010;18:606–18. Article CAS PubMed Google Scholar * Zheng X, Beissert T, Kukoc-Zivojnov N, Puccetti E,
Altschmied J, Strolz C, et al. Gamma-catenin contributes to leukemogenesis induced by AML-associated translocation products by increasing the self-renewal of very primitive progenitor cells.
Blood. 2004;103:3535–43. Article CAS PubMed Google Scholar * Xu J, Suzuki M, Niwa Y, Hiraga J, Nagasaka T, Ito M, et al. Clinical significance of nuclear non-phosphorylated beta-catenin
in acute myeloid leukaemia and myelodysplastic syndrome. Br J Haematol. 2008;140:394–401. Article CAS PubMed PubMed Central Google Scholar * Ysebaert L, Chicanne G, Demur C, De Toni F,
Prade-Houdellier N, Ruidavets JB, et al. Expression of beta-catenin by acute myeloid leukemia cells predicts enhanced clonogenic capacities and poor prognosis. Leukemia. 2006;20:1211–6.
Article CAS PubMed Google Scholar Download references ACKNOWLEDGEMENTS This work was supported by grants from the Werner und Hedy Berger-Janser Stiftung, Swiss National Science
Foundation, and the Stiftung für klinisch-experimentelle Tumorforschung (Bern). The authors thank Ursina Lüthi for excellent technical support. The authors thank the staff of the FACSLab
(Department of BioMedical Research (DBMR), University of Bern, Switzerland) for providing technical assistance. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Tumor Immunology, Department for
BioMedical Research (DBMR), University of Bern, Bern, Switzerland Ramin Radpour, Carsten Riether, Sabine Höpner & Adrian F. Ochsenbein * Department of Medical Oncology, Inselspital,
Bern University Hospital, University of Bern, Bern, Switzerland Ramin Radpour, Carsten Riether, Sabine Höpner & Adrian F. Ochsenbein * Interfaculty Bioinformatics Unit and SIB Swiss
Institute of Bioinformatics, University of Bern, Bern, Switzerland Cedric Simillion & Rémy Bruggmann Authors * Ramin Radpour View author publications You can also search for this author
inPubMed Google Scholar * Carsten Riether View author publications You can also search for this author inPubMed Google Scholar * Cedric Simillion View author publications You can also search
for this author inPubMed Google Scholar * Sabine Höpner View author publications You can also search for this author inPubMed Google Scholar * Rémy Bruggmann View author publications You
can also search for this author inPubMed Google Scholar * Adrian F. Ochsenbein View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS RR designed
and performed experiments, analyzed and interpreted data, and wrote the manuscript. CR designed experiments and interpreted data. CS and RB analyzed the data. SH performed experiments. AFO
designed experiments, wrote the manuscript, and supervised the project. CORRESPONDING AUTHOR Correspondence to Adrian F. Ochsenbein. ETHICS DECLARATIONS CONFLICT OF INTEREST The authors
declare that they have no conflict of interest. ADDITIONAL INFORMATION PUBLISHER’S NOTE: Springer Nature remains neutral with regard to jurisdictional claims in published maps and
institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY MATERIALS AND METHODS SUPPLEMENTARY FIGURES AND TABLES SUPPLEMENTARY DATASET 1 (LIST OF DIFFERENTIALLY EXPRESSED GENES)
SUPPLEMENTARY DATASET 2 (LIST OF HUB AND NODE GENES AS WELL AS THEIR CORRELATIONS) SUPPLEMENTARY NETWORKS (CORRELATION NETWORKS) RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed
under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article
are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and
your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this
license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Radpour, R., Riether, C., Simillion, C. _et al._ CD8+ T cells expand
stem and progenitor cells in favorable but not adverse risk acute myeloid leukemia. _Leukemia_ 33, 2379–2392 (2019). https://doi.org/10.1038/s41375-019-0441-9 Download citation * Received:
15 November 2018 * Revised: 08 February 2019 * Accepted: 21 February 2019 * Published: 15 March 2019 * Issue Date: October 2019 * DOI: https://doi.org/10.1038/s41375-019-0441-9 SHARE THIS
ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard
Provided by the Springer Nature SharedIt content-sharing initiative