Play all audios:
ABSTRACT Pediatric myelodysplastic syndromes (MDS) are a heterogeneous disease group associated with impaired hematopoiesis, bone marrow hypocellularity, and frequently have deletions
involving chromosome 7 (monosomy 7). We and others recently identified heterozygous germline mutations in _SAMD9_ and _SAMD9L_ in children with monosomy 7 and MDS. We previously demonstrated
an antiproliferative effect of these gene products in non-hematopoietic cells, which was exacerbated by their patient-associated mutations. Here, we used a lentiviral overexpression
approach to assess the functional impact and underlying cellular processes of wild-type and mutant _SAMD9_ or _SAMD9L_ in primary mouse or human hematopoietic stem and progenitor cells
(HSPC). Using a combination of protein interactome analyses, transcriptional profiling, and functional validation, we show that SAMD9 and SAMD9L are multifunctional proteins that cause
profound alterations in cell cycle, cell proliferation, and protein translation in HSPCs. Importantly, our molecular and functional studies also demonstrated that expression of these genes
and their mutations leads to a cellular environment that promotes DNA damage repair defects and ultimately apoptosis in hematopoietic cells. This study provides novel functional insights
into SAMD9 and SAMD9L and how their mutations can potentially alter hematopoietic function and lead to bone marrow hypocellularity, a hallmark of pediatric MDS. SIMILAR CONTENT BEING VIEWED
BY OTHERS FBXO11 IS A CANDIDATE TUMOR SUPPRESSOR IN THE LEUKEMIC TRANSFORMATION OF MYELODYSPLASTIC SYNDROME Article Open access 06 October 2020 CLINICAL EVOLUTION, GENETIC LANDSCAPE AND
TRAJECTORIES OF CLONAL HEMATOPOIESIS IN SAMD9/SAMD9L SYNDROMES Article 07 October 2021 U2AF1 IS REQUIRED FOR SURVIVAL AND FUNCTION OF HEMATOPOIETIC STEM/PROGENITOR CELLS Article 07 January
2021 INTRODUCTION Pediatric myelodysplastic syndromes (MDS) are bone marrow (BM) neoplasms that are characterized by hematopoietic cell dysfunction, an increased risk of developing acute
myeloid leukemia (AML), and a poor prognosis [1]. Unlike MDS in adults, children with MDS more commonly have BM hypocellularity, a higher frequency of chromosome 7 deletions (monosomy 7),
and a distinct set of genetic alterations, as previously shown by our group [2]. We recently reported on the genomic landscape of pediatric primary MDS and identified germline mutations in
sterile alpha motif (SAM) domain-9 (_SAMD9_) and its paralog, SAMD9-like (_SAMD9L_), in 17% of pediatric MDS patients [2], and similar findings have been reported by others [2, 3]. We
further demonstrated that several germline mutations in SAMD9 (SAMD9-E1136Q) or SAMD9L (SAMD9L-H880Q, -W1180R, and -R1281K) caused significant decreases in proliferation and cell cycle
progression in non-hematopoietic cells [3,4,5]. _SAMD9_ and _SAMD9L_ are inflammatory-inducible genes located at human 7q21 and, surprisingly, these germline mutant alleles are
preferentially absent in the cells with monosomy 7, a process known as adaptation by aneuploidy, suggesting a strong selective pressure against expression of these mutations in hematopoietic
cells [3, 4]. Similar mutations in _SAMD9_ and _SAMD9L_ have previously been observed in disorders associated with BM abnormalities, such as MIRAGE syndrome and Ataxia-Pancytopenia syndrome
(ATXPC), respectively [6,7,8,9]. Although the loss of _Samd9l_ in mice results in a myelodysplasia-like phenotype [10], the function of SAMD9 and SAMD9L, and the impact of pathogenic
germline mutations, in hematopoietic cells has yet to be determined. SAMD9 and SAMD9L are large proteins (>1500 amino acids) and share ~60% amino acid identity, with conserved functional
domains, including those involved in DNA/RNA-binding, protein binding, apoptosome formation, and NTP hydrolysis activity [11]. This complex structural feature suggests that the expression of
these genes could lead to multiple phenotypes depending on the cellular environment and context. In support of this hypothesis, SAMD9 and SAMD9L are known Poxvirus restriction factors and
are inhibited by directly binding to virally encoded proteins [12, 13]. Further, _Samd9l_ knockout mice have defects in endosomal processing and fibroblasts from SAMD9-mutant patients have
variable defects in Rab5-positive early endosome size [12,13,14]. In contrast, the mutations associated with MDS, MIRAGE, and ATXPC lead to growth arrest. Importantly, the majority of the
described pathogenic germline mutations in SAMD9 and SAMD9L occur in the C-terminal half of the protein within the highly conserved APAF-1-like domain, which contains the predicted core
functional NTPase domain [3,4,5, 7, 13]. This suggests that this domain is critical for SAMD9 and SAMD9L function in hematopoietic cells and in the development of pediatric MDS. In this
study, we used an ex vivo overexpression model to decipher the cellular function of SAMD9 and SAMD9L proteins in primary human and mouse hematopoietic cells. Using a range of proteomics,
transcriptomics, and cellular assays, we provide evidence that SAMD9 and SAMD9L regulate multiple key cellular processes, which can lead to cellular stress when dysregulated. We show that
overexpression of SAMD9 and SAMD9L regulates hematopoietic cell proliferation, cell cycle, protein translation, DNA damage response, and apoptosis. Importantly, the pathogenic mutations in
either SAMD9 or SAMD9L intensify these phenotypes. Revealing the functionality of SAMD9 and SAMD9L, as well as the impact of their pathogenic mutations, is a critical first step in
understanding the development of pediatric MDS and potentially other pediatric hematopoietic disorders characterized by BM hypocellularity, such as BM failure syndromes. RESULTS SAMD9 AND
SAMD9L REGULATE HEMATOPOIETIC CELL PROLIFERATION AND DIFFERENTIATION Our previous studies in HEK293T cells demonstrated that exogenous expression of SAMD9 or SAMD9L resulted in reduced cell
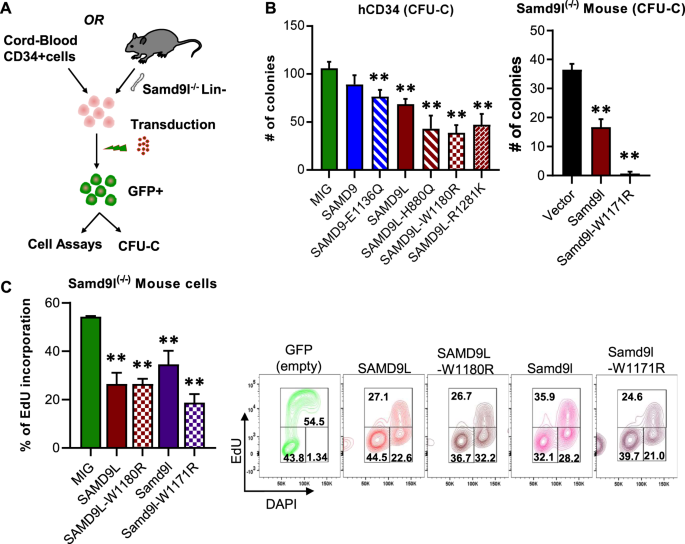
growth and that this effect was exacerbated by patient-specific mutations [3,4,5]. We developed a lentiviral overexpression model of SAMD9 and SAMD9L in primary human or mouse hematopoietic
stem and progenitor cells (HSPC) to test the effects of these mutations in a more relevant hematopoietic system (Fig. 1A). This approach uses a lentiviral MSCV-IRES-eGFP (MIG) vector to
overexpress wild-type SAMD9, SAMD9L, or mutations that we have previously identified in children with MDS (SAMD9: E1136Q; SAMD9L: H880Q, W1180R, or R1281K) [3, 4]. While human cells express
both _SAMD9_ and _SAMD9L_, the mouse genome only encodes for _Samd9l_, likely due to evolutionary conservation and functional redundancy with _SAMD9_ [14, 15]. By using lineage-depleted
(Lin−) HSPC from a previously characterized _Samd9l_−/− mouse model [10], we are able to validate our studies in a _Samd9l_-null background that both eliminates basal expression and allows
us to determine the functional and homologous redundancies between human SAMD9L, human SAMD9, and mouse Samd9l. Overexpression of wild-type or mutant SAMD9 and SAMD9L in human CD34+ cells
leads to a clear suppression in colony formation (Fig. 1B). Subtyping of the colonies shows a variable enrichment of CFU-GM colonies in the SAMD9 and SAMD9L samples relative to the GFP
vector control, accompanied by a loss in BFU-Es and the less-committed multipotential colonies, CFU-GEMM (Fig. S1A). This is also further confirmed by flow cytometry studies, where we
observed a proportional increase in CD45+CD11b+ cells over CD45−CD71+ cells in all the SAMD9 and SAMD9L groups compared to the control (Fig. S1B). Similarly, overexpression of wild-type
mouse Samd9l in _Samd9l_−/− murine HSPCs showed a significant reduction in the number of CFU-Cs relative to the control, and this effect was markedly enhanced by expressing a W1171R mutation
(similar to human W1180R) (Figs. 1B and S1C). These changes in CFU numbers were further supported by alterations in cell cycle, namely an accumulation in G2/M phase and decreases in S-phase
in both CD34+ cells and Samd9l−/− HSPCs (Fig. S1D) compared to vector controls. These data were confirmed by assessing DNA synthesis at S-phase using the EdU incorporation assay.
Overexpression of human or mouse SAMD9L and their homologous W1180R/W1171R mutations lead to a significant decrease in cells in S-phase and an accumulation in G2/M, and the mouse Samd9l
mutation intensifies this effect (Fig. 1C). Together our data suggest that overexpression of SAMD9 or SAMD9L impairs hematopoietic cell growth and that the expression of their pathogenic
mutations augments this phenotype. SAMD9 AND SAMD9L PROTEIN INTERACTOME AND THE REGULATION OF SEVERAL PATHWAYS INCLUDING RIBOSOME ASSEMBLY We next took a proteomics approach to understand
the function of the SAMD9 and SAMD9L proteins. We used APEX2, a proximity-induced labeling system that is able to effectively mark proteins within a 20 Å radius with biotin to capture both
dynamic and stable protein interacting partners [16]. HEK293T cells were co-transfected with a GFP-SAMD9 or GFP-SAMD9L fusion (including mutations) and a GFP Binding-Protein APEX2 fusion
(GBP-APEX2). Biotin labeled protein partners were isolated from cells using streptavidin resin and analyzed using mass spectrometry or western blotting. This approach revealed a significant
number of interacting proteins (_p_ value <0.05), many of which were present in all conditions compared to vector control. SAMD9 and SAMD9-E1136Q shared 66.2% interacting proteins, while
SAMD9L and SAMD9L-H880Q shared 66.9%. SAMD9 and SAMD9L shared 72.4% of interacting proteins, indicating significant redundancy in the interactome of SAMD9 and SAMD9L. There were 243
interacting proteins with a ≥5-fold increase in abundance shared amongst all four genotypes relative to vector controls (Fig. 2A). String-based KEGG pathway analysis [17] of the 243 shared
proteins revealed several enriched pathways, including ribosome, spliceosome, RNA transport, and DNA repair (Figs. 2A and S2A). Interactions with key proteins from these pathways (DHX9, DDX1
eIF3A, PRDKC, and SF3B1) were validated by IP-western blot (Fig. 2B). SAMD9 AND SAMD9L PLAY A ROLE IN RIBOSOME ASSEMBLY AND CONSEQUENTLY IN PROTEIN SYNTHESIS Given the enrichment of
ribosomal proteins (15 of the 243 common proteins) in the SAMD9/SAMD9L interactome, we sought to determine if SAMD9 and SAMD9L associate with different ribosomal subunits during polysome
formation. We performed polysome profiling in HEK293T cells and isolated proteins from sucrose gradient fractions using methanol:chloroform extractions as previously described [18].
Overexpression of SAMD9 and SAMD9L and their mutations altered ribosome assembly distribution with an accumulation in the 80S peak (Fig. 2C). Western blot analysis of sucrose gradient
fractions confirmed this observation and showed a clear association of SAMD9 and SAMD9L with the ribosomal assembly components by their co-elution with positive controls RPS6 and RPL11 [18]
and the absence of the negative control, αTubulin (Figs. 2C and S2B). The majority of SAMD9 associates with pre-polysome (subunits and monosomes) and early polysome complexes while the
SAMD9-E1136Q mutation had a strong association with pre-polysomes and late polysomes. Both SAMD9L and SAMD9L-H880Q have a majority pre-polysome assembly pattern. To further validate this
phenotype, we used CRISPR-Cas9 engineering to establish a K562 cell line with a SAMD9-E1136Q mutation. Notably, K562 cells are tetraploid for chromosome 7 and we modified two of the four
alleles, thus mimicking the heterozygous mutation state observed in patients while maintaining its physiological expression levels through its endogenous locus. We then performed polysome
profiling assays with IFNα treatment to induce SAMD9 expression. These modified isogenic K562 cells have perturbations in their polysome profile, when compared to control K562 cells, which
are similar to those observed in our overexpression models, and IFNα treated cells exacerbate this phenotype (Figs. 2D and S2C). The association of SAMD9 and SAMD9L with proteins in the
ribosome and translation pathways suggests that protein synthesis may be altered by SAMD9 or SAMD9L expression. Using the O-propargyl-puromycin (OPP) Click-it assay, which quantifies the
incorporation of a puromycin analog into newly synthesized peptides [19], we found that both wild-type SAMD9 and SAMD9L suppressed protein synthesis rate and that the pathogenic mutants
exacerbated this phenotype in HEK293T cells (Fig. S3A). Notably, overexpression of wild-type SAMD9, SAMD9L, or Samd9l did not affect protein synthesis in CD34+ and Samd9l−/− HSPCs, whereas
the pathogenic mutants suppressed protein synthesis (Figs. 2E and S3B–D), clearly demonstrating a mutant specific phenotype in primary hematopoietic cells. Similarly, CRISPR-engineered K562
cells expressing SAMD9-E1136Q show a decrease in translation compared to control cells, which is further exaggerated in IFNα treated cells compared to the control (Fig. 2F). Together, these
data show that SAMD9 and SAMD9L interact with ribosomal assembly complexes and their pathogenic mutations lead to suppression of protein translation in hematopoietic cells. FUNCTIONAL
DOMAINS OF SAMD9 AND SAMD9L INFLUENCE THEIR CELLULAR PHENOTYPES We next sought to assess how the conserved domains within SAMD9 and SAMD9L impacted these cellular phenotypes using a series
of domain-based truncations with or without patient-derived mutations (Figs. 3A and S4A, B). Interestingly, the deletion of either the amino-terminal SAM domain (protein/RNA interaction
domain) or the carboxy-terminal OB-fold domain (DNA/RNA-binding domain) of SAMD9 or SAMD9L negated the mutation-dependent cell cycle phenotype in CD34+ cells (Fig. 3B). Consistently, we
observed a similar pattern of protein synthesis rescue upon expression of each truncation in both human CD34+ cells and Samd9l−/− HSPCs (Fig. 3C, D and S4C). Collectively, these data suggest
that the SAM and OB-fold domains are critical functional domains required for cell cycle and translation regulation. To address whether these phenotypes could be associated with changes in
subcellular localization resulting from the pathogenic mutations or domain truncations, we next evaluated their expression patterns by confocal microscopy. Overexpression of SAMD9, SAMD9L,
and their variants showed distinct patterns of localization confined to the cytosol (Fig. S4D), as previously described [15]. Despite reports that SAMD9 and SAMD9L regulate endosome fusion,
colocalization with RAB5, a marker of early endosomes, was not observed [10]. Noticeably, while SAMD9 and SAMD9-E1136Q expression leads to a punctate pattern, SAMD9L and SAMD9L-H880Q
expression is exclusively diffuse throughout the cytosol. The deletion of the SAM domain of SAMD9 completely abolished puncta formation, unlike the OB-fold domain deletion (Fig. S4D).
Consistently, deletion of the SAM domain and the APAF-1-like domain of SAMD9 and SAMD9L completely rescued the cell cycle and protein synthesis phenotypes in HEK293T (Fig. S5A–D). Deletion
of the SAM or OB-fold domain alone decreased the interactions with several targets required for protein synthesis initiation, double-strand break repair, and RNA-splicing, including eIF3A,
DDX1, and SF3B1, respectively (Fig. 3F). Taken together, these data demonstrate that both the SAM domain and the OB-fold domain are required for SAMD9 and SAMD9L cellular activity.
OVEREXPRESSION OF SAMD9 AND SAMD9L PERTURBS MULTIPLE PATHWAYS IN CD34+ CELLS We next performed RNA-sequencing in human CD34+ cells overexpressing SAMD9, SAMD9L, or their mutations to further
determine the global pathways that are dysregulated by the expression of these genes (Fig. S6A–D). Differentially expressed genes with an FDR ≤0.05 were identified for the different SAMD9
and SAMD9L genotypes relative to control (Fig. 4A). Pathway-enrichment analyses of these differentially expressed genes and gene set enrichment analysis revealed consistent SAMD9- and
SAMD9L-dependent upregulation of inflammatory signaling pathways, such as TNFα via NFκB and IFN-α/β, and apoptosis signaling pathways. The downregulated pathways included DNA replication,
DNA repair, cell cycle, E2F targets, and MYC pathway targets (Figs. 4B–D and S7A–D and Table S1). Importantly, expression of mutant SAMD9 or SAMD9L leads to further enrichment in these
dysregulated pathways. Interestingly, there was consistent downregulation of the minichromosome maintenance complex (_MCM_) family, which has been previously associated with replicative
stress in hematopoietic cells, with mutant SAMD9L having the strongest downregulation (Fig. S6E) [19, 20]. Collectively, our transcriptomic analyses indicated a series of cellular stresses
and responses to cellular stress (e.g., cell cycle arrest and translation inhibition) mediated by SAMD9 and SAMD9L expression, many of which are exacerbated by pathogenic mutations
identified in pediatric MDS. SAMD9 AND SAMD9L REGULATE DNA DAMAGE REPAIR AND APOPTOSIS IN HEMATOPOIETIC CELLS Our proteomic studies (Fig. 2A, B) revealed an interaction of SAMD9 and SAMD9L
with PRDKC and DDX1, known DNA repair pathway factors [21,22,23] and our transcriptomic studies also supported a link with DNA damage (Figs. 4B–E and S8A). Therefore, we functionally
investigated the link between SAMD9/SAMD9L expression and DNA damage by analyzing gamma-H2AX (γH2AX) levels in CD34+ cells. The expression of SAMD9, SAMD9-E1136Q, SAMD9L, and SAMD9L-H880Q
significantly increased the intensity of γH2AX compared to control (Fig. 5A). Similarly, overexpression of human and mouse SAMD9L genes in Samd9l−/− HSPCs also leads to an increase in γH2AX
levels (Fig. 5B). Consistent with this DNA repair defect, live cell imaging showed nuclear condensation in _Samd9l_−/− HSPCs overexpressing wild-type or mutant _Samd9l_ relative to GFP
vector (Fig. S8B). Our transcriptomics data also pointed to apoptosis as a consequence of SAMD9 and SAMD9L expression (Figs. 4B–E and S8C). Consistent with these transcriptomic data,
expression of SAMD9, SAMD9-E1136Q, SAMD9L, and SAMD9L-H880Q caused a significant increase in CD34+ cells undergoing early and late apoptosis compared to the control (Fig. 5C), as measured by
Annexin v/DAPI staining. We also observed increased levels of apoptosis in Samd9l−/− HSPCs expressing human SAMD9L, SAMD9L-H880Q, mouse Samd9l, and Samd9l-W1171R (Fig. 5D). DISCUSSION
_SAMD9_ and _SAMD9L_ have recently been described as new germline predisposition genes in pediatric MDS and in several multisystem disorders such as MIRAGE and ATXPC syndromes
[3,4,5,6,7,8,9]. Many patients with _SAMD9_ and _SAMD9L_ mutations have outgrowth of cells with non-random loss (or inactivation) of the germline variant, presumably as a cellular adaptation
to the mutation-associated growth restriction [2, 4, 5]. The resulting haploinsufficiency of genes located on chromosome 7 can lead to MDS or AML, especially when additional cooperating
mutations are somatically acquired [5]. Despite the now strong association of germline _SAMD9_ and _SAMD9L_ variants with MDS, the cellular impact of both wild-type or mutant SAMD9 (or
SAMD9L) in hematopoietic cells has yet to be elucidated. Here, we used a series of proteomic, transcriptomic, and cell biology methodologies to show that mutant SAMD9 or SAMD9L expression
results in profound perturbations of cellular processes in hematopoietic cells, including disruption of protein synthesis and cell cycle, while also activating DNA damage responses and
apoptosis. Dysregulation of ribosomal biology and protein synthesis is a common finding in pediatric BM disorders, including Diamond-Blackfan anemia, Shwachman-Diamond syndrome, and
dyskeratosis congenital [24,25,26,27,28]. Importantly, these alterations lead to defects in translation and induce ribosomal stress, causing apoptosis [28]. The multi-omic and functional
data presented here extend these associations to SAMD9 and SAMD9L-related syndromes. Not only does expression of mutant _SAMD9_ or _SAMD9L_ impair the rate of protein synthesis, but the
SAMD9 and SAMD9L proteins are associated with components of the polysome complex and with proteins linked to ribosome assembly and translation, such as EIF3A and DHX9 [29, 30]. The
association of SAMD9 and SAMD9L to protein synthesis defects in hematopoietic cells is perhaps not surprising considering their role as Poxvirus restriction factors, in which they
specifically block cap-dependent and -independent translation of intermediate and late viral mRNAs [31]. Despite the relative lack of mutations in components of the splicing machinery in
pediatric MDS when compared to MDS in adults [2, 32, 33], the physical proximity of SAMD9 and SAMD9L to RNA helicases and splicing factors, like DDX1 and SF3B1, suggests that RNA processing
may be a more conserved feature of MDS across the age spectrum than previously recognized. The RNA-binding domains of SAMD9 and SAMD9L also suggest there may be a direct interaction with RNA
transcripts that could regulate both translation and splicing [11]. However, additional mechanistic studies are clearly required to pursue how SAMD9 and SAMD9L influence these processes. To
our knowledge, our data are the first to show that defects in the DNA repair pathway are a functional consequence of SAMD9 and SAMD9L expression. The genomic instability that results from
cell cycle arrest, ribosomal perturbations, and DNA damage is a key driver in the development of MDS [34,35,36,37,38]. Alterations of DNA repair genes drive the progression to MDS in Fanconi
anemia [39] and unrepaired DNA defects in hematopoietic cells cause remarkable long-term functional perturbations and represent a primary driving force of accrual of additional mutations,
which in turn promote clonal expansion and malignant transformation [38, 40,41,42]. We likewise showed that cells expressing SAMD9 or SAMD9L mutants accumulate in G2/M, a key checkpoint for
DNA damage [43, 44]. Consistently, our transcriptomic data further support the potential impact on cell cycle and DNA repair pathways with downregulation of notable genes, such as _MYC_,
_CDK6, E2F1_, _POLE_, and several members of the _MCM_ family. Intriguingly, all of these genes were further downregulated when pathogenic SAMD9 or SAMD9L mutations were present. Taken
together, the observed phenotypes resemble the DNA-replicative stress evoked by _MCM_ genes downregulation in aging HSPCs, which is associated with activation of γH2AX alongside cell cycle
abnormalities [19, 45]. A hallmark of pediatric MDS is BM hypocellularity and low peripheral blood counts [46]. Indeed, many of the SAMD9/SAMD9L mutations originally described by our group
in pediatric MDS were found in patients with refractory cytopenia of childhood [2], which is predominantly associated with a hypocellular BM phenotype. Likewise, the study by Bluteau et al.
also linked _SAMD9_ and _SAMD9L_ germline mutations to BM failure syndromes, which are a hypo-proliferative group of BM disorders [7]. This is in stark contrast to the hypercellular BM that
are more commonly observed in adults with MDS. These observations alone suggest a distinct pathobiology of MDS in children and adults. As shown in this study, overexpression SAMD9 or SAMD9L
in primary hematopoietic cells results in decreased proliferation and increased apoptosis, which would ultimately lead to the hypocellular phenotype observed in patients. We speculate (see
Fig. 6) that the observed effects on ribosomal biology, DNA damage, and the resulting genomic instability can drive the observed apoptotic phenotype [26, 30, 40,41,42], ultimately leading to
decrease cellularity in the BM. Unrepaired DNA defects in hematopoietic cells cause remarkable long-term functional perturbations and represent a primary driving force for accrual of
additional mutations, which in turn promote clonal expansion and malignant transformation [39, 46,47,48,49]. Clinical data suggest that there are multiple adaptive mechanisms to address this
cellular stress, including the outgrowth of cells with somatic revertant mutations and chromosome 7 deletions, all of which are potential outcomes of children harboring germline SAMD9 or
SAMD9L mutations [3, 6]. Our data also highlight an important functional redundancy between SAMD9 and SAMD9L, including both human and mouse for SAMD9L. Despite their large size (over 1500
amino acids), SAMD9 and SAMD9L share ~60% amino acid identity with multiple conserved sequence domains, and thus their functional redundancy is not surprising. Here, we show that the
expression of both proteins leads to largely identical phenotypes in hematopoietic cells, with SAMD9L consistently having the strongest effect. Notably, the human genome encodes for both
_SAMD9L_ and _SAMD9_, while murine cells only contain _Samd9l_. This lack of redundancy in the mouse genome may explain why the overall phenotypes observed in the _Samd9l-_null cells were
more pronounced than those in cord blood CD34 cells. This suggests that murine models may be more advantageous to further map the function of these proteins. Whether these two proteins in
humans are truly functionally redundant requires more extensive investigation into their structure and function. One potential source of heterogeneity may be epigenetically based. For
example, SAMD9 is considered a constitutive restriction factor for Poxvirus infection, while SAMD9L is induced under inflammatory conditions [13]. In summary, we demonstrated for the first
time the immediate impact of SAMD9 and SAMD9L overexpression in primary hematopoietic cells and our work has provided critical insights into the underlying biology that leads to the
hypocellular phenotype seen in many children with SAMD9 and SAMD9L germline mutations. Considering that many cases of pediatric MDS, like BM failure syndromes, are defined by BM
hypo-proliferation, these findings may extend to a larger percentage of pediatric myeloid disorders and may ultimately provide new therapeutic options for future investigation. METHODS
ANIMALS Samd9l−/− mice were kindly provided by the RIEKN BRC through the National Bio-Resource Project of the MEXT, Japan with approval from Dr. Hiroaki Honda [10]. Animal studies were
approved by St. Jude Children’s Research Hospital Institutional Animal Care and Use Committee. Hematopoietic cells were selected from the flushed bones using the lineage-depletion EasySep
Mouse HSPC Kit (StemCell Technologies, Canada) [50]. CELL CULTURE Human cord blood-derived CD34+ cells (Lonza, Switzerland) were cultured in expansion medium containing StemSpan SFEM-II
(StemCell Technologies, Canada) enriched with human cytokines (PeproTech, NJ) including interleukin-6 (100 ng/ml), Fms-like tyrosine kinase-3 ligand (FLT3-L, 100 ng/ml), Stem Cell Factor
(SCF, 100 ng/ml), Thrombopoietin (100 ng/ml), 1 µM Stem Regenin-1 and 35 nM UM171 (StemCell Technologies, Canada). For mouse cells, Samd9l−/− HSPCs were expanded overnight in RPMI
(ThermoFisher, MA) with 10% FBS and supplemented with the murine (6–8 weeks) cytokines including interleukin-3, interleukin-6, SCF, Thrombopoietin, and FLT3-L (PeproTech, NJ). FLOW CYTOMETRY
SORTING AND ANALYSIS Transduced cells (as measured by GFP positivity) were sorted using the FACSAria sorter (BD Biosciences, CA). Analytical flow cytometry was done using LSR FORTESSAII (BD
Biosciences, CA). For intracellular staining, transduced cells were harvested after the indicated time, fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton X-100, and stained
with the appropriate antibodies (Table S2). EdU incorporation assay was done using the Click-iT Plus EdU Kit (Invitrogen, CA) after incubating the cells with 10 µM EdU for 2 h as previously
described [2]. The translation rates were examined using Click-iT Plus OPP Protein Synthesis Kit (Invitrogen, CA) where the cells were incubated with 10 µM OPP for 30 min and assessed as
previously described [50]. DNA damage was measured using anti-phospho-H2AX(Ser139) antibody (BioLegend, CA). For DNA content, cells were stained with NuclearMask (Invitrogen, CA). For
apoptosis assessment, transduced cells were cultured for 72 h, blocked with binding buffer with 5% rat serum, and stained with Annexin-V antibody and DAPI [51]. All data were analyzed using
FlowJo software (TreeStar, OR) and presented as mean fluorescence intensities or histograms. DATA PRESENTATION AND STATISTICAL ANALYSIS All graphs were generated using GraphPad Prism 8.0
(San Diego, CA). One-way ANOVA with Bonferroni-correction was used for statistical analyses. Statistical significance was set at *_p_ < 0.05, and **_p_ < 0.01 compared to GFP vector
control unless stated differently. Additional methods are listed in the supplemental data. DATA AVAILABILITY RNA-seq data were deposited into Gene Expression Omnibus (GEO) (accession number:
GSE152420). IP-MS data were deposited into Proteomics Identification Database (PRIDE) (px-submission #428150). REFERENCES * Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau
MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–405. Article CAS PubMed Google Scholar *
Schwartz JR, Ma J, Lamprecht T, Walsh M, Wang S, Bryant V, et al. The genomic landscape of pediatric myelodysplastic syndromes. Nat Commun. 2017;8:1557. Article PubMed PubMed Central CAS
Google Scholar * Pastor VB, Sahoo SS, Boklan J, Schwabe GC, Saribeyoglu E, Strahm B, et al. Constitutional SAMD9L mutations cause familial myelodysplastic syndrome and transient monosomy
7. Haematologica. 2018;103:427–37. Article CAS PubMed PubMed Central Google Scholar * Tesi B, Davidsson J, Voss M, Rahikkala E, Holmes TD, Chiang SCC, et al. Gain-of-function SAMD9L
mutations cause a syndrome of cytopenia, immunodeficiency, MDS, and neurological symptoms. Blood. 2017;129:2266–79. Article CAS PubMed PubMed Central Google Scholar * Wong JC, Bryant V,
Lamprecht T, Ma J, Walsh M, Schwartz J, et al. Germline SAMD9 and SAMD9L mutations are associated with extensive genetic evolution and diverse hematologic outcomes. JCI Insight.
2018;3:e121086. * Schwartz JR, Wang S, Ma J, Lamprecht T, Walsh M, Song G, et al. Germline SAMD9 mutation in siblings with monosomy 7 and myelodysplastic syndrome. Leukemia. 2017;31:1827–30.
Article CAS PubMed PubMed Central Google Scholar * Bluteau O, Sebert M, Leblanc T, Peffault de Latour R, Quentin S, Lainey E, et al. A landscape of germ line mutations in a cohort of
inherited bone marrow failure patients. Blood. 2018;131:717–32. Article CAS PubMed Google Scholar * Narumi S, Amano N, Ishii T, Katsumata N, Muroya K, Adachi M, et al. SAMD9 mutations
cause a novel multisystem disorder, MIRAGE syndrome, and are associated with loss of chromosome 7. Nat Genet. 2016;48:792–7. Article CAS PubMed Google Scholar * Davidsson J, Puschmann A,
Tedgård U, Bryder D, Nilsson L, Cammenga J. SAMD9 and SAMD9L in inherited predisposition to ataxia, pancytopenia, and myeloid malignancies. Leukemia. 2018;32:1106–15. Article CAS PubMed
PubMed Central Google Scholar * Nagamachi A, Matsui H, Asou H, Ozaki Y, Aki D, Kanai A, et al. Haploinsufficiency of SAMD9L, an endosome fusion facilitator, causes myeloid malignancies in
mice mimicking human diseases with monosomy 7. Cancer Cell. 2013;24:305–17. Article CAS PubMed Google Scholar * Mekhedov SL, Makarova KS, Koonin EV. The complex domain architecture of
SAMD9 family proteins, predicted STAND-like NTPases, suggests new links to inflammation and apoptosis. Biol Direct. 2017;12:13. Article PubMed PubMed Central CAS Google Scholar * Liu J,
McFadden G. SAMD9 is an innate antiviral host factor with stress response properties that can be antagonized by poxviruses. J Virol. 2015;89:1925–31. Article PubMed CAS Google Scholar *
Meng X, Zhang F, Yan B, Si C, Honda H, Nagamachi A, et al. A paralogous pair of mammalian host restriction factors form a critical host barrier against poxvirus infection. PLoS Pathog.
2018;14:e1006884. Article PubMed PubMed Central CAS Google Scholar * Buonocore F, Kühnen P, Suntharalingham JP, Del Valle I, Digweed M, Stachelscheid H, et al. Somatic mutations and
progressive monosomy modify SAMD9-related phenotypes in humans. J Clin Invest. 2017;127:1700–13. Article PubMed PubMed Central Google Scholar * Li CF, MacDonald JR, Wei RY, Ray J, Lau K,
Kandel C, et al. Human sterile alpha motif domain 9, a novel gene identified as down-regulated in aggressive fibromatosis, is absent in the mouse. BMC Genomics. 2007;8:92. Article PubMed
PubMed Central CAS Google Scholar * Lobingier BT, Hüttenhain R, Eichel K, Miller KB, Ting AY, von Zastrow M, et al. An approach to spatiotemporally resolve protein interaction networks in
living cells. Cell. 2017;169:350–60.e12. Article CAS PubMed PubMed Central Google Scholar * Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, et al. STRING v11:
protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47:D607–13. Article CAS PubMed
Google Scholar * Karamysheva ZN, Tikhonova EB, Grozdanov PN, Huffman JC, Baca KR, Karamyshev A, et al. Polysome profiling in leishmania, human cells and mouse testis. J Vis Exp. 2018:57600.
* Flach J, Bakker ST, Mohrin M, Conroy PC, Pietras EM, Reynaud D, et al. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature.
2014;512:198–202. Article CAS PubMed PubMed Central Google Scholar * Signer RA, Magee JA, Salic A, Morrison SJ. Haematopoietic stem cells require a highly regulated protein synthesis
rate. Nature. 2014;509:49–54. Article CAS PubMed PubMed Central Google Scholar * Alvarez S, Díaz M, Flach J, Rodriguez-Acebes S, López-Contreras AJ, Martínez D, et al. Replication
stress caused by low MCM expression limits fetal erythropoiesis and hematopoietic stem cell functionality. Nat Commun. 2015;6:8548. Article CAS PubMed Google Scholar * Zhang S, Yajima H,
Huynh H, Zheng J, Callen E, Chen HT, et al. Congenital bone marrow failure in DNA-PKcs mutant mice associated with deficiencies in DNA repair. J Cell Biol. 2011;193:295–305. Article CAS
PubMed PubMed Central Google Scholar * Li L, Germain DR, Poon HY, Hildebrandt MR, Monckton EA, McDonald D, et al. DEAD Box 1 facilitates removal of RNA and homologous recombination at DNA
double-strand breaks. Mol Cell Biol. 2016;36:2794–810. Article CAS PubMed PubMed Central Google Scholar * Horos R, von Lindern M. Molecular mechanisms of pathology and treatment in
Diamond Blackfan Anaemia. Br J Haematol. 2012;159:514–27. CAS PubMed Google Scholar * Padron E, Komrokji R, List AF. Biology and treatment of the 5q- syndrome. Expert Rev Hematol.
2011;4:61–9. Article CAS PubMed Google Scholar * Ruggero D, Shimamura A. Marrow failure: a window into ribosome biology. Blood. 2014;124:2784–92. Article CAS PubMed PubMed Central
Google Scholar * Narla A, Ebert BL. Ribosomopathies: human disorders of ribosome dysfunction. Blood. 2010;115:3196–205. Article CAS PubMed PubMed Central Google Scholar * Tulpule A,
Kelley JM, Lensch MW, McPherson J, Park IH, Hartung O, et al. Pluripotent stem cell models of Shwachman-Diamond syndrome reveal a common mechanism for pancreatic and hematopoietic
dysfunction. Cell Stem Cell. 2013;12:727–36. Article CAS PubMed PubMed Central Google Scholar * Chorghade S, Seimetz J, Emmons R, Yang J, Bresson SM, Lisio M, et al. Poly(A) tail length
regulates PABPC1 expression to tune translation in the heart. Elife. 2017;6:e24139. * Lee T, Pelletier J. The biology of DHX9 and its potential as a therapeutic target. Oncotarget.
2016;7:42716–39. Article PubMed PubMed Central Google Scholar * Sivan G, Glushakow-Smith SG, Katsafanas GC, Americo JL, Moss B. Human host range restriction of the vaccinia virus C7/K1
double deletion mutant is mediated by an atypical mode of translation inhibition. J Virol. 2018;92:e01329-18. * Stankiewicz MJ, Crispino JD. AKT collaborates with ERG and Gata1s to
dysregulate megakaryopoiesis and promote AMKL. Leukemia. 2013;27:1339–47. Article CAS PubMed PubMed Central Google Scholar * Pastor V, Hirabayashi S, Karow A, Wehrle J, Kozyra EJ,
Nienhold R, et al. Mutational landscape in children with myelodysplastic syndromes is distinct from adults: specific somatic drivers and novel germline variants. Leukemia. 2017;31:759–62.
Article CAS PubMed Google Scholar * Niemeyer CM, Baumann I. Myelodysplastic syndrome in children and adolescents. Semin Hematol. 2008;45:60–70. Article CAS PubMed Google Scholar *
Corey SJ, Minden MD, Barber DL, Kantarjian H, Wang JC, Schimmer AD. Myelodysplastic syndromes: the complexity of stem-cell diseases. Nat Rev Cancer. 2007;7:118–29. Article CAS PubMed
Google Scholar * Zhou T, Chen P, Gu J, Bishop AJ, Scott LM, Hasty P, et al. Potential relationship between inadequate response to DNA damage and development of myelodysplastic syndrome. Int
J Mol Sci. 2015;16:966–89. Article CAS PubMed PubMed Central Google Scholar * Belickova M, Merkerova MD, Stara E, Vesela J, Sponerova D, Mikulenkova D, et al. DNA repair gene variants
are associated with an increased risk of myelodysplastic syndromes in a Czech population. J Hematol Oncol. 2013;6:9. Article CAS PubMed PubMed Central Google Scholar * Ribeiro HL Jr.,
de Oliveira RT, Maia AR, Pires Ferreira Filho LI, de Sousa JC, Heredia FF, et al. Polymorphisms of DNA repair genes are related to the pathogenesis of myelodysplastic syndrome. Hematol
Oncol. 2015;33:220–8. Article CAS PubMed Google Scholar * Cioc AM, Wagner JE, MacMillan ML, DeFor T, Hirsch B. Diagnosis of myelodysplastic syndrome among a cohort of 119 patients with
fanconi anemia: morphologic and cytogenetic characteristics. Am J Clin Pathol. 2010;133:92–100. Article CAS PubMed Google Scholar * Yahata T, Takanashi T, Muguruma Y, Ibrahim AA,
Matsuzawa H, Uno T, et al. Accumulation of oxidative DNA damage restricts the self-renewal capacity of human hematopoietic stem cells. Blood. 2011;118:2941–50. Article CAS PubMed Google
Scholar * Zhou T, Hasty P, Walter CA, Bishop AJ, Scott LM, Rebel VI. Myelodysplastic syndrome: an inability to appropriately respond to damaged DNA? Exp Hematol. 2013;41:665–74. Article
CAS PubMed PubMed Central Google Scholar * Bowman RL, Busque L, Levine RL. Clonal hematopoiesis and evolution to hematopoietic malignancies. Cell Stem Cell. 2018;22:157–70. Article CAS
PubMed PubMed Central Google Scholar * Stark GR, Taylor WR. Analyzing the G2/M checkpoint. Methods Mol Biol. 2004;280:51–82. CAS PubMed Google Scholar * Jeggo PA, Löbrich M.
Contribution of DNA repair and cell cycle checkpoint arrest to the maintenance of genomic stability. DNA Repair (Amst). 2006;5:1192–8. Article CAS PubMed Google Scholar *
Martín-Pardillos A, Tsaalbi-Shtylik A, Chen S, Lazare S, van Os RP, Dethmers-Ausema A, et al. Genomic and functional integrity of the hematopoietic system requires tolerance of oxidative DNA
lesions. Blood. 2017;130:1523–34. Article PubMed PubMed Central CAS Google Scholar * Locatelli F, Strahm B. How I treat myelodysplastic syndromes of childhood. Blood. 2018;131:1406–14.
Article CAS PubMed Google Scholar * Milyavsky M, Gan OI, Trottier M, Komosa M, Tabach O, Notta F, et al. A distinctive DNA damage response in human hematopoietic stem cells reveals an
apoptosis-independent role for p53 in self-renewal. Cell Stem Cell. 2010;7:186–97. Article CAS PubMed Google Scholar * Parker JE, Mufti GJ, Rasool F, Mijovic A, Devereux S, Pagliuca A.
The role of apoptosis, proliferation, and the Bcl-2-related proteins in the myelodysplastic syndromes and acute myeloid leukemia secondary to MDS. Blood. 2000;96:3932–8. Article CAS PubMed
Google Scholar * Parker JE, Fishlock KL, Mijovic A, Czepulkowski B, Pagliuca A, Mufti GJ. ‘Low-risk’ myelodysplastic syndrome is associated with excessive apoptosis and an increased ratio
of pro- versus anti-apoptotic bcl-2-related proteins. Br J Haematol. 1998;103:1075–82. Article CAS PubMed Google Scholar * Abdelhamed S, Butler JT, Doron B, Halse A, Nemecek E, Wilmarth
PA, et al. Extracellular vesicles impose quiescence on residual hematopoietic stem cells in the leukemic niche. EMBO Rep. 2019;20:e47546. Article PubMed PubMed Central CAS Google
Scholar * Doron B, Abdelhamed S, Butler JT, Hashmi SK, Horton TM, Kurre P, et al. Stress reconfigures the AML bone marrow compartment. Leukemia. 2019;33:918–30. Article CAS PubMed Google
Scholar Download references ACKNOWLEDGEMENTS This work was funded by the American Lebanese and Syrian Associated Charities of St. Jude Children’s Research Hospital and grants from the US
National Institutes of Health, including R01 HL144653 to JMK, F32HL152484-01, and the Childhood Hematological Malignancies Training Program at St. Jude T32CA236748-01 to MET. The content is
solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Support was also provided by the Edward P. Evans
Foundation (JMK). JMK holds a Career Award for Medical Scientists from the Burroughs Wellcome Fund. The authors thank Dr. Esther Obeng, Dr. Richard Kriwacki, Dr. Paul Taylor, Dr. Scott
Blanchard, and their lab members (St. Jude) for their critical feedback and/or technical support. A special thanks to the members of St. Jude core facilities, including Drs. Stacie Woolard,
and Richard Ashmun (Flow Cytometry and Cell Sorting); Drs. Jennifer Peters and Victoria Frohlich (Cell and Tissue Imaging) and Drs. Junmin Peng and Vishwajeeth Pagala (Proteomics). As well
as Klco Lab members for providing reagents and for helpful discussions. AUTHOR INFORMATION Author notes * These authors contributed equally: Melvin E. Thomas III, Sherif Abdelhamed AUTHORS
AND AFFILIATIONS * Department of Pathology, St. Jude Children’s Research Hospital, Memphis, TN, USA Melvin E. Thomas III, Sherif Abdelhamed, Ryan Hiltenbrand, Michael Walsh, Guangchun Song,
Jing Ma & Jeffery M. Klco * Department of Pediatrics, Vanderbilt University Medical Center, Nashville, TN, USA Jason R. Schwartz * Center for Advanced Genome Engineering, St. Jude
Children’s Research Hospital, Memphis, TN, USA Sadie Miki Sakurada & Shondra M. Pruett-Miller Authors * Melvin E. Thomas III View author publications You can also search for this author
inPubMed Google Scholar * Sherif Abdelhamed View author publications You can also search for this author inPubMed Google Scholar * Ryan Hiltenbrand View author publications You can also
search for this author inPubMed Google Scholar * Jason R. Schwartz View author publications You can also search for this author inPubMed Google Scholar * Sadie Miki Sakurada View author
publications You can also search for this author inPubMed Google Scholar * Michael Walsh View author publications You can also search for this author inPubMed Google Scholar * Guangchun Song
View author publications You can also search for this author inPubMed Google Scholar * Jing Ma View author publications You can also search for this author inPubMed Google Scholar * Shondra
M. Pruett-Miller View author publications You can also search for this author inPubMed Google Scholar * Jeffery M. Klco View author publications You can also search for this author inPubMed
Google Scholar CORRESPONDING AUTHOR Correspondence to Jeffery M. Klco. ETHICS DECLARATIONS CONFLICT OF INTEREST The authors declare no competing interests. ADDITIONAL INFORMATION
PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTAL RIGHTS AND
PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any
medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The
images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly
from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Thomas, M.E.,
Abdelhamed, S., Hiltenbrand, R. _et al._ Pediatric MDS and bone marrow failure-associated germline mutations in _SAMD9_ and _SAMD9L_ impair multiple pathways in primary hematopoietic cells.
_Leukemia_ 35, 3232–3244 (2021). https://doi.org/10.1038/s41375-021-01212-6 Download citation * Received: 04 July 2020 * Revised: 08 February 2021 * Accepted: 25 February 2021 * Published:
17 March 2021 * Issue Date: November 2021 * DOI: https://doi.org/10.1038/s41375-021-01212-6 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get
shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative