Play all audios:
ABSTRACT Previous studies indicate that estrogen positively regulates lung cancer progression. Understanding the reasons will be beneficial for treating women with lung cancer in the future.
In this study, we found that tumor formation was more significant in female EGFRL858R mice than in male mice. P53 expression levels were downregulated in the estradiol (E2)-treated lung
cancer cells, female mice with EGFRL858R-induced lung cancer mice, and premenopausal women with lung cancer. E2 increased DNA methyltransferase 1 (DNMT1) expression to enhance methylation in
the TP53 promoter, which led to the downregulation of p53. Overexpression of GFP-p53 decreased DNMT1 expression in lung cancer cells. TP53 knockout in mice with EGFRL858R-induced lung
cancer not only changed gene expression in cancer cells but also increased the polarization of M2 macrophages by increasing C–C motif chemokine ligand 5 (CCL5) expression and decreasing
growth differentiation factor 15 (GDF15) expression. The TP53 mutation rate was increased in females with late-stage but not early-stage lung cancer compared to males with lung cancer. In
conclusion, E2-induced DNMT1 and p53 expression were negatively regulated each other in females with lung cancer, which not only affected cancer cells but also modulated the tumor-associated
microenvironment, ultimately leading to a poor prognosis. SIMILAR CONTENT BEING VIEWED BY OTHERS FXR DEFICIENCY INDUCED FERROPTOSIS VIA MODULATION OF THE CBP-DEPENDENT P53 ACETYLATION TO
SUPPRESS BREAST CANCER GROWTH AND METASTASIS Article Open access 14 November 2024 ESTROGEN RECEPTOR BETA PROMOTES LUNG CANCER INVASION VIA INCREASING CXCR4 EXPRESSION Article Open access 21
January 2022 MACROPHAGES PROMOTE PRE-METASTATIC NICHE FORMATION OF BREAST CANCER THROUGH ARYL HYDROCARBON RECEPTOR ACTIVITY Article Open access 18 December 2024 INTRODUCTION Lung cancer,
including non-small-cell lung cancer (NSCLC) and small-cell lung cancer (SCLC), is the leading cause of cancer-related death worldwide. Approximately 85% of all new cases are NSCLC, and
adenocarcinoma is the most common subtype of NSCLC, especially in female patients. In the past three decades, the lung cancer incidence rate has decreased approximately twofold faster in men
than in women. The clinical characteristics of men and women with lung cancer are very different. Previous studies have shown that when female patients were analyzed based on hormonal
status, premenopausal women have a worse prognosis than both men and postmenopausal women, suggesting that estrogen promotes lung cancer malignancy in premenopausal women [1,2,3]. However,
the detailed mechanism(s) remains to be elucidated. TP53 is an important tumor suppressor gene that is involved in cell proliferation and metastasis through various mechanisms, such as the
cell cycle and DNA damage repair [4, 5]. Previous studies have shown that p53 and RAS are involved in the regulation of cancer cell epithelial-mesenchymal transition (EMT) [6, 7]. p53 forms
a complex with MDM2 and Slug to promote MDM2-mediated Slug degradation [8, 9]. In addition, previous studies have indicated that p53 positively regulates miR200 to silence ZEB1, subsequently
increasing E-cadherin expression and decreasing Vimentin expression to inhibit EMT [10,11,12]. Recent studies have revealed that p53 is involved in regulating the inflammatory tumor
microenvironment and maintaining cancer stem cells (CSCs) [13]. According to previous studies, p53 may be a regulator of the NF-κB signaling pathway [14, 15]. Several lines of evidence
support the involvement of p53 in the inflammatory tumor microenvironment. First, the loss of p53 augments NF-κB transcriptional activity in response to TNF-α treatment. Second, the loss of
p53 sensitizes cells to TNF-α-induced apoptosis. Third, the loss of p53 enhances inflammatory responses by inhibiting interleukin-1 receptor antagonist (sIL-1Ra) expression. Fourth, the loss
of TP53 promotes TLR3-induced cytokine and chemokine expression. Finally, the loss of p53 affects the gene expression profile [16, 17]. In this study, we found that estrogen inhibited p53
expression and induced M2-macrophage polarization. The relationship between the loss of p53 and the tumor microenvironment in lung cancer needs to be elucidated. DNA methylation is an
important modification of the genome that modulate the gene expression and is involved in physiological and pathological conditions. Several enzymes, including DNMT1, DNMT3a, and DNMT3b,
mediate DNA methylation [18,19,20]. A recent study indicates that inhibition of DNMT1 and ERα cross-talk suppresses breast cancer by inhibiting IRF4 expression [21]. In addition, DNMT1
increases the DNA methylation of TP53 and decreases p53 expression [22, 23]. DNMT1 promotes cell proliferation by methylating the hMLH1 and hMSH2 promoters in EGFR-mutated NSCLC.
SUV39H1-mediated DNMT1 participates in the epigenetic regulation of Smad3 in cervical cancer [24, 25]. In this study, we found that estrogen-induced DNMT1 increased the DNA methylation of
TP53 to repress p53 expression, which positively affected DNMT1 expression, thus promoting poor prognosis of lung cancer by modulating EMT and the microenvironment. RESULTS E2-MEDIATED
INHIBITION OF P53 IS RELATED TO A POOR PROGNOSIS IN FEMALES WITH LUNG CANCER According to a previous study, estrogen may positively regulate lung cancer development [26,27,28], but the exact
mechanisms need to be clarified. In this study, we used the tet-ON system, which is a well-known conditional transgenic system, to express EGFRL858R, which leads to cancer development [29,
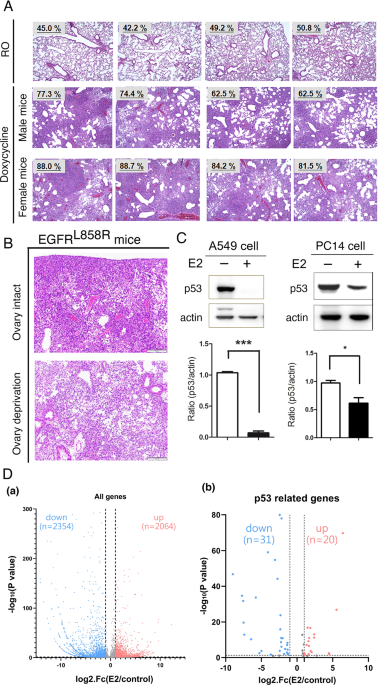
30]. Herein, we first showed that lung cancer development was worse in female EGFRL858R-induced mice than in male mice (Fig. 1A) and ovariectomized female mice (Fig. 1B). However, the
molecular mechanism by which E2 positively regulates lung cancer progression remains unclear. First, next-generation sequencing (NGS) of A549 and E2-A549 cells was performed to study the
gene expression profiles (Fig. 1D). There were 2354 genes and 2064 genes that were negatively and positively regulated respectively, by E2 treatment in A549 lung cancer cells (Fig. 1D). The
mRNA and protein levels of p53 were dramatically decreased in estrogen-treated A549 and PC14 cells (Fig. 1C,D). In addition, 51 p53-regulated genes, including 31 downregulated and 20
upregulated genes [31], were also regulated in E2-A549 cells, suggesting that E2-mediated inhibition of p53 regulated the expression of many p53 target genes during lung cancer progression
(Fig. 1D and Supplementary Fig. 1). Taken together, these findings suggest that estrogen can decrease p53 expression by inhibiting its transcriptional activity in the early period of lung
cancer progression. To study the role of p53 in sex-dependent lung cancer progression, TP53 was knocked out in mice with EGFRL858R-induced lung cancer and EGFRL858R x TP53+/− male and female
mice to delineate the role of p53 in sex-dependent lung cancer development induced by doxycycline for 1.5 months (Fig. 2A and Supplementary Fig. 2). The tumor burdens of doxycycline-induced
lung cancer mice were determined by measuring the increase in the lung area compared to that of normal mice. The tumor burden in female mice was worse than that in male mice (Fig. 2A, a),
but there was no significant difference in tumor size between male and female TP53-knockout mice, suggesting that the loss of p53 is the major reason for the poor prognosis in females with
lung cancer mice (Fig. 2A, b and Supplementary Fig. 2). Cancer development in several mice was induced by continuous doxycycline until death to study the effect of TP53 mutations on survival
rates. The survival rates of TP53-wt females were lower than that of TP53-wt male mice with EGFRL858R-induced lung cancer (Fig. 2B, a). However, there was no significant difference between
TP53-knockout male and female mice with EGFRL858R-induced lung cancer (Fig. 2B, b). According to previous studies, the TP53 mutation rate is higher in women with lung cancer than in men [32,
33]. We examined the TP53 genotype in lung cancer cohorts from the TCGA database to analyze the TP53 mutation rate in the early and late stages of lung cancer (Fig. 2C and Supplementary
Table 2). The survival rates of TP53-wt females were lower than that of TP53-wt males with EGFR mutations (Fig. 2C, a). Interestingly, in the early stage, there was no difference in the TP53
mutation rate between men and women with lung cancer. However, the TP53 mutation rate was higher in women with late-stage lung cancer than in men with late-stage men lung cancer (Fig. 2C,
b). Based on these findings, we believe that estrogen decreases p53 expression in the early stage of lung cancer progression in women, subsequently inducing genomic instability and leading
to TP53 mutation in the late stage, which ultimately results in a poor prognosis. In conclusion, these data indicate that E2-mediated inhibition of p53 in early-stage lung cancer may be
related to the poor prognosis of women with lung cancer compared with men with lung cancer. E2-INDUCED DNMT1 EXPRESSION INCREASES DNA METHYLATION OF P53 TO INHIBIT ITS TRANSCRIPTIONAL
ACTIVITY A previous study indicates that DNA methylation in the TP53 promoter region is increased during cancer progression, thereby silencing p53 expression [23, 34]. Our NGS gene
expression profile also showed that DNMT1 was increased in E2-treated A549 lung cancer cells (Supplementary Fig. 1). The mRNA and protein levels of DNMT1 were increased in E2-treated A549
and PC14 lung cancer cells (Fig. 3A, B). E2 treatment decreased the level of p53, and knockdown of DNMT1 increased the level of p53 in PC14 cells (Fig. 3C), suggesting that E2 increases
DNMT1 to inhibit p53 expression. Because p53 can repress DNMT1 expression in colon cancer cells [35], here we also showed that the overexpression of GFP-p53 in H1299L858R and A549 cells
significantly decreased the level of DNMT1. Furthermore, knockout of TP53 in mice with EGFRL858R-induced lung cancer increased the level of DNMT1 (Fig. 3D, right panel). The promoter of
DNMT1 was analyzed by software, and several estrogen receptor (ER)-binding sites were found in the promoter of DNMT1. To study the effect of E2 on the transcriptional activity of DNMT1,
luciferase activity driven by the promoter of DNMT1 was studied (Fig. 3E). The data indicated that E2 treatment significantly increased luciferase activity in A549 cells (2.0-Kbp promoter)
and the loss of one estrogen receptor (ER) binding site (1.7-Kbp promoter) decreased luciferase activity, indicating that estrogen directly binds with ER to induce the transcriptional
activity of DNMT1 (Fig. 3E). In addition, the level of p53 in male lung cancer mice was higher than that in female lung cancer mice (Fig. 3F, a). To study the role of sex in p53 levels, the
ovary was removed from female lung cancer mice by ovariectomy (Fig. 3F). The levels of p53 in male mice were higher than those in female mice. When the ovaries were removed, the levels of
p53 in ovariectomized mice were greater than that in ovary-intact female lung cancer mice, indicating that estrogen is involved in the downregulation of p53 expression (Fig. 3F). Because
DNMT1 is a DNA methyltransferase, the effect of estrogen treatment on the methylation of the TP53 promoter was studied (Fig. 3G). The data indicated that the methylation of the TP53 promoter
region was increased by E2 treatment in A549 cells (Fig. 3G). To identify the detailed methylation site(s) within the TP53 promoter, the promoter activity of TP53 was studied by a
luciferase activity assay (Fig. 3H). The data indicated that the luciferase activity of the TP53 promoter was significantly decreased by E2 treatment, but this effect was abolished by
removing the methylation motifs of the TP53 promoter, especially mutating the −1045 and −949 sites, indicating that E2-induced DNMT1 enhancement of DNA methylation within the TP53 promoter
is important for decreasing p53 expression (Fig. 3H). In summary, E2 treatment of lung cancer cells increases DNMT1 expression to enhance the DNA methylation of TP53, subsequently repressing
p53 expression. E2-MEDIATED INHIBITION OF P53 INCREASES THE DIFFERENTIATION OF M2 MACROPHAGES Because previous studies have indicated a higher TP53 mutation rate in women with lung cancer
than in men with lung cancer, this might be the major reason for the poor prognosis in women. Many p53-regulated EMT- and cancer malignancy-related genes were changed by E2 treatment
(Supplementary Fig. 1) [36], but the effect of p53 on the tumor-associated microenvironment (TME) in lung cancer needs to be addressed. To study the effect of p53 on TME during lung cancer
progression, TP53 was knocked out in the EGFRL858R-, and EGFRL858R × TP53−/−-induced lung cancer mouse models (Fig. 4). Tumor size was increased in TP53-knockout mice (Fig. 1 and
Supplementary Fig. 2). Based on these results, we hypothesized that p53 regulates not only cancer cells but also other cells in the TME, such as macrophages. The conditioned medium of
H1299L858R cells with or without GFP-p53 expression was collected and then cultured with THP-1 monocytes to study the polarization of macrophage M1/M2 macrophages (Fig. 4). The data
indicated that THP-1 cells treated with conditioned medium from H1299L858R lung cancer cells with GFP-p53 overexpression decreased the levels of CD206 and IL6 but increased the level of
IL10, suggesting that p53 in lung cancer cells can inhibit the polarization of M2 macrophages, which can promote cancer metastasis (Fig. 4A). We used the M2-macrophage marker YM1 to study
the distribution of M2 macrophages around lung tumors in EGFRL858R and EGFRL858R × TP53+/− mice (Fig. 4B). Interestingly, the tumor infiltration of M2 macrophages (TIMs) was markedly
increased in TP53-knockout mice, suggesting that the loss of p53 facilitated macrophage infiltration in the lung cancer region, which might be beneficial for cancer progression (Fig. 4B).
The effect of p53-mediated secreted factors on the migration of macrophages and cancer cells was studied (Fig. 5A–C). Conditioned medium from H1299L858R cells overexpressing GFP-p53
inhibited the migration of macrophages (Fig. 5A) and cancer cells, H1299L858R (Fig. 5B) and A549 cells (Fig. 5C), suggesting that p53-mediated secreted factors from lung cancer cells are
involved in lung cancer progression by regulating cancer cells and the tumor-associated microenvironment. To study the composition of the conditioned medium, protein arrays were used to
examine the conditioned medium of H1299 and H1299L858R cells with or without GFP-p53 expression (Fig. 5D). Because we used mice with EGFRL858R-induced lung cancer to study the effect of p53
on the TME, in the protein array, we examined the differences in H1299L858R but not in H1299 lung cancer cells (Fig. 5D). Only the proteins FGF2, GDF15, and CCL5 were regulated by p53 in
H1299L858R cells but not in H1299 cells. GFP-p53 overexpression increased the levels of FGF2 and GDF15 but decreased CCL5 in H1299L858R cells (Fig. 5D). In addition, several proteins,
including VEGF, MMP9, and PTX3, were decreased in GFP-p53-expressing H1299 and H1299L858R cells, suggesting that these proteins are regulated by p53 in an EGFR mutation-independent manner
(Fig. 5D and Supplementary Fig. 3). Furthermore, we validated the expression of CCL5, GDF15, and FGF2 in GFP-p53-expressing H1299L858R cells (Fig. 5E, a) and EGFRL858R x TP53+/− mice (Fig.
5E, b). In H1299L858R cells, GFP-p53 inhibited the mRNA level of CCL5 but increased the mRNA levels of FGF2 and GDF15, which was consistent with the conditional profile shown in Fig. 5D. In
EGFRL858R × TP53+/− mice, the loss of p53 increased the mRNA level of CCL5 and decreased the mRNA level of GDF15, which was also consistent with the conditional profile. However, the loss of
p53 increased the mRNA level of FGF2 (Fig. 5E, b, right panel), which was inconsistent with the results shown in Fig. 5D. Taken together, these results suggest that p53-mediated CCL5 and
GDF15 are secreted by cancer cells to regulate the TME. P53-MEDIATED CCL5 AND GDF15 REGULATE M2-MACROPHAGE POLARIZATION TO CONTROL CANCER PROGRESSION What is the effect of the p53-mediated
TAM on cancer progression? First, we found that E2 treatment of A549 cells dramatically decreased p53 expression and increased CD44 expression in lung cancer cells and M2 macrophages,
indicating that E2-mediated inhibition of p53 expression may lead to cancer malignancy (Fig. 6A). We used CD206 and CD68 as the M2-macrophage markers to study the effect of p53-induced CCL5
on M2-macrophage polarization (Fig. 6B). Conditioned medium collected from H1299L858R cells with GFP-p53 overexpression decreased M2-macrophage polarization, but adding recombinant CCL5
protein to the conditioned medium reversed this effect (Fig. 6B), indicating that p53-mediated inhibition of CCL5 could prevent the differentiation of M2 macrophages. In addition, the effect
of CCL5 and GDF15 on the migration of M2-macrophage and H1299 cancer cells was studied by culturing the cells with the conditioned medium collected from H1299L858R cells with or without
GFP-p53 overexpression (Fig. 6C). The data indicated that GFP-p53 overexpression inhibited the migration of M2 macrophages and H1299 lung cancer cells, and the addition of anti-GDF15
antibodies or recombinant CCL5 protein blocked the effect of GFP-p53 expression on cancer cell migration (Fig. 6C). When we used conditioned medium from H1299L858R cells with p53 expression
to treat H1299L858R cancer cells, CD44 expression in cancer cells was inhibited, suggesting that 53 mediated the TAM to inhibit cancer stemness (Fig. 6D). The effect of p53-mediated changes
in conditioned medium on the morphology of macrophages was also observed (Fig. 6E). The data indicated that lamellipodia around M2 macrophages were observed in control and GFP-overexpressing
cells but were lost in GFP-p53-overexpressing cells, suggesting that p53 in lung cancer cells regulates secreted proteins to affect the morphology of macrophages, which is related to tumor
infiltration (Fig. 6E). Finally, to study the association between M2 macrophages and sex-dependent lung cancer, samples were collected from male and female mice with EGFRL858R-induced lung
cancer and used to study M2 macrophages that were stained with anti-YM1 antibodies and then scored from 1 to 4 based on the YM1 signal (Fig. 7A, a, b). The data indicated that the scores of
female mice were higher than those of male mice, indicating that more M2 macrophages were found in female lung cancer mice (Fig. 7A). The level of CCL5 was increased in female lung cancer
mice but was decreased after the ovary was removed (Fig. 7B, left panel). In addition, the level of GDF15 was slightly decreased in female lung cancer mice but was slightly increased after
the ovary was removed (Fig. 7B, right panel). In addition, the mRNA levels of TP53 and GDF15 were increased, and the mRNA levels of DNMT1 and CCL5 were decreased in ovariectomized female
mice, which is consistent with the results of this study (Fig. 7C). Finally, the levels of p53, DNMT1, GDF15, and CCL5 in lung cancer patients were studied (Fig. 8 and Supplementary Table
3). Samples from ten lung cancer patients were used to study the levels of p53, DNMT1, CCL5, and GDF15 and then scored from 0 to 3 based on protein signals (Fig. 8A). The data indicated that
the scores of CCL5 and DNMT1 were higher than those of p53 and GDF15 (Fig. 8A) in the clinical samples from lung cancer patients, which is consistent with our study. In addition, the levels
of CCL5 and DNMT1 in female patients were higher than those in male patients, but there was no significant difference in GDF15 and p53 levels between the male and female cohorts (Fig. 8A).
DISCUSSION The poor prognosis of women with lung cancer has recently become an important clinical issue. In this study, we found that E2 inhibited p53 to control not only in cancer cells but
also cells in the tumor-associated microenvironment. We found that E2-induced DNMT1 and p53 formed a negative feedback loop, thereby increasing CCL5 and decreasing GDF15 expression,
subsequently enhancing M2-macrophage polarization and ultimately leading to poor prognosis in females with lung cancer (Fig. 8B). Previous studies have revealed that E2 treatment induces
rapid activation of the EGFR pathway, suggesting that nonnuclear ER translocation regulates the EGFR pathway to influence lung cancer progression [37]. Moreover, 67% of EGFR
mutation-positive tumors have high nuclear ERβ expression (versus 37% in EGFR wild-type tumors) [38]. Based on these previous studies, ER-mediated and EGFR-mediated signaling pathways may
positively coregulate the cancer-related gene expression during lung cancer progression. In this study, we found that E2 treatment could induce DNMT1 expression, subsequently increasing DNA
methylation in the promoter of p53. Because E2 actives EGFR signaling and the TP53 promoter sequence contains ER-binding motifs, ERs may regulate the transcriptional activity of DNMT1 by
activation of the EGFR signaling pathway and direct recruitment to its promoter. According to previous studies, the TP53 mutation rate is higher in women with lung cancer than in men and is
related to a poor prognosis in these individuals [32, 33]. In this study, we found high TP53 mutation rates only in females with late-stage lung cancer, not in those with early-stage lung
cancer. We also found that estrogen significantly inhibited p53 expression and was an important factor in inducing the high TP53 mutation rate in late-stage lung cancer. p53, which is a
tumor suppressor, maintains genomic integrity to avoid cancer heterogeneity during cancer progression [39, 40]. In this study, we found that the survival rates of EGFRL858R male mice were
higher than those of female mice, but this difference was abolished in EGFRL858R × TP53+/− mice, indicating that estrogen-mediated inhibition of p53 in female mice is critical for poor
prognosis. Although we have studied the role and mechanism by which p53 affects tumor burden in vivo, we only used eight EGFRL858R and EGFRL858R × TP53+/− mice to study the survival rate.
More mice will be used to confirm this finding in the future. We also found that many p53 target genes were altered by estrogen treatment, which is consistent with previous studies in which
p53 was revealed to be a tumor suppressor. In addition to the effect of p53 within cancer cells, when the TP53 was knocked out in mice with EGFRL858R-induced lung cancer, not only did the
size of the tumor increase, but marked immune cell infiltration was also observed around cancer cells, indicating that p53, as a tumor suppressor, inhibited not only cancer cells directly
but also M2-macrophage differentiation. The TAM is involved in cancer progression (i.e., cancer malignancy and drug resistance) [41,42,43,44]. Many proteins, such as cytokines secreted from
nearby cells (e.g., macrophages, fibroblasts, and other immune cells, including T cells and B cells), regulate cancer cell progression and macrophage polarization. Our data indicated that
estrogen-mediated inhibition of p53 in H1299L858R cancer cells changed the protein composition (FGF2, GDF15, IGFBP2, CCL5, PTX3, VEGF, and MMP9) of conditioned medium. GDF15, CCL5, and FGF2
were regulated only in H1299L858R cells but not in H1299 cancer cells, suggesting that EGFR signaling and p53 coregulate these three secreted proteins. Fibroblast growth factor type 2 (FGF2)
can bind to heparin and has broad mitogenic and angiogenic activities. Our protein array data showed that GFP-p53 positively regulated FGF2 secretion by cells, and positively regulated FGF2
in cancer cells. However, FGF2 expression was also increased in TP53-knockout mice with EGFRL858R-induced lung cancer, which is inconsistent with the results in cancer cells. p53 may
regulate FGF2 differently under different conditions, and the detailed regulatory mechanism needs to be further clarified. Growth differentiation factor 15 (GDF15) has been reported to be
involved in various physiologic or pathologic pathways including cancer [45]. GDF15 is one of the ligands that bind to TGF-β receptors to modulate the SMAD pathway and regulate gene
expression [46]. Previous studies have indicated that EZH2-mediated inhibition of GDF15 represses NSCLC proliferation [47]. In this study, we first showed that p53 positively regulated GDF15
to repress the migration abilities of M2 macrophages and lung cancer cells. In addition, C–C motif chemokine ligand 5 (CCL5) is a secreted protein involved in immunoregulatory and
inflammatory processes that functions as a chemoattractant for blood monocytes, memory T-helper cells, and eosinophils [48,49,50]. Previous studies have indicated that IL6 mediates
macrophage infiltration after irradiation by upregulating CCL5 expression in NSCLC. Our study showed that GFP-p53 increased GDF15 expression but decreased CCL5 expression in H1299L858R
cells, thereby repressing M2-macrophage differentiation and subsequently preventing cancer malignancy. E2-mediated EGFR signaling and p53 expression are critical for the poor prognosis in
females with lung cancer. DNA methylation is important for maintaining genomic integrity and gene expression [51, 52]. Disorders associated with DNA methylation are involved in various
diseases, including cancer [53]. A previous study indicated that DNMT1 could methylate the promoter of TP53 to inhibit its expression [34, 54]. We showed that estrogen treatment of lung
cancer cells could induce DNMT1 expression, thereby inducing hypermethylation of the TP53 promoter and leading TP53 downregulation. Our study also showed that other DNMTs, DNMT2, DNMT3a, and
DNMT3b were slightly increased, which might also contribute to the hypermethylation of the TP53 promoter. Although there was no significant difference in TP53 mutations between male and
female lung cancer patients in the early stage, estrogen-induced DNMT expression to inhibit p53 expression, which might be one of the critical reasons for the induction of genomic
instability, subsequently increasing TP53 mutation rates in the late stage of lung cancer in female patients. In the future, blocking estrogen or DNMT activities may be beneficial for the
survival rates of female lung cancer patients. MATERIALS AND METHODS CELL CULTURE AND TRANSFECTION Human lung adenocarcinoma epithelial cell line A549, PC14, H1299, H1299L858R, and human
monocyte THP-1 cell lines from ATCC were cultured with RPMI 1640 medium (Invitrogen, Carlsbad, CA, USA) containing 10% fetal bovine serum (Gibco™, Waltham, MA, USA), 100 μg/ml streptomycin
and 100 U/ml penicillin G sodium (Gibco). E2-A549 cells were obtained by treating A549 cells with 1 μg/ml E2 for 3 weeks. All cells were incubated at 37 °C with 5% CO2. For transfecting
plasmid, PolyJet (SignaGen Laboratories, Frederick, MD, USA) was used according to the manufacturer’s instructions. IMMUNOHISTOCHEMISTRY Paraffin-embedded human and mice lung cancer tissues
were obtained from the National Cheng Kung University Hospital Tissue Bank and doxycycline-induced EGFRL858R mice, respectively, and the tissues were cut into 5-μm sections.
Immunohistochemistry was performed using a Novolink™ Polymer Detection Systems (Leica Biosystems) following the manufacturer’s instructions. Antigen retrieval was performed using citrate
buffer (pH 6.0, Scytek). Primary antibodies, anti-p53 (A5761, ABclonal, 1:50), anti-CCL5 (A5630, ABclonal, 1:50), anti-YM1 (Abcam, 1:50), anti-DNMT1 (A16729, ABclonal, 1:50) and anti-GDF15
(A0185, ABclonal, 1:50), were used to incubate with tissue samples for overnight at 4 °C. Sections were photographed by Olympus BX-51 microscope. ANIMAL CARE AND ANIMAL MODELS The
experiments related with animals were approved by the Institutional Animal Care and Use Committee (IACUC:108005) at National Cheng Kung University (NCKU). These transgenic mice were
generated in National Laboratory Animal Center (NLAC, Taiwan, Tainan). After breeding, two-month-old transgenic mice were used to study lung cancer development. Caging was provided suitable
space and accommodates appropriate population densities that allowed animals’ sufficient freedom of movement. To provide amounts of food that must be for transgenic mice normal growth and
maintenance of normal body weight. These transgenic mice, including EGFRL858R and EGFRL858R × TP53+/− mice, were accessed to fresh and uncontaminated drinking water. Transgenic mice were
also observed and cared at least for two to three times per week. All methods involving animals were performed in accordance with the relevant guidelines and regulations. COLLECTION OF
SPECIMENS FROM LUNG CANCER PATIENTS All human study has been conducted in accordance with the guidelines and regulations. The study using human specimens was approved by the Clinical
Research Ethics Committee at National Cheng Kung University Medical Center (Tainan, Taiwan; IRB: A-ER-107-039). After surgical resection at National Cheng Kung University Hospital, specimens
of patients with lung adenocarcinomas were collected for Immunohistochemical analysis or western blotting. The pathological data were analyzed by clinical pathologists. Informed consent was
obtained from all subjects. STATISTICAL ANALYSIS All samples were used for statistical analysis. The difference between two groups was analyzed by Student’s _t_ test. The _P_ value, which
is smaller than 0.05, was considered statistically significant. SEM is used to calculate and plot error bars from raw data. Overall survivals were estimated by means of the Kaplan–Meier
method and compared using the log-rank test. REFERENCES * Hsu LH, Liu KJ, Tsai MF, Wu CR, Feng AC, Chu NM, et al. Estrogen adversely affects the prognosis of patients with lung
adenocarcinoma. Cancer Sci. 2015;106:51–59. Article CAS PubMed Google Scholar * Fidler-Benaoudia MM, Torre LA, Bray F, Ferlay J, Jemal A. Lung cancer incidence in young women vs. young
men: a systematic analysis in 40 countries. Int J Cancer. 2020;147:811–9. Article CAS PubMed Google Scholar * Jemal A, Miller KD, Ma J, Siegel RL, Fedewa SA, Islami F, et al. Higher lung
cancer incidence in young women than young men in the United States. N. Engl J Med. 2018;378:1999–2009. Article PubMed PubMed Central Google Scholar * Turgeon MO, Perry NJS,
Poulogiannis G. DNA damage, repair, and cancer metabolism. Front Oncol. 2018;8:15. Article PubMed PubMed Central Google Scholar * Lakin ND, Jackson SP. Regulation of p53 in response to
DNA damage. Oncogene. 1999;18:7644–55. Article CAS PubMed Google Scholar * Chang C-J, Chao C-H, Xia W, Yang J-Y, Xiong Y, Li C-W, et al. p53 regulates epithelial-mesenchymal transition
and stem cell properties through modulating miRNAs. Nat Cell Biol. 2011;13:317–23. Article CAS PubMed PubMed Central Google Scholar * Kim H, Choi JA, Kim JH. Ras promotes transforming
growth factor-β (TGF-β)-induced epithelial-mesenchymal transition via a leukotriene B4 receptor-2-linked cascade in mammary epithelial cells. J Biol Chem. 2014;289:22151–60. Article CAS
PubMed PubMed Central Google Scholar * Wang SP, Wang WL, Chang YL, Wu CT, Chao YC, Kao SH, et al. p53 controls cancer cell invasion by inducing the MDM2-mediated degradation of Slug. Nat
Cell Biol. 2009;11:694–704. Article CAS PubMed Google Scholar * Zhang J, Lei Y, Gao X, Liang Q, Li L, Feng J, et al. p53 attenuates the oncogenic Ras-induced epithelial-mesenchymal
transition in human mammary epithelial cells. Biochem Biophys Res Commun. 2013;434:606–13. Article CAS PubMed Google Scholar * Kim T, Veronese A, Pichiorri F, Lee TJ, Jeon YJ, Volinia S,
et al. p53 regulates epithelial-mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J Exp Med. 2011;208:875–83. Article CAS PubMed PubMed Central Google Scholar * Wang C,
Ge Q, Zhang Q, Chen Z, Hu J, Li F, et al. Targeted p53 activation by saRNA suppresses human bladder cancer cells growth and metastasis. J Exp Clin Cancer Res: CR. 2016;35:53. Article
PubMed PubMed Central CAS Google Scholar * Ye J, Wei X, Shang Y, Pan Q, Yang M, Tian Y, et al. Core 3 mucin-type O-glycan restoration in colorectal cancer cells promotes
MUC1/p53/miR-200c-dependent epithelial identity. Oncogene. 2017;36:6391–407. Article CAS PubMed Google Scholar * Uehara I, Tanaka N. Role of p53 in the regulation of the inflammatory
tumor microenvironment and tumor suppression. Cancers 2018;10:219. * Kawauchi K, Araki K, Tobiume K, Tanaka N. p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits
cell transformation. Nat Cell Biol. 2008;10:611–8. Article CAS PubMed Google Scholar * Webster GA, Perkins ND. Transcriptional cross talk between NF-kappaB and p53. Mol Cell Biol.
1999;19:3485–95. Article CAS PubMed PubMed Central Google Scholar * Weisz L, Damalas A, Liontos M, Karakaidos P, Fontemaggi G, Maor-Aloni R, et al. Mutant p53 enhances nuclear factor
kappaB activation by tumor necrosis factor alpha in cancer cells. Cancer Res. 2007;67:2396–401. Article CAS PubMed Google Scholar * Dixit D, Sharma V, Ghosh S, Mehta VS, Sen E.
Inhibition of Casein kinase-2 induces p53-dependent cell cycle arrest and sensitizes glioblastoma cells to tumor necrosis factor (TNFα)-induced apoptosis through SIRT1 inhibition. Cell Death
Dis. 2012;3:e271. Article CAS PubMed PubMed Central Google Scholar * Scala G, Federico A, Palumbo D, Cocozza S, Greco D. DNA sequence context as a marker of CpG methylation instability
in normal and cancer tissues. Sci Rep. 2020;10:1721. Article CAS PubMed PubMed Central Google Scholar * Edwards JR, Yarychkivska O, Boulard M, Bestor TH. DNA methylation and DNA
methyltransferases. Epigenetics Chromatin. 2017;10:23. Article PubMed PubMed Central CAS Google Scholar * Peng W-X, Koirala P, Zhang W, Ni C, Wang Z, Yang L, et al. lncRNA RMST Enhances
DNMT3 Expression through Interaction with HuR. Mol Ther. 2020;28:9–18. Article CAS PubMed Google Scholar * Vernier M, McGuirk S, Dufour CR, Wan L, Audet-Walsh E, St-Pierre J, et al.
Inhibition of DNMT1 and ERRα crosstalk suppresses breast cancer via derepression of IRF4. Oncogene. 2020;39:6406–20. Article CAS PubMed PubMed Central Google Scholar * Oshiro MM, Watts
GS, Wozniak RJ, Junk DJ, L Munoz-Rodriguez J, Domann FE, et al. Mutant p53 and aberrant cytosine methylation cooperate to silence gene expression. Oncogene. 2003;22:3624–34. Article CAS
PubMed Google Scholar * Nabilsi NH, Broaddus RR, Loose DS. DNA methylation inhibits p53-mediated survivin repression. Oncogene. 2009;28:2046–50. Article CAS PubMed Google Scholar * Wu
X-Y, Chen H-C, Li W-W, Yan J-D, Lv R-Y. DNMT1 promotes cell proliferation via methylating hMLH1 and hMSH2 promoters in EGFR-mutated non-small cell lung cancer. J Biochem. 2020;168:151–7.
Article CAS PubMed Google Scholar * Zhang L, Tian S, Zhao M, Yang T, Quan S, Song L, et al. SUV39H1-mediated DNMT1 is involved in the epigenetic regulation of Smad3 in cervical cancer.
Anti-cancer Agents Medicinal Chem. 2021;21:756–65. Article CAS Google Scholar * Gao X, Cai Y, Wang Z, He W, Cao S, Xu R, et al. Estrogen receptors promote NSCLC progression by modulating
the membrane receptor signaling network: a systems biology perspective. J Transl Med. 2019;17:308. Article PubMed PubMed Central CAS Google Scholar * Hsu LH, Chu NM, Kao SH. Estrogen,
estrogen receptor and lung cancer. Int J Mol Sci. 2017;18:1713. * Young M-J, Chen Y-C, Wang S-A, Chang H-P, Yang W-B, Lee C-C, et al. Estradiol-mediated inhibition of Sp1 decreases
miR-3194-5p expression to enhance CD44 expression during lung cancer progression. J Biomed Sci. 2022;29:3. Article CAS PubMed PubMed Central Google Scholar * Hsu TI, Wang MC, Chen SY,
Yeh YM, Su WC, Chang WC, et al. Sp1 expression regulates lung tumor progression. Oncogene. 2012;31:3973–88. Article CAS PubMed Google Scholar * Hsu TI, Wang YC, Hung CY, Yu CH, Su WC,
Chang WC, et al. Positive feedback regulation between IL10 and EGFR promotes lung cancer formation. Oncotarget. 2016;7:20840–54. Article PubMed PubMed Central Google Scholar * Wei C-L,
Wu Q, Vega VB, Chiu KP, Ng P, Zhang T, et al. A global map of p53 transcription-factor binding sites in the human genome. Cell. 2006;124:207–19. Article CAS PubMed Google Scholar * Gealy
R, Zhang L, Siegfried JM, Luketich JD, Keohavong P. Comparison of mutations in the p53 and K-ras genes in lung carcinomas from smoking and nonsmoking women. Cancer Epidemiol, Biomark Prev.
1999;8:297–302. CAS Google Scholar * Toyooka S, Tsuda T, Gazdar AF. The TP53 gene, tobacco exposure, and lung cancer. Hum Mutat. 2003;21:229–39. Article CAS PubMed Google Scholar *
Kang JH, Kim SJ, Noh DY, Park IA, Choe KJ, Yoo OJ, et al. Methylation in the p53 promoter is a supplementary route to breast carcinogenesis: correlation between CpG methylation in the p53
promoter and the mutation of the p53 gene in the progression from ductal carcinoma in situ to invasive ductal carcinoma. Lab Investig. 2001;81:573–9. Article CAS PubMed Google Scholar *
Peterson EJ, Bögler O, Taylor SM. p53-mediated repression of DNA methyltransferase 1 expression by specific DNA binding. Cancer Res. 2003;63:6579–82. CAS PubMed Google Scholar * Folkerd
EJ, Dowsett M. Influence of sex hormones on cancer progression. J Clin Oncol. 2010;28:4038–44. Article CAS PubMed Google Scholar * Mukku VR, Stancel GM. Regulation of epidermal growth
factor receptor by estrogen. J Biol Chem. 1985;260:9820–4. Article CAS PubMed Google Scholar * Nose N, Sugio K, Oyama T, Nozoe T, Uramoto H, Iwata T, et al. Association between estrogen
receptor-beta expression and epidermal growth factor receptor mutation in the postoperative prognosis of adenocarcinoma of the lung. J Clin Oncol. 2009;27:411–7. Article CAS PubMed Google
Scholar * Albrechtsen N, Dornreiter I, Grosse F, Kim E, Wiesmüller L, Deppert W. Maintenance of genomic integrity by p53: complementary roles for activated and non-activated p53. Oncogene.
1999;18:7706–17. Article CAS PubMed Google Scholar * Kastenhuber ER, Lowe SW. Putting p53 in context. Cell. 2017;170:1062–78. Article CAS PubMed PubMed Central Google Scholar *
Wang T, Xiao M, Ge Y, Krepler C, Belser E, Lopez-Coral A, et al. BRAF inhibition stimulates melanoma-associated macrophages to drive tumor growth. Clin Cancer Res. 2015;21:1652–64. Article
CAS PubMed PubMed Central Google Scholar * Vouri M, Hafizi S. TAM receptor tyrosine kinases in cancer drug resistance. Cancer Res. 2017;77:2775–8. Article CAS PubMed Google Scholar *
Aras S, Zaidi MR. TAMeless traitors: macrophages in cancer progression and metastasis. Br J Cancer. 2017;117:1583–91. Article CAS PubMed PubMed Central Google Scholar * Wang YC, Wu YS,
Hung CY, Wang SA, Young MJ, Hsu TI, et al. USP24 induces IL-6 in tumor-associated microenvironment by stabilizing p300 and β-TrCP and promotes cancer malignancy. Nat Commun. 2018;9:3996.
Article PubMed PubMed Central CAS Google Scholar * Wischhusen J, Melero I, Fridman WH. Growth/differentiation factor-15 (GDF-15): from biomarker to novel targetable immune checkpoint.
Front Immunol. 2020;11:951–951. Article CAS PubMed PubMed Central Google Scholar * Min KW, Liggett JL, Silva G, Wu WW, Wang R, Shen RF, et al. NAG-1/GDF15 accumulates in the nucleus and
modulates transcriptional regulation of the Smad pathway. Oncogene. 2016;35:377–88. Article PubMed CAS Google Scholar * Lu X, He X, Su J, Wang J, Liu X, Xu K, et al. EZH2-mediated
epigenetic suppression of GDF15 predicts a poor prognosis and regulates cell proliferation in non-small-cell lung cancer. Mol Ther Nucleic Acids. 2018;12:309–18. Article CAS PubMed PubMed
Central Google Scholar * Murooka TT, Rahbar R, Platanias LC, Fish EN. CCL5-mediated T-cell chemotaxis involves the initiation of mRNA translation through mTOR/4E-BP1. Blood.
2008;111:4892–901. Article CAS PubMed PubMed Central Google Scholar * Kameyoshi Y, Dörschner A, Mallet AI, Christophers E, Schröder JM. Cytokine RANTES released by thrombin-stimulated
platelets is a potent attractant for human eosinophils. J Exp Med. 1992;176:587–92. Article CAS PubMed Google Scholar * Proost P, De Meester I, Schols D, Struyf S, Lambeir AM, Wuyts A,
et al. Amino-terminal truncation of chemokines by CD26/dipeptidyl-peptidase IV. Conversion of RANTES into a potent inhibitor of monocyte chemotaxis and HIV-1-infection. J Biol Chem.
1998;273:7222–7. Article CAS PubMed Google Scholar * Meng H, Cao Y, Qin J, Song X, Zhang Q, Shi Y, et al. DNA methylation, its mediators and genome integrity. Int J Biol Sci.
2015;11:604–17. Article CAS PubMed PubMed Central Google Scholar * Petryk N, Bultmann S, Bartke T, Defossez P-A. Staying true to yourself: mechanisms of DNA methylation maintenance in
mammals. Nucleic Acids Res. 2021;49:3020–32. Article CAS PubMed Google Scholar * Ehrlich M. DNA hypermethylation in disease: mechanisms and clinical relevance. Epigenetics.
2019;14:1141–63. Article PubMed PubMed Central Google Scholar * Cheng JC, Auersperg N, Leung PCK. Inhibition of p53 represses E-cadherin expression by increasing DNA methyltransferase-1
and promoter methylation in serous borderline ovarian tumor cells. Oncogene. 2011;30:3930–42. Article CAS PubMed Google Scholar Download references ACKNOWLEDGEMENTS We are grateful for
the support of clinical specimens from the Human Biobank, Research Center of Clinical Medicine, National Cheng Kung University Hospital. This work was supported by the grants
(109-2320-B-006-025-MY3 and 109-2320-B-006-055) obtained from the Ministry of Science and Technology, Taiwan. This research was supported in part by Higher Education Sprout Project, Ministry
of Education to the Headquarters of University Advancement at National Cheng Kung University (NCKU). AUTHOR INFORMATION Author notes * These authors contributed equally: Yung-Ching Chen,
Ming-Jer Young. AUTHORS AND AFFILIATIONS * Institute of Basic Medical Sciences, National Cheng Kung University, Tainan, Taiwan Yung-Ching Chen * Department of Biotechnology and Bioindustry
Sciences, National Cheng Kung University, Tainan, Taiwan Ming-Jer Young, Hui-Ping Chang, Chia-Yu Liu & Jan-Jong Hung * Division of Thoracic Surgery, Department of Surgery, College of
Medicine National Cheng Kung University, Tainan, Taiwan Chia-Chi Lee & Yau-Lin Tseng * Department of Pharmacology, College of Medicine, National Cheng Kung University, Tainan, Taiwan
Yi-Ching Wang * The Ph.D. Program for Neural Regenerative Medicine, College of Medical Science and Technology, Taipei Medical University, Taipei, Taiwan Wen-Chang Chang * Graduate Institute
of Medical Sciences, College of Medicine, Taipei Medical University, Taipei, Taiwan Jan-Jong Hung Authors * Yung-Ching Chen View author publications You can also search for this author
inPubMed Google Scholar * Ming-Jer Young View author publications You can also search for this author inPubMed Google Scholar * Hui-Ping Chang View author publications You can also search
for this author inPubMed Google Scholar * Chia-Yu Liu View author publications You can also search for this author inPubMed Google Scholar * Chia-Chi Lee View author publications You can
also search for this author inPubMed Google Scholar * Yau-Lin Tseng View author publications You can also search for this author inPubMed Google Scholar * Yi-Ching Wang View author
publications You can also search for this author inPubMed Google Scholar * Wen-Chang Chang View author publications You can also search for this author inPubMed Google Scholar * Jan-Jong
Hung View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS CYC, YMJ, CHP, LCY, and LCC performed experiments. YMJ did the bioinformatics
analysis. TYL, WYC, and CWC provided the clinical cohorts and supervised the research. HJJ design the experiments and prepared the manuscript. CORRESPONDING AUTHOR Correspondence to Jan-Jong
Hung. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to
jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY MATERIALS RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a
Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit
to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are
included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and
your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this
license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Chen, YC., Young, MJ., Chang, HP. _et al._ Estradiol-mediated
inhibition of DNMT1 decreases p53 expression to induce M2-macrophage polarization in lung cancer progression. _Oncogenesis_ 11, 25 (2022). https://doi.org/10.1038/s41389-022-00397-4 Download
citation * Received: 11 November 2021 * Revised: 07 April 2022 * Accepted: 12 April 2022 * Published: 19 May 2022 * DOI: https://doi.org/10.1038/s41389-022-00397-4 SHARE THIS ARTICLE Anyone
you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by
the Springer Nature SharedIt content-sharing initiative