Play all audios:
DEAR EDITOR, Mucin 1 (MUC1) contains N- and C- subunit that forms a heterodimer on the apical surface of luminal epithelial cells. Nevertheless, MUC1 is aberrantly overexpressed in various
cancers, such as breast cancer, lung cancer, and prostatic cancer, which critically contributes to tumorigenesis and poor clinical outcomes.1 Several strategies have been developed for
targeted-inhibition of MUC1 including vaccines, monoclonal antibodies (MAb), and polypeptide. Vaccines usually recognize abnormal glycosylated regions in the VNTR of MUC1-N, MAb is mainly
aimed at MUC1-N with little success, polypeptide inhibitors are applied to blocking the dimerization through CQC motif in MUC1-C, among which polypeptide inhibitor GO203 has entered clinical
trials. However, none of the MUC1-targeted therapies are currently feasible in clinical treatment.2 The overexpression of MUC1 in cancer is partly due to the gene amplification and
increased transcription. But less is known about how MUC1 protein is maintained. Deciphering the mechanism underlying MUC1 degradation will provide a novel strategy to reduce MUC1 abundance
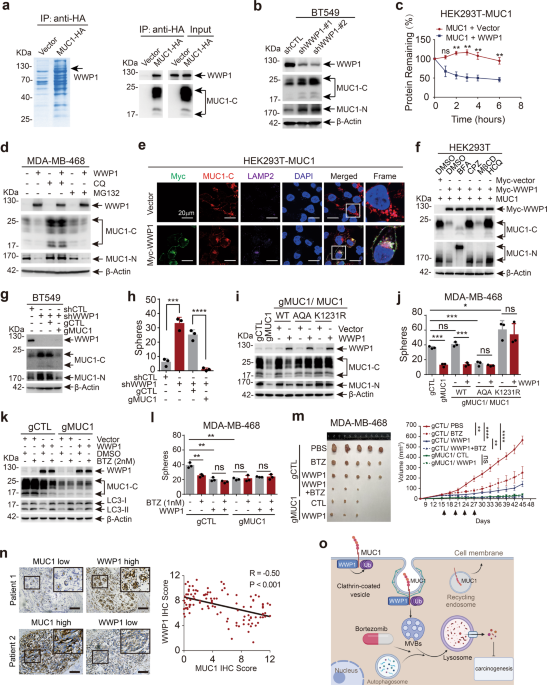
in cancer. To discover novel regulators of MUC1, we performed LC–MS/MS assay and identified WW domain-containing ubiquitin E3 ligase 1 (WWP1) that interacts with MUC1 (Supplementary Table S1
and Fig. 1a). Protein levels of MUC1 and WWP1 were detected and present an inverse correlation in cell lines from breast cancer, liver cancer and non-small cell lung cancer3 (Supplementary
Fig. S1a–c). Consistently, using complementary genetic approaches, we revealed that WWP1 negatively regulated MUC1 in protein level rather than mRNA level (Fig. 1b and Supplementary Fig.
S1d–h). WWP1 reduced MUC1 abundance in a dose-dependent manner (Supplementary Fig. S1i). Overexpression of WWP1 significantly shortened the half-life of MUC1 protein (Fig. 1c and
supplementary Fig. S1j). These results indicate that WWP1 triggers the turnover of MUC1 protein. Furthermore, WWP1-mediated degradation of MUC1 was considerably hindered by lysosome
inhibitor chloroquine (CQ) rather than proteasome inhibitor MG132, suggesting WWP1-induced degradation of MUC1 through lysosome pathway (Fig. 1d and Supplementary Fig. S2a). This result was
further corroborated by WWP1-induced co-localization of MUC1 and LAMP2 (Fig. 1e). Inhibitors were employed to examine the subcellular location where WWP1 degraded MUC1. The result validated
that only CPZ (an inhibitor for clathrin-dependent endocytosis) prominently blocked WWP1-mediated MUC1 degradation (Fig. 1f), suggesting a crucial role of clathrin-dependent endocytosis.
Immunofluorence staining revealed that WWP1 increased MUC1 level in early- and late- endosomes and reduced MUC1 in circulating endosomes (Supplementary Fig. S2b–d). Activation of autophagy
by rapamycin augmented WWP1-dependent degradation of MUC1 in BT549 cell line (Supplementary Fig. S2e). Collectively, these results demonstrate that WWP1 degrades MUC1 through
clathrin-mediated endosome/lysosome pathway. A positive correlation of WWP1 level and growth inhibition was observed in all of 4 cell lines (Supplementary Fig. S3a–d). Ablation of MUC1 using
CRISPR-Cas9 (gRNAs) in WWP1-silenced BT549 cells led to a diminished cell proliferation (Fig. 1g and Supplementary Fig. S3e), indicating that WWP1 targeted MUC1 for growth inhibition.
Similar results were obtained in colony formation (Supplementary Fig. S3f), migration (Supplementary Fig. S3g, h) and mammospheres formation assay (Fig. 1h and Supplementary Fig. S3i). These
results illustrate that WWP1 alleviates cell malignance through reduction of MUC1 level. The WWP1C890A mutant lacking of E3 ligase activity had little, if any effect on MUC1 turnover
(Supplementary Fig. S4a, b), indicating the E3 ligase activity is essential for WWP1 to degrade MUC1. Co-IP experiments with WWP1 truncation mutants demonstrated that WWP1 binds to MUC1
through its WW1/2 domains (Supplementary Fig. S4c, d). Accordingly, WWP1 wildtype but not C890A mutant ubiquitinates MUC1 (Supplementary Fig. S4e). K48- and K63-linked polyubiquitination are
the canonical signals to flag substrate proteins for degradation, our result revealed that K63 linkage rather than K48 linkage was mainly responsible for MUC1 ubiquitination by WWP1
(Supplementary Fig. S4f). We then verified the potential ubiquitination sites of MUC1 by bringing mutations in possible lysine residues. Mutants in MUC1-C but not MUC1-N were refractory to
WWP1-induced degradation. Notably, MUC1-K1231R was almost completely resistant to degradation by WWP1 (Supplementary Fig. S4g–j). These results suggest that WWP1 promotes K63-linked
ubiquitination of MUC1 primarily at the K1231 residue. The interaction of endogenous MUC1 and WWP1 was confirmed in BT549 cells (Supplementary Fig. S5a). We further mapped the motif in MUC1
required for the binding to WWP1. Interestingly, deletion of either of two potential PY motifs that WWP1 may recognizes4 had little, if any effect on WWP1-induced degradation of MUC1
(Supplementary Fig. S5b). Assessing on MUC1-CD truncation mutants displayed that WWP1 interacted with MUC1-CD1–20 (Supplementary Fig. S5c, d). Deletion of CD1–6 in MUC1 considerably
attenuated WWP1-induced MUC1 degradation (Supplementary Fig. S5e). The MUC1-CQC was finally proved to be indispensable to interact with WWP1 (Supplementary Fig. S5f, g). Consistently,
MUC1-AQA and MUC1-K1231R mutants that were resistant to ubiquitination by WWP1 cannot be degraded by WWP1 (Fig. 1i) and failed to rescue either cell proliferation (Supplementary Fig. S5h) or
mammosphere formation (Fig. 1j) in MDA-MB-468/gMUC1 cells. Our results also simplify a necessity of membrane localization for MUC1 turnover by WWP1 (Supplementary Fig. S5i–m). These results
clarify that WWP1 suppress cancer cell growth by promoting ubiquitination and degradation of MUC1. Unlike the known substrates of WWP1 that are all degraded through the proteasome pathway,4
WWP1 directed MUC1 degradation through the lysosome pathway. Given that proteasome inhibitors can also induce anti-tumor effects through activating the autophagy-lysosome pathway,5 we
hypothesized that the proteasome inhibitor might enhance WWP1-mediated lysosomal degradation of MUC1 via hampering WWP1 activity in the proteasome pathway. Indeed, bortezomib treatment
significantly reduced MUC1 with WWP1 existent in both BT549 and MDA-MB-468 cells (Fig. 1k and Supplementary Fig. S6a). Management with bortezomib was associated with inhibition of cell
proliferation and mammospheres formation in MDA-MB-468/gCTL rather than MDA-MB-468/gMUC1 cells (Fig. 1l and Supplementary Fig. S6b, c). Finally, we employed xenograft models to assess the
anti-tumor activity of bortezomib under the condition of WWP1 overexpression or MUC1 ablation. Bortezomib treatment markedly delayed tumor growth in WWP1-expressing MDA-MB-468/gCTL but not
in MDA-MB-468/gMUC1 cells without affecting mice body weight (Fig. 1m and Supplementary Fig. S6d, e). IHC staining assay on xenograft tissues confirmed that the expression of MUC1 was
considerably decreased on the treatment of bortezomib or overexpression of WWP1 (Supplementary Fig. S6f). In summary, these results demonstrate that bortezomib prominently stimulates
WWP1-induced MUC1 degradation and tumor destruction. In order to test the clinical relevance for the connection between MUC1 and WWP1, we analyzed patient samples in cBioPortal database and
found that gene amplification of MUC1 and WWP1 presents noteworthy mutual exclusiveness (Supplementary Fig. S7a–c), we further analyzed 108 human breast cancer samples and found a reverse
correlation between the levels of MUC1 and WWP1 (Fig. 1n and Supplementary Fig. S7d). Furthermore, out of 17 specimens that had both high expression of MUC1 and WWP1, 14 of them (82.4%)
displayed different distribution of MUC1 and WWP1 in tumor cells (Supplementary Fig. S7e). Collectively, these results show an inverse association of MUC1 level with WWP1 expression in
cancer tissues. The MUC1 overexpressed on cell membrane involves in multiple oncogenic signaling pathways (for example EGFR and β-catenin, etc.)2 and promotes tumorigenesis. Our study
reveals that WWP1 can degrade the membrane MUC1 through the lysosomal pathway, thereby inhibiting tumor growth. Importantly, we validate the function of proteasomal inhibitor-bortezomib in
promoting WWP1-mediated MUC1 degradation, providing a novel strategy for management of WWP1/MUC1 positive cancers (Fig. 1o). DATA AVAILABILITY All the data used for the current study are
available from the corresponding author upon reasonable request. REFERENCES * Lv, Y. et al. Erlotinib overcomes paclitaxel-resistant cancer stem cells by blocking the EGFR-CREB/GRbeta-IL-6
axis in MUC1-positive cervical cancer. _Oncogenesis_ 8, 70 (2019). Article CAS Google Scholar * Kufe, D. W. MUC1-C in chronic inflammation and carcinogenesis; emergence as a target for
cancer treatment. _Carcinogenesis_ 41, 1173–1183 (2020). Article CAS Google Scholar * Siegel, R. L., Miller, K. D., Fuchs, H. E. & Jemal, A. Cancer Statistics, 2021. _CA Cancer J.
Clin._ 71, 7–33 (2021). Article Google Scholar * Zhi, X. & Chen, C. WWP1: a versatile ubiquitin E3 ligase in signaling and diseases. _Cell. Mol. Life Sci._ 69, 1425–1434 (2012).
Article CAS Google Scholar * Sha, Z., Schnell, H. M., Ruoff, K. & Goldberg, A. Rapid induction of p62 and GABARAPL1 upon proteasome inhibition promotes survival before autophagy
activation. _J. Cell Biol._ 217, 1757–1776 (2018). Article CAS Google Scholar Download references ACKNOWLEDGEMENTS This work was supported by National Natural Science Foundation of China
(grant numbers 82073111 and 81874197) to L.H. and Chinese Universities Scientific Fund; CAMS Innovation Fund for Medical Sciences (2019-I2M-5-051) to G.C. We would like to appreciate Prof.
Yingli Wu and Prof. Jing Yi at Shanghai Jiao Tong University School of Medicine for construction suggestions; to appreciate Dr. Kewen Zhao and Dr. Bing Liao at Shanghai Jiao Tong University
School of Medicine for kindly provide us plasmids and reagents. Our proteomics and mass spectrometry work was performed at the Proteomics Platform of Core Facility of Basic Medical Sciences,
Shanghai Jiao Tong University School of Medicine (SJTU-SM). AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Histoembryology, Genetics and Developmental Biology, Key Laboratory
of Cell Differentiation and Apoptosis of Chinese Ministry of Education, Shanghai Key Laboratory of Reproductive Medicine, Shanghai Jiao Tong University School of Medicine, Innovative
research team of high-level local universities in Shanghai, Shanghai, P. R. China Chunhua Liao, Zhi Pang, Huayun Deng, Xiaodong Liao, Shengze Li, Jinke Cheng, Guoqiang Chen & Lei Huang *
Department of Plastic Surgery, Xiangya Hospital, Central South University, Changsha, P. R. China Liping Yu & Min Qi * Liver Cancer Institute, Zhongshan Hospital, Key Laboratory of
Carcinogenesis and Cancer Invasion, Ministry of Education, Fudan University, Shanghai, P. R. China Zhi Pang Authors * Chunhua Liao View author publications You can also search for this
author inPubMed Google Scholar * Liping Yu View author publications You can also search for this author inPubMed Google Scholar * Zhi Pang View author publications You can also search for
this author inPubMed Google Scholar * Huayun Deng View author publications You can also search for this author inPubMed Google Scholar * Xiaodong Liao View author publications You can also
search for this author inPubMed Google Scholar * Shengze Li View author publications You can also search for this author inPubMed Google Scholar * Jinke Cheng View author publications You
can also search for this author inPubMed Google Scholar * Min Qi View author publications You can also search for this author inPubMed Google Scholar * Guoqiang Chen View author publications
You can also search for this author inPubMed Google Scholar * Lei Huang View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS L.H., G.C., C.L.,
Z.P., and L.Y. designed experiments; C.L., L.Y., Z.P., H.D., X.L., and S.L. performed experiments; L.H., G.C., C.L., and J.C. analyzed data; C.L., L.H., and G.C. wrote the manuscript; L.Y.
and M.Q. participated in clinical experiments. CORRESPONDING AUTHORS Correspondence to Min Qi, Guoqiang Chen or Lei Huang. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no
competing interests. SUPPLEMENTARY INFORMATION WWP1 TARGETING MUC1 FOR UBIQUITIN-MEDIATED LYSOSOMAL DEGRADATION TO SUPPRESS CARCINOGENESIS RIGHTS AND PERMISSIONS OPEN ACCESS This article is
licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give
appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in
this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative
Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a
copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Liao, C., Yu, L., Pang, Z. _et al._ WWP1 targeting MUC1
for ubiquitin-mediated lysosomal degradation to suppress carcinogenesis. _Sig Transduct Target Ther_ 6, 297 (2021). https://doi.org/10.1038/s41392-021-00660-x Download citation * Received:
25 February 2021 * Revised: 17 May 2021 * Accepted: 23 May 2021 * Published: 18 August 2021 * DOI: https://doi.org/10.1038/s41392-021-00660-x SHARE THIS ARTICLE Anyone you share the
following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer
Nature SharedIt content-sharing initiative