Play all audios:
This study aimed to develop a pan-genotypic and multifunctional small interfering RNA (siRNA) against hepatitis B virus (HBV) with an efficient delivery system for treating chronic hepatitis
B (CHB), and explore combined RNA interference (RNAi) and immune modulatory modalities for better viral control. Twenty synthetic siRNAs targeting consensus motifs distributed across the
whole HBV genome were designed and evaluated. The lipid nanoparticle (LNP) formulation was optimized by adopting HO-PEG2000-DMG lipid and modifying the molar ratio of traditional
polyethylene glycol (PEG) lipid in LNP prescriptions. The efficacy and safety of this formulation in delivering siHBV (tLNP/siHBV) along with the mouse IL-2 (mIL-2) mRNA (tLNP/siHBVIL2) were
evaluated in the rAAV-HBV1.3 mouse model. A siRNA combination (terms “siHBV”) with a genotypic coverage of 98.55% was selected, chemically modified, and encapsulated within an optimized LNP
(tLNP) of high efficacy and security to fabricate a therapeutic formulation for CHB. The results revealed that tLNP/siHBV significantly reduced the expression of viral antigens and DNA (up
to 3log10 reduction; vs PBS) in dose- and time-dependent manners at single-dose or multi-dose frequencies, with satisfactory safety profiles. Further studies showed that tLNP/siHBVIL2
enables additive antigenic and immune control of the virus, via introducing potent HBsAg clearance through RNAi and triggering strong HBV-specific CD4+ and CD8+ T cell responses by expressed
mIL-2 protein. By adopting tLNP as nucleic acid nanocarriers, the co-delivery of siHBV and mIL-2 mRNA enables synergistic antigenic and immune control of HBV, thus offering a promising
translational therapeutic strategy for treating CHB.
Hepatitis B virus (HBV) infection can lead to chronic hepatitis B (CHB), liver fibrosis, cirrhosis, and hepatocellular carcinoma (HCC); however, therapeutic options are limited.1
Nucleot(s)ide analogues efficiently suppress viral replication but fail to reduce viral antigen levels, PEGylated interferons possess both direct antiviral and immunomodulating effects,
whereas the efficacy is moderate, and response rate in clinical patients is low. The HBV genome is a compact 3.2 kb circular, partially double-stranded, relaxed circular DNA (rcDNA). Once
entering the nucleus, rcDNA is repaired by endogenous enzymes into covalently closed circular DNA (cccDNA), which encodes four overlapping open reading frames (ORFs), including the core
antigen (HBcAg), surface antigen (HBsAg), e antigen (HBeAg), and X protein (HBx).2 All HBV transcripts have a common 3′ end and share the same polyadenylation signal (PAS). Besides,
replication-incompetent HBV linear fragments can integrate into the host genome (intHBV), serving as the predominant source of HBsAg in HBeAg-negative patients.3,4,5 High-circulating HBV
antigen levels, especially HBsAg, contribute to immune tolerance and viral persistence.6,7 Since complete cure (clearance of cccDNA and intHBV) is unattainable, the evolving treatment goal
for CHB is to achieve HBsAg seroclearance with therapy of finite duration, that is, “functional cure”.8,9,10
RNA interference (RNAi) technology has shown promises for treating genetic disease and virus-infection diseases. It is also considered as an attractive therapeutic modality to achieve
functional cure of CHB by inducing antigen inhibition, viraemia reduction, and cccDNA silence.11,12,13 siRNA-based therapies can also relieve high viral antigen-induced immune tolerance,
offering opportunity to gain immune control of the viruses by subsequent immune-stimulation. However, designing functional small interfering RNAs (siRNAs) against complicated viruses like
HBV is challenging considering the extraordinarily high genetic diversity among the 10 different genotypes (A to J).14,15,16 siRNA designed for targeting conserved regions among HBV
genotypes is achievable by computational prediction and is anticipated to resist potential viral mutational escape, whereas siRNA targeting the common 3′-end sequences shared by
cccDNA-derived transcripts may loss targets on intHBV-derived transcripts.16,17,18 Further studies using siRNA triggers designed to address both cccDNA- and intHBV-driven synthesis are
expected to surpass those of previous agents in terms of efficacy and functionality.
The clinical application of RNAi therapeutics is hindered by challenges associated with liver-targeted delivery systems. With advantages of newly developed GalNAc ((N-acetylgalactosamine)
technology and lipid nanoparticles for liver-specific delivery, siRNA-based drugs such as Partisiran [Onpattro®] and Inclisiran [Leqvio®] are clinically available. Recently, ionizable
cationic lipid nanoparticles (LNPs) were designed for the efficient delivery of therapeutic nucleic acids in vivo.19 LNPs are typically composed of ionizable cationic lipids, cholesterol,
polyethylene glycol (PEG) lipids, and auxiliary lipids. PEG lipids provide a neutral hydrophilic exterior, stabilize the particle, and prevent rapid clearance in the circulation. However,
there is also “PEG dilemma”. PEG ratio affects the performance of LNP in vivo. Despite the weak immunogenicity of PEG, a certain proportion of humans may develop low levels of PEG-specific
antibodies, leading to accelerated clearance of PEGylated nanomedicines and reduced efficacy.20,21 The terminal PEGylation group influences PEG immunogenicity, with hydroxy-PEG being less
immunogenic than methoxy-PEG.22 Moreover, our group recently uncovered that hydroxy-PEG has weaker antigenicity than methoxy-PEG. Hydroxy-PEG can avoid the binding of pre-existing anti-PEG
antibodies in human blood, thereby reducing complement activation, which allows LNPs to escape antibody recognition and rapid clearance.23
Although RNAi- and antibody-mediated HBsAg clearance may withdraw immune dysfunction by reducing the viral burden, they cannot activate viral-specific T cells to realize long-term viral
control.24,25 Only tiny minority of patients produce antibodies against HBsAg in the clinic, and viral antigens recover after drug withdrawal.26,27,28 Recently, pre-clinical and clinical
studies have been investigating novel combination strategies for attaining improved therapeutic outcomes, such as combined RNAi therapeutics with therapeutic vaccines, interferons, and
anti-HBs antibodies.29,30,31 Studies indicated that IL-2 is a critical mediator for proper viral antigen presentation and activation and differentiation of virus-specific CD4+ and CD8+ T
cells.32,33,34,35 Sequential low-dose IL-2 along with IFN-α can increase the frequency and restore the function of HBV-specific CD8+ T cell responses in patients,36 indicating the potential
of constructing viral-specific immune control to complement RNAi. We propose that concurrent suppression of the viral load by RNAi and enhancement of host immunity by IL-2 may be sufficient
to break immune tolerance and reconstruct antiviral immunity.
In the present study, we screened and chemically modified a pan-genotypic and multifunctional siRNA combination (siHBV) against all forms of cccDNA- and intHBV-derived transcripts, and
validated its efficacy and safety in multiple cell culture and murine models. An optimized LNP platform (tLNP) with low antigenicity and high efficacy was utilized to define the role of
siHBV in controlling HBV transcription and replication and to evaluate its safety profile. By utilizing the feasibility of tLNP to encapsulate different forms of nucleic acids, co-delivery
of siHBV and mouse IL-2 (mIL-2) mRNA within a single tLNP formulation was applied to obtain both antigenic and immune control of the virus. We anticipate that tLNP-based siHBV and mIL-2 mRNA
co-delivery may provide a feasible approach for the treatment of CHB.
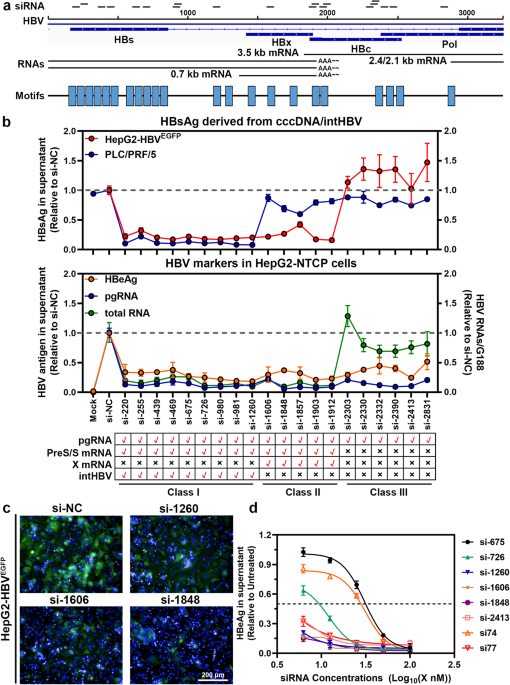
To design highly conservative siRNA against HBV, a collection of 11,185 HBV genome sequences were downloaded from the Hepatitis Virus Database (HVDB) website
(http://s2as02.genes.nig.ac.jp/index.html) and analyzed for highly conserved motifs via motif-based sequence analysis tools (MEME Suite 5.4.1) (https://meme-suite.org/meme/tools/meme). HBV
typical sequences were put into to professional siRNA design websites, including siDirect, siDESIGN Center, DSIR et al. Twenty siRNAs targeting HBV conserved motifs were selected
distributing throughout the HBV whole genome. The criteria used for siRNA design were as follows: 1. Accordance with the basic design principle provided by online software; 2. Have limited
homology with any known sequence in the human, mouse and rat genomes. Unmodified and chemically modified siRNA used in this study were then synthesized by RiboBio (Guangzhou, China) and
GenePharma (Shanghai, China) for experiments.
We defined “conservation” as the percentage of sequence entries out of the 11,185 HBV sequences that showed perfect identity with the 19/2-18-mer.
HepG2-HBVEGFP cell line was constructed in-house as previous reported.37,38 In brief, pSBbi-HBVEGFP was created by fusion of the loxP sites flanking monomeric linear HBV sequence (GenBank
accession no. V01460.1) with the EGFP sequence and the splicing acceptor sequence inserted into the core region in the HBV genome to the pSBbi vector (Addgene plasmid #60525). The
pSBbi-HBVEGFP and pCMV-SB100 plasmids were then co-transfected into HepG2 cells to pick neomycin-resistance colonies and to construct stable cell lines.
HepG2, HepAD38, HepG2-NTCP, and HepG2-HBVEGFP cells were maintained in DMEM supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. PLC/PRF/5 cells were maintained in
RPMI-1640 medium supplemented with 10% FBS and 1% penicillin-streptomycin. Cells were seeded in 96-, 48-, or 24- well plates, then treated with indicated doses of siRNA. Transfection was
carried out using Lipofectamine RNAiMAX transfection reagents (Invitrogen, USA) according to the manufacturer’s protocol or using LNPs. Supernatants and cells were collected at 72 h
post-transfection.
Pan-genotypic antiviral activity was conducted in HepG2 cells transfected with plasmid DNA harboring 1.3 × overlength sequence of HBV genotype A, B, C, and D isolates. HepG2 cells were
transfected with HBV genotype A-D encoding plasmids, then treated with varying doses of siRNA. Culture supernatants were collected at 72 h post-transfection and assayed for HBsAg and HBeAg
levels using ELISA (Autobio Diagnostics Co., Ltd., China).
siRNA was transfected with RNAiMAX at the concentration of 0, 6.25, 12.5, 25, 50, and 100 nM, respectively. For HBV antigen and mRNA examination, the cells and supernatants were harvested
separately at 72 h post-transfection.
C57BL/6 mice (5–6 weeks old) were purchased from GemPharmatech company (Jiangsu, China). Animals were maintained under specific pathogen-free conditions in Fudan University Laboratory Animal
Center or in the ABSL-2 animal facility at the Key Laboratory of Medical Molecular Virology (MOE/NHC/CAMS), School of Basic Medical Sciences, Shanghai Medical College, Fudan University. All
procedures involving experimental animals were performed according to the protocols approved by the Animal Ethics Committee of School of Basic Medical Science, Fudan University (approval
No. 20211115-004).
The rAAV-HBV1.3 mouse model39 was employed to evaluate the gene silencing efficiency of siHBV encapsulating tLNP (terms “tLNP/siHBV”). In brief, male C57BL/6 mice were inoculated with 2.5 ×
1010 vector genomes (v.g.)/mice of rAAV-HBV1.3 virus carrying a 1.3 × overlength HBV genome (ayw, Five Plus, Beijing, China). Animals were bled one day before start of treatment and divided
into groups to obtain similar HBsAg and HBeAg levels. Six groups of mice (eight animals per group) were dosed with 1× phosphate-buffered saline (PBS), tLNP/siNC, tLNP/siHBV, and ETV,
respectively. Both siNC and siHBV were modified by 2′-O-methylation to improve their stability. The dose of tLNP/siNC was 1 mg/kg. The doses of tLNP/siHBV were 0.1, 0.3 and 1 mg/kg,
respectively. A single dose of tLNP/siHBV formula was intravenously injected into mice at indicated doses. Blood samples were collected weekly during the experiments. Liver specimens were
collected for histological and molecular analyses.
For multidose experiments, tLNP/siHBV were weekly (five doses in total) or biweekly (three doses in total) injected into the mice. The serum samples were collected weekly, and were applied
for HBsAg, HBeAg and HBV DNA analysis according to the manufacturer’s instructions. The mice were sacrificed at the end of experiment, and tissue samples were collected for further
detection.
Viral titers in the serum were quantified with HBV-specific primers and probes using commercial kits (Sansure Biotech, Changsha, China) with a standard curve for absolute quantification.
HBsAg, HBeAg, anti-HBs and anti-HBc levels were quantified via ELISA analysis using commercial kits (AutoBio Diagnostics Co., Ltd., Zhengzhou, China), respectively.
As for HBV-derived mRNA, such as pgRNA and total RNAs, cell or liver RNA were extracted and reverse transcribed into cDNA using PrimeScriptTM RT reagent Kit with gDNA Eraser (Takara, Japan),
and real-time quantitative PCR was performed using SYBR® Premix Ex TaqTM (Tli RNaseH Plus), Bulk (Takara, Japan). HBV-specific primers were used as follows: HBV pgRNA (forward:
5′-GCCTTAGAGTCTCCTGAGCA-3′; reverse: 5′-GAGGGAGTTCTTCTTCTAGG-3′) and the HBV total RNA (forward: 5′-GCTTTCACTTTCTCGCCAAC-3′; reverse: 5′-GAGTTCCGCAGTATGGATCG-3′). The expression of human or
mouse housekeeping genes (GAPDH, β-Actin) were used for normalization.
As for intrahepatic HBsAg levels, liver sections were homogenized with PBS, centrifuged, and applied for HBsAg determination and protein quantification using commercial kits. Intrahepatic
cccDNA, core particle DNA and total RNA were extracted as we previous reported,40 then applied for southern blot, northern blot and real-time quantitative PCR analysis with specific primers.
Organs were collected and fixed in 10% formaldehyde, embedded in paraffin, and then sectioned for H&E staining. Histopathologic evaluation was performed by an experienced liver pathologist
in a blinded manner. For immunochemistry, anti-CD8 (Servicebio, Wuhan, China), anti-CD4 (Servicebio, Wuhan, China) and anti-HBc (Long Island Antibody, Shanghai, China) antibodies were used.
For flow cytometry analysis, isolated cells were suspended in FACS buffer. Cell-surface staining was performed using anti-mouse CD3, anti-mouse CD8 (Alexa Fluor 700, eBioscience) and
anti-mouse CD4 (eFluor™ 506, eBioscience) antibodies. Dead cells were excluded from analysis by fixable viability dye eF780 staining. Intracellular cytokine staining was performed using
indicated antibody after fixation and permeabilization. Data were collected using Attune flow cytometer (Thermo Fisher, USA) and analyzed using FlowJo software version 10 (Tree Star,
Ashland, OR). H-2Kb tetramer conjugated with HBV-derived peptides S109 (VWLSVIWM) and C93 (MGLKFRQL) were purchased from Creative Biosciences (Guangzhou, China), and were subjected for
detecting HBsAg-specific and core-specific CD4+ and CD8+ T cells.
The data were expressed as the means ± standard deviations (SDs) or means ± standard error of means (SEMs). One-way or two-way analysis of variance (ANOVA) was applied for multiple
comparisons. Significance was defined as *P