Play all audios:
Dear Editor, While chemoimmunotherapy has been the standard treatment for chronic lymphocytic leukemia (CLL), it has been supplanted by targeted therapies, including the Bruton’s tyrosine
kinase inhibitor, ibrutinib, and the bcl-2 inhibitor, venetoclax [1]. The prognosis and prediction of response to chemotherapy are intimately linked to the leukemic cell _IGHV_ mutational
status and fluorescent in situ hybridization abnormalities [2]. Patients with _IGHV_ unmutated CLL have more aggressive disease with a preponderance of deletion 17p13 (del 17p; loss of
_TP53_ gene) or del 11q22-23 (del 11q; loss of ataxia telangiectasia mutated (_ATM_) gene) cases. Del 11q occurs in 20% of CLL patients and is associated with a short time to first treatment
(TTFT) and early relapse following chemotherapy [3]. However, these patients have an excellent response to ibrutinib [4]. Two-thirds of patients have an isolated del 11q and one-third an
_ATM_ mutation on the other allele, which can produce an additional loss of ATM function [5, 6]. However, an isolated del 11q has biological effects as telomere length is extremely short in
del 11q cells, even without an additional _ATM_ mutation [7]. Moreover, the fraction of the cell population with del 11q is important, with shortening of TTFT only occurring when >25–58%
of cells have the deletion [8, 9]. These data suggest that loss of one ATM gene can have biological effects. To determine if ATM protein levels are altered in CLL samples with del 11q, we
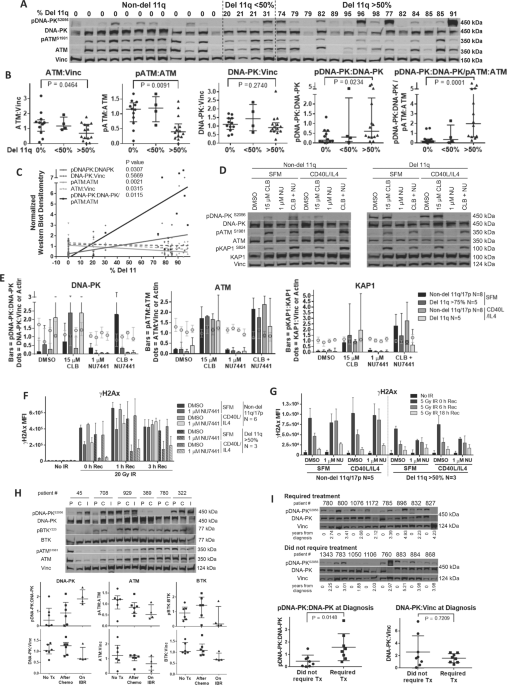
examined the levels of total and phosphorylated (activated) ATM (pATMS1981) and DNA-PK (pDNA-PKS2056) by western blot in 30 patient samples with varying numbers of cells with del 11q (from 0
to 91%, Fig. 1A, B and Tables S1 and S2). Materials and Methods are in Supplementary. Twelve cases did not have del 11q or del 17 p (0%), 4 cases had a low del 11q fraction (<50%) while
14 cases had had a high del 11q fraction (>50%). While the levels of DNA-PK and ATM protein varied, ATM levels and the pATM:ATM ratio decreased as the fraction of del 11q cells increased
(_p_ = 0.009). These changes likely represent a dose-dependent loss of ATM activity as the fraction of cells with a del 11q increases, as mutations on the other allele do not necessarily
correlate with further loss of ATM function [6]. In contrast, the pDNA-PK:DNA-PK ratio increased as the fraction of del 11q cells increased (_p_ = 0.023). Similarly, the ratio of activated
DNA-PK relative to activated ATM (pDNA-PK:DNA-PK/pATM:ATM) increased significantly (_p_ = 0.0001) as the fraction of del 11q cells (Fig. 1A–C and Table S2) increased. Thus, DNA-PK is
activated in del 11q cases with a high fraction of del 11 cells, to compensate for loss of ATM. We then assessed the biological function of the DNA damage response (DDR) proteins in del 11q
samples to determine if ATM function is lost and compensated for by an increase in DNA-PK activity. We thus compared non-del 11q and del 11q (>75% deletion) CLL samples following
treatment with the DNA-cross-linking drug, chlorambucil, in the presence and absence of the DNA-PK inhibitor, NU7441. We also assessed the induction of phosphorylated KAP1 (pKAP1S824) as a
measure of ATM function [6]. Chlorambucil increased DNA-PK activation to a 2.5-fold greater degree in del 11q samples, as compared to non-del 11q samples, and pDNA-PK formation was
eliminated by NU7441 (Figs. 1D, E and S1 and Tables S1 and S3). pATM and pKAP1 levels were similarly increased by chlorambucil in all cases. pATM and pKAP1 increased further in non-del 11q
samples following NU7441 but not in del 11q cells. To evaluate the DDR, we compared the effects of NU7441 by γH2AX foci formation (measures DDR signaling at breaksites) and alkali comet
assay (measures DNA strand breaks) in irradiated samples [10]. Del 11q cells (>50%) developed lower γH2AX levels compared to non-del 11q cells following irradiation (Fig. 1F and Table
S1). Co-treatment with NU7441 delayed γH2AX formation and clearance, especially in del 11q samples (Fig. 1G and Table S1). Although ATM kinase is typically involved in γH2AX formation,
DNA-PK is invoked with sub-optimal ATM function [11]. Furthermore, DNA strand break repair did not appear to be affected by del 11q status or DNA-PK activity (Fig. S2 and Table S1). Thus,
ATM mediates DDR signaling in non-del 11q samples but these activities may be replaced by DNA-PK in del 11q cells [11]. DNA-PK activity may also be increased in CLL patients who have
received prior chlorambucil [12]. We thus examined DNA-PK and ATM activation in six patients who received chemoimmunotherapy and then ibrutinib following relapse (Fig. 1H and Table S4).
Activation of DNA-PK was significantly increased in four patients relapsing after chemoimmunotherapy (45, 708, 929, 322) paralleled by a decrease in activated ATM. In contrast, two patients
had low pDNA-PK prior to therapy with little change following chemotherapy. DNA-PK activation increased further following ibrutinib, with a decline in activated ATM. In contrast to ATM,
enhanced DNA-PK supresses homologous recombination, induces non-homologous end-joining (NHEJ) and is associated with aggressive disease in multiple cancers [13]. To determine if this was
true in CLL, pDNA-PK levels were compared in 16 new CLL patients, 8 requiring therapy in a median of 3 years, while 8 did not require treatment in this time period (Fig. 1I and Table S5).
The baseline pDNA-PK:DNA-PK level was higher in those that required therapy suggesting that increased DNA-PK may explain the short TTFT in del 11q (_p_ = 0.015). To determine whether the
elevated DNA-PK activity in del 11q cells influenced drug sensitivity, CLL samples were treated with NU7441. Interestingly, del 11q CLL cells were more sensitive to NU7441 than non-del 11q
cases [14]. In total, 1 µM NU7441 decreased cell viability by 30% at 72 h (Fig. 2A and Table S1). In contrast, NU7441 had little activity against normal B (CD19+) and T (CD3+) cells,
demonstrating that this is a tumor specific activity. Inhibition of DNA-PK has been shown to enhance drug sensitivity in CLL, so we assessed whether this was greater in del 11q cells (Fig.
2B, C and Tables S1 and S6) [14]. Six samples had >50% del 11q, 4 < 50% del 11q, 17 were non-del 11q and 1 del 17p. Following 72 h treatment, cell death was measured by Annexin V/7AAD
staining and the drug concentration required to reduce cell viability by 50% (EC50) was measured. Samples with >50% del 11q cells were more resistant to chemotherapy than non-del 11q
patients (median chlorambucil EC50, 38.1 μM versus 15.8 μM for del 11q and non-del 11q, respectively; Fig. 2B, _p_ = 0.013). In contrast, the median ibrutinib EC50 for del 11q and non-del
11q cells were similar, at 6.1 and 3.9 μM, respectively (Fig. 2C, _p_ = 0.3124). However, NU7441 sensitized both del 11q and non-del 11q cells to the chemotherapies chlorambucil, fludarabine
or bendamustine to a similar extent (Tables S1 and S6). Resistant cells were also sensitized, including a sample with del 17p. NU7441 did not sensitize CLL cells to ibrutinib or idelalisib.
NU7441 produced little sensitization of normal B and T cells to chlorambucil (Tables S1 and S6). As NU7441 is not for clinical use, we evaluated M3814 and CC-115 in CLL cells, as both
agents are being evaluated clinically [13, 15]. Initially, CLL cells from two patients were treated with CC-115 (inhibitor of DNA-PK/mTOR/PI3K) or M3814 (inhibitor of DNA-PK; Fig. S3) and
chlorambucil or ibrutinib. Synergy was greater between chlorambucil and M3814 than between chlorambucil and CC-115; however, ibrutinib showed synergy with CC-115 but not M3814 (Fig. S4),
likely a result of the effect of CC-115 on mTOR/PI3K activity. Further studies with M3814 showed it to be strongly synergistic with chlorambucil in CLL cells from 14 untreated patients
without del 17p (Figs. 2D and S5), independent of del 11q status or anti-IgM treatment. In patients on long-term ibrutinib, we observed enhanced pDNA-PK levels over time, which inversely
correlated with pATM levels (Fig. 2E and Table S7). Importantly, sensitization between chlorambucil and M3814 was observed in cells from three of five patients on long-term ibrutinib (505,
190, and 847) including one (505) who was resistant to ibrutinib (Fig. 2D, F). Synergy increased throughout 1 year of ibrutinib treatment in patient 190 (Fig. S3). The combination of
ibrutinib with M3814 in cells from patients on long-term ibrutinib showed additive or slightly synergistic cytotoxicity in three of four patients (505, 830, and 847) while the del 17p sample
(171) showed antagonism (Fig. 2D, F). These studies show that DNA-PK is activated and responsible for DDR in del 11q CLL cells, and this may explain the short TTFT and resistance to
chemotherapy, but not ibrutinib, in these patients. DNA-PK is not activated by disease duration or progression but increases following ibrutinib and chemotherapy, suggesting that it is an
effect of the DNA damage signaling response/NHEJ. Del 11q cells were more sensitive to DNA-PK inhibition than non-del 11q cells or normal lymphocytes, but inhibition sensitized all CLL types
(but not normal lymphocytes) to chemotherapy, including those that were both chemotherapy- and ibrutinib-resistant. Hyperactivated DNA-PK is associated with aggressive disease in a variety
of hematologic and solid tumors; likely reflecting enhanced mutation-prone NHEJ activity with genomic instability [13]. Thus, combining M3814 with chemotherapy may be a useful approach for
treating multidrug-resistant CLL patients. DATA AVAILABILITY Please e-mail the corresponding author for any data requests. REFERENCES * Kay NE, Hampel PJ, van Dyke DL, Parikh SA. CLL update
2022: a continuing evolution in care. Blood Rev. 2022;54:100930. https://doi.org/10.1016/j.blre.2022.100930. Article Google Scholar * Parikh SA, Strati P, Tsang M, West CP, Shanafelt TD.
Should IGHV status and FISH testing be performed in all CLL patients at diagnosis? A systematic review and meta-analysis. Blood. 2016;127:1752–60.
https://doi.org/10.1182/blood-2015-10-620864. Article CAS Google Scholar * Stankovic T, Skowronska A. The role of ATM mutations and 11q deletions in disease progression in chronic
lymphocytic leukemia. Leuk Lymphoma. 2014;55:1227–39. http://www.ncbi.nlm.nih.gov/pubmed/23906020. Article CAS Google Scholar * Kipps TJ, Fraser G, Coutre SE, Brown JR, Barrientos JC,
Barr PM, et al. Long-term studies assessing outcomes of ibrutinib therapy in patients with del(11q) chronic lymphocytic leukemia. Clin Lymphoma Myeloma Leuk. 2019;19:715–22.e6. Article
Google Scholar * Austen B, Skowronska A, Baker C, Powell JE, Gardiner A, Oscier D, et al. Mutation status of the residual ATM allele is an important determinant of the cellular response to
chemotherapy and survival in patients with chronic lymphocytic leukemia containing an 11q deletion. J Clin Oncol. 2007;25:5448–57. Article CAS Google Scholar * Jiang Y, Chen HC, Su X,
Thompson P, Liu X, Do KA, et al. ATM function and its relationship with ATM gene mutations in chronic lymphocytic leukemia with the recurrent deletion. Blood Cancer J. 2016;6:e465. Article
CAS Google Scholar * Britt-Compton B, Lin TT, Shmed G, Weston V, Jones R, Fegan C, et al. Extreme telomere erosion in ATM-mutated and 11q-deleted CLL patients is independent of disease
stage. Leukemia. 2011;26:1–4. Google Scholar * Jain P, Keating M, Thompson PA, Trinh L, Wang X, Wierda W, et al. High fluorescence in situ hybridization percentage of deletion 11q in
patients with chronic lymphocytic leukemia is an independent predictor of adverse outcome. Am J Hematol. 2015;90:471–7. Article CAS Google Scholar * Marasca R, Maffei R, Martinelli S,
Fiorcari S, Bulgarelli J, Debbia G, et al. Clinical heterogeneity of de novo 11q deletion chronic lymphocytic leukaemia: prognostic relevance of extent of 11q deleted nuclei inside leukemic
clone. Hematol Oncol. 2013;31:348–55. Article CAS Google Scholar * Mariotti LG, Pirovano G, Savage KI, Ghita M, Ottolenghi A, Prise KM, et al. Use of the γ-H2AX assay to investigate DNA
repair dynamics following multiple radiation exposures. PLoS ONE. 2013;8:1–12. http://www.ncbi.nlm.nih.gov/pubmed/24312182. Article Google Scholar * Enriquez-Rios V, Dumitrache LC, Downing
SM, Li Y, Brown EJ, Russell HR, et al. DNA-PKcs, ATM, and ATR interplay maintains genome integrity during neurogenesis. J Neurosci. 2017;37:893–905. Article CAS Google Scholar * Muller
C, Christodoulopoulos G, Salles B, Panasci L. DNA-dependent protein kinase activity correlates with clinical and in vitro sensitivity of chronic lymphocytic leukemia lymphocytes to nitrogen
mustards. Blood. 1998;92:2213–20. http://www.bloodjournal.org/content/92/7/2213.short. Article CAS Google Scholar * Dylgjeri E, Knudsen KE. DNA-PKcs: a targetable protumorigenic protein
kinase. Cancer Res. 2022;82:523–33. Article CAS Google Scholar * Willmore E, Elliott SL, Mainou-Fowler T, Summerfield GP, Jackson GH, O’Neill F, et al. DNA-dependent protein kinase is a
therapeutic target and an indicator of poor prognosis in B-cell chronic lymphocytic leukemia. Clin Cancer Res. 2008;14:3984–92. Article CAS Google Scholar * Tsuji T, Sapinoso LM, Tran T,
Gaffney B, Wong L, Sankar S, et al. CC-115, a dual inhibitor of mTOR Kinase and DNA-PK, blocks DNA damage repair pathways and selectively inhibits ATM-deficient cell growth in vitro.
Oncotarget. 2017;8:74688–702. Article Google Scholar Download references ACKNOWLEDGEMENTS The Manitoba Tumor Bank (Winnipeg, Manitoba) is supported by the CancerCare Manitoba Foundation
and is a member of the Canadian Tissue Repository Network. We are thankful to Drs. Jody Haigh (U. Manitoba) and Roseline Godbout (U. Alberta) for their comprehensive pre-submission review of
the final manuscript. FUNDING Supported by Research Manitoba (Manitoba CLL Cluster Grant), Canadian Institutes of Health Research, Canadian Foundation for Innovation, and CancerCare
Manitoba Foundation. AUTHOR INFORMATION Author notes * These authors contributed equally: James B. Johnston, Sachin Katyal. AUTHORS AND AFFILIATIONS * CancerCare Manitoba Research Institute,
CancerCare Manitoba, Winnipeg, MB, R3E 0V9, Canada Sara E. F. Kost, Ali Saleh, Shek H. Yuan, Bozena Kuzio, Spencer B. Gibson, Versha Banerji, James B. Johnston & Sachin Katyal *
Department of Pharmacology and Therapeutics, Max Rady College of Medicine, Rady Faculty of Health Sciences, University of Manitoba, Winnipeg, MB, R3E 3P4, Canada Ali Saleh, Shek H. Yuan,
Bozena Kuzio & Sachin Katyal * Department of Oncology, University of Alberta, Edmonton, AB, Canada Spencer B. Gibson * Department of Internal Medicine, Max Rady College of Medicine, Rady
Faculty of Health Sciences, University of Manitoba, Winnipeg, MB, R3E 3P4, Canada Lin Yang, Versha Banerji & James B. Johnston * Department of Biochemistry and Medical Genetics Max Rady
College of Medicine, Rady Faculty of Health Sciences, University of Manitoba, Winnipeg, MB, R3E 3P4, Canada Versha Banerji Authors * Sara E. F. Kost View author publications You can also
search for this author inPubMed Google Scholar * Ali Saleh View author publications You can also search for this author inPubMed Google Scholar * Shek H. Yuan View author publications You
can also search for this author inPubMed Google Scholar * Bozena Kuzio View author publications You can also search for this author inPubMed Google Scholar * Spencer B. Gibson View author
publications You can also search for this author inPubMed Google Scholar * Lin Yang View author publications You can also search for this author inPubMed Google Scholar * Versha Banerji View
author publications You can also search for this author inPubMed Google Scholar * James B. Johnston View author publications You can also search for this author inPubMed Google Scholar *
Sachin Katyal View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS SEFK, AS, SHY, and LY carried out the experiments with technical assistance
from BK. SEFK, SBG, JBJ, and SK designed the research, collected, analyzed and interpreted data. VB, LY, and JBJ cared for patients contributing samples. SEFK, JBJ, and SK wrote the
manuscript. All authors approved the manuscript in its final format. CORRESPONDING AUTHORS Correspondence to James B. Johnston or Sachin Katyal. ETHICS DECLARATIONS CONFLICT OF INTEREST VB
and LY: advisory board-members for Abbvie, Janssen and AstraZeneca. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published
maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY MATERIAL RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0
International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the
source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Kost, S.E.F., Saleh, A., Yuan, S.H. _et al._ DNA-PK hyperactivation occurs in
deletion 11q chronic lymphocytic leukemia and is both a biomarker and therapeutic target for drug-resistant disease. _Blood Cancer J._ 13, 20 (2023).
https://doi.org/10.1038/s41408-022-00781-8 Download citation * Received: 16 August 2022 * Revised: 20 December 2022 * Accepted: 22 December 2022 * Published: 27 January 2023 * DOI:
https://doi.org/10.1038/s41408-022-00781-8 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative