Play all audios:
ABSTRACT BACKGROUND Chimeric antigen receptor (CAR) T cell therapy has been successfully translated to clinical practice for the treatment of B cell malignancies. The suppressive
microenvironment of many malignancies is a bottleneck preventing treatment success of CAR T cells in a broader range of tumours. Among others, the immunosuppressive metabolite adenosine is
present in high concentrations within many tumours and dampens anti-tumour function of immune cells and consequently therapeutic response. METHODS Here, we present the impact of the
selective adenosine A2A and A2B receptor antagonist AB928/etrumadenant on CAR T cell cytokine secretion, proliferation, and cytotoxicity. Using phosphorylation-specific flow cytometry, we
evaluated the capability of AB928 to shield CAR T cells from adenosine-mediated signalling. The effect of orally administered AB928 on CAR T cells was assessed in a syngeneic mouse model of
colon carcinoma. RESULTS We found that immunosuppressive signalling in CAR T cells in response to adenosine was fully blocked by the small molecule inhibitor. AB928 treatment enhanced CAR T
cell cytokine secretion and proliferation, granted efficient cytolysis of tumour cells in vitro and augmented CAR T cell activation in vivo. CONCLUSIONS Together our results suggest that
combination therapy with AB928 represents a promising approach to improve adoptive cell therapy. SIMILAR CONTENT BEING VIEWED BY OTHERS ENHANCING IMMUNOTHERAPY IN CANCER BY TARGETING
EMERGING IMMUNOMODULATORY PATHWAYS Article 27 September 2021 INHIBITION OF ENT1 RELIEVES INTRACELLULAR ADENOSINE-MEDIATED T CELL SUPPRESSION IN CANCER Article Open access 12 May 2025
CRISPR/CAS9 MEDIATED DELETION OF THE ADENOSINE A2A RECEPTOR ENHANCES CAR T CELL EFFICACY Article Open access 28 May 2021 BACKGROUND Immunotherapy has become a new pillar of cancer therapy
improving the clinical outcome of many patients with solid and haematological malignancies. Immune checkpoint blockade (ICB) has changed clinical practice and demonstrated the clinical
utility of T cells in oncology [1]. ICB can lead to durable clinical responses in a variety of cancer types by reactivating suppressed or exhausted T effector cells [2, 3]. Currently,
approved checkpoint inhibitors target mainly the programmed cell death protein 1 (PD-1) axis or cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) to reinvigorate anti-tumour immunity [1].
However, a significant number of patients will either fail to respond or relapse after an initial response. Frequently, this can be attributed to an immunosuppressive environment, which is
not overcome by conventional ICB [4]. Extracellular adenosine acts as soluble immune checkpoint and has emerged as a promising target for immunotherapy [5,6,7]. Within solid tumours,
extracellular ATP levels are elevated due to high cellular turnover and active secretion [8, 9]. In the canonical pathway of extracellular adenosine generation, the ectonucleotidases CD39
and CD73 lead to the sequential dephosphorylation of extracellular ATP [10]. Expression of CD39 and CD73 on tumour cells, immune cells, fibroblasts, endothelial cells, and stromal cells is
upregulated by hypoxia and TGF-β in the tumour microenvironment (TME) [6, 11, 12]. Other mechanisms such as extracellular AMP generation from NAD+ via CD38 and dysregulation of adenosine
consuming pathways further contribute to extracellular adenosine accumulation within solid tumours [7, 13]. High concentrations of extracellular adenosine dampen anti-tumour immunity [5, 6,
14]. Upon binding of extracellular adenosine, the G-protein coupled adenosine A2A and A2B receptors (A2AR and A2BR) (Kd of 100 nM and 15 µM, respectively) [15] mediate an intracellular
build-up of cAMP that compromises T cell effector functions [14, 16,17,18,19]. Owing to the predominant expression and higher affinity of A2AR, T cell suppression is primarily mediated by
signalling downstream of A2AR [20, 21]. Besides direct suppression of effector cells, A2AR and A2BR activation promotes the generation and suppressive capacity of myeloid cells and
regulatory T cells (Tregs) [22,23,24,25,26]. Several agents counteracting the immunosuppressive adenosine axis have been developed and have shown promising preclinical anti-tumour activity
[5,6,7]. The small molecule AB928/etrumadenant (for short, AB928) is a highly selective antagonist, targeting the A2A and A2B receptor [27, 28]. Importantly, results from early clinical
trials in healthy volunteers and in patients demonstrated safety and favourable pharmacological properties of the new drug, potentially hinting at its potential for combinatorial treatments
[27, 29, 30]. Another immunotherapeutic axis leveraging T cell function at the forefront of development in oncology are chimeric antigen receptor (CAR) T cells. These are autologous T cells,
genetically engineered to stably express a synthetic receptor targeting a specific antigen [31]. While CAR T cell therapy is highly efficacious in the treatment of some haematological
malignancies [32,33,34], it still lacks efficacy in the vast majority of tumours [35, 36]. Three major mechanisms for CAR T cell failure in solid tumours have been identified: Lack of T cell
access to tumour sites, antigen heterogeneity and importantly immune suppression [37]. In fact, the immunosuppressive TME limits CAR T cell responses against solid tumours [31] and
anti-tumour CAR T cell responses are suppressed by adenosine. Recent preclinical evidence suggests that pharmacological as well as genetic targeting of A2AR may improve CAR T cell efficacy
[20, 21, 38,39,40]. However, targeting has thus far been limited to the A2AR pathway and mainly to anti-CD19, anti-mesothelin and anti-Her2 CAR T cells [20, 21, 38,39,40]. Whether A2BR
co-targeting yields similar or better results is unclear. Given the high complexity of CAR function and design [36], it also remains to be determined if the approach is broadly applicable
across different models. These considerations and the advanced clinical development stage of AB928 become important when considering implementing adenosine receptor blockade into cell
therapy trials. Thus, we asked the question if AB928-mediated blockade of A2AR and A2BR synergises both with murine and human CAR T cells for optimised functionality against a range of
murine and human cancer cell lines. MATERIALS AND METHODS MICE Wild-type C57BL/6 and BALB/c mice were purchased from Janvier (St. Bertevin, France) or Charles River (Sulzfeld, Germany).
ANIMAL EXPERIMENTS All experimental studies were approved and performed in accordance with guidelines and regulations of the local regulatory agency (Regierung von Oberbayern). The
experiments were randomised and conducted with adequate controls. The investigators were not blinded during the experiments. Tumours were induced by subcutaneous injection of 106 CT26-EpCAM
tumour cells. Daily oral treatment with 10 mg AB928 formulated in 100 µl PEG/solutol (70/40) or control treatment was initiated, once the tumour was palpable. Mice were injected
intravenously with 107 T cells the following day. In accordance with the animal experiment application, tumour growth and health status of mice were checked at least every other day. CELL
LINES Murine Panc02-EpCAM, 4T1, T110299 and CT26-EpCAM have been previously described [41,42,43]. Murine LL/2 were purchased from the European Collection of Authenticated Cell Cultures
(ECACC). The T110299 and LL/2 cell lines were modified to stably express full-length murine EpCAM (UNIPROT entry Q99JW5), to generate the cell lines T110299-EpCAM and LL/2-EpCAM. Human
SUIT-2-MSLN have been previously described [41]. 293Vec-Galv, 293Vec-Eco, and 293Vec-RD114 have been previously described [44]. The virus producing cell lines 293Vec-Eco for
anti-EpCAM-CAR-28z and 293Vec-RD114 for anti-MSLN-CAR-28z have been previously described [45]. 293Vec-RD114 for anti-MSLN-CAR-4-1BBz were generated as previously described [46]. All cell
lines were cultured as previously described [41, 42]. All cell lines were periodically tested for mycoplasma contamination with the commercial testing kit MycoAlertTM (Lonza, Basel,
Switzerland). Authentication of human cell lines by short tandem repeat DNA profiling was conducted in-house. MURINE T CELL CULTURE AND TRANSDUCTION The transduction using the retroviral
vector pMP71 and culture of primary murine T cells has been previously described [47]. In brief, 1.2 × 106 virus producing 293Vec-Eco cells were seeded into a 6-well plate 24 h prior to
splenocyte isolation. After 48 and 72 h, the virus-containing supernatant was used to transduce murine T cells. Murine T cells were expanded from murine splenocytes by activation with
anti-CD3 and anti-CD28 antibodies (clones 145-2C11 and 37.51, Thermo Fisher Scientific, Waltham, MA, USA) and human IL-2 (10 IU/ml, Novartis, Basel, Switzerland) for 24 h. Subsequently
murine T cells were stimulated Dynabeads™ Mouse T-Activator CD3/CD28 (Thermo Fisher Scientific) and human IL-15 (50 ng/ml, Peprotech). Prior to some experiments Dynabeads™ were removed.
HUMAN T CELL CULTURE AND TRANSDUCTION The transduction using the retroviral vector pMP71 and culture of primary human T cells has been previously described [48]. In brief, 1.2 × 106 virus
producing 293Vec-RD114 cells were seeded into a 6-well plate and virus containing supernatant was harvested and used for transduction after 48 and 72 h. After approval by the institutional
review board of the Ludwig-Maximilians-Universität (Munich, Germany), peripheral blood samples were collected from healthy donors. Peripheral blood mononuclear cells (PBMC) were isolated by
density gradient centrifugation using Histopaque®-1077 (Sigma-Aldrich, St. Louis, MO, USA). T cells were isolated from PBMCs by magnetic cell separation with CD3 Microbeads (Miltenyi Biotec,
Bergisch Gladbach, Germany). T cells were activated with Dynabeads™ Human T-Activator CD3/CD28 (Thermo Fisher Scientific) and T cell culture was supplemented with human IL-2 (200 IU/ml) and
human IL-15 (5 ng/ml). Prior to experiments Dynabeads™ were removed. FLOW CYTOMETRY Multicolour flow cytometry was carried out according to previously published protocols [49]. Samples were
analysed with a CytoFLEX LX flow cytometer (Beckmann Coulter, Brea, CA, USA) and BD FACSCanto™ II and BD LSRFortessa™ II flow cytometers (BD Biosciences, Franklin Lakes, NJ, USA). Cells
were stained with Fixable Viability Dye eFluor™ 780 (Thermo Fisher Scientific) to exclude dead cells. Surface staining of murine T cells was performed with the following antibodies: anti-CD3
(BV510 or BV421, clone 17A2, Biolegend San Diego, CA, USA), anti-CD4 (BV785 or AF700, clone GK1.5, Biolegend), anti-CD8a (Pacific Blue or FITC, clone 53-6.7, Biolegend), anti-CD25 (APC,
clone PC61, Biolegend), anti-CD44 (PerCP/Cy5.5, clone IM7, Biolegend), anti-CD69 (PE-Cy7 or BV510, clone H1.2F3, Biolegend), anti-CD62L (Pacific Blue or PE/Cy5, clone MEL-14, Biolegend),
anti-PD-1 (BV421 or BV650, clone 29F.1A12, Biolegend), anti-TIM3 (BV605 or APC, clone RMT3-23, Biolegend), anti-LAG3 (PerCP/Cyanine5.5, clone C9B7W, Biolegend), anti-TIGIT (APC, clone 1G9,
Biolegend). Anti-EpCAM CAR expression on T cells was confirmed by mCherry tag detection. Surface staining of human T cells was performed with the following antibodies: anti-CD3 (PerCP, clone
OKT3, Biolegend), anti-CD4 (AF700, clone A161A1, Biolegend), anti-CD8 (BV785, clone RPA-T8, Biolegend), anti-CD45RO (PE-Cy7, clone UCHL1, Biolegend), anti-CCR7 (BV412, clone G043H7,
Biolegend), anti-PD-1 (APC, clone EH12.2H7, Biolegend), anti-TIM3 (PE/Dazzle™ 594, clone F38-2E2, Biolegend), anti-LAG3 (BV605, 11C3C65, Biolegend). Anti-MSLN-28z CAR or Anti-MSLN-4-1BBz CAR
expression on T cells was detected by staining for the c-myc tag included in the receptors using anti-c-myc (FITC, clone SH1-26E7.1.3, Miltenyi Biotec). For intracellular staining,
anti-EpCAM CAR T cells were stimulated by plate bound recombinant EpCAM-Fc chimera protein (1 µg/ml coated overnight, R&D Systems, Minneapolis, MN, USA) for 18 h. For the last 4 h BD
GolgiStop™ (BD Biosciences) was added. Cells were fixed and permeabilised using BD Cytofix/Cytoperm™ (BD Biosciences) and subsequently stained for IFN-γ (PE-Cy7, clone XMG1.2, Biolegend).
For phospho-specific flow cytometry of p-CREB, T cells were pretreated with AB928 (titration from 10 nM to 10 µM) for 1 h, then 5 µM NECA was added for 1 h. Cells were fixed and
permeabilised using BD Cytofix™ and BD Phosflow™ Perm Buffer III (both BD Biosciences) according to the manufacturer’s instructions. Cells were then stained for CREB (pS133) / ATF-1 (pS63)
(AF647, Clone J151-21, BD Biosciences). CYTOTOXICITY ASSAY Impedance-based real-time killing assays were performed using an xCELLigence system (ACEA Biosciences, San Diego, CA, USA), as
previously described [41]. Briefly, 2.5 ×104 Panc02-EpCAM, 4T1, or SUIT-2-MSLN tumour cells were seeded per well in a 96-well plate. A total of 5 ×104 anti-EpCAM CAR T cells or 2.5 ×104
anti-MSLN-28z CAR T cells and the indicated treatments were added to the tumour cells when the cell index reached approximately 1. The cell index is a measure of the relative change in the
electrical impedance to represent the cell status and was normalised to the timepoint of treatment. CYTOKINE AND GRANZYME B RELEASE ASSAY CAR T cells were treated and stimulated as indicated
in the figure legends. Protein concentrations in the supernatant were determined by commercially available ELISA (human and murine IFN-γ and IL-2 by BD Biosciences and murine TNF-α and
murine granzyme B (GzmB) by R&D Systems). PROLIFERATION ASSAY In a 96-well plate, 105 anti-EpCAM CAR T cells per well were activated by plate bound recombinant EpCAM-Fc chimera protein
(0.5 µg/ml coated overnight, R&D Systems) over a period of 48 h. Before the experiment Dynabeads™ were removed. Cell numbers were determined by flow cytometry with CountBright™ Absolute
Counting Beads (Thermo Fisher Scientific) at the beginning and end of the assay to calculate fold proliferation of T cells. STATISTICAL ANALYSIS The flow cytometry data were analysed with
FlowJo V10.3 software. Statistical analysis was performed with the GraphPad Prism 9 software. Data are presented as indicated in the figure legends. Statistical analysis was performed as
indicated in the figure legends. The Bonferroni correction was used to account for multiple comparisons. _P_ < 0.05 was considered statistically significant and represented as *_P_ <
0.05, **_P_ < 0.01, and ***_P_ < 0.001. No statistical methods were used to predetermine sample size. RESULTS ADENOSINE INHIBITS CAR T CELL ACTIVATION Extracellular adenosine
suppresses T cell and CAR T cell activation [5, 7, 38]. We hypothesised that combination therapy with AB928 may enhance CAR T cell function by blocking immunosuppressive signalling in
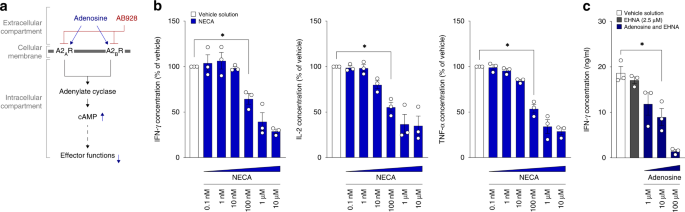
response to extracellular adenosine, thereby maintaining effective CAR T cell responses (Fig. 1a). To confirm the suppressive effect of adenosine in our murine anti-EpCAM CAR T cell model,
we cocultured said CAR T cells with tumour cells of the pancreatic ductal adenocarcinoma cell line Panc02-EpCAM. Cocultures were performed in the presence or absence of the stable adenosine
receptor agonist 5’-_N_-ethylcarboxamide adenosine (NECA) or adenosine (combined with erythron-9-(2-hydroxy-3-nonyl)adenine (EHNA) to prevent adenosine deaminase mediated degradation of
adenosine) in serial titrations to mimic high concentrations of extracellular adenosine in the TME. After 24 h the coculture supernatants, representing T cell activation and degranulation,
were collected, and subjected to ELISA readouts. As hypothesised, NECA dampened IFN-γ, IL-2 and TNF-α release in a dose dependent manner (Fig. 1b). Also, adenosine itself resulted in a
decreased protein concentration of IFN-γ in the supernatants (Fig. 1c). These results are in concordance with previously published data and highlight the susceptibility of CAR T cells to
adenosine-mediated suppression, supporting the rationale for studying the combination therapy with AB928 [20, 21, 38,39,40]. AB928 PROTECTS CAR T CELL ACTIVATION FROM ADENOSINE-MEDIATED
SUPPRESSION To investigate whether AB928 can shield CAR T cells from adenosine-mediated suppression, we cocultured anti-EpCAM CAR T cells and Panc02-EpCAM tumour cells in the presence or
absence of inhibiting concentrations of NECA and serially titrated AB928. While NECA impaired IFN-γ, IL-2 and TNF-α secretion as described above, addition of AB928 in concentrations ranging
from 100 nM to 10 μM fully restored cytokine secretion (Fig. 2a). To rule out cell line-specific effects, cocultures were performed with a panel of murine cancer cell lines (namely the
mammary carcinoma cell line 4T1, the lung carcinoma cell line LL/2-EpCAM, the pancreatic ductal adenocarcinoma cell line T110299-EpCAM and the colon carcinoma cell line CT26-EpCAM). The data
(Fig. 2b) are consistent among all cell lines, corroborating the overarching principle of adenosine suppression and CAR T cell disinhibition by AB928. Next, we used flow cytometry to
analyse the effect of NECA and AB928 on anti-EpCAM CAR T activation upon stimulation with recombinant EpCAM. We found that NECA reduced the amount of IFN-γ+ cells in the population of CD4+
and CD8+ CAR T cells, whereas AB928 reversed NECA-mediated suppression (Figs. 2c and S1a). Activation markers CD25 and CD69 were downregulated after NECA treatment and AB928 reversed this
effect, both for cocultures with antigen expressing tumour cells or recombinant EpCAM stimulation. Interestingly, upregulation of CD25 and CD69 in the AB928 containing condition was more
pronounced than in the vehicle control condition when CAR T cells were activated by antigen-expressing tumour cells, but not when activated with recombinant protein (Fig. S1b). Overall,
these findings demonstrate the capability of AB928 to counteract adenosine-mediated suppression of CAR T cells. AB928 ENABLES EFFICIENT CAR T CELL EFFECTOR RESPONSES To test if AB928 can
improve the anti-tumour efficacy of CAR T cells, we performed cytotoxicity assays using real time cell analysis (RTCA). In the control condition, anti-EpCAM CAR T cells efficiently lysed
Panc02-EpCAM tumour cells. Addition of inhibiting concentrations of NECA resulted in diminished tumour cell lysis, whereas AB928 rescued CAR T cell-mediated cytotoxicity (Fig. 3a). In line
with this, AB928 also enabled the efficient cytolysis of 4T1 breast cancer cells, despite inhibiting concentrations of adenosine being present (Fig. S2a). Granzymes are important mediators
of CAR T cell killing [50]. Consistent with our data on cytotoxicity, we observed that AB928 augmented GzmB release from CAR T cells in the presence of NECA (Fig. 3b). To determine the
effect of NECA and AB928 on CAR T cell phenotype and proliferation, we performed flow cytometry. Staining for CD44 and CD62L expression, we observed a transition towards an effector-like
(CD44+/CD62L−) CAR T cell phenotype upon stimulation. NECA-mediated drifting of this phenotype was blocked by AB928 (Figs. 3c and S2b). Activation-induced upregulation of the inhibitory
receptors PD-1 and TIM3 was inhibited by NECA and restored by AB928. LAG3 and TIGIT expression was not influenced by NECA or AB928 (Fig. 3d). CAR T cell proliferation in the presence of
inhibiting NECA concentrations was also augmented by AB928 (Fig. 3e). Thus, AB928 efficiently blocks adenosine-mediated suppression of crucial CAR T cell effector functions. AB928 SHIELDS
CAR T CELLS FROM IMMUNOSUPPRESSIVE SIGNALLING IN RESPONSE TO ADENOSINE A2AR and A2BR signalling promotes the activity of adenylyl cyclases, leading to elevated levels of cAMP. Intracellular
cAMP accumulation then activates protein kinase A, resulting in the phosphorylation of cAMP response element-binding protein (CREB) which promotes FoxP3 expression and thus Treg generation
[51, 52]. To investigate the effect of AB928 on adenosine-mediated signalling in anti-EpCAM CAR T cells, we performed phosphorylation-specific flow cytometry, staining for p-CREB. Likewise,
basal p-CREB levels were increased in CD4+ and CD8+ CAR T cells upon incubation with saturating concentrations of NECA (5 μM). When preincubated with AB928 at concentrations higher than 1
μM, the addition of NECA did not result in any detectable increase in CREB phosphorylation (Fig. 4a, b). These data indicate that AB928 is capable of effectively shielding CAR T cells from
immunosuppressive signalling even in the presence of high concentrations of extracellular adenosine. ORALLY ADMINISTERED AB928 AUGMENTS CAR T CELL ACTIVATION IN VIVO We next evaluated the
effect of AB928 on CAR T cells in vivo. BALB/c mice were subcutaneously injected with 106 CT26-EpCAM tumour cells. Once tumours were established, daily oral treatment with 10 mg AB928 or
control treatment was initiated. In all, 107 anti-EpCAM CAR T cells were intravenously injected the following day. Forty-eight hours later, the CAR T cell phenotype was assessed by flow
cytometry (Fig. 5a). In line with our previous findings, AB928 treatment resulted in increased expression of CD69, and CAR T cells presented a more effector-like (CD44+/CD62L−) phenotype
(Fig. 5b, c). Expression of the inhibitory receptors PD-1, TIM3, and LAG3 was not influenced by AB928 treatment (Fig. 5d). These results indicate that orally administered AB928 boosts CAR T
cell activation in vivo. AB928 AMELIORATES ACTIVATION OF HUMAN CAR T CELLS IN THE PRESENCE OF ADENOSINE We generated human CAR T cells expressing a second-generation anti-mesothelin (MSLN)
CAR with an intracellular CD3ζ domain and either a CD28 (anti-MSLN-28z CAR) or 4-1BB (anti-MSLN-4-1BBz CAR) costimulatory domain (Fig. S3) to confirm AB928 effects in different CAR designs
in the human system. Anti-MSLN CAR T cells were cocultured with SUIT-2-MSLN tumour cells in the presence or absence of NECA and serially titrated AB928. NECA dampened IFN-γ and IL-2 release
of both anti-MSLN-28z (Fig. 6a) and anti-MSLN-4-1BBz (Fig. 6b) CAR T cells. Interestingly, IFN-γ release was affected by NECA to a lesser extent than IL-2. AB928 in turn restored cytokine
release (Fig. 6a, b). Thus, AB928 blocks adenosine mediated suppression of cytokine production by human CAR T cells, independently of CAR design. To further analyse the effect of NECA and
AB928 on human anti-MSLN-28z CAR T cells, we determined the CAR T cell phenotype by flow cytometry and performed RTCA-based cytotoxicity assays. AB928 restored activation dependent
upregulation of PD-1, whereas TIM3, LAG3 and CD45RO/CCR7 expression were not influenced by NECA or AB928 (Figs. 6c and S4b). Neither NECA nor AB928 modulated CAR T cell cytotoxicity of
anti-MSLN-28z CAR T cells (Fig. S4a). DISCUSSION Among other aspects, insufficient T cell trafficking to the tumour [53, 54], antigen heterogeneity [46] and the immunosuppressive TME [49]
are major factors preventing treatment success of CAR T cell therapy in a broader range of tumours [35, 37]. Extracellular adenosine acts as a soluble immune suppressant in the TME, limiting
effective anti-tumour T cell responses [14]. Many strategies targeting the adenosine axis have been developed and investigated. Among others, targeting the adenosine-producing
ectonucleotidases CD39 and CD73 and the adenosine receptors A2AR and A2BR to improve the efficacy of immunotherapies or conventional chemotherapies has been explored [5, 6]. Blockade of A2AR
has been brought forward as a strategy to enhance CAR T cells but it remained unclear if co-blockade of A2BR would yield similar or better results and if the effect would be conserved
across different CAR designs. In the present study, the impact of the A2AR and A2BR antagonist AB928 on efficacy of different second-generation CAR T cells was assessed. It has been
described that extracellular adenosine specifically acts by dampening T cell receptor-mediated signalling [55]. This lack of adequate T cell stimulation is partly responsible for diminished
effector functions, T cell anergy and generation of Tregs in the presence of high concentrations of adenosine [22]. In our model of murine anti-EpCAM CAR T cells, cytokine secretion,
upregulation of activation markers and proliferation in response to antigen-dependent stimulation were markedly impaired in the presence of adenosine or its analogue NECA. In contrast,
addition of AB928 led to improved IFN-γ, IL-2 and TNF-α secretion as well as restored upregulation of PD-1, TIM3, CD25 and CD69 in response to antigen-stimulus, indicating adequate T cell
activation. Beyond being a surrogate marker for T cell functionality, this is of critical importance as IFN-γ and TNF-α have been described to play a critical role for treatment success of
ACT against solid tumours by inducing anti-tumour immunity from bystander immune cells and by evoking antigen-independent destruction of tumour and stroma cells [56,57,58,59]. Similarly,
IL-2 promotes T cell proliferation and effector functions [60, 61]. Consistently, AB928 also reversed NECA mediated inhibition of T cell proliferation. We observed that IFN-γ production of
both CD4+ and CD8+ CAR T cells was enhanced in the presence of AB928. This finding is of interest considering recent findings highlighting the significant role played by CD4+ T cell in
establishing and sustaining anti-tumour immunity [61,62,63,64]. While the effect on PD-1 and TIM3 expression is in line with results by Giuffrida et al. [21], it contrasts some previously
reported data suggesting that adenosine signalling enhances exhaustion and anergy of T cells, evidenced by the upregulation of checkpoint molecules [20, 65]. However, in our experimental
setting, there was no chronic stimulation, thus anergy and exhaustion were unlikely to occur. Instead, upregulation of PD-1 and TIM3 is a physiological consequence of acute T cell activation
[66, 67] and in consequence of adenosine-mediated inhibition of T cell activation any markers associated with activation will be reduced and likewise reinstalled upon inhibition of
adenosine signalling. Previous studies have extensively characterised the effect of adenosine on adoptively transferred T cells [14, 21, 38]. Importantly, genetic ablation of adenosine
receptors leads to an effector-like phenotype, enhanced activation, and effector function of CAR T cells, ultimately resulting in better survival [21, 38]. Here we found that daily oral
dosing of AB928 is efficacious in improving CAR T cell activation and promotes an effector-like phenotype of CAR T cells in tumour bearing mice, confirming previous findings, and
demonstrating functionality of the adenosine receptor inhibitor on CAR T cells in an in vivo setting. Moreover, AB928 also protected second-generation human anti-MSLN-28z and
anti-MSLN-4-1BBz CAR T cells from adenosine-mediated suppression of cytokine release. Of note, cells bearing second-generation CARs with different costimulatory motives for the same antigen
were comparably suppressed by adenosine, indicating that neither CD28 nor 4-1BB costimulatory domains can overcome adenosine-mediated effects. The effect of adenosine on direct CAR T cell
killing is controversial. Masoumi et al. observed reduced cytolytic function of human CAR T cells in the presence of NECA in a flow cytometry-based cytotoxicity assay [39]. Interestingly
A2AR knockdown, but not SCH58261, a small molecule A2AR antagonist, protected CAR T cells. Here we used RTCA to assess CAR T cell killing. We observed impaired cytotoxic function of murine
anti-EpCAM CAR T cells in the presence of NECA or adenosine. Of note, AB928 protected CAR T cell killing capacity from inhibition, highlighting its potential utility. This also calls
attention to the advantages that dual targeting of adenosine receptors might have over single targeting, although further work is still required to fully demonstrate this observation. In
contrast, we did not observe suppression of human anti-MSLN-28z CAR T cell cytotoxicity by NECA. Importantly, this is in line with previously published evidence by Beavis et al. and
Giuffrida et al. reporting that NECA had negligible impact on murine and human CAR T cell cytotoxicity in a chromium release assays [21, 38]. Thus, it seems, that adenosine does not hamper
the killing abilities of CAR T cells in certain settings, possibly because of the strength of CAR T cells in vitro. However, we cannot disregard the possibility that in other settings, such
as long-term exposure to adenosine, the cytotoxic potential of these CAR T cells would be suppressed by adenosine. Overall, the factors determining to what extend adenosine influences CAR T
cell cytotoxicity have yet to be defined. T cells are highly sensitive to adenosine-mediated suppression, with Giuffrida et al. recently suggesting that genetic or pharmacological targeting
of A2AR should prevent more than 50% of the adenosine-mediated effect on T cells [21]. A2AR and A2BR activation leads to signalling via the cAMP-PKA-CREB axis [51]. We have shown that
NECA-induced CREB phosphorylation in anti-EpCAM CAR T cells was abrogated in the presence of 1 µM AB928. This result demonstrates that AB928 can effectively and efficiently block
immunosuppressive signalling in response to adenosine. Importantly, AB928 plasma levels of 1 µM and higher are feasible and safe in patients [27]. We found that signalling was blocked in
both CD4+ and CD8+ CAR T cells. This is important, as it has been previously shown that A2AR and A2BR activation on CD4+ T cells may promote the generation of Tregs [23, 26]. Putatively
CAR-transduced Tregs [68] could be potentially boosted by adenosine but not so if AB928 is present, although this would need to be formally demonstrated. It remains to be determined how
AB928 compares to other strategies targeting adenosine receptors to overcome suppression of adoptively transferred T cells. The immunosuppressive effect of adenosine on T cells is primarily
mediated by the predominantly expressed A2AR, making it an attractive target to improve T cell-mediated anti-tumour immunity [20, 21]. The small molecule A2AR antagonists SCH58261
[38,39,40], CPI-444 [65] and KW6002 [69], as well as approaches genetically targeting A2AR with shRNA knockdown [38, 39] or CRISPR/Cas9-mediated knockout [20, 21] have successfully been used
to enhance ACT in preclinical studies. However, growing evidence suggests that A2BR also plays an important role in adenosine-mediated suppression of anti-tumour responses by indirectly
suppressing T cell function. It has been shown, that A2BR antagonism reduces differentiation and suppressive capacity of Tregs and suppressive myeloid cells, leading to an increased presence
of tumour-infiltrating CD8+ T cells in vivo [23, 25, 26, 70]. Recently, Chen et al. showed that pharmacological A2BR antagonism prior to adoptive T cell transfer improves treatment efficacy
[71]. Among small molecule inhibitors targeting adenosine receptors, AB928 is the first dual A2AR and A2BR antagonist in clinical development. However, we have not yet formally proven the
advantage of dual over single targeting. Genetic targeting is an elegant way to render CAR T cells resistant to one or more immunosuppressive factors [72]. It provides continuous protection
from suppression and therefore allows single dosing of the T cell product. However, safety concerns regarding off-target editing and the administration of CAR T cells with permanent deletion
of immune checkpoints remain [72]. Small molecule inhibitors in turn may suffer from variable pharmacokinetics (PK) and require repeated dosing, which in turn can be beneficial in case of
unwanted serious adverse events [73]. Further, they may also enable improved recruitment of endogenous anti-tumour responses by acting on other immune cells [74, 75]. The combination therapy
of CAR T cells with cell intrinsic or extrinsic ICB has already been explored in more detail for the PD-1 axis and is currently being investigated in clinical trials. Thus far it is unclear
which approach provides the best results regarding clinical outcome and safety [76]. AB928 is currently under evaluation in phase 1 and 2 clinical trials testing the efficacy, safety, PK,
and pharmacodynamics (PD) of AB928-based combination therapies for tumour indications (NCT03846310, NCT04262856, NCT04381832, NCT03720678, NCT04660812). AB928 can be orally administered by
daily dosing. Promising preliminary results and results from previous studies suggest beneficial safety, PK, and PD profiles of the drug in patients [27, 29, 30]. Overall, AB928 reliably
protected murine and human CAR T cells from all suppressive adenosine-mediated effects observed in this study, and when administered similarly to regimens currently under investigation in
clinical trials, AB928 improved CAR T cell activation in vivo. Thus, we reason that the combination therapy with AB928 has high translational potential and may be a promising approach to
enhance CAR T cell efficacy. However, given the fact that other limitations of CAR T cell therapy such as limited trafficking to the tumour remain, we believe that multimodal approaches
targeting more than one bottleneck of CAR T cell therapy are necessary to enable treatment of a broader range of tumours. We recently found that combined tumour-directed trafficking and
expression of a dominant-negative receptor (DNR) to shield CAR T cells from TGF-β in the immunosuppressive TME synergistically improves CAR T cell efficacy in solid tumours [45]. Similar
approaches combining tumour-directed recruitment and protection from the immunosuppressive TME may also be applicable for the combination therapy of AB928 and CAR T cells in the future. DATA
AVAILABILITY The data sets generated and/or analysed during the current study available from the corresponding author on reasonable request. REFERENCES * Postow MA, Callahan MK, Wolchok JD.
Immune checkpoint blockade in cancer therapy. J Clin Oncol. 2015;33:1974–82. Article CAS PubMed PubMed Central Google Scholar * Leach Dana R, Krummel Matthew F, Allison James P.
Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–6. Article Google Scholar * Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on
tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci USA. 2002;99:12293. Article CAS PubMed PubMed Central Google Scholar *
Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer. 2018;118:9–16. Article CAS PubMed PubMed Central Google Scholar * Allard B,
Allard D, Buisseret L, Stagg J. The adenosine pathway in immuno-oncology. Nat Rev Clin Oncol. 2020;17:611–29. Article CAS PubMed Google Scholar * Vigano S, Alatzoglou D, Irving M,
Ménétrier-Caux C, Caux C, Romero P, et al. Targeting adenosine in cancer immunotherapy to enhance T-cell function. Front Immunol. 2019;10:925. Article CAS PubMed PubMed Central Google
Scholar * Boison D, Yegutkin GG. Adenosine metabolism: emerging concepts for cancer therapy. Cancer Cell. 2019;36:582–96. Article CAS PubMed PubMed Central Google Scholar * Di Virgilio
F, Sarti AC, Falzoni S, De Marchi E, Adinolfi E. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat Rev Cancer. 2018;18:601–18. Article PubMed Google
Scholar * Pellegatti P, Raffaghello L, Bianchi G, Piccardi F, Pistoia V, Di Virgilio F. Increased level of extracellular ATP at tumor sites: in vivo imaging with plasma membrane luciferase.
PLoS ONE. 2008;3:e2599. Article PubMed PubMed Central Google Scholar * Zimmermann H. Extracellular metabolism of ATP and other nucleotides. Naunyn Schmiedebergs Arch Pharmacol.
2000;362:299–309. Article CAS PubMed Google Scholar * Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, et al. Ecto-5′-nucleotidase (CD73) regulation by
hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Investig. 2002;110:993–1002. Article CAS PubMed PubMed Central Google Scholar * Ryzhov SV,
Pickup MW, Chytil A, Gorska AE, Zhang Q, Owens P, et al. Role of TGF-β signaling in generation of CD39+CD73+ myeloid cells in tumors. J Immunol. 2014;193:3155–64. Article CAS PubMed
Google Scholar * Howard M, Grimaldi JC, Bazan JF, Lund Frances E, Santos-Argumedo L, Parkhouse RME, et al. Formation and hydrolysis of cyclic ADP-ribose catalyzed by lymphocyte antigen
CD38. Science. 1993;262:1056–9. Article CAS PubMed Google Scholar * Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MKK, et al. A2A adenosine receptor protects tumors from
antitumor T cells. Proc Natl Acad Sci USA. 2006;103:13132–7. Article CAS PubMed PubMed Central Google Scholar * Müller CE, Jacobson KA. Recent developments in adenosine receptor ligands
and their potential as novel drugs. Biochim Biophys Acta. 2011;1808:1290–308. Article PubMed Google Scholar * Wolberg G, Zimmerman TP, Hiemstra K, Winston M, Chu LC. Adenosine inhibition
of lymphocyte-mediated cytolysis: possible role of cyclic adenosine monophosphate. Science. 1975;187:957. Article CAS PubMed Google Scholar * Ohta A, Sitkovsky M. Role of
G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature. 2001;414:916–20. Article CAS PubMed Google Scholar * Hinz S, Navarro G,
Borroto-Escuela D, Seibt BF, Ammon Y-C, de Filippo E, et al. Adenosine A(2A) receptor ligand recognition and signaling is blocked by A(2B) receptors. Oncotarget. 2018;9:13593–611. Article
PubMed PubMed Central Google Scholar * Raskovalova T, Lokshin A, Huang X, Su Y, Mandic M, Zarour HM, et al. Inhibition of cytokine production and cytotoxic activity of human antimelanoma
specific CD8+ and CD4+ T lymphocytes by adenosine-protein kinase A type I signaling. Cancer Res. 2007;67:5949. Article CAS PubMed Google Scholar * Li N, Tang N, Cheng C, Hu T, Wei X, Han
W, et al. Improving the anti-solid tumor efficacy of CAR-T cells by inhibiting adenosine signaling pathway. Oncoimmunology. 2020;9:1824643. Article PubMed PubMed Central Google Scholar
* Giuffrida L, Sek K, Henderson MA, Lai J, Chen AXY, Meyran D, et al. CRISPR/Cas9 mediated deletion of the adenosine A2A receptor enhances CAR T cell efficacy. Nat Commun. 2021;12:3236.
Article CAS PubMed PubMed Central Google Scholar * Zarek PE, Huang C-T, Lutz ER, Kowalski J, Horton MR, Linden J, et al. A2A receptor signaling promotes peripheral tolerance by inducing
T-cell anergy and the generation of adaptive regulatory T cells. Blood. 2008;111:251–9. Article CAS PubMed PubMed Central Google Scholar * Ohta A, Kini R, Ohta A, Subramanian M, Madasu
M, Sitkovsky M. The development and immunosuppressive functions of CD4(+) CD25(+) FoxP3(+) regulatory T cells are under influence of the adenosine-A2A adenosine receptor pathway. Front
Immunol. 2012;3:190. Article CAS PubMed PubMed Central Google Scholar * Ryzhov S, Novitskiy SV, Zaynagetdinov R, Goldstein AE, Carbone DP, Biaggioni I, et al. Host A(2B) adenosine
receptors promote carcinoma growth. Neoplasia. 2008;10:987–95. Article CAS PubMed PubMed Central Google Scholar * Ryzhov S, Novitskiy SV, Goldstein AE, Biktasova A, Blackburn MR,
Biaggioni I, et al. Adenosinergic regulation of the expansion and immunosuppressive activity of CD11b+Gr1+ cells. J Immunol. 2011;187:6120–9. Article CAS PubMed Google Scholar *
Nakatsukasa H, Tsukimoto M, Harada H, Kojima S. Adenosine A2B receptor antagonist suppresses differentiation to regulatory T cells without suppressing activation of T cells. Biochem Biophys
Res Commun. 2011;409:114–9. Article CAS PubMed Google Scholar * Seitz L, Jin L, Leleti M, Ashok D, Jeffrey J, Rieger A, et al. Safety, tolerability, and pharmacology of AB928, a novel
dual adenosine receptor antagonist, in a randomized, phase 1 study in healthy volunteers. Invest N Drugs. 2019;37:711–21. Article CAS Google Scholar * Walters MJ, Tan JB, Becker A, Yi F,
Park T, Leleti MR, et al. Characterization of the potent and selective A2AR antagonist AB928 for the treatment of cancer. Cancer Res. 2017;77:45722017. * Powderly J, Spira A, Gutierrez R,
DiRenzo D, Udyavar A, Karakunnel JJ, et al. 1206P - Phase I evaluation of AB928, a novel dual adenosine receptor antagonist, combined with chemotherapy or AB122 (anti-PD-1) in patients (pts)
with advanced malignancies. Ann Oncol. 2019;30:v493. Article Google Scholar * Spira AI, Conkling PR, Johnson ML, Gardner O, Gilbert HN, Scharville M, et al. ARC-4 study: efficacy and
safety of AB928 plus carboplatin, pemetrexed and a PD-1 antibody in participants with metastatic non-small cell lung cancer (mNSCLC). J Clin Oncol. 2020;38:e21659. Article Google Scholar *
June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359:1361. Article CAS PubMed Google Scholar * Maude SL, Frey N, Shaw
PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–17. Article PubMed PubMed Central Google
Scholar * Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl J Med.
2017;377:2531–44. Article CAS PubMed PubMed Central Google Scholar * Turtle CJ, Hay KA, Hanafi L-A, Li D, Cherian S, Chen X, et al. Durable molecular remissions in chronic lymphocytic
leukemia treated with CD19-specific chimeric antigen receptor-modified T cells after failure of ibrutinib. J Clin Oncol. 2017;35:3010–20. Article CAS PubMed PubMed Central Google Scholar
* Newick K, Moon E, Albelda SM. Chimeric antigen receptor T-cell therapy for solid tumors. Mol Ther Oncolytics. 2016;3:16006. Article CAS PubMed PubMed Central Google Scholar *
Stoiber S, Cadilha BL, Benmebarek M-R, Lesch S, Endres S, Kobold S. Limitations in the design of chimeric antigen receptors for cancer therapy. Cells. 2019;8:472. Article CAS PubMed
PubMed Central Google Scholar * Lesch S, Benmebarek MR, Cadilha BL, Stoiber S, Subklewe M, Endres S, et al. Determinants of response and resistance to CAR T cell therapy. Semin Cancer
Biol. 2020;65:80–90. Article CAS PubMed Google Scholar * Beavis PA, Henderson MA, Giuffrida L, Mills JK, Sek K, Cross RS, et al. Targeting the adenosine 2A receptor enhances chimeric
antigen receptor T cell efficacy. J Clin Investig. 2017;127:929–41. Article PubMed PubMed Central Google Scholar * Masoumi E, Jafarzadeh L, Mirzaei HR, Alishah K, Fallah-Mehrjardi K,
Rostamian H, et al. Genetic and pharmacological targeting of A2a receptor improves function of anti-mesothelin CAR T cells. J Exp Clin Cancer Res. 2020;39:49. Article PubMed PubMed Central
Google Scholar * Fallah-Mehrjardi K, Mirzaei HR, Masoumi E, Jafarzadeh L, Rostamian H, Khakpoor-Koosheh M, et al. Pharmacological targeting of immune checkpoint A2aR improves function of
anti-CD19 CAR T cells in vitro. Immunol Lett. 2020;223:44–52. Article CAS PubMed Google Scholar * Karches CH, Benmebarek M-R, Schmidbauer ML, Kurzay M, Klaus R, Geiger M, et al.
Bispecific antibodies enable synthetic agonistic receptor-transduced T cells for tumor immunotherapy. Clin Cancer Res. 2019;25:5890–900. Article CAS PubMed PubMed Central Google Scholar
* Voigt C, May P, Gottschlich A, Markota A, Wenk D, Gerlach I, et al. Cancer cells induce interleukin-22 production from memory CD4(+) T cells via interleukin-1 to promote tumor growth.
Proc Natl Acad Sci USA. 2017;114:12994–9. Article CAS PubMed PubMed Central Google Scholar * Demel UM, Böger M, Yousefian S, Grunert C, Zhang L, Hotz PW, et al. Activated SUMOylation
restricts MHC class I antigen presentation to confer immune evasion in cancer. J Clin Investig. 2022;132:e152383. * Ghani K, Wang X, de Campos-Lima PO, Olszewska M, Kamen A, Rivière I, et
al. Efficient human hematopoietic cell transduction using RD114- and GALV-pseudotyped retroviral vectors produced in suspension and serum-free media. Hum Gene Ther. 2009;20:966–74. Article
CAS PubMed PubMed Central Google Scholar * Cadilha BL, Benmebarek M-R, Dorman K, Oner A, Lorenzini T, Obeck H, et al. Combined tumor-directed recruitment and protection from immune
suppression enable CAR T cell efficacy in solid tumors. Sci Adv. 2021;7:eabi5781. Article CAS PubMed PubMed Central Google Scholar * Benmebarek M-R, Cadilha BL, Herrmann M, Lesch S,
Schmitt S, Stoiber S, et al. A modular and controllable T cell therapy platform for acute myeloid leukemia. Leukemia. 2021;35:2243–57. Article CAS PubMed PubMed Central Google Scholar *
Rapp M, Grassmann S, Chaloupka M, Layritz P, Kruger S, Ormanns S, et al. C-C chemokine receptor type-4 transduction of T cells enhances interaction with dendritic cells, tumor infiltration
and therapeutic efficacy of adoptive T cell transfer. Oncoimmunology. 2015;5:e1105428. Article PubMed PubMed Central Google Scholar * Kobold S, Steffen J, Chaloupka M, Grassmann S,
Henkel J, Castoldi R, et al. Selective bispecific T cell recruiting antibody and antitumor activity of adoptive T cell transfer. J Natl Cancer Inst. 2015;107:364. * Kobold S, Grassmann S,
Chaloupka M, Lampert C, Wenk S, Kraus F, et al. Impact of a new fusion receptor on PD-1-mediated immunosuppression in adoptive T cell therapy. J Natl Cancer Inst. 2015;107:djv146. Article
PubMed PubMed Central Google Scholar * Benmebarek M-R, Karches CH, Cadilha BL, Lesch S, Endres S, Kobold S. Killing mechanisms of chimeric antigen receptor (CAR) T cells. Int J Mol Sci.
2019;20:1283. Article CAS PubMed PubMed Central Google Scholar * Sheth S, Brito R, Mukherjea D, Rybak LP, Ramkumar V. Adenosine receptors: expression, function and regulation. Int J Mol
Sci. 2014;15:2024–52. Article PubMed PubMed Central Google Scholar * Kim H-P, Leonard WJ. CREB/ATF-dependent T cell receptor-induced FoxP3 gene expression: a role for DNA methylation. J
Exp Med. 2007;204:1543–51. Article CAS PubMed PubMed Central Google Scholar * Zhang J, Endres S, Kobold S. Enhancing tumor T cell infiltration to enable cancer immunotherapy.
Immunotherapy. 2019;11:201–13. Article PubMed Google Scholar * Lesch S, Blumenberg V, Stoiber S, Gottschlich A, Ogonek J, Cadilha BL, et al. T cells armed with C-X-C chemokine receptor
type 6 enhance adoptive cell therapy for pancreatic tumours. Nat Biomed Eng. 2021;5:1246–60. * Linnemann C, Schildberg FA, Schurich A, Diehl L, Hegenbarth SI, Endl E, et al. Adenosine
regulates CD8 T-cell priming by inhibition of membrane-proximal T-cell receptor signalling. Immunology. 2009;128:e728–37. Article PubMed PubMed Central Google Scholar * Zhang B, Karrison
T, Rowley DA, Schreiber H. IFN-gamma- and TNF-dependent bystander eradication of antigen-loss variants in established mouse cancers. J Clin Investig. 2008;118:1398–404. Article CAS PubMed
PubMed Central Google Scholar * Baer C, Squadrito ML, Laoui D, Thompson D, Hansen SK, Kiialainen A, et al. Suppression of microRNA activity amplifies IFN-γ-induced macrophage activation
and promotes anti-tumour immunity. Nat Cell Biol. 2016;18:790–802. Article CAS PubMed Google Scholar * Briesemeister D, Sommermeyer D, Loddenkemper C, Loew R, Uckert W, Blankenstein T,
et al. Tumor rejection by local interferon gamma induction in established tumors is associated with blood vessel destruction and necrosis. Int J Cancer. 2011;128:371–8. Article CAS PubMed
Google Scholar * Larson RC, Kann MC, Bailey SR, Haradhvala NJ, Llopis PM, Bouffard AA, et al. CAR T cell killing requires the IFNγR pathway in solid but not liquid tumours. Nature.
2022;604:563–70. Article CAS PubMed Google Scholar * Boyman O, Sprent J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol.
2012;12:180–90. Article CAS PubMed Google Scholar * Bos R, Sherman LA. CD4+ T-cell help in the tumor milieu is required for recruitment and cytolytic function of CD8+ T lymphocytes.
Cancer Res. 2010;70:8368. Article CAS PubMed PubMed Central Google Scholar * Wang D, Aguilar B, Starr R, Alizadeh D, Brito A, Sarkissian A, et al. Glioblastoma-targeted CD4+ CAR T cells
mediate superior antitumor activity. JCI Insight. 2018;3:e99048. * Oh DY, Kwek SS, Raju SS, Li T, McCarthy E, Chow E, et al. Intratumoral CD4(+) T cells mediate anti-tumor cytotoxicity in
human bladder cancer. Cell. 2020;181:1612.e13–25.e13. Article Google Scholar * Melenhorst JJ, Chen GM, Wang M, Porter DL, Chen C, Collins MA, et al. Decade-long leukaemia remissions with
persistence of CD4+ CAR T cells. Nature. 2022;602:503–9. Article CAS PubMed PubMed Central Google Scholar * Leone RD, Sun I-M, Oh M-H, Sun I-H, Wen J, Englert J, et al. Inhibition of
the adenosine A2a receptor modulates expression of T cell coinhibitory receptors and improves effector function for enhanced checkpoint blockade and ACT in murine cancer models. Cancer
Immunol Immunother. 2018;67:1271–84. Article CAS PubMed Google Scholar * Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubat T, Yagita H, et al. Expression of the PD-1 antigen on the
surface of stimulated mouse T and B lymphocytes. Int Immunol. 1996;8:765–72. Article CAS PubMed Google Scholar * Hastings WD, Anderson DE, Kassam N, Koguchi K, Greenfield EA, Kent SC, et
al. TIM-3 is expressed on activated human CD4+ T cells and regulates Th1 and Th17 cytokines. Eur J Immunol. 2009;39:2492–501. Article CAS PubMed PubMed Central Google Scholar * Lee JC,
Hayman E, Pegram HJ, Santos E, Heller G, Sadelain M, et al. In vivo inhibition of human CD19-targeted effector T cells by natural T regulatory cells in a xenotransplant murine model of B
cell malignancy. Cancer Res. 2011;71:2871–81. Article CAS PubMed PubMed Central Google Scholar * Kjaergaard J, Hatfield S, Jones G, Ohta A, Sitkovsky M. A(2A) adenosine receptor gene
deletion or synthetic A(2A) antagonist liberate tumor-reactive CD8(+) T cells from tumor-induced immunosuppression. J Immunol. 2018;201:782–91. Article CAS PubMed Google Scholar *
Iannone R, Miele L, Maiolino P, Pinto A, Morello S. Blockade of A2b adenosine receptor reduces tumor growth and immune suppression mediated by myeloid-derived suppressor cells in a mouse
model of melanoma. Neoplasia. 2013;15:1400–9. Article PubMed PubMed Central Google Scholar * Chen S, Akdemir I, Fan J, Linden J, Zhang B, Cekic C. The expression of adenosine A2B
receptor on antigen-presenting cells suppresses CD8+ T-cell responses and promotes tumor growth. Cancer Immunol Res. 2020;8:1064. Article CAS PubMed PubMed Central Google Scholar * Liu
J, Zhou G, Zhang L, Zhao Q. Building potent chimeric antigen receptor T cells with CRISPR genome editing. Front Immunol. 2019;10:456. Article CAS PubMed PubMed Central Google Scholar *
van Waterschoot RAB, Parrott NJ, Olivares-Morales A, Lavé T, Rowland M, Smith DA. Impact of target interactions on small-molecule drug disposition: an overlooked area. Nat Rev Drug Discov.
2018;17:299. Article PubMed Google Scholar * Huck BR, Kötzner L, Urbahns K. Small molecules drive big improvements in immuno-oncology therapies. Angew Chem Int Ed Engl. 2018;57:4412–28.
Article CAS PubMed PubMed Central Google Scholar * Stock S, Kluever A-K, Endres S, Kobold S. Enhanced chimeric antigen receptor T cell therapy through co-application of synergistic
combination partners. Biomedicines. 2022;10:307. Article CAS PubMed PubMed Central Google Scholar * Grosser R, Cherkassky L, Chintala N, Adusumilli PS. Combination immunotherapy with
CAR T cells and checkpoint blockade for the treatment of solid tumors. Cancer Cell. 2019;36:471–82. Article CAS PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS
The authors acknowledge the Core Facility Flow Cytometry of the University Hospital, LMU Munich, funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – 499549016,
for assistance with the generation of flow cytometry data and thank M. Caruso (Québec, Canada), L. von Baumgarten (Munich, Germany) and J. Sieveke (Essen, Germany) for providing cell lines
used in this study. FUNDING This study was supported by grants from the international doctoral programme “i-Target: Immunotargeting of cancer” funded by the Elite Network of Bavaria (to SK
and SE), the Melanoma Research Alliance (grant number 409510 to SK), the Marie-Sklodowska-Curie “Training Network for the Immunotherapy of Cancer (IMMUTRAIN)” funded by the H2020 programme
of the European Union (to SE and SK), the Marie-Sklodowska-Curie Program Training Network for Optimizing Adoptive T Cell Therapy of Cancer funded by the H2020 Program of the European Union
(Grant 955575, to S.K.), the Else Kröner-Fresenius-Stiftung (to SK), the German Cancer Aid (to SK), the Ernst-Jung-Stiftung (to SK) by LMU Munich’s Institutional Strategy LMUexcellent within
the framework of the German Excellence Initiative (to SE and SK), by the Go-Bio-initiative (to SK), by the m4-award of the Bavarian Ministry of Economical Affairs (to SK and SE), by the
Bundesministerium für Bildung und Forschung (to SE and SK), by the European Research Council Starting Grant (grant number 756017 to SK), by the Deutsche Forschungsgemeinschaft (DFG, German
Research Foundation) and –SFB- TRR 338/1 2021 –452881907 (to SK), the Fritz-Bender-Foundation (to SK), the Wilhelm-Sander-Stiftung (to SK), the Deutsche José-Carreras Leukämie-Stiftung (to
SK) and the Hector Foundation (to SK). MS received a grant from the Förderprogramm für Forschung und Lehre (FöFoLe) of the LMU Munich and was supported by the Studienstiftung des deutschen
Volkes. BLC received an endowment for cancer research from the foundation of the medical faculty of the LMU Munich. SS was supported by the Else Kröner-Fresenius Clinician Scientist Program
Cancer Immunotherapy, the Munich Clinician Scientist Program (MCSP) and the DKTK School of Oncology. Open Access funding enabled and organized by Projekt DEAL. AUTHOR INFORMATION AUTHORS AND
AFFILIATIONS * Center of Integrated Protein Science Munich (CIPS-M) and Division of Clinical Pharmacology, Department of Medicine IV, University Hospital, Ludwig-Maximilians-Universität
München, Munich, Germany Matthias Seifert, Mohamed-Reda Benmebarek, Daria Briukhovetska, Florian Märkl, Janina Dörr, Bruno L. Cadilha, Jakob Jobst, Sophia Stock, David Andreu-Sanz, Theo
Lorenzini, Ruth Grünmeier, Arman Oner, Hannah Obeck, Lina Majed, Dario Dhoqina, Manouk Feinendegen, Adrian Gottschlich, Jin Zhang, Stefan Endres & Sebastian Kobold * National Cancer
Institute (NCI), Bethesda, MD, USA Mohamed-Reda Benmebarek * Department of Medicine III, University Hospital, Ludwig-Maximilians-Universität München, Munich, Germany Sophia Stock * German
Cancer Consortium (DKTK), Partner Site Munich, Munich, Germany Sophia Stock & Sebastian Kobold * Independent Consultant, Freiburg, Germany Ulrike Schindler * Einheit für Klinische
Pharmakologie (EKLiP), Helmholtz Zentrum München, Research Center for Environmental Health (HMGU), Neuherberg, Germany Sebastian Kobold Authors * Matthias Seifert View author publications
You can also search for this author inPubMed Google Scholar * Mohamed-Reda Benmebarek View author publications You can also search for this author inPubMed Google Scholar * Daria
Briukhovetska View author publications You can also search for this author inPubMed Google Scholar * Florian Märkl View author publications You can also search for this author inPubMed
Google Scholar * Janina Dörr View author publications You can also search for this author inPubMed Google Scholar * Bruno L. Cadilha View author publications You can also search for this
author inPubMed Google Scholar * Jakob Jobst View author publications You can also search for this author inPubMed Google Scholar * Sophia Stock View author publications You can also search
for this author inPubMed Google Scholar * David Andreu-Sanz View author publications You can also search for this author inPubMed Google Scholar * Theo Lorenzini View author publications You
can also search for this author inPubMed Google Scholar * Ruth Grünmeier View author publications You can also search for this author inPubMed Google Scholar * Arman Oner View author
publications You can also search for this author inPubMed Google Scholar * Hannah Obeck View author publications You can also search for this author inPubMed Google Scholar * Lina Majed View
author publications You can also search for this author inPubMed Google Scholar * Dario Dhoqina View author publications You can also search for this author inPubMed Google Scholar * Manouk
Feinendegen View author publications You can also search for this author inPubMed Google Scholar * Adrian Gottschlich View author publications You can also search for this author inPubMed
Google Scholar * Jin Zhang View author publications You can also search for this author inPubMed Google Scholar * Ulrike Schindler View author publications You can also search for this
author inPubMed Google Scholar * Stefan Endres View author publications You can also search for this author inPubMed Google Scholar * Sebastian Kobold View author publications You can also
search for this author inPubMed Google Scholar CONTRIBUTIONS MS, MRB, DB, FM, BLC, JD, JJ, SS, DS, LM, TL, HO, RG, AÖ, DD, MF, AG, JZ, and US performed or assisted with experiments, analysed
data, and supported the project. SK and SE supervised the project and did the funding acquisition. MS, MRB, DB and SK designed the experiments. MS and BLC designed the figures. MS, MRB and
SK wrote the manuscript. All authors critically read and approved the final manuscript. CORRESPONDING AUTHOR Correspondence to Sebastian Kobold. ETHICS DECLARATIONS COMPETING INTERESTS Parts
of this work have been performed for the doctoral thesis of MS at the Ludwig-Maximilians-Universität München. SE and SK are inventors of several patent applications filed by the
Ludwig-Maximilians-Universität München in the field of immunooncology. SE and SK received research support from TCR2 Inc and Tabby Therapeutics for work unrelated to the present manuscript.
SK received research support from Arcus Biosciences to perform parts of the present study. US is a former employee of Arcus Biosciences and holds stocks from Arcus Biosciences and Amgen. The
authors declare no other competing interests. ETHICS APPROVAL AND CONSENT TO PARTICIPATE Collection of peripheral blood samples from healthy donors were approved by the institutional review
board of the Ludwig-Maximilians-Universität (Munich, Germany). The study was performed in accordance with the Declaration of Helsinki. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer
Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY LEGEND REVISED SUPPLEMENTARY FIGURE 1
REVISED SUPPLEMENTARY FIGURE 2 REVISED SUPPLEMENTARY FIGURE 3 REVISED SUPPLEMENTARY FIGURE 4 REVISED AGREEMENT OF COAUTHORS TO CHANGES IN THE AUTHORLIST RIGHTS AND PERMISSIONS OPEN ACCESS
This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as
long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third
party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the
article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright
holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Seifert, M., Benmebarek, MR.,
Briukhovetska, D. _et al._ Impact of the selective A2AR and A2BR dual antagonist AB928/etrumadenant on CAR T cell function. _Br J Cancer_ 127, 2175–2185 (2022).
https://doi.org/10.1038/s41416-022-02013-z Download citation * Received: 07 January 2022 * Revised: 13 September 2022 * Accepted: 04 October 2022 * Published: 20 October 2022 * Issue Date:
07 December 2022 * DOI: https://doi.org/10.1038/s41416-022-02013-z SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a
shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative