Play all audios:
Cell division cycle associated 7 (CDCA7) plays a role in various malignancies, especially pancreatic cancer (PC). However, its expression pattern and functional significance in PC require
further research. Therefore, this study aimed to investigate CDCA7 expression levels and biological functions in PC using in vitro and in vivo experiments. Western blotting,
immunohistochemistry, and real-time polymerase chain reaction were performed to detect CDCA7 expression in PC cells and tissues. Additionally, the biological functions of CDCA7 were assessed
using cell proliferation, wound healing, and Transwell assays. CDCA7 overexpression promoted PC cell proliferation, migration, and invasion, and increased resistance to the chemotherapy
drug gemcitabine, possibly through enhanced aerobic glycolysis. Additionally, immunoprecipitation assay showed that CDCA7 interacted with STAT3 protein and affected the transcriptional
regulation of hexokinase 2. Conclusively, targeting CDCA7 might be a promising therapeutic strategy to increase gemcitabine sensitivity by inhibiting glycolysis in PC cells.
Pancreatic cancer (PC) remains a serious challenge in oncology due to its high mortality rate, with only a 12% 5-year survival rate [1]. Major contributors to the poor prognosis of PC
include difficulties in early detection and limited effectiveness of drug therapies [2]. Notably, due to the limited treatment options for pancreatic cancer, only approximately 20% of
patients are eligible for surgical resection [3]. In addition to surgery, chemotherapy is an important treatment strategy for PC. Currently, gemcitabine is the most commonly used drug for PC
chemotherapy. However, the efficacy of gemcitabine treatment in PC remains modest, with an overall response rate of less than 20%. Importantly, the low efficacy of gemcitabine is primarily
due to the rapid onset of drug resistance in tumor cells, leading to inadequate therapeutic responses [4, 5]. Therefore, it is crucial to investigate the mechanisms underlying PC development
and chemotherapy resistance, as well as develop efficacious targeted therapies.
Cell division cycle associated protein 7 (CDCA7) belongs to the family of cell cycle regulatory proteins. It has been identified as a c-Myc responsive gene, contributing to MYC-mediated
tumorigenesis by acting as a transcriptional regulator [6, 7]. As an oncogenic gene with copy number amplification, CDCA7 exhibits high expression in various tumors and is correlated with
unfavorable prognosis [8, 9]. CDCA7 promotes tumor cell proliferation, invasion, and metastasis in various cancers, including esophageal, gastric, and ovarian cancers, and regulates
malignant biological processes, including inflammatory factors [10,11,12]. CDCA7 is a drug resistance-associated cell cycle protein that is highly expressed in paclitaxel-resistant subgroup
of non-small cell lung cancer [13, 14]. However, the precise role of CDCA7 in PC progression remains unclear.
Recently, the role of metabolic reprogramming in the development of drug resistance in PC has attracted considerable attention [15]. Research findings indicate that stroma formation and the
complex tumor microenvironment, including aberrant glycolytic metabolism reprogramming, are crucial factors contributing to chemotherapy resistance in PC [16]. Additionally, an increasing
number of preclinical and clinical trials are being performed to develop metabolic-targeted therapies for PC [15]. Moreover, therapeutic approaches targeting metabolism holds great potential
for improving unfavorable prognosis in patients with PC [15]. Overall, metabolism research not only aids in understanding cancer initiation and progression but also offers novel
perspectives for PC treatment.
Signal transducer and activator of transcription 3 (STAT3) is a member of the STAT protein family. STAT3 can be phosphorylated by specific kinases, resulting in the formation of heterodimers
that migrate to the nucleus where they function as transcription factors critical in various cellular processes, including cell growth and apoptosis [17,18,19]. For example, induction of
STAT3 signaling increases tumor glycolysis and proliferation, prevents apoptosis, and promotes drug resistance [20, 21]. Additionally, FBP1 binds to STAT3, blocks the binding of STAT3 to
STAT3-mediated gene promoters, inhibits glycolysis in ovarian cancer cells, and improves cisplatin resistance in ovarian cancer [22]. Based on these findings, it could be speculated that
regulating glycolysis-related enzymes via STAT3 inhibition may ameliorate chemotherapy resistance [23, 24].
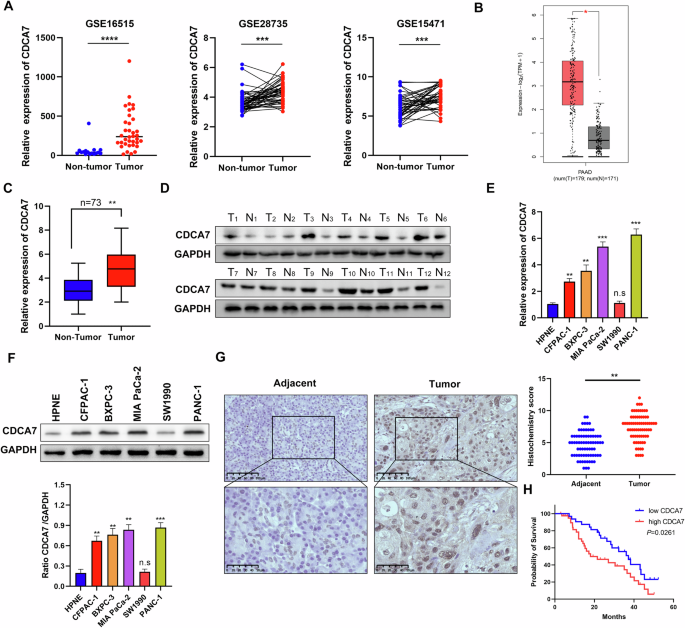
Therefore, this study aimed to investigate CDCA7 expression levels and functional significance in PC, focusing on its effect on STAT3. CDCA7 expression level was upregulated in PC and
closely correlated with adverse clinical outcomes. Additionally, CDCA7 expression was related to the glycolysis pathway in PC cells. Moreover, high expression of CDCA7 enhanced the
transcriptional activity of STAT3, which promoted hexokinase 2 (HK2) expression. Collectively, these changes increased aerobic glycolysis, ultimately promoting PC cell proliferation and
invasion and gemcitabine resistance. Based on these findings, we believe that targeting CDCA7 may be a potential chemotherapy sensitization strategies for PC.
Bioinformatics analysis was performed to verify the transcription levels of CDCA7 in the GTEx and TCGA databases using the Gene Expression Profiling Interactive Analysis (GEPIA).
Additionally, we examined CDCA7 expression in cancerous and adjacent non-tumor tissues using datasets (GSE15471, GSE28735, and GSE16515) from the Gene Expression Omnibus database.
Cancer and para-cancerous specimens from 73 patients with PC were collected from the Department of Hepatobiliary Surgery, Affiliated Hospital of Guizhou Medical University. This study was
approved by the Ethics Committee of the Affiliated Hospital of Guizhou Medical University, and each patient provided written informed consent.
Human pancreatic cell lines HPNE, CFPAC-1, BxPC-3, MIA PaCa-2, SW1990, and PANC-1 were obtained from the American Type Culture Collection. HPNE and BxPC-3 were grown in RPMI-1640 medium
supplemented with 10% FBS, whereas CFPAC-1, MIA PaCa-2, SW1990, and PANC-1 were cultivated in DMEM with 10% FBS. The cultures were maintained in a humidified atmosphere of 5% CO2 at 37 °C.
Authentication of the cell lines was performed through STR profiling, and mycoplasma contamination was ruled out, ensuring the reliability of the experimental outcomes.
Total RNA was extracted from clinical pancreatic tissues, adjacent non-tumor tissues, and PC cells subjected to different treatments using the RNA rapid extraction kit (YISHAN, Shanghai,
China). Reverse transcription was performed to generate cDNA using PrimeScript RT (YISHEN, Shanghai, China). qRT-PCR was performed to quantify gene expression levels. The primer sequences
are provided in Supplementary Table 1.
Proteins were extracted using a lysis buffer, separated using SDS-PAGE, and transferred to Millipore membranes. Thereafter, the membranes were blocked with 5% non-fat milk for a minimum of 1
h and incubated with the corresponding antibodies overnight at 4 °C. After washing with water, the membranes were incubated with affinity-labeled goat anti-mouse/rabbit antibody at 20 °C
for 2 h. Protein bands were visualized using ECL reagents (Boster Biotechnology Co., Ltd.) and a chemiluminescence imaging system (Tanon). The antibody information is provided in
Supplementary Table 2.
Cell proliferation activity was detected using cell counting kit-8 (CCK-8), 5-ethyl-2’deoxyuridine (EdU), and plate cloning experiments. In the CCk8 experiment, cells were seeded in 96-well
cells (six replicate wells per group) followed by the addition of CCK-8 reagent and further incubation for 3 h. Finally, the OD value was measured using a spectrophotometer. In the EdU
experiment, cells were seeded into 24-well plates, followed by the addition of EdU reagent (Beyotime, Shanghai, China) to each well and further incubation. Finally, the ratio of edu-positive
nuclei in six microscopic fields (three independent replicates) was analyzed. In the plate cloning experiment, cells were seeded in a 6-well plate and cultured. After 14 days, cells were
fixed with 4% paraformaldehyde (Biosharp, Hefei, Anhui, China) for 30 min and stained with 0.25% crystal violet solution (Biosharp, Hefei, Anhui, China) for 30 min. Images of the stained
cells were captured after washing with PBS.
PC cells were seeded into a 6-well plate and cultured on growth density reached 90%. Thereafter, a wound was carefully inflicted on the cell layer using a 200 μL pipette tip, followed by the
replacement of the culture medium with serum-free medium and incubation for 48 h. Images of each wound were meticulously captured at both the start (0 h) and end (48 h) of the incubation
period using an inverted microscope (Olympus, Tokyo, Japan).
Briefly, PC cells were starved and seeded in the upper chamber, followed by the addition of 600 μL of 20% FBS DMEM medium to the lower chamber. After 24 h, the cells were washed with PBS,
fixed with 4% paraformaldehyde for 30 min, and stained with a 0.25% crystal violet solution for another 30 min. Images were captured using an inverted microscope (Olympus, Tokyo, Japan).
After fixation, embedding, sectioning, and deparaffinization, PC tissue sections were blocked with 3% H2O2 and 5% BSA. Therefore, the sections were incubated with primary antibodies
targeting CDCA7, anti-Ki67, and PCNA overnight at 4 °C, followed by incubation with secondary antibodies for 2 h at room temperature. Positively stained cells and signal intensity were
assessed in three randomly selected areas by two independent observers, who were blinded to the treatments.
All animal experiments were approved by the Ethics Committee of Guizhou Medical University. Forty-eight female BALB/c nude mice (6-week-old) were randomly divided into 8 groups (n = 6
mice/group). PC cells (2 × 106) were injected into the left axilla of each mouse. Tumor volume was assessed every 7 days and calculated using the formula: Tumor volume = (length × width2)/2.
Mice were euthanized at 42 days post-injection, and tumor tissues were harvested, weighed, and subjected to immunohistochemical analysis.
HK2 promoter was cloned into pGL4.10[luc2] vector. The indicated reporter vectors were transfected into PC cells together with pGL4.74[hRluc/TK] vector. After 48 hours cells were harvested.
Finally, the fluorescence signals of firefly and Renilla luciferases were detected using the Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA). This process was independently
repeated three times.
The CDCA7-overexpressing plasmid was constructed using the GV341 vector from GeneChem (Shanghai, China). Short hairpin RNA lentiviruses targeting CDCA7, STAT3, and HK2 were also acquired
from GeneChem. The lentiviral particles were produced by co-transfecting the plasmids with packaging vectors and Lipo3000 (Invitrogen, MA, USA) into 293 T cells. The virus-containing
supernatant was collected, filtered through a 0.45 μm filter, concentrated using PEG6000 (Sigma, #81253), and then resuspended in PBS before being aliquoted for future transfections.
Infected cells were selected with puromycin for 72 h. The shRNA sequences are listed in Supplementary Table 3.
After exposure to various treatments, PC cells were seeded into a 24-well plate, fixed, permeabilized, blocked, and incubated with primary antibodies against CDCA7 and STAT3 at 4 °C for 10
h. Thereafter, the cells were incubated with secondary antibodies in the dark at room temperature for 2 h. After washing with PBS, cell nuclei were counterstained with
4’,6-diamidino-2-phenylindole, and visualization and quantification of target protein expression were performed using fluorescence microscopy.
Proteins were extracted from PC cells and incubated overnight at 4 °C with anti-CDCA7 antibody. Thereafter, protein A + G beads (Beyotime, Shanghai, China) were added and incubated for 2 h
under constant rotation. The beads were collected using a magnetic rack and boiled after adding 1× loading buffer. Protein analysis was conducted using western blotting, silver staining, or
mass spectrometry (MS). Silver staining was performed according to the manufacturer’s instructions.
PC cells at the log-phase were plated in 96-well plates. Glucose uptake, ATP levels, and lactate production were measured using the Glucose Assay Kit (Absin, abs580025), ATP Assay Kit
(Sigma, MAK190), and Lactate Assay Kit (Sigma, MAK064), respectively, according to the manufacturers’ protocols.
The orthotopic pancreatic cancer tumor studies were conducted using C57BL/6 J mice. Female mice were randomly divided into 8 groups, with 5 mice per group. At the age of 8 weeks, the mice
underwent orthotopic pancreatic cancer tumor induction. A suspension of 1 × 106 PANC-02-Luc cells was harvested and injected into the pancreatic tail via a minimal laparotomy. Twenty-one
days following the injection, high-resolution ultrasound imaging was conducted on each mouse to verify tumor establishment.
To assess PC cell metabolism, cells were seeded in Seahorse XF96 plates (Seahorse, Cat No. 101085-004) at a density of 3.5 × 104 cells/well until cell confluence reached 80%. After rinsing
with PBS, glycolysis and extracellular acidification rate (ECAR) were measured using Seahorse Glycolytic Stress Test Kit (Seahorse, Cat No. 103020-100) and Seahorse XFe96 Analyzer,
respectively. The analysis was performed on three independent replicates.
All statistical analyses were performed using SPSS (version 23.0; IBM Corp., Armonk, NY, USA). Data are expressed as mean ± standard deviation. Significant differences were determined using
Student’s t-test for two group comparison or one-way analysis of variance for multi-group comparison. Overall survival was determined using the Kaplan-Meier method. Statistical significance
was set at p