Play all audios:
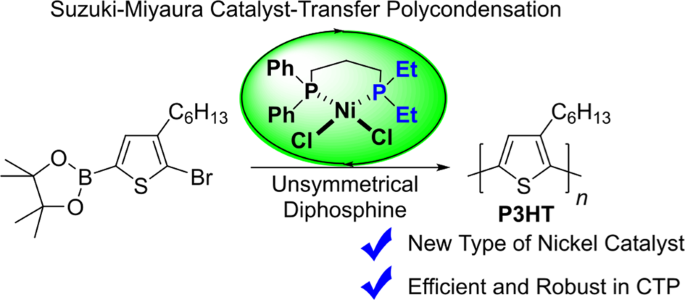
Original Article Published: 03 September 2019 A robust nickel catalyst with an unsymmetrical propyl-bridged diphosphine ligand for catalyst-transfer polymerization Matthew A. Baker1, Josué
Ayuso-Carrillo1, Martin R. M. Koos ORCID: orcid.org/0000-0002-7829-47291, Samantha N. MacMillan ORCID: orcid.org/0000-0001-6516-18232, Anthony J. Varni1, Roberto R. Gil ORCID:
orcid.org/0000-0002-8810-50471 & …Kevin J. T. Noonan ORCID: orcid.org/0000-0003-4061-75931 Show authors Polymer Journal volume 52, pages 83–92 (2020)Cite this article
973 Accesses
7 Citations
3 Altmetric
Metrics details
Subjects Catalyst synthesisConjugated polymers AbstractA new nickel diphosphine catalyst has been synthesized in which the bidentate ligand has two different phosphine donors, a typical PPh2 group and a stronger σ-donating PEt2 group. The
catalyst was highly effective for the chain-growth polymerization of a 3-alkylthiophene monomer using a Suzuki–Miyaura cross-coupling. The catalyst is particularly effective for this
polymerization in the presence of excess free ligand. The unsymmetrical diphosphine nickel complex reported here represents a new approach to tuning metal-ligand reactivity in the
chain-growth polymerization of aromatic monomers. In addition, this new nickel catalyst exhibited increased hydrolytic resistance in the polymerization as compared to commercially available
1,3-bis(diphenylphosphino)propane nickel dichloride.
You have full access to this article via your institution.
Download PDF Similar content being viewed by others Nickel-catalysed hydrodimerization of unactivated terminal alkenes Article02 February 2023 Identification of a potent palladium-aryldiphosphine catalytic system for high-performance carbonylation of alkenes Article Open access 05 March 2024 1-octene polymerization
catalyzed by titanium and zirconium complexes supported by [PN] or [NPN] ligands Article Open access 18 February 2025 Introduction
Metal-catalyzed cross-couplings continue to be among the most powerful strategies for forming C–C bonds, and they are commonly employed to synthesize π-conjugated polymers. These reactions
typically proceed by a step-growth mechanism, but an interaction between the metal catalyst and the π-system [1, 2] of the growing macromolecule can result in a chain-growth process (known
as catalyst-transfer polymerization or CTP). This affords well-defined polymers with controllable molecular weights and narrow molecular weight distributions [3,4,5,6,7,8,9,10,11]. The
ancillary ligand bound to the metal is critical to the chain-growth process because it governs both the metal-polymer π-interactions and the cross-coupling efficiency [3,4,5,6,7,8,9,10,11].
Of the cross-coupling reactions used for CTP, the Suzuki–Miyaura reaction is advantageous due to the functional group compatibility of organoboron moieties and the relatively mild reaction
conditions needed to promote C–C bond formation. While palladium complexes are often used to catalyze this transformation [12,13,14,15,16,17,18,19,20,21,22,23,24], nickel catalysts such as
Ni(dppp)Cl2 and Ni(IPr)(PPh3)Cl2 can also be employed to polymerize X-Ar-B(OR)2 monomers [25, 26]. Herein, we prepared a nickel diphosphine precatalyst with two unique phosphine donors
(Scheme 1) for CTP.
Scheme 1Synthesis of the Ni(sepp)Cl2 precatalyst for CTP
Full size imageThe desymmetrization of the diphosphine ligand enables specific modifications of the steric and electronic environments on each donor atom bound to the metal. In this report, the bidentate
ligand is composed of diaryl (PPh2) and dialkyl (PEt2) phosphines with a bridging propyl linker. This diphosphine was synthesized according to a published procedure [27,28,29], and it is
abbreviated “split” diethyl diphenyl phosphine or sepp for simplicity. The ligand strongly binds nickel and offers improved hydrolytic resistance under basic conditions compared to
commercially available 1,3-bis(diphenylphosphino)propane nickel dichloride (Ni(dppp)Cl2). Most importantly, Ni(sepp)Cl2 can catalyze the controlled polymerization of a 3-hexylthiophene
monomer using Suzuki-Miyaura coupling.
The polymerization catalyst is particularly effective when the reaction is conducted in the presence of excess free ligand. Remarkably, although the catalyst is unsymmetric, a single
catalyst resting state was identified using 31P NMR spectroscopy, indicating that oxidative addition to the Ni0 occurs preferentially with the growing chain trans to the PEt2 group. An
externally initiated variant of this catalyst was also synthesized and shown to be highly effective for controlling the polymer end groups. Altogether, the data suggest that individual
modification of the donor ligands in bidentate structures may be a valuable ligand design strategy for chain-growth polycondensation and, more broadly, for cross-coupling
reactions.
Materials and methodsAll synthetic manipulations of air-sensitive compounds were carried out under an N2 atmosphere using standard Schlenk techniques or in an N2-filled glovebox. All glassware was dried
overnight in an oven and heated under vacuum prior to use. Solvents were dried and degassed prior to use. Deionized water for the polymerizations was thoroughly degassed by a continuous flow
of N2 for at least 30 min. The sepp ligand was prepared according to a literature procedure [27] though a different reducing agent was used for the final step [30]. The 1H and 31P NMR
spectra of sepp and the ligand precursors are provided in the Supporting Information (Figs. S1–S3). Monomer 1 (2-(5-bromo-4-hexylthiophen-2-yl)−4,4,5,5-tetramethyl-1,3,2-dioxaborolane) was
synthesized from literature procedures with minor modifications [23, 31]. All other compounds were purchased from commercial vendors and used as received.
NMR analysisAll NMR spectra were recorded on a 500 Bruker Avance III or a 500 Bruker Avance Neo (1H, 500 MHz; 13C, 125.8 MHz; 31P, 202.5 MHz) spectrometer. NMR signals were referenced to residual
solvent for 1H and deuterated solvents for 13C{1H}. The proton-decoupled 31P{1H} NMR spectra were electronically referenced using internal Bruker software according to a universal scale
determined from the precise ratio, Ξ , of the resonance frequency of the 31P nuclide to the 1H resonance of TMS in a dilute solution (φ < 1%) [32]. All polymer samples subjected to 31P{1H}
NMR analysis were prepared by removing 0.5 mL of the polymer solution and transferring it into an NMR tube housed in a Schlenk tube under N2. Aliquots were removed from the polymerization at
specific time points and analyzed.
Gel-permeation chromatographyGel-permeation chromatography (GPC) measurements were performed on a Waters Instrument equipped with a 717 plus autosampler, a Waters 2414 refractive index (RI) detector, and two SDV columns
(Porosity 1000 and 100,000 Å; Polymer Standard Services) with THF as the eluent (~1 mg/mL, flow rate 1 mL/min, 40 °C). A 10-point calibration based on polystyrene standards (Polystyrene,
ReadyCal Kit, Polymer Standard Services) was used to determine the molecular weights.
MALDI-TOF analysisMALDI-TOF analyses were performed using an Applied Biosystems Voyager DE-STR mass spectrometer. One microliter of a solution of the matrix
(trans-2-[3-(4-t-butyl-phenyl)-2-methyl-2-propenylidene]malonitrile, DCTB) in THF (10 mg/mL) was spotted onto a well of the MALDI plate, and the solvent was allowed to evaporate. Polymer
sample solutions (2 mg/mL in THF) were prepared, and 1 μL of this solution was spotted onto the well by a layering method. The solvent was evaporated prior to analysis. Data were collected
in positive polarity mode in either linear or reflection mode.
X-ray analysisLow-temperature X-ray diffraction data for (sepp)Ni(o-C6H4CO2Me)Br and Ni(sepp)Cl2 were collected on a Rigaku XtaLAB Synergy diffractometer coupled to a Rigaku Hypix detector with Cu Kα
radiation (λ = 1.54184 Å) from a PhotonJet microfocus X-ray source at 100 K. The diffraction images were processed and scaled using CrysAlisPro software [33]. The structures were solved
through intrinsic phasing using SHELXT [34] and refined against F2 on all data by full-matrix least squares with SHELXL [35] following established refinement strategies [36]. All nonhydrogen
atoms were refined anisotropically. All hydrogen atoms bound to carbon were included in the model at geometrically calculated positions and refined using a riding model. The isotropic
displacement parameters of all hydrogen atoms were fixed to 1.2 times the Ueq value of the atoms they are linked to (1.5 times for methyl groups). Details of the data quality and a summary
of the residual values of the refinements are listed in the Supporting Information (Tables S1 and S2). The crystallographic data for (sepp)Ni(o-C6H4CO2Me)Br and Ni(sepp)Cl2 have been
deposited with the Cambridge Crystallographic Data Center (CCDC) under the reference numbers CCDC 1911056 and 1911057, respectively. These data can be obtained free of charge via
www.ccdc.cam.ac.uk/data_request/cif.
End-group analysisThe degree of polymerization (DP) for each poly(3-hexylthiophene) (P3HT) sample was estimated from the alkyl region of the 1H NMR spectrum [37,38,39]. For polymers prepared using
Ni(sepp)Cl2, the α-CH2 signal of the hexyl tail on the H-terminated end group (triplet at 2.62 ppm) [37, 39] was compared to the α-CH2 signal of the polymer (triplet centered at 2.81 ppm,
integrated from 2.90 to 2.65 ppm). The H-terminated end group signal was set to 2, and the DP was calculated as follows: DP = (integration of polymer CH2 signal/2) + 1 for the Br-terminated
end group + 1 for the H-terminated group. For polymers prepared using (sepp)Ni(o-C6H4CO2Me)Br, the CH3 signal of the o-C6H4CO2Me end group (singlet at 3.80 ppm) [38] was compared to the
α-CH2 signal of the polymer (triplet centered at 2.81 ppm, integration from 2.90 to 2.65 ppm). The end group signal was set to 3, and the DP was calculated as follows: DP = (integration of
polymer CH2 signal/2) + 1 for the H-terminated end group.
Computational studiesDensity functional theory calculations were performed for all compounds using a mixed basis set with B3LYP 6–31 G(d) for all atoms in the ligand and SDD for nickel in the Gaussian 09 suite
[40]. The Cartesian coordinates of the optimized geometries are available as xyz files, and the total energies of the two isomers are included in Table S3 (ESI).
ExperimentalproceduresNi(sepp)Cl2
To an oven-dried 100 mL Schlenk flask were added NiCl2 (0.118 g, 0.91 mmol), sepp (0.300 g, 0.95 mmol) and degassed ethanol (15 mL) under an inert atmosphere. The suspension was stirred at
90 °C for 1 h and then cooled to 8 °C to facilitate precipitation of the target compound. The nickel dihalide complex was isolated as an air-stable, red-orange solid by vacuum filtration and
washed with ethanol and diethyl ether (0.140 g, 35% yield). Crystals were grown from a concentrated solution in CH2Cl2 (~20 mg/mL) layered with n-hexane or diethyl ether.
31P{1H} NMR (202 MHz, CD2Cl2) δ 8 (br s, 2P). 1H NMR (500 MHz, CD2Cl2) δ 7.97 (d, J = 7.4 Hz, 4H), 7.56 (t, J = 7.5 Hz, 4H), 7.50 (t, J = 7.4 Hz, 2H), 3.07 (br s, 2H), 2.48 (br s, 2H), 2.07
(br s, 2H), 1.91 (s, 2H), 1.70 (p, J = 5.8 Hz, 2H), 1.43 (t, J = 7.4 Hz, 6H). 13C{1H} NMR (126 MHz, CD2Cl2): δ 135.4, 134.1 (br), 131.8, 129.2, 26.1, 23.6 (br), 20.2, 19.1, 10.5. HRMS
(DART-MS) (m/z): [2M−Cl]+ calculated for C38H52Cl3Ni2P4, 853.0792; found, 853.0789.
(PPh3)2Ni(o-C6H4CO2Me)BrIn a glovebox, a scintillation vial equipped with a magnetic stir bar was charged with bis(1,5-cyclooctadiene)nickel (0.165 g, 0.6 mmol), triphenylphosphine (0.315 g, 1.20 mmol), and toluene
(6 mL). After complete dissolution of the reagents in toluene, methyl-2-bromobenzoate (0.142 g, 0.66 mmol) was added into the reaction mixture, and the solution was vigorously stirred at 23
°C for 18 h, during which time a solid precipitated from the reaction mixture. The suspended solids were precipitated by hexanes (~60 mL), isolated by filtration and washed with hexanes
(~200 mL) and methanol (~200 mL). The resulting bright-yellow solid was dried under reduced pressure at 50 °C, affording 0.420 g (88% yield) of the nickel complex.
31P{1H} NMR (202 MHz, CD2Cl2): δ 20.8 (s). 1H NMR (500 MHz, CD2Cl2): δ 7.75–7.53 (br s, 12H), 7.50 (d, J = 7.5 Hz, 1H), 7.41–7.10 (br m, 18H), 6.68 (d, J = 7.9 Hz, 1H), 6.48 (t, J = 7.6 Hz,
1H), 6.32 (t, J = 7.4 Hz, 1H), 3.74 (s, 3H). 13C{1H} NMR (126 MHz, CD2Cl2): δ 169.5, 166.2, 136.8, 136.6, 135.1, 132.5, 131.8, 129.9, 128.9, 128.0, 121.5, 51.9. HRMS (DART-MS) (m/z): [M +
H]+ calculated for C44H38BrNiO2P2, 797.0884; found, 797.6169.
(sepp)Ni(o-C6H4CO2Me)BrIn a glovebox, a scintillation vial equipped with a stir bar was charged with (PPh3)2Ni(o-C6H4CO2Me)Br (0.237 g, 0.30 mmol), sepp (0.103 g, 0.33 mmol), and THF (4.8 mL). The solution was
vigorously stirred at 23 °C for 18 h, during which time a precipitate formed in the reaction mixture. The suspended solids were precipitated by hexanes (~50 mL), isolated by filtration and
washed with hexanes (~150 mL). The resulting bright-yellow solid was dried under reduced pressure at 23 °C, affording 0.170 g (97% yield) of the nickel complex. Crystals were grown from a
concentrated solution in CH2Cl2 (~20 mg/mL) layered with n-hexane. 31P{1H} (202 MHz, THF-d8) δ 18.0 (d, JPP = 70.3 Hz, 1P), −5.7 (d, JPP = 70.2 Hz, 1P). 1H NMR (500 MHz, THF-d8) δ 8.55 (br
s, 2H), 7.59 (br s, 3H), 7.49–7.41 (m, 1H), 6.98 (dt, J = 7.7, 1.8 Hz, 2H), 6.80 (t, J = 7.7 Hz, 2H), 6.62 (tt, J = 7.4, 1.6 Hz, 1H), 6.55 (t, J = 8.9 Hz, 2H), 6.32 (t, J = 7.4 Hz, 1H), 3.81
(s, 3H), 2.53–2.31 (m, 2H; note: this signal overlaps with H2O), 2.28–2.15 (m, 1H), 2.11–1.99 (m, 2H), 1.89–1.76 (m, 1H), 1.71–1.66 (m, 2H; note: this signal overlaps with the solvent
signal), 1.66–1.55 (m, 1H), 1.40 (dt, J = 15.8, 7.6 Hz, 3H), 1.30–1.19 (m, 1H), 1.12 (dt, J = 14.6, 7.6 Hz, 3H). 13C{1H} NMR (126 MHz, THF-d8) δ 174.6 (dd, JPC = 79.7, 35.7 Hz), 172.3 (d,
JPC = 5.8 Hz), 139.2 (d, JPC = 2.6 Hz), 136.8 (d, JPC = 11.9 Hz), 135.7, 135.5 (dd, JPC = 47.2, 4.6 Hz), 132.7 (d, JPC = 44.9 Hz), 132.2, 131.6 (d, JPC = 7.5 Hz), 130.0 (dd, JPC = 4.5, 2.5
Hz), 129.7 (d, JPC = 5.5 Hz), 129.5 (d, JPC = 10.0 Hz), 128.8 (d, JPC = 2.2 Hz), 127.7 (d, JPC = 9.5 Hz), 121.1, 51.6, 28.7 (dd, JPC = 21.7, 7.9 Hz), 20.6 (dd, JPC = 17.2, 3.5 Hz), 19.5 (d,
JPC = 3.5 Hz), 19.3 (d, JPC = 24.2 Hz), 18.8 (d, JPC = 28.1 Hz), 8.6, 8.2 (d, JPC = 3.8 Hz). HRMS (DART-MS) (m/z): [M−Br]+ calculated for C27H33NiO2P2, 509.1309; found,
509.1307.
Representative polymerization procedureIn a glovebox, a scintillation vial equipped with a stir bar and an open-top Teflon screw cap was charged with a calculated amount of catalyst and K3PO4‧H2O (0.070 g, 0.30 mmol). In several
cases (entries 3–4 and 7–8 in Table 1), a calculated amount of sepp ligand (1.0 equiv relative to catalyst) was also added to the reaction. Monomer 1 (0.056 g, 0.15 mmol) and then THF (6 mL)
were added to the vial, which was then sealed and removed from the glovebox. The reaction was heated to 50 °C, and water (0.09 mL) was injected. Two hours after the addition of water, the
polymerization was quenched by the addition of 6 N methanolic HCl (50 mL) with stirring for 30 min. The resulting precipitate was isolated by filtration, washed with methanol (100 mL), water
(50 mL), and then methanol (200 mL) before drying. The experiment to obtain Mn versus conversion and the semilogarithmic plot was conducted according to a previous report [25].
Table 1Preparation of P3HT using a Suzuki–Miyaura cross-couplingFull size tableResults and discussion
The combination of the previously reported diphosphine [27,28,29] with NiCl2 in refluxing ethanol (Scheme 1) afforded Ni(sepp)Cl2 in a low yield (35%). The NMR signals (1H and 13C{1H}) of
the precatalyst were broad due to the very short T2 relaxation times induced by the quadrupolar chlorides [41]. An extremely broad, nearly imperceptible signal was present in the 31P{1H} NMR
spectrum, presumably due to this quadrupolar relaxation. Single crystal X-ray diffraction (Fig. 1) was then employed to unambiguously assign the structure.
Fig. 1Solid-state molecular structure of Ni(sepp)Cl2 (thermal ellipsoids at 50% probability) with H atoms omitted. Only one of the two independent molecules is shown
Full size imageThere are two independent molecules in the unit cell of Ni(sepp)Cl2. The average nickel–phosphine bond lengths for the two different phosphines are nearly identical (2.1609(4) Å for the
Ni−PPh2 moiety and 2.1712(4) Å for the Ni−PEt2 unit). Interestingly, the bite angles in the two independent precatalyst molecules differ by over 2° and average to 95°. This suggests a
slightly larger steric constraint imposed by the two unique phosphines in Ni(sepp)Cl2 compared to that of Ni(dppp)Cl2, in which the bite angle of the diphosphine is 91.8° [42].
Upon complete characterization of the Ni(sepp)Cl2 complex, its performance in CTP was evaluated. This precatalyst was used to initiate the polymerization of a 3-alkylthiophene monomer
bearing a bromine at the 2-position and a pinacol boronate at the 5 position (1) in THF with K3PO4·H2O as the base and minimal water at 50 °C. The concentrations of base and water used in
these experiments were identical to our optimized conditions for Ni(dppp)Cl2. Using 4 mol% Ni(sepp)Cl2, P3HT with a DP of 27, an Mn of 7.9 kg/mol, and Ð of 1.07 (Table 1, Entry 1) was
obtained.
The molecular weight and distribution of the P3HT sample suggested that Ni(sepp)Cl2 was a promising catalyst for CTP, but it also raised some questions. Prior reports on the mechanism of
Suzuki–Miyaura cross-coupling have indicated that ligand substitution reactions of metal-halogen bonds to form metal-hydroxo species are critical for the transmetalation with the aryl
boronate [43,44,45,46,47,48,49]. Considering this, activation of the nickel dihalide precatalyst likely requires exchange of the −Cl ligands for −OH ligands prior to transmetalation and
reductive elimination (depicted in Scheme 2). Consequently, this hydroxide exchange can potentially impact the efficiency of the precatalyst reduction. In our prior work on the
polymerization of 1 with Ni(dppp)Cl2 [25], NMR spectroscopy revealed a significant portion of free dppp in the reaction mixture. The presence of dppp likely stemmed from metal-hydroxide
formation involving the precatalyst and subsequent dissociation of the diphosphine ligand to afford the catalytically inactive nickel hydroxide (proposed pathway in Scheme 2). Miyaura and
coworkers have documented sensitivity of dihalide precatalysts for Suzuki-Miyaura cross-coupling in their previous work [50,51,52]. Although a portion of the Ni(dppp)Cl2 precatalyst was lost
to hydrolysis, this event was critical to the overall chain-growth polymerization process since the combination of the remaining active catalyst and free ligand were necessary to achieve
the controlled polymerization, as evidenced by control experiments [25]. The hydrolysis of Ni(dppp)Cl2 led us to consider whether Ni(sepp)Cl2 with the stronger PEt2 donor would show
different precatalyst initiation behavior.
Scheme 2Proposed pathway of precatalyst reduction and hydrolysis
Full size imageOur interrogation of the polymerization reaction using 31P{1H} NMR spectroscopy revealed that Ni(sepp)Cl2 was significantly more resistant to precatalyst hydrolysis when compared to
Ni(dppp)Cl2 (Fig. 2). For comparison, free dppp and its oxide account for nearly 60% of the integration intensity in the spectrum of an aliquot of the Ni(dppp)Cl2-initiated polymerization,
while for Ni(sepp)Cl2, the free ligand and its oxide account for only 10% of the mixture. In addition to hydrolytic resistance, only one pair of doublets was observed from the major product
in the Ni(sepp)Cl2-initiated polymerization (Fig. 2). The chemical shifts are close to those of the pair of doublets observed when polymerizing with Ni(dppp)Cl2.
Fig. 2Crude 31P{1H} NMR spectra recorded in THF of the polymerization reaction conducted with Ni(dppp)Cl2 (top) and Ni(sepp)Cl2 (bottom). Aliquots were removed 60 min after initiation from 25 mM
polymerizations of monomer 1 with 10 mol % catalyst, 2 equiv of K3PO4·H2O, and 33.3 equiv of degassed water. * indicates unidentified side-product
Full size imageTransmetalation has been identified as the rate-limiting step when using Ni(dppp)Cl2 as the catalyst in both Kumada–Corriu- and Suzuki–Miyaura-initiated CTPs [25, 53, 54]. The resting state
of the catalyst, (dppp)Ni(P3HT)Br, can be observed using 31P{1H} NMR spectroscopy [53, 54]. The two doublets in the top of Fig. 2 correspond to this Ni2+ species that has undergone oxidative
addition. The downfield signal near 20 ppm (2JPP = 64.4 Hz) corresponds to the PPh2 group trans to the halogen, and the upfield signal near −3 ppm corresponds to the PPh2 group trans to the
growing chain (2JPP = 65.8 Hz) [55]. A similar pattern is observed for the polymerization with Ni(sepp)Cl2. One doublet appeared at 19.1 ppm (2JPP = 71.0 Hz), and the other appeared further
upfield at −4.5 ppm (2JPP = 70.9 Hz).
The similarity to the Ni(dppp)Cl2 polymerization suggested that the oxidative addition product is also the resting state of the catalyst in this case. Moreover, the upfield shift of the
second signal was indicative that the more electron-rich PEt2 is trans to the growing chain. If the oxidative addition was not selective, then one would expect to see two pairs of doublets
for the two possible stereoisomers. Some minor doublets are indeed observed in the spectrum, so it is likely that the other stereoisomer forms (along with several other minor species), but
the major signals, are the result of the selective arrangement of the P3HT and Br around the unsymmetrical nickel catalyst.
To confirm the positions of the phosphino groups relative to the corresponding aryl bromide, a model compound was synthesized. Luscombe and co-workers [38, 56] previously synthesized an
externally initiated (dppp)Ni(o-tolyl)Br catalyst for cross-coupling polymerizations. Unfortunately, the isolation and characterization of the analogous (sepp)Ni(o-tolyl)Br was unsuccessful.
Methylbenzoate was considered as a replacement for the o-tolyl ligand since the methyl ester should be a suitable substituent to block the axial site of the nickel [57], and it can
potentially coordinate to the metal via the oxygen atoms. Bis(1,5-cyclooctadiene)nickel, PPh3, and methyl 2-bromobenzoate were combined to form an air-stable Ni2+ complex prior to ligand
exchange with sepp (Scheme 3).
Scheme 3Ligand exchange of sepp with PPh3 to prepare an externally initiated Ni2+ complex
Full size imageThe 31P{1H} NMR signals for (sepp)Ni(o-C6H4CO2Me)Br were nearly identical to those observed in the spectrum obtained from the polymerization reactions. For (sepp)Ni(o-C6H4CO2Me)Br in THF-d8,
signals were observed at 18.0 ppm (2JPP = 70.3 Hz) and −5.7 ppm (2JPP = 70.2 Hz). Two minor doublets that exchange with the major species are also observed at 4.8 and 1.9 ppm. The identity
of this species is still under investigation. A 2D 1H−31P HMQC experiment optimized for long-range coupling (J = 8 Hz) confirmed that the downfield signal near 19 ppm corresponds to the PPh2
group of sepp. The correlations of the P atom to the broad protons of the phenyl rings are indicated in Fig. 3.
Fig. 3Selected region of an 1H−31P HMQC experiment (500 MHz) recorded for (sepp)Ni(o-C6H4CO2Me)Br in THF-d8 at 22 °C
Full size imageThe signal near −5 ppm corresponds to the PEt2 group, as evidenced by the correlations to the methylene and methyl groups (marked by the blue box in Fig. 3). Notably, the phosphorus atom of
the PEt2 group also correlates strongly with nearly all the signals of the propyl backbone of the 6-membered metallacycle and the trans-2-methylbenzoate group. The assignments for
(sepp)Ni(o-C6H4CO2Me)Br are consistent with those of the spectra acquired during the polymerizations (Fig. 2, bottom) and suggest an energetic preference for the reactive arene ligand being
trans to the PEt2 group.
The formation of a single stereoisomer was also confirmed for (sepp)Ni(o-C6H4CO2Me)Br using single crystal X-ray diffraction (Fig. 4). There are two independent molecules in the unit cell
(similar to Ni(sepp)Cl2). In the solid-state molecular structure, the nickel–phosphine bond lengths are significantly different, and the PPh2−Ni bond trans to the Br is nearly 0.1 Å shorter
than the PEt2−Ni bond (2.1384(4) versus 2.2277(4) Å, respectively). This was expected considering the strong σ-donating properties of the reactive arene ligand opposite the PEt2 group. The
average bite angle of the ligands was nearly 98° for this complex. The oxygen atom (O12) and nickel are within the sum of their Van der Waals radii but there is not a strong bonding
interaction (Ni1–O12 distance is 2.545 Å).
Fig. 4Solid-state molecular structure of (sepp)Ni(o-C6H4CO2Me)Br (thermal ellipsoids at 50% probability) with H atoms omitted. Only one of the two independent molecules is shown
Full size imageThe formation of the major isomer during the polymerization (Fig. 2), and the observation of the same preference when synthesizing a complex by ligand exchange (Figs. 3 and 4), suggests a
significant energy difference with the relative orientation of the reactive arene and the bromide. The total energies of the two possible stereoisomers were calculated (3-methylthiophene was
used to mimic the polymer chain), and as expected, the trans relationship of the PEt2 group and the reactive ligand is favored by 3.7 kcal/mol (Fig. 5—computed structures A and B). We
suspect the energy difference is due to steric constraints as more distortion from square planar geometry around the nickel is observed in A. A mean plane was constructed from four atoms for
both the optimized geometries: the nickel, the two phosphines from the ligand, and the carbon atom of the 2-position for the thiophene ring. In A (the higher energy structure), the bromine
atom is distorted by 0.55 Å from the plane, while this value is only 0.25 Å in B (lower energy structure). A picture illustrating these distortions from the mean planes is provided in Fig.
S26.
Fig. 5DFT-optimized structures of the two possible stereoisomers of (sepp)Ni(3-methylthiophene)Br (H atoms omitted). Optimization and total energy calculations were conducted with a mixed basis
set using B3LYP 6–31 G(d) for all ligand atoms and SDD for nickel
Full size imageOnce the energy preference for the Ni2+ species generated by oxidative addition was established, the two catalysts, Ni(sepp)Cl2 and (sepp)Ni(o-C6H4CO2Me)Br, were used to polymerize monomer
1. The MALDI-TOF mass spectra for the P3HT samples generated using these two catalysts (4 mol %) revealed some interesting features. For Ni(sepp)Cl2, a tail was observed in the
lower-molecular-weight region (along with a minor second distribution) and for (sepp)Ni(o-C6H4CO2Me)Br, a second mass distribution was observed at lower molecular weights (Fig. 6a, b,
respectively). The appearance of these lower-molecular-weight species suggested unproductive side-reactions (chain-transfer or chain-chain coupling) during the polymerization. In the GPC
traces, we observed higher-molecular-weight shoulders in some cases, which is indicative of chain–chain coupling. This can occur when two growing chains ((sepp)Ni(polymer)Br) exchange
reactive ligands producing (sepp)Ni(polymer)2 and (sepp)NiBr2. Reductive elimination from (sepp)Ni(polymer)2 can then produce higher-molecular-weight materials, and the resultant metal
species can begin growing new chains, affording lower-molecular-weight species.
Fig. 6MALDI-TOF mass spectra for P3HT samples. a Ni(sepp)Cl2 and b (sepp)Ni(o-C6H4CO2Me)Br. The spectra in c and d correspond to the same catalysts with an additional 1.0 equiv of sepp added to
the reaction mixture (relative to [Ni]). The reaction conditions were as described in Table 1, entries 1, 5, 3, and 7 for a, b, c, and d, respectively
Full size imageIn our previous report on Suzuki–Miyaura CTPs using Ni(dppp)Cl2, catalyst hydrolysis was critical to controlling the polymerization process, as it released free ligand into the reaction
mixture [25]. The free ligand does not have a significant impact on the rate of the polymerization, but it was necessary to obtain high-molecular-weight P3HT with good control over the end
groups. Since Ni(sepp)Cl2 and (sepp)Ni(o-C6H4CO2Me)Br are more resistant to hydrolysis, minimal free ligand is present in the reaction during the polymerization. To evaluate whether
additional free ligand could improve the polymerization, sepp was added to Ni(sepp)Cl2- and (sepp)Ni(o-C6H4CO2Me)Br-initiated polymerizations (Fig. 6c, d).
The change in the MALDI-TOF mass spectra for the as-obtained P3HT samples prepared using these two different catalyst systems was remarkable. When the polymerization was conducted in the
presence of additional sepp ligand, the distribution of chain lengths was in line with a living polymerization (Poisson distribution) (Fig. 6c, d). Very minor secondary distributions were
observed in both spectra, indicating that although the polymerization is not perfect, additional ligand greatly improves the control over the end groups (see the integrations of the P3HT
samples in the ESI) and the molecular weight distribution. For reference, the molecular weight data from GPC and NMR are included in Table 1.
A molecular weight versus conversion plot and a semilogarithmic plot were generated for (sepp)Ni(o-C6H4CO2Me)Br in the presence of additional sepp ligand. This experiment was completed in an
identical manner to our previous report though different time intervals were used [25]. The collected data were then compared to those of (dppp)Ni(o-tolyl)Br with additional dppp (Fig. 7)
[25]. As expected, the profile is very similar to that of the dppp system, but the newly synthesized catalyst produces the polymer at a slower rate. In both semilog plots, deviations from
linearity were observed, similar to the 3-hexylthiophene polymerization using a Kumada coupling [58]. As such, no rate constants were extracted; however, qualitatively, it is clear that
(dppp)Ni is faster. As expected, the Mn values increase for both catalysts with conversion, although they both begin to level off at higher conversions. This is indicative of chain-transfer
towards the end of the polymerization, which could be a result of repeated sampling because the dispersity of the final polymer samples was higher (~1.15) than in the isolated
polymerizations presented in Table 1 (entries 7 and 8).
Fig. 7Left: semilogarithmic plot of concentration versus time. Right: number-average molecular weight versus conversion. Black circles correspond to 2 mol% (dppp)Ni(o-C6H4Me)Br with dppp, and blue
squares correspond to 2 mol% (sepp)Ni(o-C6H4CO2Me)Br with sepp. For both polymerizations, [1] was 25 mM in THF with 0.5 mM catalyst and ligand. The (dppp)Ni experimental data was taken
directly from ref. [25]
Full size imageUltimately, the additional ligand clearly has a positive impact on the polymerization. Miyaura proposed in prior work that free ligand stabilizes Ni(0) [50,51,52, 59], and Van Der Boom has
illustrated that additional phosphine ligand can suppress intermolecular pathways in aryl halide bond activations with platinum species [60]. These reports highlight the importance of free
ligands for improving catalyst lifetime and chain fidelity, respectively. Future work will explore this effect in more detail to better elucidate how free ligand exerts this positive
effect.
ConclusionsIn conclusion, we have prepared a new, unsymmetrical nickel diphosphine catalyst for CTP. The diphosphine comprises two electronically and sterically distinct moieties, one PPh2 group and
one stronger σ-donating PEt2 group. The nickel catalyst retains one ligand site that is identical to the highly successful Ni(dppp) system, while offering another donor in the scaffold as a
tunable site. Ni(sepp)Cl2 proved to be an excellent catalyst for the controlled polymerization of an organoboron precursor if excess free ligand is present in the reaction mixture. The P3HT
obtained using this catalyst system is essentially identical to that obtained using Ni(dppp)Cl2.
This work, as well as our prior efforts, are consistent with the reports from Yokozawa et al. [61], McNeil and co-workers [62], and Choi and co-workers [14] who have noted the benefits of
free ligand in CTP reactions. Lee and Luscombe [63] have also noted the importance of additional pyridine in recent studies on chain-growth polymerizations via direct arylation. In the
future, we will explore other classes of unsymmetrical diphosphines for CTP. Additionally, the Ni(sepp)Cl2 system and the externally initiated variant will be evaluated as catalysts for the
polymerization of other monomers using Suzuki–Miyaura CTPs.
References He WY, Patrick BO, Kennepohl P. Identifying the missing link in catalyst transfer polymerization. Nat Commun. 2018;9:3866.
PubMed PubMed Central Google Scholar
Leone AK, Goldberg PK, McNeil AJ. Ring-walking in catalyst-transfer polymerization. J Am Chem Soc. 2018;140:7846–50.
CAS PubMed Google Scholar
Baker MA, Tsai CH, Noonan KJT. Diversifying cross-coupling strategies, catalysts and monomers for the controlled synthesis of conjugated polymers. Chem Eur J. 2018;24:13078–88.
CAS Google Scholar
Verheyen L, Leysen P, Van den Eede M-P, Ceunen W, Hardeman T, Koeckelberghs G. Advances in the controlled polymerization of conjugated polymers. Polymer. 2017;108:521–46.
CAS Google Scholar
Yokozawa T, Ohta Y. Transformation of step-growth polymerization into living chain-growth polymerization. Chem Rev. 2016;116:1950–68.
CAS PubMed Google Scholar
Leone AK, McNeil AJ. Matchmaking in catalyst-transfer polycondensation: optimizing catalysts based on mechanistic insight. Acc Chem Res. 2016;49:2822–31.
CAS PubMed Google Scholar
Bryan ZJ, McNeil AJ. Conjugated polymer synthesis via catalyst-transfer polycondensation (CTP): mechanism, scope, and applications. Macromolecules. 2013;46:8395–405.
CAS Google Scholar
Okamoto K, Luscombe CK. Controlled polymerizations for the synthesis of semiconducting conjugated polymers. Polym Chem. 2011;2:2424–34.
CAS Google Scholar
Yokozawa T, Yokoyama A. Chain-growth condensation polymerization for the synthesis of well-defined condensation polymers and π-conjugated polymers. Chem Rev. 2009;109:5595–619.
CAS PubMed Google Scholar
Grisorio R, Suranna GP. Intramolecular catalyst transfer polymerisation of conjugated monomers: from lessons learned to future challenges. Polym Chem. 2015;6:7781–95.
CAS Google Scholar
Leone AK, Mueller EA, McNeil AJ. The history of palladium-catalyzed cross-couplings should inspire the future of catalyst-transfer polymerization. J Am Chem Soc. 2018;140:15126–39.
CAS PubMed Google Scholar
Sugita H, Nojima M, Ohta Y, Yokozawa T. Unstoichiometric Suzuki–Miyaura cyclic polymerization of extensively conjugated monomers. Polym Chem. 2019;10:1182–5.
CAS Google Scholar
Kosaka K, Uchida T, Mikami K, Ohta Y, Yokozawa T. AmPhos Pd-catalyzed Suzuki Miyaura catalyst-transfer condensation polymerization: narrower dispersity by mixing the catalyst and base prior
to polymerization. Macromolecules. 2018;51:364–9.
CAS Google Scholar
Seo K-B, Lee I-H, Lee J, Choi I, Choi T-L. A rational design of highly controlled Suzuki–Miyaura catalyst-transfer polycondensation for precision synthesis of polythiophenes and their block
copolymers: marriage of palladacycle precatalysts with MIDA-boronates. J Am Chem Soc. 2018;140:4335–43.
CAS PubMed Google Scholar
Sugita H, Nojima M, Ohta Y, Yokozawa T. Unusual cyclic polymerization through Suzuki–Miyaura coupling of polyphenylene bearing diboronate at both ends with excess dibromophenylene. Chem
Commun. 2017;53:396–9.
CAS Google Scholar
Grisorio R, Suranna GP. Impact of precatalyst activation on Suzuki-Miyaura catalyst-transfer polymerizations: new mechanistic scenarios for pre-transmetalation events. ACS Macro Lett.
2017;6:1251–6.
CAS Google Scholar
Zhang H-H, Peng W, Dong J, Hu Q-S. t-Bu3P-coordinated 2-Phenylaniline-based palladacycle complex/ArBr as robust initiators for controlled Pd(0)/t-Bu3P-catalyzed Suzuki cross-coupling
polymerization of AB-type monomers. ACS Macro Lett. 2016;5:656–60.
CAS Google Scholar
Zhang H-H, Xing C-H, Hu Q-S, Hong K. Controlled Pd(0)/t-Bu3P-catalyzed Suzuki cross-coupling polymerization of AB-type monomers with ArPd(t-Bu3P)X or Pd2(dba)3/t-Bu3P/ArX as the initiator.
Macromolecules. 2015;48:967–78.
CAS Google Scholar
Kosaka K, Ohta Y, Yokozawa T. Influence of the boron moiety and water on Suzuki-Miyaura catalyst-transfer condensation polymerization. Macromol Rapid Commun. 2015;36:373–7.
CAS PubMed Google Scholar
Sui A, Shi X, Tian H, Geng Y, Wang F. Suzuki-Miyaura catalyst-transfer polycondensation with Pd(IPr)(OAc)2 as the catalyst for the controlled synthesis of polyfluorenes and polythiophenes.
Polym Chem. 2014;5:7072–80.
CAS Google Scholar
Grisorio R, Mastrorilli P, Suranna GP. A Pd(AcO)2/t-Bu3P/K3PO4 catalytic system for the control of Suzuki cross-coupling polymerisation. Polym Chem. 2014;5:4304–10.
CAS Google Scholar
Zhang H-H, Xing C-H, Hu Q-S. Controlled Pd(0)/t-Bu3P-catalyzed Suzuki cross-coupling polymerization of AB-type monomers with PhPd(t-Bu3P)I or Pd2(dba)3/t-Bu3P/ArI as the initiator. J Am Chem
Soc. 2012;134:13156–9.
CAS PubMed Google Scholar
Yokozawa T, Suzuki R, Nojima M, Ohta Y, Yokoyama A. Precision synthesis of poly(3-hexylthiophene) from catalyst-transfer Suzuki−Miyaura coupling polymerization. Macromol Rapid Commun.
2011;32:801–6.
CAS PubMed Google Scholar
Yokoyama A, Suzuki H, Kubota Y, Ohuchi K, Higashimura H, Yokozawa T. Chain-growth polymerization for the synthesis of polyfluorene via Suzuki−Miyaura coupling reaction from an externally
added initiator unit. J Am Chem Soc. 2007;129:7236–7.
CAS PubMed Google Scholar
Baker MA, Zahn SF, Varni AJ, Tsai CH, Noonan KJT. Elucidating the role of diphosphine ligand in nickel-mediated Suzuki-Miyaura polycondensation. Macromolecules. 2018;51:5911–7.
CAS Google Scholar
Qiu Y, Worch JC, Fortney A, Gayathri C, Gil RR, Noonan KJT. Nickel-catalyzed Suzuki polycondensation for controlled synthesis of ester-functionalized conjugated polymers. Macromolecules.
2016;49:4757–62.
CAS Google Scholar
Benn FR, Briggs JC, McAuliffe CA. Unsymmetrical bis(phosphorus) compounds: synthesis of unsymmetrical ditertiary phosphines, phosphine oxides, and diquaternary phosphonium salts. J Chem Soc
Dalton Trans. 1984:293–5.
Briggs JC, McAuliffe CA, Hill WE, Minahan DMA, Dyer G. Unsymmetrical bisphosphorus ligands - Phosphorus-31 and Carbon-13 nuclear magnetic-resonance and mass-spectral measurements. J Chem Soc
Perkin Trans 2. 1982:321–5.
Google Scholar
Briggs JC, Dyer G. New synthesis of unsymmetrical bidendate phosphine ligands. Chem Ind. 1982:163–4.
Petit C, Favre-Reguillon A, Albela B, Bonneviot L, Mignani G, Lemaire M. Mechanistic Insight into the reduction of tertiary phosphine oxides by Ti(OiPr)4/TMDS. Organometallics.
2009;28:6379–82.
CAS Google Scholar
Liversedge IA, Higgins SJ, Giles M, Heeney M, McCulloch I. Suzuki route to regioregular polyalkylthiophenes using Ir-catalysed borylation to make the monomer, and Pd complexes of bulky
phosphanes as coupling catalysts for polymerisation. Tetrahedron Lett. 2006;47:5143–6.
CAS Google Scholar
Harris RK, Becker ED, De Menezes SMC, Goodfellow R, Granger P. NMR nomenclature. Nuclear spin properties and conventions for chemical shifts (IUPAC recommendations 2001). Pure Appl Chem.
2001;73:1795–818.
CAS Google Scholar
CrysAlisPro; The Woodlands, TX: Rigaku OD; 2015.
Sheldrick G. SHELXT—integrated space-group and crystal-structure determination. Acta Cryst Sec A. 2015;71:3–8.
Google Scholar
Sheldrick G. A short history of SHELX. Acta Cryst Sec A. 2008;64:112–22.
CAS Google Scholar
Müller P. Practical suggestions for better crystal structures. Crystallogr Rev. 2009;15:57–83.
Google Scholar
Kohn P, Huettner S, Komber H, Senkovskyy V, Tkachov R, Kiriy A, et al. On the role of single regiodefects and polydispersity in regioregular poly(3-hexylthiophene): defect distribution,
synthesis of defect-free chains, and a simple model for the determination of crystallinity. J Am Chem Soc. 2012;134:4790–805.
CAS PubMed Google Scholar
Bronstein HA, Luscombe CK. Externally initiated regioregular P3HT with controlled molecular weight and narrow polydispersity. J Am Chem Soc. 2009;131:12894–5.
CAS PubMed Google Scholar
Iovu MC, Sheina EE, Gil RR, McCullough RD. Experimental evidence for the quasi-“living” nature of the Grignard metathesis method for the synthesis of regioregular poly(3-alkylthiophenes).
Macromolecules. 2005;38:8649–56.
CAS Google Scholar
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, et al. Gaussian 09. Gaussian, Inc., Wallingford, CT: Gaussian, Inc.; 2009.
Pople JA. The effect of quadrupole relaxation on nuclear magnetic resonance multiplets. Mol Phys. 1958;1:168–74.
CAS Google Scholar
Bomfim JAS, de Souza FP, Filgueiras CAL, de Sousa AG, Gambardella MTP. Diphosphine complexes of nickel: analogies in molecular structures and variety in crystalline arrangement. Polyhedron.
2003;22:1567–73.
CAS Google Scholar
Thomas AA, Zahrt AF, Delaney CP, Denmark SE. Elucidating the role of the boronic esters in the Suzuki–Miyaura reaction: structural, kinetic, and computational investigations. J Am Chem Soc.
2018;140:4401–16.
CAS PubMed PubMed Central Google Scholar
Thomas AA, Denmark SE. Pre-transmetalation intermediates in the Suzuki-Miyaura reaction revealed: the missing link. Science. 2016;352:329–32.
CAS PubMed Google Scholar
Lennox AJJ, Lloyd-Jones GC. Transmetalation in the Suzuki–Miyaura coupling: the fork in the trail. Angew Chem Int Ed. 2013;52:7362–70.
CAS Google Scholar
Carrow BP, Hartwig JF. Distinguishing between pathways for transmetalation in Suzuki-Miyaura reactions. J Am Chem Soc. 2011;133:2116–9.
CAS PubMed PubMed Central Google Scholar
Amatore C, Jutand A, Le Duc G. Kinetic data for the transmetalation/reductive elimination in palladium-catalyzed Suzuki-Miyaura reactions: unexpected triple role of hydroxide ions used as
base. Chem Eur J. 2011;17:2492–503.
CAS PubMed Google Scholar
Schmidt AF, Kurokhtina AA, Larina EV. Role of a base in Suzuki-Miyaura reaction. Russ J Gen Chem. 2011;81:1573.
CAS Google Scholar
Christian AH, Muller P, Monfette S. Nickel hydroxo complexes as intermediates in nickel-catalyzed Suzuki-Miyaura cross-coupling. Organometallics. 2014;33:2134–7.
CAS Google Scholar
Inada K, Miyaura N. Synthesis of biaryls via cross-coupling reaction of arylboronic acids with aryl chlorides catalyzed by NiCl2/triphenylphosphine complexes. Tetrahedron. 2000;56:8657–60.
CAS Google Scholar
Ueda M, Saitoh A, Oh-tani S, Miyaura N. Synthesis of biaryls via nickel-catalyzed cross-coupling reaction of arylboronic acids and aryl mesylates. Tetrahedron. 1998;54:13079–86.
CAS Google Scholar
Saito S, Oh-tani S, Miyaura N. Synthesis of biaryls via a nickel(0)-catalyzed cross-coupling reaction of chloroarenes with arylboronic acids. J Org Chem. 1997;62:8024–30.
CAS PubMed Google Scholar
Lanni EL, McNeil AJ. Evidence for ligand-dependent mechanistic changes in nickel-catalyzed chain-growth polymerizations. Macromolecules. 2010;43:8039–44.
CAS Google Scholar
Senkovskyy V, Sommer M, Tkachov R, Komber H, Huck WTS, Kiriy A. Convenient route to initiate Kumada catalyst-transfer polycondensation using Ni(dppe)Cl2 or Ni(dppp)Cl2 and sterically
hindered Grignard compounds. Macromolecules. 2010;43:10157–61.
CAS Google Scholar
Senkovskyy V, Tkachov R, Beryozkina T, Komber H, Oertel U, Horecha M, et al. “Hairy” poly(3-hexylthiophene) particles prepared via surface-initiated Kumada catalyst-transfer
polycondensation. J Am Chem Soc. 2009;131:16445–53.
CAS PubMed Google Scholar
Boyd SD, Jen AK-Y, Luscombe CK. Steric stabilization effects in nickel-catalyzed regioregular poly(3-hexylthiophene) synthesis. Macromolecules. 2009;42:9387–9.
CAS Google Scholar
Doubina N, Ho A, Jen AK-Y, Luscombe CK. Effect of initiators on the Kumada catalyst-transfer polycondensation reaction. Macromolecules. 2009;42:7670–7.
CAS Google Scholar
Sheina EE, Liu J, Iovu MC, Laird DW, McCullough RD. Chain growth mechanism for regioregular nickel-initiated cross-coupling polymerizations. Macromolecules. 2004;37:3526–8.
CAS Google Scholar
Saito S, Sakai M, Miyaura N. A synthesis of biaryls via nickel(0)-catalyzed cross-coupling reaction of chloroarenes with phenylboronic acids. Tetrahedron Lett. 1996;37:2993–6.
CAS Google Scholar
Zenkina O, Altman M, Leitus G, Shimon LJW, Cohen R, van der Boom ME. From azobenzene coordination to aryl-halide bond activation by platinum. Organometallics. 2007;26:4528–34.
CAS Google Scholar
Yokoyama A, Kato A, Miyakoshi R, Yokozawa T. Precision synthesis of poly (N-hexylpyrrole) and its diblock copolymer with poly(p-phenylene) via catalyst-transfer polycondensation.
Macromolecules. 2008;41:7271–3.
CAS Google Scholar
Hall AO, Lee SR, Bootsma AN, Bloom JWG, Wheeler SE, McNeil AJ. Reactive ligand influence on initiation in phenylene catalyst-transfer polymerization. J Polym Sci Part A Polym Chem.
2017;55:1530–5.
CAS Google Scholar
Lee JA, Luscombe CK. Dual-catalytic Ag-Pd system for direct arylation polymerization to synthesize poly(3-hexylthiophene). ACS Macro Lett. 2018;7:767–71.
CAS Google Scholar
Download references
AcknowledgementsK.J.T.N. is grateful to the NSF for a CAREER Award (CHE-1455136). The NMR instrumentation at Carnegie Mellon University is partially supported by the NSF (CHE-0130903, CHE-1039870, and
CHE-1726525). The authors would also like to thank both Prof. Tsutomu Yokozawa and Prof. Anne McNeil for helpful discussions.
Author informationAuthors and Affiliations Department of Chemistry, Carnegie Mellon University, 4400 Fifth Avenue, Pittsburgh, PA, 15213-2617, USA
Matthew A. Baker, Josué Ayuso-Carrillo, Martin R. M. Koos, Anthony J. Varni, Roberto R. Gil & Kevin J. T. Noonan
Department of Chemistry and Chemical Biology, Baker Laboratory, Cornell University, Ithaca, NY, 14853-1301, USA
Samantha N. MacMillan
AuthorsMatthew A. BakerView author publications You can also search for this author inPubMed Google Scholar
Josué Ayuso-CarrilloView author publications You can also search for this author inPubMed Google Scholar
Martin R. M. KoosView author publications You can also search for this author inPubMed Google Scholar
Samantha N. MacMillanView author publications You can also search for this author inPubMed Google Scholar
Anthony J. VarniView author publications You can also search for this author inPubMed Google Scholar
Roberto R. GilView author publications You can also search for this author inPubMed Google Scholar
Kevin J. T. NoonanView author publications You can also search for this author inPubMed Google Scholar
Corresponding author Correspondence to Kevin J. T. Noonan.
Ethics declarationsConflict of interestThe authors declare that they have no conflict of interest.
Additional informationPublisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary informationSupporting Information FileCartesian Coordinates For Computed Nickel ComplexesRights and permissions Reprints and permissions
About this articleCite this article Baker, M.A., Ayuso-Carrillo, J., Koos, M.R.M. et al. A robust nickel catalyst with an unsymmetrical propyl-bridged diphosphine ligand for
catalyst-transfer polymerization. Polym J 52, 83–92 (2020). https://doi.org/10.1038/s41428-019-0259-3
Download citation
Received: 07 June 2019
Revised: 09 July 2019
Accepted: 30 July 2019
Published: 03 September 2019
Issue Date: January 2020
DOI: https://doi.org/10.1038/s41428-019-0259-3
Share this article Anyone you share the following link with will be able to read this content:
Get shareable link Sorry, a shareable link is not currently available for this article.
Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative
This article is cited by An isolable, chelating bis[cyclic (alkyl)(amino)carbene] stabilizes a strongly bent, dicoordinate Ni(0) complex Braulio M. Puerta LombardiMorgan R. FaasRoland
Roesler Nature Communications (2024)