Play all audios:
ABSTRACT Myasthenia gravis (MG) is one of the most well characterised autoimmune disorders affecting the neuromuscular junction with autoantibodies targeting the acetylcholine receptor
(AChR) complex. The vast majority of patients present with ocular symptoms including double vision and ptosis, but may progress on to develop generalised fatiguable muscle weakness. Severe
involvement of the bulbar muscles can lead to dysphagia, dysarthria and breathing difficulties which can progress to myasthenic crisis needing ventilatory support. Given the predominant
ocular onset of the disease, it is important that ophthalmologists are aware of the differential diagnosis, investigations and management including evolving therapies. When the disease
remains localised to the extraocular muscles (ocular MG) IgG1 and IgG3 antibodies against the AChR (including clustered AChR) are present in nearly 50% of patients. In generalised MG this is
seen in nearly 90% patients. Other antibodies include those against muscle specific tyrosine kinase (MuSK) and lipoprotein receptor related protein 4 (LRP4). Even though decremental
response on repetitive nerve stimulation is the most well recognised neurophysiological abnormality, single fibre electromyogram (SFEMG) in experienced hands is the most sensitive test which
helps in the diagnosis. Initial treatment should be using cholinesterase inhibitors and then proceeding to immunosuppression using corticosteroids and steroid sparing drugs. Patients
requiring bulbar muscle support may need rescue therapies including plasma exchange and intravenous immunoglobulin (IVIg). Newer therapeutic targets include those against the B lymphocytes,
complement system, neonatal Fc receptors (FcRn) and various other elements of the immune system. 摘要 重症肌无力(MG)是影响神经肌肉接头的最典型的自身免疫性疾病之一, 自身抗体以乙酰胆碱受体(AChR)复合体为靶点。绝大多数患者的眼部症状包括复视和上睑下垂,
但可能会发展为全身性疲劳性肌无力。严重的延髓肌受累可导致吞咽困难、构音障碍和呼吸困难, 进而发展为需呼吸机支持的肌无力危象。鉴于该病以眼部起病为主, 眼科医生必须掌握鉴别诊断、调查和对其的管理, 以及不断更新的治疗方法。当病情仍局限于眼外肌(眼MG)时, 近50%的患者存在抗AChR(包括簇状AChR)的IgG1和IgG3抗体。在全身性MG中,
近90%的患者存在以上情况。其他抗体包括针对肌肉特异性酪氨酸激酶(Musk)和脂蛋白受体相关蛋白4(LRP4)的抗体。尽管对重复神经刺激的反应减弱是最常见的神经生理学异常, 但对有经验的医生来说, 单纤维肌电(SFEMG)是最敏感的有助于诊断的检查。最初的治疗应使用胆碱酯酶抑制剂, 随后使用皮质类固醇和类固醇保留药物进行免疫抑制治疗。需延髓肌支持的患者可能需抢救治疗,
包括血浆置换和静脉注射免疫球蛋白(IVIg)。新的治疗靶点包括针对B淋巴细胞、补体系统、新生儿Fc受体(FcRN)和免疫系统其它靶点的治疗。 SIMILAR CONTENT BEING VIEWED BY OTHERS DEMOGRAPHICS AND OCULAR FINDINGS IN CHILDREN WITH MYASTHENIA Article 25 March
2022 MYASTHENIA GRAVIS: THE CHANGING TREATMENT LANDSCAPE IN THE ERA OF MOLECULAR THERAPIES Article 08 January 2024 UNUSUAL PRESENTATIONS OF MYASTHENIA GRAVIS AND MISDIAGNOSIS Article Open
access 04 March 2025 INTRODUCTION Myasthenia gravis (MG) is the most common autoimmune disease affecting the neuromuscular junction (NMJ) clinically characterised by fatigable weakness of
the ocular, limb and bulbar (speech, swallowing and respiratory) muscles. The incidence of MG varies between 1.7 to 21.3 per million person-years. The estimated United Kingdom (UK)
prevalence of MG is 15 per 100,000 population [1, 2]. There is significant social, economic and emotional burden for patients with MG, which can often be more troublesome than the disabling
symptoms and hospitalisations [3]. Most patients with MG present with ocular symptoms (mainly ptosis and ophthalmoplegia) and in ~20% of patients the disease remain localised to the eye
muscles (ocular MG). In the remaining patients the disease can cause generalised symptoms which can range from being mild fatigue to those needing ventilatory support for respiratory crisis.
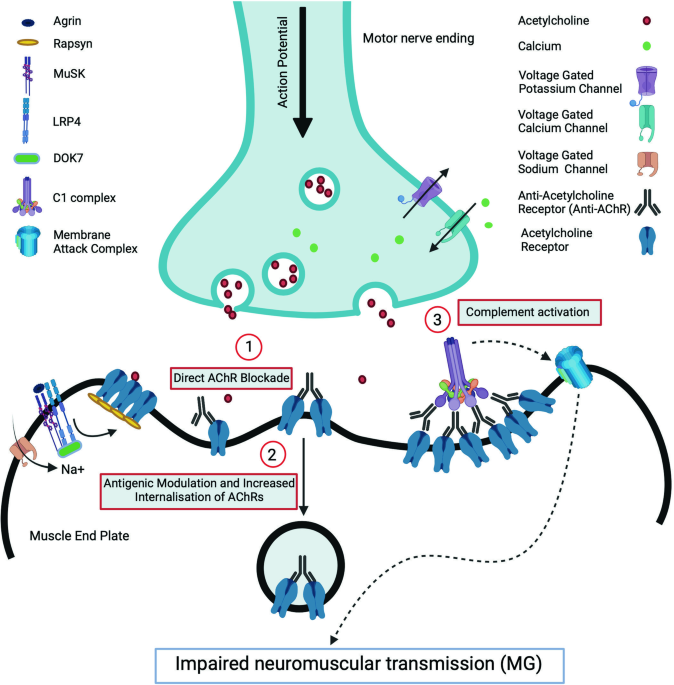
The most commonly described antibodies are against the acetylcholine receptor (AChR) or muscle specific tyrosine kinase (MuSK). PATHOPHYSIOLOGY AT THE NEUROMUSCULAR JUNCTION In the NMJ,
nerve action potential provokes the activation of presynaptic voltage gated calcium channels (VGCC) which initiates a cascade of events eventually causing the release of acetylcholine (ACh)
into the synaptic cleft. Acetylcholine binds to its receptor (AChR) in the synaptic folds of the postsynaptic membrane thereby opening the central pore of the receptors and subsequently the
voltage gated sodium channels (VGSC), which then initiate the muscle action potential [4]. Normal NMJ function requires clustering of AChR in the crests of the synaptic folds which is
achieved by a tyrosine kinase enzyme, MuSK. Activation of MuSK occurs during the development of NMJ by the interaction of agrin released from the developing motor axons with a post-synaptic
protein, lipoprotein receptor related protein 4 (LRP4). Binding of this complex is crucial for the dimerisation and activation of MuSK. MuSK activation induces a series of phosphorylation
reactions recruiting DOK7 and rapsyn and finally inducing clustering and stabilisation of AChR [5]. The postsynaptic neuromuscular dysfunction in MG is produced by IgG autoantibodies which
act by three main mechanisms: (1) direct functional blockade of the receptors, (2) antigenic modulation leading to receptor internalisation and degradation and (3) complement activation and
destruction of the membrane [6]. AChR antibodies are predominantly of the complement fixing IgG1 and IgG3 subtypes. Complement fixation and generation of the membrane attack complex result
in damage to AChR and voltage gated sodium channels in the post-synaptic membrane which reduces the safety factor and increases the threshold for excitation (Fig. 1). MuSK antibodies which
are the IgG4 isotype do not significantly activate the complement pathway. Both IgG4 MuSK and IgG1/G2 LRP4 antibodies reduces neuromuscular transmission by inhibiting the clustering of AChR
in the post-synaptic membrane [7]. ROLE OF THYMUS IN MG All patients with suspected MG should undergo imaging of the thymus gland. Combination of genetic susceptibility and environmental
triggers such as viral infections have been implicated in the origin of MG leading to intra-thymic changes which play a pivotal role in the pathogenesis of AChR-MG [8]. The most accepted
hypothesis centres around the failure of self-tolerance which occurs inside the thymus in AChR-MG. Self-tolerance is a balance between immune cell generation and the removal of auto-reactive
lymphocytes in a timely fashion. During the T-cell maturation and differentiation in the thymus, the T regulatory cells (Tregs) are exposed to thymic myoid cells which express AChR and
autoimmune regulatory (AIRE) medullary epithelial cells. The latter play an important role in the clonal deletion of T cells sensitised to auto-antigens during the development of central
tolerance. The lymphocytes which display auto-reactivity are removed within the thymus whereas those that escape this culling are suppressed in the periphery by the Treg cells [9]. For each
of the antibody subtypes of MG distinctive clinical phenotypes have been described [10]. There are three subtypes for AChR antibody positive MG: early onset, EOMG (<50 years of age), late
onset, LOMG (>50 years) and thymoma associated MG, TAMG. EOMG predominantly occurs in women and have strong associations with human leucocyte antigen (HLA) DR3-D8 and thymic follicular
hyperplasia. In contrast, LOMG often occurs in males, has no HLA association and may display anti-striational antibodies even though the thymus can be atrophic [11]. Up to 30% of MG have
associated thymoma and often have co-existing anti-ryanodine receptor and anti-titin antibodies [12]. MuSK-MG tends to present with a bulbar-dominant phenotype and thymus is usually normal
[13]. In EOMG, the germinal centres in the hyperplastic thymic follicles are implicated as the site of origin of autoimmunity. The mechanisms include aberrant production of cytokines and
imbalance in the function of T effector and Treg cells. There is increase in the pro-inflammatory Th17 and T follicular helper cells which promote B cell activation and generate
autoantibodies [9, 14]. In addition, the regulatory B cells which suppress autoimmunity are also functionally abnormal. In TAMG, pathological abnormalities consistent with immune
proliferation in the areas adjacent to the tumour have been reported without any follicular hyperplasia. Thymomas express loss of AIRE making the cells susceptible for mounting autoimmune
responses. LOMG on the other hand is associated with involuted thymus with paucity of the myoid cells and regulatory thymic cells which prevent autoimmunity. Thymus is usually normal and may
not play a pathogenetic role in MuSK-MG [11]. OCULAR MYASTHENIA GRAVIS Clinically, ptosis (droopy eyelids) may be unilateral or bilateral, is usually asymmetric, and patients often complain
of diplopia (double vision) [15]. Ocular MG typically has wide variation in its severity and patients may also complain of dizziness, unsteady gait or blurred vision [16]. On examination,
the less affected lid may be hyper-retracted in concordance with Hering’s law of equal innervation. One of the commonly observed signs, which is seen fairly commonly in ocular MG, is the
drooping of the contralateral lid with passive elevation of a ptotic lid. When the patient looks down for 15 s and then rapidly looks up, the ptotic eyelid overshoots and then slowly droops
to the previous ptotic position. This sign, named the Cogan’s lid twitch sign, is due to transient improvement of lid strength after resting of the levator palpebrae superioris muscle.
Another commonly used technique is the Ice pack test where ice is filled in a thin surgical glove and kept against the closed eyelid of a seated patient for 2–5 min. The sensitivity of a
5-min icepack application ranged from 76.9% (when assessing the changes in strabismus deviation) to 92.3% (testing degree of ptosis as quantified by the change in marginal reflex distance,
MRD) with a very high specificity (98.3%) [17]. The common bedside tests used to diagnose ocular myasthenia are shown in Box 1. Weakness of the extraocular muscles (ophthalmoparesis) in
combination with ptosis and normal pupillary responses is highly suggestive of ocular MG. The main differential diagnoses of ocular myasthenia are mentioned in Table 1. BOX 1 COMMON BED-SIDE
TESTS TO DIAGNOSE OCULAR MYASTHENIA * (1) Assess fatiguability Ask patient to hold an upgaze for 2–3 min and assess fatigue of the levator palpebare superioris, by assessing the change in
the interpalpebral fissure distance or the marginal reflex distance, MRD. * (2) Ice Pack test Although a commercial ice pack can be used, practically this is done by holding an ice-filled
thin surgical glove over the closed eye for 2 min. When assessing ophthalmoplegia, this may need to be kept over the eye for up to 10 min. A change in the interpalpebral distance or marginal
reflex distance of \(\ge\) 2 mm is considered to be a positive response. Improvement in ophthalmoplegia is considered significant when there is a change in prism dioptre (PD) by 50% or at
least 10 PD, when pre-test deviation is more than 20 PD. Test is unlikely to be positive in Horner’s syndrome and cranial nerve palsies and can be positive even in myasthenia which is
co-existent thyroid eye disease. * (3) Cogan’s lid twitch sign This is elicited when there is visible ptosis. Patient is asked to look down for 15 s followed by changing to primary gaze
(looking straight ahead) and observing a twitch in the droopy lid. * (4) Tensilon test Historically injection of short acting cholinesterase inhibitors like edrophonium was used to assess
the change in ptosis or ophthalmoplegia, but this is very rarely used nowadays due to safety concerns. WHY ARE OCULAR MUSCLES MORE SUSCEPTIBLE TO MG WEAKNESS? Several reasons have been
suggested for the predilection of extraocular muscles (EOM) in MG [18, 19]: * (1) EOM twitch fibres have higher firing frequencies, lower density of AChR and less number of packets of ACh
released, thereby increasing their susceptibility to fatigue. * (2) 20% of EOM contain multi-terminal fibres, which have sparse junctional folds, lower AChR density and do not have a safety
factor. * (3) There have been suggestions that antibodies in ocular MG patients’ serum react more strongly with ocular muscle antigens [20, 21], although further studies have refuted this
argument [22]. It is likely that the previous findings were due to the high proportion of normal adult AChRs in ocular muscles compared to fetal AChRs in denervated leg muscles. EOMs (but
probably not the levator muscles) have been thought to contain both the fetal (γ) and adult (ε) form of AChRs as opposed to the extremity muscles which contain exclusive adult isoforms [23].
Ocular MG patients’ serum reacts more strongly with adult than fetal AChRs [24]. * (4) EOM differentially express immune-related genes making them more susceptible to autoimmune diseases
[25]. * (5) Diminished complement regulatory activity could render the EOM more susceptible to autoimmune attack [26]. * (6) Lastly, but perhaps most importantly, minor weakness of EOM can
misalign the visual axis to produce symptoms. EOM may be disproportionately sensitive to the disease. Indeed, many ocular MG patients show subclinical neurophysiological defects in the
peripheral limb muscles. Previously, an animal model for ocular MG has been developed using mice transgenic for HLA-DQ8 and HLA-DR3 with deficient MHC-Class II after immunising with a
recombinant AChR α-subunit [27]. It is not clear why this model should develop ocular signs and careful comparisons with other models have not been done. These and other similar models could
prove a valuable tool in studying the pathophysiological mechanisms underlying ocular MG. ANTIBODIES IN MG Autoantibodies are seen in ~50% of patients with ocular MG [28] whereas nearly 95%
of generalised MG patients have antibodies against various components of the post-synaptic membrane [10]. 85% of antibodies are targeted against the AChR and antibodies against other parts
of the NMJ, namely MuSK, LRP4 and agrin have been reported in 5%, 2% and less than 1% respectively (Fig. 1). This leaves ~5–7% of patients where a specific antibody cannot be identified
using the currently available assays and these are referred to have seronegative MG [11]. As mentioned earlier, in the pure ocular MG the seronegative status can be closer to 50%. Antibodies
against several other molecules have been described including acetylcholinesterase, ColQ, titin, ryanodine, Kv1.4 and cortactin, but their exact pathophysiological role is unknown [29, 30].
NEUROPHYSIOLOGY IN MG The two most commonly employed techniques for the diagnosis of ocular MG are repetitive nerve stimulation (RNS) and single fibre EMG (SFEMG) studies. Although the
diagnostic yield of RNS is increased when the orbicularis oculi, orbicularis oris or nasalis muscles are tested, patients often find these examinations difficult to tolerate. Less than 50%
of ocular MG patients demonstrate decremental response as opposed to 75% of generalised MG patients [16]. SFEMG has been described as the most sensitive test to identify a defect in
neuromuscular transmission, especially when done by experienced neurophysiologists [31,32,33]. SFEMG has been found to be a reliable and sensitive technique for the diagnosis of
neuromuscular transmission failure [32, 34, 35]. In SFEMG, the action potentials from two adjacent muscle fibres arrive at the recording electrode at varying time intervals. This represents
the combined jitter at the two end-plates. The standard deviation of these variations can be used to express jitter. In order to assess the status of neuromuscular transmission, usually 20
potential pairs are examined [32]. Increased jitter is a sign of impaired neuromuscular transmission which can be sub-clinical (i.e. without apparent weakness) in the early stages, but can
lead to blockage of impulse transmission (also called jitter blocking), which is always associated with clinical weakness. In other words, a normal jitter in an apparently weak muscle (e.g.:
ptosis) will be highly unlikely to be due to myasthenia gravis or another neuromuscular transmission disorder. MANAGEMENT OF MYASTHENIA GRAVIS The therapeutic strategy in MG is guided by
the clinical pattern (ocular versus generalised), autoantibody subtype, and disease severity. Current therapy of MG revolves around symptomatic treatment (cholinesterase inhibitors), long
term steroids and steroid-sparing immunomodulation including thymectomy and rescue therapy in acute crisis. A summary of the various pharmacological therapies currently used in MG (including
usual dosing) is shown in Table 2. With the advent of newer therapies, myasthenia patients are often systematically assessed in clinic using standardised scoring systems which include
patient-reported outcomes like the Myasthenia Gravis Activities of Daily Living (MG-ADL) or Myasthenia Gravis Quality of Life (MG-QoL-15) scores. In addition, MG Composite, a combined
patient-reported outcome and physician examination score is also relatively easy to perform. All of these scores are usually done regularly in many specialist myasthenia clinics. More
detailed assessment can be done using Quantitative of Myasthenia Gravis (QMG) scale, which is more often done in a clinical trial setting along with the other scores. A detailed discussion
of these scores is outside the remit of this article. CHOLINESTERASE INHIBITORS Cholinesterase inhibitors such as pyridostigmine (usual dose 30–60 mg three to four times a day, titrating up
if needed up to 360 mg/day in divided doses) can produce rapid relief of symptoms in mild MG and are usually prescribed as the initial therapy. Patients may develop cholinergic side effects
like abdominal and limb muscle cramps, diarrhoea etc and these can be counteracted by drugs like Propantheline. Some patients with MuSK-MG may show a paradoxical worsening with
cholinesterase inhibitors. CORTICOSTEROIDS Majority of patients require suppression of autoantibody production with immunosuppressive treatment which remains the cornerstone of MG therapy.
Oral corticosteroids are initially used to induce remission while long-term maintenance is achieved with either low dose oral corticosteroids or non-steroidal immunosuppressants. Even though
highly effective, the beneficial effects have to be balanced with the morbidity associated with long term corticosteroid usage. Worsening of bulbar symptoms leading to myasthenic crisis can
occasionally be precipitated by high dose steroids, although is less likely to occur with gradual titration of the dose with close monitoring. STEROID SPARING IMMUNOSUPPRESSANTS
Azathioprine, mycophenolate mofetil, methotrexate, ciclosporin and tacrolimus are the most frequently used non-steroidal agents. Agents like cyclophosphamide are usually reserved for poorly
responsive patients. Azathioprine is probably the safest of these in women of childbearing age, but the onset of action is often delayed by several months (sometimes even up to 18 months).
It is essential that levels of Thiopurine Methyl Transferase (TPMT) are checked before starting Azathioprine since TPMT deficiency leads to accumulation of active 6-mercaptopurine and its
toxic metabolites, 6-thioguanine nucleotide analogues causing bone marrow suppression. For faster onset of action, many myasthenia specialists prefer Mycophenolate (especially where there is
no teratogenic risk) but even this does not become effective for at least 3–6 months. PLASMA EXCHANGE AND INTRAVENOUS IMMUNOGLOBULINS Rapidly acting, but short-lasting agents such as
therapeutic plasma exchange (PLEX) or intravenous immunoglobulin (IVIg) are used in patients with impending respiratory crisis and severe MG [36]. Even though there is limited randomised
control trial evidence, short term benefit of PLEX in myasthenic crisis has been shown in several case series [37]. Both PLEX and IVIg have rapid onset of action but are generally considered
unsuitable for long term maintenance therapy due to potential side effects [38, 39]. Nevertheless in treatment-refractory MG it is not unusual for patients to receive regular IVIg, PLEX or
immunoadsorption [40, 41]. Subcutaneously administered immunoglobulin (SCIg) is an attractive alternative to IVIg providing not only more persistent blood levels and fewer systemic adverse
effects, but also reducing health care resource utilisation, preserving patient autonomy and providing better quality of life [42, 43]. THYMECTOMY In all MG patients who have a thymoma or
those who are AChR antibody positive, therapeutic thymectomy is recommended. The beneficial effect of thymectomy in older patients and antibody negative MG is not fully established, and it
is generally recommended in onset of disease less than 50 years of age [36, 44, 45]. Practically however, it is often used until the age of 65 if the patients are otherwise fit and healthy.
Thymectomy is not recommended in MuSK-MG since there are no consistent thymic abnormalities in this sub-group [46]. RITUXIMAB Rituximab is a chimeric mouse/human antiCD20 mAb which rapidly
depletes the mature and memory B cells in the peripheral blood while largely sparing the pre-B cells and plasma cells located in the bone marrow and secondary lymphoid organs. This effect is
achieved by target cell apoptosis, antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity, with the effect lasting up to 6 months [47]. Rituximab spares the
long-lived plasma cells which do not express CD20 while it depletes the short-lived plasma cells and B regulatory cells (interleukin-10 producing cells) which have CD20 expression [48]. AChR
antibodies are produced by the long-lived plasma cells whereas short-living plasma blasts are thought to be the source of the MuSK antibodies [49]. This might be the reason that some
systematic reviews suggest lower corticosteroid requirements, better remission rates (47% vs. 16%) and minimal manifestation status (72% vs. 30%) in patients with MuSK-MG as opposed to
AChR-MG [50, 51]. The standard Rituximab regimen includes two infusions of 1 g 2 weeks apart (some clinicians use 375 mg/m2 weekly infusions for 4 weeks). Reinfusions are usually guided by
clinical symptoms or B cell repopulation [50]. Two systematic reviews showed unequivocal clinical improvement in 68–77% of AChR-MG patients [52, 53]. When routine and low dose Rituximab (171
and 89 subjects respectively) were compared in refractory AChR-MG no significant difference in the therapeutic outcomes or side effects were noted. Newer retrospective studies (albeit
without a clear comparator arm) have shown good efficacy and safety profile using low dose Rituximab regimens as monotherapy or add-on immunotherapy in new-onset and early MG [54,55,56].
When used early (the mean time of initiation since MG diagnosis of 1.6 months with a median dose of 183 mg) Rituximab has been shown to produce sustained clinical remission and also
discontinuation of steroids in nearly 80% of patients by 22 months [54, 56]. A recent double-blind placebo controlled trial used low dose Rituximab (single IV infusion of 500 mg) at
diagnosis and has shown efficacy in lowering the MG scores at week 16 needing low (<10 mg) Prednisolone dose without any rescue therapies (71% vs. 29%, _p_ = 0.007) [57]. Adverse effects
have been documented in 4 to 26% of patients, most commonly infusion reactions, reduced blood cell counts, opportunistic infections and prolonged hypogammaglobulinemia [50, 53]. ROLE OF
EARLY FAST ACTING THERAPIES IN MG The traditional treatment plan in MG used to be to start low and titrate up slowly. This can cause significant morbidity from mainly steroid-related side
effects affecting the quality of life. In recent years, increasing evidence is now gathering on the use of early and fast acting therapies in achieving minimal manifestation earlier and
reducing the dose and duration of steroid use. In a large study looking at 700 patients, half of the group were treated with intravenous steroids, PLEX, IVIg or a combination of these within
the first few days of diagnosis and there is significant (_p_ < 0.0001) difference in the proportion of patients achieving minimal manifestations (74.3% vs. 58.9%), disease duration (8.6
vs. 14.2 years), maximum prednisolone dose (20.2 vs. 25.0 mg) and duration of use of Prednisolone dose >10 mg (2.0 vs. 3.5 years) when compared with the more “conventional” regimens
[58]. NEWER IMMUNOTHERAPIES IN MG In the last few years, various novel treatment options are being studied in MG and these predominantly target various cells and immune pathways implicated
in the pathogenesis of MG (Fig. 2) [59]. Inhibition of complement pathway and neonatal Fc receptor (FcRn) are two of the most successful mechanisms identified among them. Inhibition of B and
T cells by direct depletion or through cytokines are also being studied. A few of the drugs, namely, Eculizumab, Ravulizumab, and Efgartigimod, have obtained approval from regulatory
agencies while the majority are in various phases of development [60, 61]. A summary of the various newer therapeutic options is shown in Table 3 (adapted from [59]). COMPLEMENT PATHWAY
INHIBITORS An overview of the complement system and the potential therapeutic targets are shown in Fig. 3. In the classical complement pathway, antigen-antibody complexes bind to the six
globular heads of C1q, activating C2 and C4 to form C3 convertase (C2a4b), which converts C3 to the active C3b. C3b cleaves C5 to its active form C5b, which in turn forms the membrane attack
complex (MAC) with C6, C7, C8 and C9. The MAC forms lytic pores on the cell surface leading to cell destruction and thereby neuromuscular transmission failure. There are various direct and
indirect evidence for the role of complement in the pathogenesis of MG: * (1) NMJs from myasthenia gravis patients and experimental models of myasthenia show deposition of C3 fragments and
membrane attack complex (MAC) [62]. * (2) Depletion of complement protects animals from experimental myasthenia [63]. * (3) Experimental myasthenia can be prevented by anti-complement
component C6 [64] or by soluble complement receptor 1 (sCR1), a complement inhibitor [65]. * (4) C5 gene influences the development of murine myasthenia gravis [66]. * (5) Mice lacking the
complement regulator gene, decay accelerating factor (DAF), are more susceptible to experimental myasthenia [67]. * (6) Knocking out two complement regulator genes, DAF and CD59, produces
severe respiratory weakness in an active immunisation model of AChR-myasthenia [68, 69]. Complement cascade can be disrupted by various molecules with the prime targets being C5, along with
components of C3 and C1 [70]. C5 inhibitors like Eculizumab, Ravulizumab, and Zilucoplan have shown to be the most beneficial in clinical trials while more studies with other molecules are
in the pipeline [71]. ECULIZUMAB Eculizumab is a humanised monoclonal antibody (mAb) directed against the C5 component of the complement pathway. Eculizumab prevents the C5 cleavage to C5a
and C5b and thus inhibits the C5a-induced chemotaxis of inflammatory cells and the formation of MAC. Eculizumab has been used in paroxysmal nocturnal haemoglobinuria (PNH) [72] and atypical
haemolytic uraemic syndrome (HUS) [73] and more recently for aquaporin IgG positive neuromyelitis optica spectrum disorder (NMOSD) [74]. Eculizumab has been found to be safe and effective in
MG in a placebo-controlled randomised clinical trial (REGAIN). Even though the study failed to attain significance for the primary endpoint (mean rank of 56.6 vs. 68.3, _p_ = 0.0698), the
pre-specified secondary outcomes (changes in QMG and MGQOL-15 scores and the responder analysis for MG-ADL and QMG) were significantly better in the Eculizumab group starting from week 1 and
sustained through week 26, and fewer patients in the active group needed rescue therapy [75]. In a post hoc analysis, 25% of refractory MG on Eculizumab had attained minimal manifestations
status at 26 weeks, which was double that of the placebo group [76]. The side effects were mild to moderate, including headache, upper respiratory infection and nasopharyngitis, with no
difference between the groups. No patients developed meningococcal infection. The open label extension (OLE) phase of the Eculizumab study showed a reduction of 75% in the episodes of
myasthenic worsening compared to the baseline and rapid improvements in all the myasthenia specific scores. No safety issues were reported [77]. At the end of the OLE, 84.7% showed
clinically meaningful improvement in MG-ADL scores (≥3 points) and 71.4% patients showed improvement in QMG scores (≥5 points). A significantly higher proportion of Eculizumab-treated
patients attained minimal symptom expression (defined as MG-ADL of 0–1 or MG-QoL15 score of 0–3) at week 26 of REGAIN [78]. In addition, Eculizumab has been shown to be beneficial in
patients who were previously receiving chronic IVIg [79], Rituximab [80] or were ventilator-dependent [81]. The clinical trials, various subgroup studies and case series have clearly
established the role of Eculizumab as a rescue therapy in refractory MG, but its role as a first-line agent and duration of therapy are undefined. Even though it was licensed to use in MG in
2019, the annual cost of therapy which exceeds half a million US dollars has been a major deterrent to the wider use of this drug [82, 83]. RAVULIZUMAB Ravulizumab is a long-acting C5
complement inhibitor with a mechanism of action similar to eculizumab, which only needs fewer infusions. In the phase 3 randomised placebo-controlled CHAMPION-MG study (NCT03920293), 175
adults with symptomatic AChR antibody positive gMG were recruited to receive ravulizumab infusion versus placebo. The primary efficacy endpoint of significant improvement in MG-ADL and for
the secondary outcomes were achieved in the treatment group at 26 weeks, without any significant side effects [84]. ZILUCOPLAN Zilucoplan is a small macrocyclic peptide molecule which
prevents the terminal activation of the complement cascade by binding to C5 complement component. It also binds to the existing C5b to prevent its attachment to C6. The advantages of this
molecule include its small size which ensures good NMJ penetration, ability to concomitantly administer IVIg therapy since this is not an antibody and the potential for self-administration
(given the once daily subcutaneous route of delivery) [85]. POZELIMAB WITH CEMDISIRAN Pozelimab is a human mAb which acts on the C5 complement whereas Cemdisiran is a small synthetic
interfering ribonucleic acid (siRNA) which can suppresses the production of C5 in the liver. Both molecules are given as subcutaneous injections and are generally safe and well-tolerated at
different doses. In animal studies, combination of Pozelimab with Cemdisiran allowed lower doses and decreased dosing frequency compared to use of the individual agents separately [86]. The
phase 3 randomised controlled trial of the combination (intravenous Pozelimab loading followed by 4-weekly subcutaneous injections along with Cemdisiran 200 mg subcutaneous 4-weekly) versus
placebo in gMG is ongoing (NCT05070858). FCRN INHIBITORS Neonatal Fc receptors (FcRn) are widely distributed in various cells, particularly endothelial and myeloid cells. FcRn helps in the
recycling of IgG and increases its longevity in circulation. The binding of FcRn to IgG is pH dependent and occurs only in an acidic pH. After the uptake of IgG by the cells, it is
transported to the acidic environment of endosomes where FcRn binds to IgG and prevents it from degraded by the lysosomal enzymes. FcRn transports IgG back to the surface and releases it in
the neutral physiological pH [87]. This pathway prolongs the half-life of IgG antibodies which typically remain in circulation nearly 4 times longer than the other immunoglobulin subtypes
like IgM and IgA. Serum albumin shares the long half-life and FcRn-mediated recycling by binding to a site distinct from that of IgG [88]. Serum proteins and IgG molecules which do not bind
to FcRn are degraded within the lysosomes [89]. Blocking of FcRn can interfere with recycling of IgG antibodies associated with MG and many authors consider this technique similar to a
“medical plasma exchange”, given the rapid reduction in IgG levels. While reducing the autoreactive IgG antibodies, these agents keep the concentrations of IgM and IgA antibodies stable
[90]. A proportion (~25%) of the normal IgG response is retained and recovery of IgG levels occur faster compared to B cell depleting therapies [91]. Hence these therapies mount an effective
immune response without increasing the risk of infections. The various molecules studied in MG include engineered Fc fragments (Efgartigimod), mAb against FcRn (Rozanolixizumab,
Nipocalimab, Batoclimab, and Orilanolimab), and peptide fragments. EFGARTIGIMOD Efgartigimod is an engineered Fc domain of human IgG1 with increased affinity for FcRn receptors than the
endogenous IgG, and thus competitively inhibits IgG recycling [89]. The phase 3 multicentric randomised controlled ADAPT trial of efgartigimod studied 167 patients and proved its efficacy
against placebo as an add-on therapy for generalised MG. Compared to placebo, Efgartigimod treated AChR-MG had significant improvement in MG-ADL score at 4 weeks (primary outcome). The other
efficacy scores (QMG, MGC and MGQoL15 revised) showed a similar pattern with the maximum improvement noted by 4–5 weeks and sustained for 7 weeks. In total, 40–70% reduction in total IgG
and AChR antibody levels were noted within the first week of the initial dose, recovering by 12 weeks and the levels corresponded inversely with clinical improvement. The drug was tolerated
well and adverse effects including serious ones were no more common than in placebo [92]. NIPOCALIMAB Nipocalimab is a fully human aglycosylated IgG1 mAb against FcRn which strongly binds to
FcRn independent of pH. In the phase 2 study (NCT03772587), 68 patients with gMG were found to have dose dependent reduction in IgG and anti-AChR antibody levels which correlated with
clinical improvement [93]. Results are awaited from the ongoing phase 3 randomised controlled study in adults (NCT04951622) and phase 2/3 study open-label study in children (NCT05265273).
ROZANOLIXIZUMAB Rozanolixizumab is a high affinity humanised IgG4 mAb against FcRn which is administered subcutaneously. This drug completed phase 2 trial in 43 patients with moderate to
severe gMG who were positive for either of AChR or MuSK antibodies. The primary efficacy outcome of improvement in QMG score from day 1 to 29 was not significant but overall data including
antibody measurements suggested potential efficacy for the drug in moderate to severe gMG, without any significant side effects [94]. The phase 3 study (NCT03971422) is ongoing. B CELL AND
PLASMA CELL DEPLETING THERAPIES In the presence of follicular dendritic cells, pro-inflammatory T helper cells and cytokines, there is proliferation of the autoreactive B cells within the
thymic germinal centres and in the periphery, driving the pathogenesis of MG. The autoreactive B cells differentiate into antibody secreting plasma cells [95, 96]. In MG the B cells can be
directly depleted or inhibited or indirectly by targeting their facilitators like cytokines or other immune cells [97]. Rituximab, a chimeric mouse/human antiCD20 mAb is already a
well-established treatment in MG, especially with MuSK antibodies (produced by short-living plasma cells) and has been described earlier. INEBILIZUMAB Inebilizumab is a humanised mAb
targeting CD19 which is expressed in a broader group of B lineage cells compared to CD20, especially on the early pro-B cells and the majority of plasma cells in blood and secondary lymphoid
organs and about half of the plasma cells in the bone marrow [98]. Antibody dependent cell mediated cytotoxicity is the primary mechanism of action of Inebilizumab which is currently
undergoing phase 3 clinical studies in MG (NCT04524273). BORTEZOMIB Bortezomib, targets plasma cells by a potent and reversible proteasome inhibition. Proteasomes are ubiquitous
intracellular protein complexes and play a central role in protein homoeostasis by regulating the cell turnover and mediating the degradation of pro-apoptotic factors. As plasma cells are
terminally differentiated and non-dividing, they are usually resistant to radiotherapy, glucocorticoids, standard oral immunosuppressants, and CD19/20 inhibitors. The rapid intracellular
synthesis of antibodies in these cells render them highly susceptible to mechanisms which deter the protein degradation pathways like proteasome inhibitors. In addition, Bortezomib also
targets nuclear factor κB (NF-κB) signalling pathway which has important anti-apoptotic functions and is often upregulated in inflammatory diseases. Bortezomib, though primarily used for
myeloma and mantle cell lymphoma, is a potential therapeutic option in a variety of refractory autoimmune diseases including MG, although the trial was terminated due to poor recruitment
[99]. There are experimental data and case reports suggesting potential benefits in MG, but its use is limited by dose-dependent neurotoxicity in the form of polyneuropathy. The propensity
for neuropathy is diminished by restricting the dose to a single cycle of bortezomib which appears sufficient to induce remission in autoimmune diseases and by using subcutaneous rather than
intravenous route of administration [100]. TOLEBRUTINIB Bruton’s Tyrosine Kinase (BTK) belongs to the protein kinase family and is important in the signalling pathways involved in B cell
proliferation and function including production of antibodies and cytokines, and antigen presentation. It is also expressed in myeloid cells (components of innate immunity) and influences
the production of inflammatory cytokines and adhesion molecules which promote inflammation [101]. The suppression of these functions by BTK inhibitors like Tolebrutinib, results in their
efficacy in autoimmune diseases and B cell malignancies. This orally administered irreversible covalent BTK inhibitor, is currently being evaluated in a phase 3 placebo-controlled randomised
trial in MG (NCT05132569). CYTOKINE AND CHEMOKINE TARGETING THERAPIES Cytokines are small signalling molecules which help in the coordination of immune system function and communication
between the various immune and inflammatory cells. These are secreted by stimulated cells (chiefly T helper cells and macrophages) and act on a variety of target cells which harbour their
specific receptors. Chemokines are a specific subgroup of cytokines with chemoattractant properties that attract leucocytes to sites of inflammation.94 The targeting of various cytokines
have been attempted or is currently underway in MG, including Belimumab a human recombinant neutralising mAb against the B-cell activating factor (BAFF) [102, 103], Iscalimab a fully human
non-depleting antiCD40 mAb which targets the activation and signalling pathways mediated by CD40 (which is a co-stimulatory molecule expressed in B cells and other antigen presenting cells
which interacts with its ligand CD154 (CD40L) located on activated T cells, essential for the T cell dependent antibody responses, and the differentiation, survival and activation of memory
B cells) [104] and IL-6 receptor antagonists like Tocilizumab and Satralizumab [105, 106]. CHIMERIC AUTOANTIBODY RECEPTOR (CAAR) T CELL THERAPY Chimeric antigen receptor (CAR) T cell therapy
has shown marked success in the treatment of B cell malignancies and involves harvesting of T cells from the patient and genetically modifying them by attaching an artificially engineered
receptor referred to as chimeric antigen receptor (CAR). When the CAR T cells are infused into the circulation, the modified receptors bind to a specific antigen on the target cells (e.g.,
cancerous cells in B cell malignancies) and destroy them. The effect of CAR T cells is sustained long-term by their in vivo multiplication [107]. This can be modified to be used in
autoimmune diseases where a specific pathogenic antibody is identified, by using an engineered T cell which can bind the specific autoreactive B-cell. This technique is referred to as
Chimeric autoantibody receptor (CAAR) T cell therapy and by specifically targeting B cells which express the autoantibody, a general depletion of the B cell lineage cells is avoided. The
ongoing studies in MG include engineered T cells directed against B cell Maturation Antigen (BCMA) (NCT04146051) and MuSK antibody (NCT05451212). STEM CELL TRANSPLANTATION Autologous
hematopoietic stem cell transplantation (HSCT) is being increasingly used for eliminating disease activity in various autoimmune neurological conditions including MG. The procedure involves
stimulating the production of hematopoietic cells, harvesting them from circulation, ablating and resetting the immune system and then re-infusing the treated cells from the patient. The
evidence for HSCT in MG is limited to a handful of patients [108]. The high risk for debilitating side effects including infections, secondary autoimmune diseases, and neoplasms makes HSCT a
harder choice among the various treatment options, but nevertheless may need to be considered in selected refractory patients. CONCLUSIONS A good proportion of patients with MG will present
to the eye clinic with ptosis and double vision. It is important to be aware of the differential diagnosis and the bedside tests as well as neurophysiological and immunological tests which
will aid the diagnosis. A stepwise approach to diagnosis and treatment with clinical clues to looks out for impending myasthenic crisis is shown in Fig. 4. Traditional treatments with
cholinesterase inhibitors, corticosteroids and steroid sparing immunosuppressants are still the mainstay in the management of vast majority of patients with MG. However, ophthalmologists
would need to be aware of the vast array of newer treatments which are being used in MG and where possible, work in collaboration with specialist neuroimmunology clinics. REFERENCES * Carr
AS, Cardwell CR, McCarron PO, McConville J. A systematic review of population based epidemiological studies in myasthenia gravis. BMC Neurol. 2010;10:46. Article PubMed PubMed Central
Google Scholar * Robertson NP, Deans J, Compston DA. Myasthenia gravis: a population based epidemiological study in Cambridgeshire, England. J Neurol Neurosurg Psychiatry. 1998;65:492–6.
Article CAS PubMed PubMed Central Google Scholar * Lehnerer S, Jacobi J, Schilling R, Grittner U, Marbin D, Gerischer L, et al. Burden of disease in myasthenia gravis: taking the
patient’s perspective. J Neurol. 2022;269:3050–63. Article PubMed Google Scholar * Rodriguez Cruz PM, Cossins J, Beeson D, Vincent A. The neuromuscular junction in health and disease:
molecular mechanisms governing synaptic formation and homeostasis. Front Mol Neurosci. 2020;13:610964. Article PubMed PubMed Central Google Scholar * Borges LS, Richman DP.
Muscle-specific kinase myasthenia gravis. Front Immunol. 2020;11:707. Article CAS PubMed PubMed Central Google Scholar * Phillips WD, Vincent A. Pathogenesis of myasthenia gravis:
update on disease types, models, and mechanisms. F1000Res. 2016;5:F1000 Faculty Rev-1513. Article PubMed PubMed Central Google Scholar * Huda R. New approaches to targeting B cells for
myasthenia gravis therapy. Front Immunol. 2020;11:240. Article CAS PubMed PubMed Central Google Scholar * Levinson AI, Song D, Gaulton G, Zheng Y. The intrathymic pathogenesis of
myasthenia gravis. Clin Dev Immunol. 2004;11:215–20. PubMed PubMed Central Google Scholar * Alahgholi-Hajibehzad M, Kasapoglu P, Jafari R, Rezaei N. The role of T regulatory cells in
immunopathogenesis of myasthenia gravis: implications for therapeutics. Expert Rev Clin Immunol. 2015;11:859–70. Article CAS PubMed Google Scholar * Gilhus NE, Verschuuren JJ. Myasthenia
gravis: subgroup classification and therapeutic strategies. Lancet Neurol. 2015;14:1023–36. Article CAS PubMed Google Scholar * Huijbers MG, Marx A, Plomp JJ, Le Panse R, Phillips WD.
Advances in the understanding of disease mechanisms of autoimmune neuromuscular junction disorders. Lancet Neurol. 2022;21:163–75. Article CAS PubMed Google Scholar * Romi F. Thymoma in
myasthenia gravis: from diagnosis to treatment. Autoimmune Dis. 2011;2011:474512. PubMed PubMed Central Google Scholar * Guptill JT, Sanders DB, Evoli A. Anti-MuSK antibody myasthenia
gravis: clinical findings and response to treatment in two large cohorts. Muscle Nerve. 2011;44:36–40. Article PubMed Google Scholar * Leite MI, Jones M, Strobel P, Marx A, Gold R, Niks
E, et al. Myasthenia gravis thymus: complement vulnerability of epithelial and myoid cells, complement attack on them, and correlations with autoantibody status. Am J Pathol.
2007;171:893–905. Article PubMed PubMed Central Google Scholar * Elrod RD, Weinberg DA. Ocular myasthenia gravis. Ophthalmol Clin North Am. 2004;17:275–309. Article PubMed Google
Scholar * Kusner LL, Puwanant A, Kaminski HJ. Ocular myasthenia: diagnosis, treatment, and pathogenesis. Neurologist. 2006;12:231–9. Article PubMed Google Scholar * Chatzistefanou KI,
Kouris T, Iliakis E, Piaditis G, Tagaris G, Katsikeris N, et al. The ice pack test in the differential diagnosis of myasthenic diplopia. Ophthalmology. 2009;116:2236–43. Article PubMed
Google Scholar * Kaminski HJ, Maas E, Spiegel P, Ruff RL. Why are eye muscles frequently involved in myasthenia gravis? Neurology. 1990;40:1663–9. Article CAS PubMed Google Scholar *
Porter JD, Baker RS. Muscles of a different ‘color’: the unusual properties of the extraocular muscles may predispose or protect them in neurogenic and myogenic disease. Neurology.
1996;46:30–7. Article CAS PubMed Google Scholar * Tindall RS. Humoral immunity in myasthenia gravis: clinical correlations of anti-receptor antibody avidity and titer. Ann N Y Acad Sci.
1981;377:316–31. Article CAS PubMed Google Scholar * Vincent A, Newsom Davis J. Anti-acetylcholine receptor antibodies. J Neurol Neurosurg Psychiatry. 1980;43:590–600. Article CAS
PubMed PubMed Central Google Scholar * Hayashi M, Kida K, Yamada I, Matsuda H, Tsuneishi M, Tamura O. Differences between ocular and generalized myasthenia gravis: binding characteristics
of anti-acetylcholine receptor antibody against bovine muscles. J Neuroimmunol. 1989;21:227–33. Article CAS PubMed Google Scholar * Kaminski HJ, Kusner LL, Nash KV, Ruff RL. The
gamma-subunit of the acetylcholine receptor is not expressed in the levator palpebrae superioris. Neurology. 1995;45:516–8. Article CAS PubMed Google Scholar * MacLennan C, Beeson D,
Buijs AM, Vincent A, Newsom-Davis J. Acetylcholine receptor expression in human extraocular muscles and their susceptibility to myasthenia gravis. Ann Neurol. 1997;41:423–31. Article CAS
PubMed Google Scholar * Porter JD, Khanna S, Kaminski HJ, Rao JS, Merriam AP, Richmonds CR, et al. Extraocular muscle is defined by a fundamentally distinct gene expression profile. Proc
Natl Acad Sci USA. 2001;98:12062–7. Article CAS PubMed PubMed Central Google Scholar * Kaminski HJ, Li Z, Richmonds C, Lin F, Medof ME. Complement regulators in extraocular muscle and
experimental autoimmune myasthenia gravis. Exp Neurol. 2004;189:333–42. Article CAS PubMed Google Scholar * Yang H, Wu B, Tuzun E, Saini SS, Li J, Allman W, et al. A new mouse model of
autoimmune ocular myasthenia gravis. Invest Ophthalmol Vis Sci. 2007;48:5101–11. Article PubMed Google Scholar * Evoli A, Iorio R. Controversies in ocular myasthenia gravis. Front Neurol.
2020;11:605902. Article PubMed PubMed Central Google Scholar * Takamori M. Myasthenia gravis: from the viewpoint of pathogenicity focusing on acetylcholine receptor clustering,
trans-synaptic homeostasis and synaptic stability. Front Mol Neurosci. 2020;13:86. Article CAS PubMed PubMed Central Google Scholar * Koneczny I, Herbst R. Myasthenia gravis: pathogenic
effects of autoantibodies on neuromuscular architecture. Cells. 2019;8:671. Article CAS PubMed PubMed Central Google Scholar * Sanders DB, Stalberg EV. AAEM minimonograph #25:
single-fiber electromyography. Muscle Nerve. 1996;19:1069–83. Article CAS PubMed Google Scholar * Stalberg E, Trontelj JV. The study of normal and abnormal neuromuscular transmission
with single fibre electromyography. J Neurosci Methods. 1997;74:145–54. Article CAS PubMed Google Scholar * Ukachoke C, Ashby P, Basinski A, Sharpe JA. Usefulness of single fiber EMG for
distinguishing neuromuscular from other causes of ocular muscle weakness. Can J Neurol Sci. 1994;21:125–8. Article CAS PubMed Google Scholar * Trontelj JV, Stalberg E. Single motor
end-plates in myasthenia gravis and LEMS at different firing rates. Muscle Nerve. 1991;14:226–32. Article CAS PubMed Google Scholar * Trontelj JV, Stalberg EV. Multiple innervation of
muscle fibers in myasthenia gravis. Muscle Nerve. 1995;18:224–8. Article CAS PubMed Google Scholar * Narayanaswami P, Sanders DB, Wolfe G, Benatar M, Cea G, Evoli A, et al. International
consensus guidance for management of myasthenia gravis: 2020 update. Neurology. 2021;96:114–22. Article PubMed PubMed Central Google Scholar * Gajdos P, Chevret S, Toyka K. Plasma
exchange for myasthenia gravis. Cochrane Database Syst Rev. 2002;2002:CD002275. PubMed PubMed Central Google Scholar * Bershad EM, Feen ES, Suarez JI. Myasthenia gravis crisis. South Med
J. 2008;101:63–9. Article PubMed Google Scholar * Sanders DB, Wolfe GI, Benatar M, Evoli A, Gilhus NE, Illa I, et al. International consensus guidance for management of myasthenia gravis:
executive summary. Neurology. 2016;87:419–25. Article PubMed PubMed Central Google Scholar * Hoffmann S, Meisel A. Strategies in the treatment of refractory myasthenia gravis. Neurol
Int Open. 2018;O2:E56–9. Google Scholar * Mantegazza R, Antozzi C. When myasthenia gravis is deemed refractory: clinical signposts and treatment strategies. Ther Adv Neurol Disord.
2018;11:1756285617749134. Article PubMed PubMed Central Google Scholar * Abolhassani H, Sadaghiani MS, Aghamohammadi A, Ochs HD, Rezaei N. Home-based subcutaneous immunoglobulin versus
hospital-based intravenous immunoglobulin in treatment of primary antibody deficiencies: systematic review and meta analysis. J Clin Immunol. 2012;32:1180–92. Article CAS PubMed Google
Scholar * Hadden RD, Marreno F. Switch from intravenous to subcutaneous immunoglobulin in CIDP and MMN: improved tolerability and patient satisfaction. Ther Adv Neurol Disord. 2015;8:14–9.
Article PubMed PubMed Central Google Scholar * Wolfe GI, Kaminski HJ, Aban IB, Minisman G, Kuo HC, Marx A, et al. Randomized trial of thymectomy in myasthenia gravis. N Engl J Med.
2016;375:511–22. Article PubMed PubMed Central Google Scholar * Wolfe GI, Kaminski HJ, Aban IB, Minisman G, Kuo HC, Marx A, et al. Long-term effect of thymectomy plus prednisone versus
prednisone alone in patients with non-thymomatous myasthenia gravis: 2-year extension of the MGTX randomised trial. Lancet Neurol. 2019;18:259–68. Article CAS PubMed PubMed Central
Google Scholar * Clifford KM, Hobson-Webb LD, Benatar M, Burns TM, Barnett C, Silvestri NJ, et al. Thymectomy may not be associated with clinical improvement in MuSK myasthenia gravis.
Muscle Nerve. 2019;59:404–10. Article CAS PubMed Google Scholar * Stathopoulos P, Dalakas MC. Evolution of anti-B cell therapeutics in autoimmune neurological diseases.
Neurotherapeutics. 2022;19:691–710. Article CAS PubMed PubMed Central Google Scholar * Zografou C, Vakrakou AG, Stathopoulos P. Short- and long-lived autoantibody-secreting cells in
autoimmune neurological disorders. Front Immunol. 2021;12:686466. Article CAS PubMed PubMed Central Google Scholar * Fichtner ML, Jiang R, Bourke A, Nowak RJ, O’Connor KC. Autoimmune
pathology in myasthenia gravis disease subtypes is governed by divergent mechanisms of immunopathology. Front Immunol. 2020;11:776. Article CAS PubMed PubMed Central Google Scholar *
Tandan R, Hehir MK 2nd, Waheed W, Howard DB. Rituximab treatment of myasthenia gravis: a systematic review. Muscle Nerve. 2017;56:185–96. Article CAS PubMed Google Scholar * Hehir MK,
Hobson-Webb LD, Benatar M, Barnett C, Silvestri NJ, Howard JF Jr, et al. Rituximab as treatment for anti-MuSK myasthenia gravis: multicenter blinded prospective review. Neurology.
2017;89:1069–77. Article CAS PubMed Google Scholar * Di Stefano V, Lupica A, Rispoli MG, Di Muzio A, Brighina F, Rodolico C. Rituximab in AChR subtype of myasthenia gravis: systematic
review. J Neurol Neurosurg Psychiatry. 2020;91:392–5. Article PubMed Google Scholar * Li T, Zhang GQ, Li Y, Dong SA, Wang N, Yi M, et al. Efficacy and safety of different dosages of
rituximab for refractory generalized AChR myasthenia gravis: a meta-analysis. J Clin Neurosci. 2021;85:6–12. Article CAS PubMed Google Scholar * Du Y, Li C, Hao YF, Zhao C, Yan Q, Yao D,
et al. Individualized regimen of low-dose rituximab monotherapy for new-onset AChR-positive generalized myasthenia gravis. J Neurol. 2022;269:4229–40. Article CAS PubMed Google Scholar
* Brauner S, Eriksson-Dufva A, Hietala MA, Frisell T, Press R, Piehl F. Comparison between rituximab treatment for new-onset generalized myasthenia gravis and refractory generalized
myasthenia gravis. JAMA Neurol. 2020;77:974–81. Article PubMed Google Scholar * Li H, Huang Z, Jia D, Xue H, Pan J, Zhang M, et al. Low-dose rituximab treatment for new-onset generalized
myasthenia gravis. J Neuroimmunol. 2021;354:577528. Article CAS PubMed Google Scholar * Piehl F, Eriksson-Dufva A, Budzianowska A, Feresiadou A, Hansson W, Hietala MA, et al. Efficacy
and safety of rituximab for new-onset generalized myasthenia gravis: the RINOMAX randomized clinical trial. JAMA Neurol. 2022;79:1105–12. Article PubMed PubMed Central Google Scholar *
Uzawa A, Suzuki S, Kuwabara S, Akamine H, Onishi Y, Yasuda M, et al. Effectiveness of early cycles of fast-acting treatment in generalised myasthenia gravis. J Neurol Neurosurg Psychiatry.
2023;94:467–73. Article PubMed Google Scholar * Nair SS, Jacob S. Novel immunotherapies for myasthenia gravis. Immunotargets Ther. 2023;12:25–45. Article CAS PubMed PubMed Central
Google Scholar * Menon D, Barnett C, Bril V. Novel treatments in myasthenia gravis. Front Neurol. 2020;11:538. Article PubMed PubMed Central Google Scholar * Schneider-Gold C, Gilhus
NE. Advances and challenges in the treatment of myasthenia gravis. Ther Adv Neurol Disord. 2021;14:17562864211065406. Article CAS PubMed PubMed Central Google Scholar * Sahashi K, Engel
AG, Lambert EH, Howard FM Jr. Ultrastructural localization of the terminal and lytic ninth complement component (C9) at the motor end-plate in myasthenia gravis. J Neuropathol Exp Neurol.
1980;39:160–72. Article CAS PubMed Google Scholar * Lennon VA, Seybold ME, Lindstrom JM, Cochrane C, Ulevitch R. Role of complement in the pathogenesis of experimental autoimmune
myasthenia gravis. J Exp Med. 1978;147:973–83. Article CAS PubMed Google Scholar * Biesecker G, Gomez CM. Inhibition of acute passive transfer experimental autoimmune myasthenia gravis
with Fab antibody to complement C6. J Immunol. 1989;142:2654–9. Article CAS PubMed Google Scholar * Piddlesden SJ, Jiang S, Levin JL, Vincent A, Morgan BP. Soluble complement receptor 1
(sCR1) protects against experimental autoimmune myasthenia gravis. J Neuroimmunol. 1996;71:173–7. Article CAS PubMed Google Scholar * Christadoss P. C5 gene influences the development of
murine myasthenia gravis. J Immunol. 1988;140:2589–92. Article CAS PubMed Google Scholar * Lin F, Kaminski HJ, Conti-Fine BM, Wang W, Richmonds C, Medof ME. Markedly enhanced
susceptibility to experimental autoimmune myasthenia gravis in the absence of decay-accelerating factor protection. J Clin Invest. 2002;110:1269–74. Article CAS PubMed PubMed Central
Google Scholar * Kaminski HJ, Kusner LL, Richmonds C, Medof ME, Lin F. Deficiency of decay accelerating factor and CD59 leads to crisis in experimental myasthenia. Exp Neurol.
2006;202:287–93. Article CAS PubMed Google Scholar * Morgan BP, Chamberlain-Banoub J, Neal JW, Song W, Mizuno M, Harris CL. The membrane attack pathway of complement drives pathology in
passively induced experimental autoimmune myasthenia gravis in mice. Clin Exp Immunol. 2006;146:294–302. Article CAS PubMed PubMed Central Google Scholar * Dalakas MC. Role of
complement, anti-complement therapeutics, and other targeted immunotherapies in myasthenia gravis. Expert Rev Clin Immunol. 2022;18:691–701. Article CAS PubMed Google Scholar * San PP,
Jacob S. Role of complement in myasthenia gravis. Front Neurol. 2023;14:1277596. Article PubMed PubMed Central Google Scholar * Zhou S, Dong X, Chen C, Ma L, Wu Y, Zhou Y, et al.
Efficacy and safety of eculizumab for paroxysmal nocturnal hemoglobinuria: a systematic review and meta-analysis. J Pediatr Hematol Oncol. 2021;43:203–10. Article CAS PubMed Google
Scholar * Mahat U, Matar RB, Rotz SJ. Use of complement monoclonal antibody eculizumab in Shiga toxin producing Escherichia coli associated hemolytic uremic syndrome: a review of current
evidence. Pediatr Blood Cancer. 2019;66:e27913. Article PubMed Google Scholar * Levy M, Fujihara K, Palace J. New therapies for neuromyelitis optica spectrum disorder. Lancet Neurol.
2021;20:60–7. Article CAS PubMed Google Scholar * Howard JF Jr, Utsugisawa K, Benatar M, Murai H, Barohn RJ, Illa I, et al. Safety and efficacy of eculizumab in anti-acetylcholine
receptor antibody-positive refractory generalised myasthenia gravis (REGAIN): a phase 3, randomised, double-blind, placebo-controlled, multicentre study. Lancet Neurol. 2017;16:976–86.
Article CAS PubMed Google Scholar * Mantegazza R, Wolfe GI, Muppidi S, Wiendl H, Fujita KP, O’Brien FL, et al. Post-intervention status in patients with refractory myasthenia gravis
treated with eculizumab during REGAIN and its open-label extension. Neurology. 2021;96:e610–8. Article CAS PubMed PubMed Central Google Scholar * Muppidi S, Utsugisawa K, Benatar M,
Murai H, Barohn RJ, Illa I, et al. Long-term safety and efficacy of eculizumab in generalized myasthenia gravis. Muscle Nerve. 2019;60:14–24. Article CAS PubMed PubMed Central Google
Scholar * Vissing J, Jacob S, Fujita KP, O’Brien F, Howard JF, REGAIN Study Group. ‘Minimal symptom expression’ in patients with acetylcholine receptor antibody-positive refractory
generalized myasthenia gravis treated with eculizumab. J Neurol. 2020;267:1991–2001. Article CAS PubMed PubMed Central Google Scholar * Jacob S, Murai H, Utsugisawa K, Nowak RJ, Wiendl
H, Fujita KP, et al. Response to eculizumab in patients with myasthenia gravis recently treated with chronic IVIg: a subgroup analysis of REGAIN and its open-label extension study. Ther Adv
Neurol Disord. 2020;13:1756286420911784. Article CAS PubMed PubMed Central Google Scholar * Siddiqi ZA, Nowak RJ, Mozaffar T, O’Brien F, Yountz M, Patti F, et al. Eculizumab in
refractory generalized myasthenia gravis previously treated with rituximab: subgroup analysis of REGAIN and its extension study. Muscle Nerve. 2021;64:662–9. Article CAS PubMed Google
Scholar * Usman U, Chrisman C, Houston D, Haws CC, Wang A, Muley S. The use of eculizumab in ventilator-dependent myasthenia gravis patients. Muscle Nerve. 2021;64:212–5. Article CAS
PubMed Google Scholar * Mantegazza R, Cavalcante P. Eculizumab for the treatment of myasthenia gravis. Expert Opin Biol Ther. 2020;20:991–8. Article CAS PubMed Google Scholar * Tice
JA, Touchette DR, Lien PW, Agboola F, Nikitin D, Pearson SD. The effectiveness and value of eculizumab and efgartigimod for generalized myasthenia gravis. J Manag Care Spec Pharm.
2022;28:119–24. PubMed Google Scholar * Vu T, Meisel A, Mantegazza R, Annane D, Katsuno M, Aguzzi R, et al. Summary of research: terminal complement inhibitor ravulizumab in generalized
myasthenia gravis. Neurol Ther. 2023;12:1435–8. Article PubMed PubMed Central Google Scholar * Howard JF Jr, Vissing J, Gilhus NE, Leite MI, Utsugisawa K, Duda PW, et al. Zilucoplan: an
investigational complement C5 inhibitor for the treatment of acetylcholine receptor autoantibody-positive generalized myasthenia gravis. Expert Opin Investig Drugs. 2021;30:483–93. Article
CAS PubMed Google Scholar * Devalaraja-Narashimha K, Huang C, Cao M, Chen YP, Borodovsky A, Olson WC, et al. Pharmacokinetics and pharmacodynamics of pozelimab alone or in combination
with cemdisiran in non-human primates. PLoS ONE. 2022;17:e0269749. Article CAS PubMed PubMed Central Google Scholar * Lunemann JD. Getting specific: targeting Fc receptors in myasthenia
gravis. Nat Rev Neurol. 2021;17:597–8. Article PubMed Google Scholar * Ward ES, Gelinas D, Dreesen E, Van Santbergen J, Andersen JT, Silvestri NJ, et al. Clinical significance of serum
albumin and implications of FcRn inhibitor treatment in IgG-mediated autoimmune disorders. Front Immunol. 2022;13:892534. Article PubMed PubMed Central Google Scholar * Wolfe GI, Ward
ES, de Haard H, Ulrichts P, Mozaffar T, Pasnoor M, et al. IgG regulation through FcRn blocking: a novel mechanism for the treatment of myasthenia gravis. J Neurol Sci. 2021;430:118074.
Article CAS PubMed Google Scholar * Ulrichts P, Guglietta A, Dreier T, van Bragt T, Hanssens V, Hofman E, et al. Neonatal Fc receptor antagonist efgartigimod safely and sustainably
reduces IgGs in humans. J Clin Invest. 2018;128:4372–86. Article PubMed PubMed Central Google Scholar * Ling LE, Hillson JL, Tiessen RG, Bosje T, van Iersel MP, Nix DJ, et al. M281, an
anti-FcRn antibody: pharmacodynamics, pharmacokinetics, and safety across the full range of IgG reduction in a first-in-human study. Clin Pharm Ther. 2019;105:1031–9. Article CAS Google
Scholar * Howard JF Jr, Bril V, Vu T, Karam C, Peric S, Margania T, et al. Safety, efficacy, and tolerability of efgartigimod in patients with generalised myasthenia gravis (ADAPT): a
multicentre, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2021;20:526–36. Article CAS PubMed Google Scholar * Guptill J, Antozzi C, Bril V, Gamez J, Meuth S, Blanco J,
et al. Vivacity-MG: a phase 2, multicenter, randomized, double-blind, placebo-controlled study to evaluate the safety, tolerability, efficacy, pharmacokinetics, pharmacodynamics, and
immunogenicity of nipocalimab administered to adults with generalized myasthenia gravis. Neurology. 2021;96. * Bril V, Benatar M, Andersen H, Vissing J, Brock M, Greve B, et al. Efficacy and
safety of rozanolixizumab in moderate to severe generalized myasthenia gravis: a phase 2 randomized control trial. Neurology. 2021;96:e853–65. Article CAS PubMed PubMed Central Google
Scholar * Stathopoulos P, Kumar A, Heiden JAV, Pascual-Goni E, Nowak RJ, O’Connor KC. Mechanisms underlying B cell immune dysregulation and autoantibody production in MuSK myasthenia
gravis. Ann N Y Acad Sci. 2018;1412:154–65. Article CAS PubMed PubMed Central Google Scholar * Yi JS, Guptill JT, Stathopoulos P, Nowak RJ, O’Connor KC. B cells in the pathophysiology
of myasthenia gravis. Muscle Nerve. 2018;57:172–84. Article PubMed Google Scholar * Behin A, Le Panse R. New pathways and therapeutic targets in autoimmune myasthenia gravis. J
Neuromuscul Dis. 2018;5:265–77. Article PubMed PubMed Central Google Scholar * Chen D, Gallagher S, Monson NL, Herbst R, Wang Y. Inebilizumab, a B cell-depleting anti-CD19 antibody for
the treatment of autoimmune neurological diseases: insights from preclinical studies. J Clin Med. 2016;5:107. Article PubMed PubMed Central Google Scholar * Kohler S, Marschenz S,
Grittner U, Alexander T, Hiepe F, Meisel A. Bortezomib in antibody-mediated autoimmune diseases (TAVAB): study protocol for a unicentric, non-randomised, non-placebo controlled trial. BMJ
Open. 2019;9:e024523. Article PubMed PubMed Central Google Scholar * Klimas R, Sgodzai M, Motte J, Mohamad N, Renk P, Blusch A, et al. Dose-dependent immunomodulatory effects of
bortezomib in experimental autoimmune neuritis. Brain Commun. 2021;3:fcab238. Article PubMed PubMed Central Google Scholar * Robak E, Robak T. Bruton’s kinase inhibitors for the
treatment of immunological diseases: current status and perspectives. J Clin Med. 2022;11:2807. Article CAS PubMed PubMed Central Google Scholar * Ragheb S, Lisak RP. B-cell-activating
factor and autoimmune myasthenia gravis. Autoimmune Dis. 2011;2011:939520. PubMed PubMed Central Google Scholar * Hewett K, Sanders DB, Grove RA, Broderick CL, Rudo TJ, Bassiri A, et al.
Randomized study of adjunctive belimumab in participants with generalized myasthenia gravis. Neurology. 2018;90:e1425–34. Article CAS PubMed PubMed Central Google Scholar * Elgueta R,
Benson MJ, de Vries VC, Wasiuk A, Guo Y, Noelle RJ. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev. 2009;229:152–72. Article CAS PubMed Google
Scholar * Aricha R, Mizrachi K, Fuchs S, Souroujon MC. Blocking of IL-6 suppresses experimental autoimmune myasthenia gravis. J Autoimmun. 2011;36:135–41. Article CAS PubMed Google
Scholar * Jonsson DI, Pirskanen R, Piehl F. Beneficial effect of tocilizumab in myasthenia gravis refractory to rituximab. Neuromuscul Disord. 2017;27:565–8. Article PubMed Google Scholar
* June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med. 2018;379:64–73. Article CAS PubMed PubMed Central Google Scholar * Bryant A, Atkins H, Pringle CE, Allan D,
Anstee G, Bence-Bruckler I, et al. Myasthenia gravis treated with autologous hematopoietic stem cell transplantation. JAMA Neurol. 2016;73:652–8. Article PubMed Google Scholar Download
references ACKNOWLEDGEMENTS No direct help for writing this manuscript was obtained from any external sources, except for the images which were drawn using Biorender.com. The author would
like to thank his doctoral fellow Pyae Phyo San who helped to draw some of the original figures. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * University Hospitals Birmingham, Birmingham, UK
Saiju Jacob * Institute of Immunology and Immunotherapy, University of Birmingham, Birmingham, UK Saiju Jacob Authors * Saiju Jacob View author publications You can also search for this
author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to Saiju Jacob. ETHICS DECLARATIONS COMPETING INTERESTS SJ has served as an international advisory board member for
Alexion, Alnylam, Argenx, Janssen, Immunovant, Regeneron and UCB pharmaceuticals, is currently an expert panel member of Myasthenia Gravis consortium for Argenx pharmaceuticals and has
received speaker fees from Argenx, Eisai, Terumo BCT and UCB pharmaceuticals. He is also a board member (trustee) of the UK myasthenia patient charity, Myaware. ADDITIONAL INFORMATION
PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. RIGHTS AND PERMISSIONS OPEN ACCESS This article is
licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give
appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in
this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative
Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a
copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Jacob, S. Treating myasthenia gravis beyond the eye
clinic. _Eye_ 38, 2422–2436 (2024). https://doi.org/10.1038/s41433-024-03133-x Download citation * Received: 02 January 2024 * Revised: 17 February 2024 * Accepted: 07 May 2024 * Published:
24 May 2024 * Issue Date: August 2024 * DOI: https://doi.org/10.1038/s41433-024-03133-x SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get
shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative