Play all audios:
ABSTRACT Investigating the impact of landscape features on patterns of genetic variation is crucial to understand spatially dependent evolutionary processes. Here, we assess the population
genomic variation of two bird species (_Conopophaga cearae_ and _Sclerurus cearensis_) through the Caatinga moist forest enclaves in northeastern Brazil. To infer the evolutionary dynamics
of bird populations through the Late Quaternary, we used genome-wide polymorphism data obtained from double-digestion restriction-site-associated DNA sequencing (ddRADseq), and integrated
population structure analyses, historical demography models, paleodistribution modeling, and landscape genetics analyses. We found the population differentiation among enclaves to be
significantly related to the geographic distance and historical resistance across the rugged landscape. The climate changes at the end of the Pleistocene to the Holocene likely triggered
synchronic population decline in all enclaves for both species. Our findings revealed that both geographic distance and historical connectivity through highlands are important factors that
can explain the current patterns of genetic variation. Our results further suggest that levels of population differentiation and connectivity cannot be explained purely on the basis of
contemporary environmental conditions. By combining historical demographic analyses and niche modeling predictions in a historical framework, we provide strong evidence that climate
fluctuations of the Quaternary promoted population differentiation and a high degree of temporal synchrony among population size changes in both species. You have full access to this article
via your institution. Download PDF SIMILAR CONTENT BEING VIEWED BY OTHERS IDIOSYNCRATIC RESPONSES TO DRIVERS OF GENETIC DIFFERENTIATION IN THE COMPLEX LANDSCAPES OF ISTHMIAN CENTRAL AMERICA
Article 13 October 2020 LOCAL ADAPTATION HAS A ROLE IN REDUCING VULNERABILITY TO CLIMATE CHANGE IN A WIDESPREAD AMAZONIAN FOREST LIZARD Article 05 May 2025 GENOME-WIDE GENETIC VARIATION
COUPLED WITH DEMOGRAPHIC AND ECOLOGICAL NICHE MODELING OF THE DUSKY-FOOTED WOODRAT (_NEOTOMA FUSCIPES_) REVEAL PATTERNS OF DEEP DIVERGENCE AND WIDESPREAD HOLOCENE EXPANSION ACROSS NORTHERN
CALIFORNIA Article 15 December 2020 INTRODUCTION Disentangling the drivers of spatial genetic variation is a central theme in evolution. This is because genetic variation is a major factor
in a species’ ability to adapt and persist in the face of climate and habitat changes (Manel et al. 2003; Barrett and Schluter 2008). Examining the relationship between genetic
differentiation and different landscape variables while accounting for the geographic distance among populations has often been the primary approach used to study the factors and processes
of a population’s genetic differentiation and its spatial structure. The pattern of isolation by distance (IBD) proposed by Wright (1943) is the simplest pattern that causes genetic
differentiation among populations. According to this pattern, geographic isolation leads to gene-flow restrictions and drift, implying a positive relationship between genetic differentiation
and geographic distance. However, this “pure geographic distance pattern” might not often be the best predictor of geographic variation in some organisms since it does not incorporate other
factors that may affect gene flow, such as landscape features, range boundaries, or environmental conditions (McRae 2006; Storfer et al. 2007; Lee and Mitchell-Olds 2011). In this sense,
IBD should be contrasted with more complex patterns (Jenkins et al. 2010), such as isolation by resistance (IBR; McRae 2006) and/or isolation by environment (IBE; Wang and Bradburd 2014).
IBR posits that genetic differentiation is related to the resistance distance, which is obtained by incorporating landscape variables that can constrain the dispersal among populations, such
as anthropogenic barriers, mountains, rivers, soil, and vegetation type, among others (McRae 2006). Likewise, IBE considers environmental heterogeneity and local adaptation to explain the
patterns of genetic differentiation, but accounts for populations’ dependency on specific environmental conditions across the landscape (Wang and Bradburd 2014). Herein we seek to understand
how geographic and landscape features influence spatial genetic variation, population structure, and gene flow through the Neotropical highland forest enclaves embedded in the Caatinga
ecoregion. These forest enclaves are viewed as an archipelago of moist forest islands surrounded by an extensive semiarid tropical sea of seasonally dry forest and shrub woodlands with an
average temperature of 27 °C and an annual precipitation <800 mm (Andrade-Lima 1982; Tabarelli and Santos 2004). Locally referred to as “_brejos de altitude_”, these highland forest
enclaves are found mostly in plateaus of 500–1100 m elevation (Andrade-Lima 1982; Tabarelli and Santos 2004), and are characterized by a humid tropical or subtropical climate with mild
temperatures and annual rainfall >1200 mm (Andrade et al. 2017). A growing body of evidence supports the hypothesis that the current distribution of these forest enclaves reflects past
biome dynamics, resulting from multiple episodes of forest expansions and contractions during the Pleistocene (Oliveira et al. 1999; Behling et al. 2000; Auler et al. 2004; Wang et al. 2004;
Silveira et al. 2019). Distinct palaeoindicators and niche models suggest that moister climates have occurred throughout much of the Pleistocene (Oliveira et al. 1999; Behling et al. 2000;
Auler et al. 2004; Wang et al. 2004; Silveira et al. 2019), which would have facilitated the connection between the Amazon and Atlantic rainforests across the Caatinga (Batalha-Filho et al.
2013; Ledo and Colli 2017). Paleopalynological records also suggested the expansion of humid forests through highlands (elevations higher than 400 m) during the Heinrich Stadial 1 Event
(18.1–14.7 kya), which might have allowed migration routes that connected currently isolated forest enclaves (Pinaya et al. 2019). Finally, at the beginning of the Holocene, the drier and
colder climate seems to have induced the contraction of forests, confining them to highland humid patches, while the current dry conditions of Caatinga dominate the surrounding areas
(Oliveira et al. 1999; Behling et al. 2000; Auler et al. 2004; Wang et al. 2004; Ledru et al. 2016; Silveira et al. 2019). Recent studies have emphasized the role of these complex past
regional dynamics in shaping the current patterns of distribution and diversification of many organisms (Carnaval and Bates 2007; d’Horta et al. 2011; Batalha-Filho et al. 2014; Buainain et
al. 2020; Bocalini et al. 2021). While some isolated populations in these enclaves have accumulated enough differentiation to be recognized as separate species (e.g., d’Horta et al. 2011;
Batalha-Filho et al. 2014; Cabanne et al. 2014), others are considered isolated populations from geographically structured species with wider distributions across the Atlantic and/or Amazon
rainforests (e.g., Buainain et al. 2020; Bocalini et al. 2021). Two forest-dependent passerines distributed across these forest patches at high elevations of the Caatinga in northeastern
Brazil, the Ceara Gnateater (_Conopophaga cearae_) and the Ceara Leaftosser (_Sclerurus cearensis_), are ideally placed to evaluate the role of past biome dynamics in shaping current
patterns of genetic variation (Fig. 1). While _S. cearensis_ is restricted to the state of Ceará (del Hoyo et al. 2020b), _C. cearae_ has a wider distribution, occurring in the states of
Ceará, Pernambuco, and Paraíba, as well as in the Chapada Diamantina in the state of Bahia (del Hoyo et al. 2020a). Here, we assess the population genetic variation of these two bird species
using genome-wide single-nucleotide polymorphism (SNP) data generated from double-digest restriction-site-associated DNA sequencing (ddRADseq). By integrating landscape genomics, historical
demography, and ecological niche modeling tools, we depict the spatiotemporal dynamics of bird populations distributed through the Caatinga moist forest enclaves. Specifically, we aim to
test whether population differentiation across enclaves can be explained by three non-mutually exclusive patterns of spatial genetic variation: (1) the IBD pattern—positive relationship
between the genetic and geographic distances, (2) the IBR pattern—positive relationship between the genetic distance and landscape resistance to lower altitudes and climatic suitability
areas in the past, and (3) the IBE pattern—positive relationship between the genetic and environmental distances due to regional climatic heterogeneity. We also address the impact of the
Late Quaternary climate change on the demography of these bird populations by using a statistical phylogeographic approach. If the hypothesis of a larger distribution of the forest enclaves
during the last glacial maximum (LGM) followed by subsequent fragmentation during the Holocene (Silveira et al. 2019) is true, a signal of recent population size decline in both species is
expected. By combining these approaches, we were able to illuminate the effect of Late Quaternary climatic oscillations on the demography and isolation of these forest enclaves. MATERIALS
AND METHODS SAMPLING, RAD LIBRARY PREPARATION, AND SEQUENCING We gathered samples from fieldwork during 2015 and 2018 and loans from Brazilian collections (Table S1). We obtained samples
from 61 individuals of _C. cearae_ and 50 individuals of _S. cearensis_ across seven forest enclaves of both species in northeastern Brazil (Fig. 1 and Table S1). Genomic DNA was extracted
from muscle tissues using the DNeasy Blood & Tissue kit (QIAGEN, Hilden, Germany). Samples were processed using the ddRAD library approach (Parchman et al. 2012; Peterson et al. 2012).
The libraries were constructed following the protocols of Parchman et al. (2012) and Peterson et al. (2012) with modifications added by A. Brelsford, A. Mastretta-Yanes, J. Leuenberger, and
R. Sermier. The modifications included the use of the restriction enzyme _Sbf_I instead of _Eco_RI (recommended for large genomes or more individuals per library); dual-index barcoding to
allow multiplexing >96 samples per library; Y-adapter for _Mse_I from Peterson et al. (2012) to prevent amplification of _Mse_I-_Mse_I fragments; CutSmart buffer to simplify restriction
and ligation mixes; purification post ligation using Agencourt AMPure beads (Beckman Coulter, California, USA); PCR using Q5 Hot Start Polymerase (New England BioLabs, Massachusetts, USA);
modified PCR to decrease the number of cycles by increasing the number of replicates (four separate 10 µl reactions per restriction-ligation product, which were later combined) and starting
DNA volume; addition of primers and dNTPs for a final thermal cycle to reduce the production of single-stranded or heteroduplex PCR products; and final cleanup and size selection with AMPure
beads. The DNA was double digested with _Sbf_I and _Mse_I. Next, specific adapters and unique barcodes were ligated to the digested DNA, and a purification step (to remove short fragments)
was carried out using AMPure beads. After that, we performed PCR amplifications using Illumina PCR primers to amplify fragments that had both adapters and barcodes ligated onto the ends,
followed by size selection of 400–500 bp fragments by gel extraction, and another purification step with the MinElute Gel Extraction kit (QIAGEN, Hilden, Germany) and magnetic AMPure beads.
All libraries were quantified using the Qubit Fluorometer and subjected to paired-end sequencing using 150 bp reads on an Illumina platform at Macrogen Inc. (Seoul, Republic of Korea).
PROCESSING RADSEQ DATA The Sabre tool (https://github.com/najoshi/sabre) was used to demultiplex the barcoded reads into separate fastq files. The interactive toolkit _ipyrad_ v0.7.29 (Eaton
and Overcast 2020) was then used for processing the restriction-site-associated genomic datasets generated for each species (see Table S2 for _ipyrad_ parameters). Specifically, we
performed the following steps: filtering of low-quality reads, clustering within samples, joint estimation of heterozygosity and error rate, consensus base calling, clustering among samples,
and formatting output files. During the processing, we used a de novo assembly method, minimum depth for both statistical and majority-rule base calling set to 10, strict filter for
adapters, and trimming of the first five and the last five bases from all reads. A maximum of 10% missing data per individual was allowed for each species’ dataset. We used default values
for the remaining parameters. For the final dataset, sequences from two individuals of _S. cearensis_ (USP/LGEMA13614 and UFBA1144) were discarded due to the low coverage, which resulted in
only 4420 and 120 loci in the assembly, respectively, instead of 14,612 loci recovered for the remaining samples (see “Results”). For most analyses, we considered only one randomly chosen
SNP per locus, except for the _Dxy_ calculations, for which we used all sites (variant and invariants). GENETIC DIVERSITY, POPULATION STRUCTURE, AND MIGRATION TESTS General measures of
genetic diversity were obtained in _DnaSP_ v.6.12 (Rozas et al. 2017) through calculations of haplotype (gene) diversity (Nei 1987), nucleotide diversity (Nei 1987), and average number of
nucleotide differences (Tajima 1993). The genetic structure of each species across the forest enclaves was investigated by principal component analysis (PCA) using the _glPca_ function from
the package _adegenet_ 2.1.2 (Jombart 2008) in R v4.0.6 (R Core Team 2018). We also evaluated population genetic structure using the sparse non-negative matrix factorization approach
(_sNMF_) in the LEA package (Frichot and François 2015), which is based on likelihood methods, PCA, and NMF algorithms, showing robustness to departures from traditional population genetic
model assumptions (Frichot et al. 2014). We tested the number of genetically distinct clusters (_K_) between 1 and 10. To test the accuracy of the results, two hundred replicates were run
for each _K_ under different alpha regularization parameter values (1, 10, 100, and 1000). The optimal value of _K_ was determined using the cross-entropy criterion, which uses the
imputation of masked genotypes to evaluate the capability of the algorithm to estimate ancestry coefficients (Frichot et al. 2014). To examine the patterns of spatial population structure
and connectivity, we used the estimated effective migration surface method (EEMS; Petkova et al. 2016). The EEMS approach is robust in untangling the contribution of pure IBD from that of
historical barriers or heterogeneous landscapes in driving population differentiation (see Petkova et al. 2016). Similar to an IBR approach, EEMS delineates the process by which genetic
differentiation accrues in landscapes lacking homogeneity, achieved through the integration of all potential migration routes between two specific points (Petkova et al. 2016). This analysis
measures effective migration as representing historical rates of gene flow and relates it to expected pairwise genetic dissimilarities, resulting in a spatial representation of patterns of
genetic variation, and the identification of migration corridors and/or barriers to gene flow (Petkova et al. 2016). The average genetic dissimilarity matrix was computed with the
_bed2diffs_ program (https://github.com/dipetkov/eems/tree/master/bed2diffs), after converting the VCF file into BED format using PLINK v1.9 (Chang et al. 2015). The habitat polygons were
constructed based on the distribution of the sampling points at http://www.birdtheme.org/useful/v3largemap.html, an online Google Maps tool where it is possible to draw polylines and
polygons around the areas of interest from geographic coordinates. For _C. cearae_, the area covered by the polygon comprised the Brazilian states of Ceará, Piauí, Bahia, Sergipe, Alagoas,
Pernambuco, Paraíba, and Rio Grande do Norte, while for _S. cearensis_, the area of interest was only the state of Ceará. Analyses were run using the _runeems_snps_ version 1 of EEMS for 30
× 106 iterations, with the first 5 × 106 excluded as burn-in, and deme sizes of 400 and 200 for _C. cearae_ and _S. cearensis_, respectively. For each species, three independent analyses
were performed to ensure convergence, which was verified by comparing the posterior trace plots generated by the R package _reemsplots_
(https://github.com/dipetkov/eems/tree/master/plotting). This same package was used to visualize the final combined results. ISOLATION BY DISTANCE, ISOLATION BY RESISTANCE, AND ISOLATION BY
ENVIRONMENT We applied the generalized dissimilarity modeling (GDM; Ferrier et al. 2007) approach to quantify the effects of IBD, IBR, and IBE on population differentiation across forest
enclaves for both species. This approach models biological variation as a function of environment and geography using distance matrices, and takes into account nonlinear relationships
between response and predictor variables (Ferrier et al. 2007). We obtained the population differentiation between sampled localities by calculating pairwise _Dxy_ genetic distances (Tables
S3 and S4) for each species using the _PopGenome_ package (Pfeifer et al. 2014) in R v4.0.4. For the IBD, we obtained a pairwise geographic distance matrix (in Km) between localities (Tables
S5 and S6) using the Geographic Distance Matrix Generator 1.2.3 (available at http://biodiversityinformatics.amnh.org/open_source/gdmg/index.php). In IBR, we considered lower altitudes
(current resistance) and lower suitability areas derived by paleodistribution (historical resistance) models (see below) as the resistance for landscape dispersal. Both bird species are
currently restricted to the moist forest enclaves at higher elevations (at least 500 m) encrusted in the dry vegetation of the Caatinga ecoregion. As paleoecological records suggest past
connections between currently isolated forest enclaves through highlands during the Late Pleistocene (Pinaya et al. 2019; Piacsek et al. 2021), we considered higher elevations as indicating
the conductance among localities. Elevation data were obtained from the EarthEnv database at a 1-km resolution (http://www.earthenv.org/; Amatulli et al. 2018). We also used the output of
ecological niche modeling from the Bølling-Allerød Interstadial epoch (14.7–12.9 kya), considering higher-suitability areas as indicating conductance among localities. We used this temporal
profile because of its largest paleodistribution for both species (see the ecological niche modeling results). Then, we used the circuit theory to produce the spatial resistance distances
between localities using the _Circuitscape_ v4.0.5 (McRae et al. 2008) to obtain resistance surfaces and pairwise resistance matrices between localities (Tables S7–S10). We obtained
environmental data for IBE using the 19 bioclimatic variables of current conditions available in _Paleoclim_ (Brown et al. 2018) derived from CHELSA 2.1 (Karger et al. 2017) at 2.5
arc-minutes resolution. We used the _Raster_ package to extract the climate values per locality for each species. To minimize the collinearity among variables, we selected uncorrelated
(Pearson’s _r_ < 0.7) bioclimatic variables for each species based on the occurrence data: (a) _C. cearae_—mean diurnal range (bio2), isothermality (bio3), mean temperature of the warmest
quarter (bio10), annual precipitation (bio12), precipitation of the driest month (bio14), precipitation seasonality (bio15), precipitation of the warmest quarter (bio18), and precipitation
of the coldest quarter (bio19); (b) _S. cearensis_—isothermality (bio3), temperature seasonality (bio4), mean temperature of the wettest quarter (bio8), precipitation of the wettest month
(bio13), precipitation seasonality (bio15), precipitation of the driest quarter (bio17), and precipitation of the coldest quarter (bio19). Bioclimatic variables used in this analysis are
different from those used in the ecological niche modeling as the occurrence points are different in each analysis (here we must use localities with genetic sampling). The consideration of
different occurrences can produce distinct environmental spaces and affect the collinearity among variables. Finally, we used the _gdm_ package (Ferrier et al. 2007;
https://github.com/fitzLab-AL/GDM) in R v4.0.5 to calculate the generalized dissimilarity model. We considered the _Dxy_ pairwise distance matrix as the response variable. We fit the model
using the following predictor variables (bioformat = 3): bioclimatic variables (eight for _C. cearae_ and seven for _S. cearensis_), geographic distance matrix (IBD), resistance matrix
derived from elevation data (IBRtopo), and resistance matrix derived from past distribution of ecological niche modeling (Bølling-Allerød epoch; IBRpaleo). We selected the best predictors
using a stepwise matrix permutation and backward elimination approach. At each step, the least important predictor was dropped (backward elimination) and the process continued until all
nonsignificant predictors were removed. We used 500 permutations and defined the best model as the one that retained significant predictors (_p_ ≤ 0.05). We also calculated the variance
partitioning of significant predictors of the best model using _gdm.partition.deviance_ function. DEMOGRAPHIC INFERENCES Significant breakthroughs in the development of methods based on
population genomics have aided in the reconstruction of historical demography. Despite the popularity of some methods, there is some concern about the accuracy of the inferred demographic
models because they are mostly based on a set of assumptions that are commonly ignored and/or violated in natural systems. For example, numerous studies have shown that population structure
and gene flow could generate spurious signs of population size changes over time (expansion and/or bottleneck), even when the populations remained stationary through time (Wakeley 1999;
Nielsen and Beaumont 2009; Chikhi et al. 2010, 2018; Mazet et al. 2016). Here, we used the _Stairway plot_ v2.1.1 (Liu and Fu 2020), a model-flexible method based on SFS (site frequency
spectrum), to investigate the impact of Quaternary climatic changes on the demographic history of both species. As both bird species are structured in three genetic clusters (see “Results”,
Fig. 1), we used two different sampling approaches to reduce the potential conflicting effect of population structure (e.g., Heller et al. 2013; Chikhi et al. 2018; Wang et al. 2021; Violi
et al. 2023): (i) all individuals across the whole set of populations were pooled (hereafter, species demographic analysis); and (ii) individuals from the different genetic clusters
uncovered for both the species were analyzed separately (hereafter, cluster-based demographic analysis). In this case, the results should be interpreted by assuming that each cluster
represents a single isolated population with an independent evolutionary history. For species demographic analysis, we obtained the SFS for the whole individuals per species (full datasets).
For cluster-based demographic analysis, individuals from each cluster (Fig. 1) were separated into different _ipyrad_ runs using the same parameters as the full datasets and retaining only
the loci present in all individuals. Due to potential issues with admixture affecting demographic inference (e.g., Heller et al. 2013), individuals of mixed ancestry (_q_-values lower than
0.8 for _C. cearae_ and 0.75 for _S. cearensis_) (Fig. 1; samples from Maciço de Baturité of _C. cearae_ and Serra de Uruburetama of _S. cearensis_), as revealed by the _sNMF_ results, were
removed in the cluster-based demographic analysis. The folded SFS of each cluster was obtained using the Python script _easySFS_ (http://github.com/isaacovercast/easySFS). To run the
_Stairway plot_, we assumed a mutation rate of 2.5 × 10−9 (Nadachowska-Brzyska et al. 2015) and a generation time of 2.03 years for _C. cearae_ and 2.19 years for _S. cearensis_ according to
Bird et al. (2020). The estimation (median) and 95% pseudo-confidence interval (CI) were generated based on 200 replications. For cluster-based demographic analysis, we also tested a set of
alternative historical demographic scenarios (models). The three models included (model 1) constant population size (“null model”), in which the current effective population size (_Ncur_)
was the only parameter estimated; (model 2) population decline, where we estimated the _Ncur_, ancestral population size (_Nanc_), and the time at which population decline began (_Tdec_);
and (model 3) population growth, where _Ncur_, _Nanc_, and time when population growth started (_Tgro_) were estimated. For the last two scenarios, the range of the resize parameter was
bounded by a factor of 1–5 and 0.5–0.9 to simulate population decline and growth, respectively, under a uniform prior distribution. We applied the same mutation rate and generation times
used for the _Stairway plot_ analysis. We used the coalescent-based modeling package _fastsimcoal26_ (Excoffier and Foll 2011; Excoffier et al. 2013) to evaluate each model based on the
observed SFS. Fastsimcoal runs were performed with 40 replicates for each scenario, with 250,000 simulations per likelihood estimate, a stopping criterion of 0.001, and 10–40
expectation-conditional cycles (ECM). We obtained the 95% CI for each parameter by performing 100 parametric bootstraps of simulated SFS from the maximum likelihood estimates and
re-estimating the parameters with 40 runs for each of the 100 simulated SFS. Model comparisons were performed based on their likelihoods using the Akaike information criteria (AIC; Akaike
1974). ECOLOGICAL NICHE MODELING To infer the effects of Quaternary climatic oscillations on species range dynamics, we modeled the distribution of both species in the present conditions and
along different periods of the Late Quaternary. Considering the forest dependency of these two bird species, we expected larger distribution ranges during warmer and wetter periods,
especially during the deglaciation (17–11 kya) at Heinrich, Bølling-Allerød, and Young Dryas interstadial epochs, and range contraction during warmer periods of the Holocene (last 8.3 kya).
Species occurrences were obtained from the following online resources: Global Biodiversity Information Facility (GBIF, https://www.gbif.org/), speciesLink (https://splink.cria.org.br/),
Sistema de Informação sobre a Biodiversidade Brasileira (SiBBr, https://www.sibbr.gov.br/), and our genetic sampling (Table S1). Occurrences were filtered by excluding points outside the
known species range, which resulted in 79 and 36 points for _C. cearae_ and _S. cearensis_, respectively (Tables S11 and S12). To reduce bias in the modeling, we used a thinning filter to
exclude locations within a radius of up to 10 km using _spThin_ package (Aiello-Lammens et al. 2015). We generated ecological niche models (ENMs) under the maximum entropy approach (Phillips
et al. 2006) along with bioclimatic variables available in _Paleoclim_ (Brown et al. 2018) at a 2.5 arc-minutes resolution. As for ENM analysis, we used more records than genetic sampling
points and we selected new uncorrelated (Pearson’s _r_ < 0.7) bioclimatic variables for each species based on the occurrence data: (a) _C. cearae_—isothermality (bio3), temperature
seasonality (bio4), mean temperature of the wettest quarter (bio8), mean temperature of the driest quarter (bio9), precipitation of the driest month (bio14), precipitation seasonality
(bio15), and precipitation of the warmest quarter (bio18); (b) _S. cearensis_—temperature seasonality (bio4), mean temperature of the wettest quarter (bio8), precipitation of the driest
month (bio14), precipitation of the warmest quarter (bio18), and precipitation of the coldest quarter (bio19). We used the following climatic models to uncover nine periods from the Late
Pleistocene to the present (Brown et al. 2018): current conditions; late-Holocene, Meghalayan (4.2–0.3 kya); mid-Holocene, Northgrippian (8.326–4.2 kya); early-Holocene, Greenlandian
(11.7–8.326 kya); Younger Dryas Stadial (12.9–11.7 kya); Bølling-Allerød (14.7–12.9 kya); Heinrich Stadial 1 (17.0–14.7 kya); Last Glacial Maximum (ca. 21 kya); and Last Interglacial (ca.
130 kya). For _C. cearae_, the layers were cropped to a geographic extent between the coordinates −44.71 (maximum longitude), −33.77 (minimum longitude), −20.00 (maximum latitude), and −2.20
(minimum latitude), and for _S. cearensis_, they were cropped to −44.71 (maximum longitude), −33.77 (minimum longitude), −10.00 (maximum latitude), and −2.20 (minimum latitude) using the
_raster_ package (https://rspatial.org/raster/). The background environment for each species was obtained by randomly generating 1000 points through the minimum convex polygon. We ran and
evaluated models using the ENM evaluate function from the _ENMeval_ package (Muscarella et al. 2014). We tested regularization multiplier values from 1 to 5, with increments of 0.5, as well
as the following combinations of feature classes: linear, linear-quadratic, hinge, and linear-quadratic-hinge. To avoid overfitting, we performed a spatial cross-validation with the
checkerboard2 method, which splits our data into four evaluation sets. For each set, the model was trained in the remaining three sets and tested using the evaluation set. The best model was
chosen based on the omission rate and area under the curve (AUC) values (i.e., we chose the model with the lowest omission rate and highest AUC). The final best-fitted models were generated
using the maxent function in the dismo package (https://github.com/rspatial/dismo). RESULTS GENETIC DIVERSITY AND POPULATION STRUCTURE From our ddRAD sequencing, we obtained 156,543,279 raw
reads for 61 samples of _C. cearae_ and 164,824,866 raw reads for 48 samples of _S. cearensis_. After the _ipyrad_ filtering steps (see the parameters used in Table S2), we retained
1,690,372–3,201,267 reads per sample for _C. cearae_ and 2,506,901–4,718,625 reads per sample for _S. cearensis_. The consensus base calling and clustering among samples considering a
minimum depth coverage of 10 recovered 14,612 loci with 161,748 SNPs for _C. cearae_ in a matrix with 6.47% of missing sites, and 17,789 loci with 61,301 SNPs for _S. cearensis_ in a matrix
with 5.12% of missing sites. For _C. cearae_ and _S. cearensis_, we found an average heterozygosity of 0.010 and 0.006, respectively, with an error rate of 0.002. Using all samples from each
species, the haplotype (gene) diversity, nucleotide diversity, and average number of nucleotide differences were 0.015, 0.00009, and 0.022 for _C. cearae_ and 0.002, 0.00001, and 0.005 for
_S. cearensis_, respectively. Regarding the _Dxy_ distance, the values varied among pairs of localities for both species (Tables S3 and S4). In _C. cearae_, the values ranged from 0.00805
between Morro do Chapéu and Itatira to 0.00123 between Brejo da Madre de Deus and Cruz do Espírito Santo, while in _S. cearensis_ they ranged from 0.00242 between Crato and Guaramiranga to
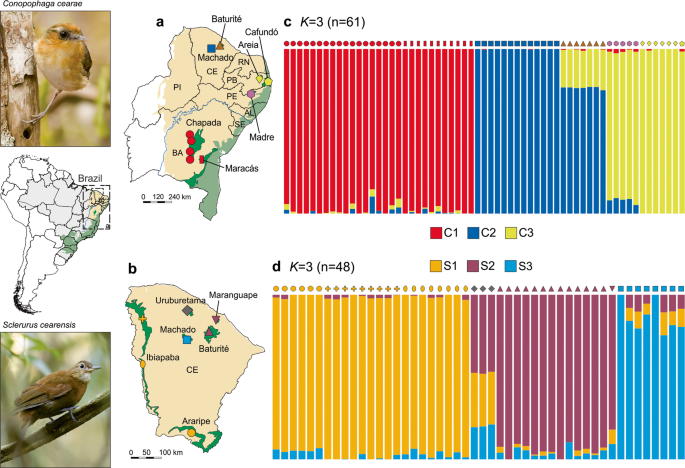
0.00001 between Uruburetama and Itapipoca (Tables S1, S3, and S4). Population clustering using _sNMF_ revealed that both species are subdivided into three distinct genetic clusters (_K_ = 3;
Fig. 1 and S1). For _C. cearae_, the clusters were (i) south of São Francisco River (C1); (ii) the northern region of Ceará state (C2), including individuals from Serra do Machado and
Maciço de Baturité; and (iii) the northeastern region (C3), including Areia, Brejo da Madre de Deus, and Açude Cafundó (Fig. 1a, c). In _S. cearensis_, the clusters uncovered were (i) the
western region of Ceará state (S1), including Chapada do Araripe and the north and south enclaves of Serra do Ibiapaba; (ii) the central region of Ceará state (S2), constituted by
individuals from Serra do Machado; and (iii) the northeastern part of Ceará state (S3), composed of Maciço de Baturité and Serra do Maranguape (Fig. 1b, d). Additionally, samples of _S.
cearensis_ from the Serra de Uruburetama enclave showed a significant admixture among those three clusters (Fig. 1d). The population structure as revealed by _sNMF_ is also evident in the
PCA results (Fig. S2), which in turn indicates further population substructure within the three main clusters, corresponding to populations with mixed ancestry (Fig. 1 and S2). The map of
EEMS for _C. cearae_ was generally congruent with the results inferred by _sNMF_, showing a clear barrier to gene flow between north and south forest enclaves, and a barrier separating east
and west forest enclaves in the north (Fig. 2a). The only exception was within the cluster C2, in which the forest enclave of Maciço de Baturité appeared isolated from Serra do Machado (Fig.
2a). Interestingly, samples from Maciço de Baturité exhibited a moderate admixture with cluster C3 (Fig. 1c). For _S. cearensis_, the EEMS map showed a strong barrier to gene flow between
the east and west enclaves, while Serra do Machado and Serra de Uruburetama appeared isolated from the remaining populations (Fig. 2b). ISOLATION BY DISTANCE, ISOLATION BY RESISTANCE, AND
ISOLATION BY ENVIRONMENT The overall GDM modeling explained 61.66% and 60.48% of the genetic differentiation for _C. cearae_ and _S. cearensis_, respectively. However, three predictors for
_C. cearae_ (bio3, bio14, and IBRtopo) and one predictor for _S. cearensis_ (IBRtopo) did not show significance on genetic differentiation, showing I-spline values equal to zero. In _C.
cearae_, predictors for IBD (I-splines = 0.531), IBE (bio10 = 0.357, bio19 = 0.318, bio18 = 0.183, bio2 = 0.159, bio15 = 0.149, bio12 = 0.008), and IBR (paleo = 0.153) had unique
contributions and explained 44.12% of the genetic differentiation between populations (Fig. S3). For _S. cearensis_, IBD (I-splines = 0.008), IBE (bio13 = 0.055, bio4 = 0.042, bio19 = 0.037,
bio15 = 0.028, bio17 = 0.025, bio8 = 0.007), and IBR (paleo = 0.056) had unique contributions and explained 40.37% of the genetic differentiation between populations (Fig. S3).
Notwithstanding, after permutations, the best model was the one where only geographic distance (IBD) contributed significantly (_p_ < 0.001) to the genetic differentiation in _C. cearae_,
and only ENM (IBRpaleo) showed significant contribution (_p_ < 0.001) for _S. cearensis_ (Fig. 3). Therefore, the geographic distance alone explained 38.14% of the variation for _C.
cearae_, while IBRpaleo alone explained 18.23% of the variation for _S. cearensis_. HISTORICAL DEMOGRAPHY MODEL TESTING Demographic histories inferred in the _Stairway plot_ showed signs of
recent and synchronic declines of effective population size (_N_e) in both the species (Fig. S4) and cluster-based demographic analyses (Fig. 4a, b). In species demographic analysis,
population decline started around 1 kya (Fig. S4), and in the cluster-based demographic analysis, the more pronounced decline occurred in the last 10 kya (Holocene), which was preceded by
population growth (Fig. 4a, b). Historical alternative demographic scenarios (models) suggested population decline as the best-fitting model for each differentiated cluster for both species
(Fig. 4c and Table 1), showing a synchronic population shrinking (Table 1). According to this scenario, clusters C1 and C2 of _C. cearae_ showed a current population size (_Ncur_) of around
1 million genes, whereas the ancestral population size (_Nanc_) was estimated to be ~5.1 million genes (Table 1). For these clusters, the time when the population started to decline (_Tdec_)
was estimated at c.a. 1 kya, during the Holocene (Table 1). For the cluster C3 of _C. cearae_, _Ncur_ and _Nanc_ were estimated at around 31 and 82 thousand genes, respectively, with
population decline also starting at c.a. 1 kya, during the Holocene (Table 1). In _S. cearensis_, we recovered _Ncur_ and _Nanc_ of around 1 and 5.1 million genes, respectively, and _Tdec_
of c.a. 1 kya, during the Holocene, for all clusters (Table 1). ECOLOGICAL NICHE MODELING Model selection by _ENMeval_ fitted the hinge model with a regularization multiplier of five for
both species (see Table S13 for parameters of the best-fitted models). ENM across all evaluated periods from the Late Quaternary indicated a potential range variation through time for both
the species (Fig. 5) with periods of range contraction and range expansion. For the present conditions (Fig. 5a), our modeling is congruent with the known distribution of both species, but
substantial changes in the potential distribution range were uncovered for the last 130 kya (Fig. 5). Overall, the projection to past conditions showed a general trend of potential range
expansion during wetter periods, especially in the Bølling-Allerød Interstadial epoch (14.7–12.9 kya), and a potential range contraction during warmer periods of the Holocene (last 8.3 kya).
DISCUSSION DRIVERS OF POPULATION GEOGRAPHIC VARIATION Contemporary genetic variation of species harbors information that can help reconstruct their evolutionary history (Ellegren and
Galtier 2016). This kind of inference can be further improved by explicitly comparing the demographic dynamics and population structure of co-distributed and ecologically similar species
that evolved under common historical biogeographic drivers (e.g., Lourenço et al. 2018). By examining the spatial patterns of genetic variation in two ecologically related endemic bird
species restricted to highland forest enclaves in northeastern Brazil, we found an overall similar pattern of genetic structure. Consistent with the strong dependency on forest habitats
(Sick 1997; del Hoyo et al. 2020a, 2020b) and low vagility among fragmented forest landscapes, as previously demonstrated in their congeners (_C. lineata_ and _S. scansor_) endemic to the
Atlantic Forest (Marini 2010), we found high levels of population structure in both _C. cearae_ and _S. cearensis_. This likely results from the effects of genetic drift as a consequence of
the isolation of forest enclaves (Fig. 1 and S1). Also, genetic drift and the reduced gene flow associated with a small distribution range may be factors that can help explain the lower
values of genetic diversity in _S. cearensis_ when compared to _C. cearae_. For both species, results from the generalized dissimilarity modeling showed that environmental conditions (IBE)
did not predict the genetic differentiation of bird populations through forest enclaves. This result likely reflects a relative environmental homogeneity or a nonadaptive genetic variation
among sampled localities for each species. On the other hand, we found a general trend toward an increase in genetic differentiation with the geographic distance between populations of _C.
cearae_, suggesting that distance between forest enclaves plays a key role in shaping the current patterns of genetic structure. This result is in agreement with the ubiquity of the IBD
pattern found in a myriad of organisms, including bird species (e.g., Cabanne et al. 2007; Maldonado-Coelho 2012; for a review, see Jenkins et al. 2010; Perez et al. 2018). Our analysis
further identified historical resistance distance (IBRpaleo) as a predictor of genetic divergence for _S. cearensis_. In contrast, the current IBRtopo did not play a significant role in
population differentiation for both species, which agrees with paleoecological data (Pinaya et al. 2019; Piacsek et al. 2021) that suggest the role of an interplay between elevation and past
climate conditions in driving montane forest expansion during the Late Quaternary. Although the reasons for the unbalanced contribution of IBD and IBR between the two species are unclear,
our findings clearly suggest that dispersal limitation due to geographical distance and historical habitat resistance together had a compound effect on the current patterns of population
differentiation in both species. Also, the broader distribution of _C. cearae_ might have contributed to the strongest IBD effect on genetic differentiation when compared with _S.
cearensis_. We found additional support for this conclusion from the EEMS analysis (Fig. 2), which showed patterns of unevenly distributed gene flow for both species. For example, this
analysis showed geographically close population pairs with very restricted or lack of migration (e.g., Serra do Machado and Serra do Baturité in _C. cearae_, Fig. 2a) and, on the contrary,
geographically distant population pairs that present high migration rates (e.g., Serra do Ibiapaba and Chapada do Araripe in _S. cearensis_, Fig. 2b). The differential habitat occupancy and
biogeographic history of these species can also reflect the patterns observed here. _Conopophaga cearae_ has a broader range across enclaves in the states of Bahia, Ceará, Rio Grande do
Norte, Paraíba, Pernambuco, and Alagoas, whereas _S. cearensis_ occurs only in the state of Ceará (Fig. 1). In this state, _S. cearensis_ occupies more enclaves than _C. cearae_ (Fig. 1).
These distinct patterns of enclaves’ occupancy might reflect distinct historical dispersal or extinction rates between species during the Late Quaternary. Interestingly, _S. cearensis_ is
more conspicuous in dry forest and rainforest enclaves, whereas _C. cearae_ is apparently less frequent in dry forest enclaves (personal observation). Additionally, species competition could
also play a role in enclaves’ occupancy. The absence of _C. cearae_ in the enclaves of the western state of Ceará (northern Serra da Ibiapaba) might be explained by the occurrence of
another _Conopophaga_ species in this region, the _C. roberti_, which occurs in western Amazonia and exhibits forest dependency (Greeney 2020). Likewise, the absence of _S. cearensis_ in the
enclaves of the state of Bahia is likely explained by the presence of _S. scansor_ in Chapada Diamantina and Maracás. Further studies that relate species traits and habitat requirements
with genetic variation should test these hypotheses. The effective migration surfaces suggest that key geographical barriers have restricted gene flow (Fig. 2), such as the case of São
Francisco River valley for _C. cearae_. This result is consistent with previous studies that have found this river as an important barrier for many organisms (e.g., lizards: Lanna et al.
2020; frogs: Bruschi et al. 2019; Thomé et al. 2021; birds: Capelli et al. 2020), including _C. cearae_ (Batalha-Filho et al. 2014). Our results also suggest that past patterns of highland
corridors during wetter periods of the Quaternary may have played an important role as dispersion routes between different forest enclaves (see Fig. S5) (Auler et al. 2004; Wang et al. 2004;
Silveira et al. 2019; Piacsek et al. 2021). This is consistent with the role of IBRpaleo, which significantly predicted patterns of genetic differentiation among populations, particularly
considering the historically suitable areas as a key variable for population migration during historically wetter periods. The Montane rainforest expansion during the Heinrich Stadial 1
Event (18–14 kya), as suggested by paleopollen and speleothem data (Pinaya et al. 2019), may have allowed the formation of dispersal corridors across the highlands (Pinaya et al. 2019;
Piacsek et al. 2021). Thus, enclaves located in the same mountain chain or plateaus were more likely to exchange genes during the Quaternary than isolated enclaves in highlands surrounded by
valleys or lowlands (Fig. S5). While further analysis with denser sampling within the same mountain chain or plateaus would provide the opportunity to test this hypothesis, our results
emphasize the importance of accounting for current and past landscape features as drivers of intraspecific differentiation. DEMOGRAPHIC HISTORY THROUGH THE LATE QUATERNARY Results from
_Stairway plot_ analyses revealed that the historical demographic profiles of both bird species were similar when the entire set of populations was assumed to behave as a single population
or when it was assumed that the population was structured into three distinct genetic clusters. This suggests that sampling design has no bearing on the demographic reconstruction for both
bird species. Importantly, a parallel temporal pattern of population size decline across all genetic clusters of both species is supported by the best-fit model inferred by _fastsimcoal_.
This is consistent with the results of _Stairway plot_, suggesting that both species were impacted by a similar process leading to population decline during the last 10 to 1 kya (Figs. 3 and
4; Table 1). Although these results should be interpreted with caution, overall they are concordant with previous expectations of population size decline (Cabanne et al. 2008; Bocalini et
al. 2021) due to reductions in the distribution of forest enclaves after the LGM (Silveira et al. 2019). The classical refuge model for the Atlantic Forest postulates refuge areas for
enclaves in the Chapada Diamantina hills during the LGM, but did not recover stability areas for enclaves at the northern São Francisco River (Carnaval and Moritz 2008; Carnaval et al.
2009). However, more recent ENM-based analyses of past spatial dynamics suggest forest expansion for all enclaves into areas currently occupied by the Caatinga seasonally dry woodlands
during the LGM (Silveira et al. 2019). These results concur with independent evidence provided by our ENM results (Fig. 5), which inferred a generalized range contraction for both species
after warmer and moister conditions of the Bølling-Allerød interstadial epoch (14.7–12.9 kya; Köhler et al. 2011) that might have triggered population growth and connection of currently
isolated enclaves. These findings along with abundant evidence from the literature, such as speleothems (Auler et al. 2004; Wang et al. 2004), paleopalynology (Oliveira et al. 1999; Behling
et al. 2000; Bouimetarhan et al. 2018; Pinaya et al. 2019; Piacsek et al. 2021), and sediments (Jacob et al. 2007; Pessenda et al. 2010), support the hypothesis that populations of both
passerine species experienced a recent decline with the increase of dry conditions during the Holocene (Fig. 4). Despite demographic analyses only depicting the last change of population
size, the collection of data regarding historical dynamics of forest enclaves allowed us to hypothesize that these areas may have experienced cyclical episodes of expansion and contraction
along the Quaternary climate changes (Oliveira et al. 1999; Behling et al. 2000; Auler et al. 2004; Wang et al. 2004; Silveira et al. 2019). For example, a comparative demographic analysis
of squamates and amphibian species endemic to the Caatinga dry vegetation revealed signs of synchronic population expansions in the Late Pleistocene (Gehara et al. 2017), which is the
opposite of the pattern observed here for our forest-dependent bird species. This strongly suggests that the Caatinga and forest enclaves might have had alternating episodes of expansion and
retraction throughout the Quaternary climate changes. CONCLUSIONS This study provides important insights into how isolation and reduction of population sizes due to historical fragmentation
of the habitat affected current patterns of genetic variation of forest enclaves from northeastern Brazil. For this, we explored the landscape genetic variation of two endemic
forest-dependent bird species by combining the population genomics and spatiotemporal dynamics of the habitat within a comparative framework. We have shown that both current geographic
distances and historical connectivity through highlands are important factors explaining the current patterns of genetic variation. Overall, our results suggest that levels of population
differentiation and connectivity cannot be explained based purely on contemporary environmental conditions. Combining historical demographic analyses and niche modeling predictions in a
historical framework, we provided strong evidence that climate fluctuations of the Quaternary promoted population differentiation and a high degree of temporal synchrony among population
size changes in both species. Our study highlights the importance of an integrative framework in disentangling the drivers of geographic variation for other multiple endemic species of these
unique highland forest enclaves, adding new insights regarding the conservation value of this ecosystem for long-term biodiversity persistence, despite being intrinsically unstable.
Moreover, recognizing the role of genetic isolation of populations distributed through the enclaves is of utmost importance for prioritizing measures of conservation that can prevent the
fragmentation and destruction of this ecosystem in an era of accelerating deforestation and exploitation of natural resources. DATA ARCHIVING Input files for population genetic analyses have
been made available on Dryad (https://doi.org/10.5061/dryad.7d7wm37w1). REFERENCES * Aiello-Lammens ME, Boria RA, Radosavljevic A, Vilela B, Anderson RP (2015) spThin: an R package for
spatial thinning of species occurrence records for use in ecological niche models. Ecography 38:541–545. https://doi.org/10.1111/ecog.01132 Article ADS Google Scholar * Akaike H (1974) A
new look at the statistical model identification. IEEE Trans Autom Contr 19:716–723. https://doi.org/10.1109/TAC.1974.1100705 Article MathSciNet ADS Google Scholar * Amatulli G, Domisch
S, Tuanmu M-N, Parmentier B, Ranipeta A, Malczyk J et al. (2018) A suite of global, cross-scale topographic variables for environmental and biodiversity modeling. Sci Data 5:180040.
https://doi.org/10.1038/sdata.2018.40 Article PubMed PubMed Central Google Scholar * Andrade EM, Aquino D, Chaves LCG, Lopes FB (2017) Water as capital and its uses in the Caatinga. In:
Silva JMC, Leal IR, Tabarelli M (eds) Caatinga. Springer, Cham Google Scholar * Andrade-Lima D (1982) Present day forest refuges in northeastern Brazil. In: Prance GT (ed.) Biological
diversification in the tropics. Columbia University Press, New York, pp. 245–254 Google Scholar * Auler AS, Wang X, Edwards RL, Cheng H, Cristalli PS, Smart PL et al. (2004) Quaternary
ecological and geomorphic changes associated with rainfall events in present semi-arid northeastern Brazil. J Quat Sci 19:693–701. https://doi.org/10.1002/jqs.876 Article Google Scholar *
Barrett RDH, Schluter D (2008) Adaptation from standing genetic variation. Trends Ecol Evol. 23:38–44. https://doi.org/10.1016/j.tree.2007.09.008 Article PubMed Google Scholar *
Batalha-Filho H, Fjeldså J, Fabre P-H, Miyaki CY (2013) Connections between the Atlantic and the Amazonian forest avifaunas represent distinct historical events. J Ornithol 154:41–50.
https://doi.org/10.1007/s10336-012-0866-7 Article Google Scholar * Batalha-Filho H, Pessoa RO, Fabre P-H, Fjeldså J, Irestedt M, Ericson PGP et al. (2014) Phylogeny and historical
biogeography of gnateaters (Passeriformes, Conopophagidae) in the South America forests. Mol Phylogenet Evol 79:422–432. https://doi.org/10.1016/j.ympev.2014.06.025 Article PubMed Google
Scholar * Behling H, Arz HW, Pätzold, Wefer G (2000) Late Quaternary vegetational and climate dynamics in northeastern Brazil, inferences from marine core GeoB 3104-1. Quat Sci Rev
19:981–994. https://doi.org/10.1016/S0277-3791(99)00046-3 Article ADS Google Scholar * Bird JP, Martin R, Akçakaya HR, Gilroy J, Burfield IJ, Garnett ST et al. (2020) Generation lengths
of the world’s birds and their implications for extinction risk. Conserv Biol 34:1252–1261. https://doi.org/10.1111/cobi.13486 Article PubMed Google Scholar * Bocalini F,
Bolívar-Leguizamón SD, Silveira LF, Bravo GA (2021) Comparative phylogeographic and demographic analyses reveal a congruent pattern of sister relationships between bird populations of the
northern and south-central Atlantic Forest. Mol Phylogenet Evol 154:106973. https://doi.org/10.1016/j.ympev.2020.106973 Article PubMed Google Scholar * Bouimetarhan I, Chiessi CM,
González-Arango C, Dupont L, Voigt I, Prange M et al. (2018) Intermittent development of forest corridors in northeastern Brazil during the last deglaciation: climatic and ecologic evidence.
Quat Sci Rev 192:86–96. https://doi.org/10.1016/j.quascirev.2018.05.026 Article ADS Google Scholar * Brown JL, Hill DJ, Dolan AM, Carnaval AC, Haywood AM (2018) PaleoClim, high spatial
resolution paleoclimate surfaces for global land areas. Sci Data 5:180254. https://doi.org/10.1038/sdata.2018.254 Article PubMed PubMed Central Google Scholar * Bruschi DP, Peres EA,
Lourenço LB, Bartoleti LFDM, Sobral-Souza T, Recco-Pimentel SM (2019) Signature of the paleo-course changes in the São Francisco river as source of genetic structure in neotropical
Pithecopus nordestinus (Phyllomedusinae. Anura) Treefrog. Front Gen 10:728 * Buainain N, Canton R, Zuquim G, Tuomisto H, Hrbek T, Sato H et al. (2020) Paleoclimatic evolution as the main
driver of current genomic diversity in the widespread and polymorphic Neotropical songbird _Arremon taciturnus_. Mol Ecol 29:2922–2939. https://doi.org/10.1111/mec.15534 Article PubMed
Google Scholar * Cabanne GS, Santos FR, Miyaki CY (2007) Phylogeography of _Xiphorhynchus fuscus_ (Passeriformes, Dendrocolaptidae): vicariance and recent demographic expansion in southern
Atlantic forest. Biol J Linn Soc 91:73–84. https://doi.org/10.1111/j.1095-8312.2007.00775.x Article Google Scholar * Cabanne GS, d’Horta FM, Sari EHR, Santos FR, Miyaki CY (2008) Nuclear
and mitochondrial phylogeography of the Atlantic forest endemic _Xiphorhynchus fuscus_ (Aves: Dendrocolaptidae): biogeography and systematics implications. Mol Phylogenet Evol 49:760–773.
https://doi.org/10.1016/j.ympev.2008.09.013 Article CAS PubMed Google Scholar * Cabanne GS, Trujillo-Arias N, Calderón L, d'Horta FM, Miyaki CY (2014) Phenotypic evolution of an
Atlantic Forest passerine (_Xiphorhynchus fuscus_): biogeographic and systematic implications. Biol J Linn Soc 113:1047–106. https://doi.org/10.1111/bij.12362 Article Google Scholar *
Capelli D, Batalha-Filho H, Japyassú HF (2020) Song variation in the Caatinga suboscine Silvery-cheeked Antshrike (_Sakesphorus cristatus_) suggests latitude and São Francisco River as
drivers of geographic variation. J Ornithol 161:873–884. https://doi.org/10.1007/s10336-020-01779-4 Article Google Scholar * Carnaval AC, Bates JM (2007) Amphibian DNA shows marked genetic
structure and tracks Pleistocene climate change in northeastern Brazil. Evolution 61:2942–2957. https://doi.org/10.1111/j.1558-5646.2007.00241.x Article CAS PubMed Google Scholar *
Carnaval AC, Moritz C (2008) Historical climate modeling predicts patterns of current biodiversity in the Brazilian Atlantic forest. J Biogeogr 35:1187–1201.
https://doi.org/10.1111/j.1365-2699.2007.01870.x Article Google Scholar * Carnaval AC, Hickerson MJ, Haddad CFB, Rodrigues MT, Moritz C (2009) Stability predicts genetic diversity in the
Brazilian Atlantic forest hotspot. Science 323:785–789. https://doi.org/10.1126/science.1166955 Article CAS PubMed ADS Google Scholar * Chang CC, Chow CC, Tellier LCAM, Vattikuti S,
Purcell SM, Lee JJ (2015) Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience 4:s13742-015-0047-8. https://doi.org/10.1186/s13742-015-0047-8 Article
CAS Google Scholar * Chikhi L, Sousa VC, Luisi P, Goossens B, Beaumont MA (2010) The confounding effects of population structure, genetic diversity and the sampling scheme on the detection
and quantification of population size changes. Genetics 186:983–995. https://doi.org/10.1534/genetics.110.118661 Article PubMed PubMed Central Google Scholar * Chikhi L, Rodríguez W,
Grusea S, Santos P, Boitard S, Mazet O (2018) The IICR (inverse instantaneous coalescence rate) as a summary of genomic diversity: insights into demographic inference and model choice.
Heredity 120:13–24. https://doi.org/10.1038/s41437-017-0005-6 Article PubMed Google Scholar * del Hoyo J, Collar N, Kirwan GM (2020a) Ceara Gnateater (_Conopophaga cearae_), version 1.0.
In: del Hoyo J, Elliott A, Sargatal J, Christie DA, de Juana E (eds) Birds of the world. Cornell Lab of Ornithology, Ithaca Google Scholar * del Hoyo J, Remsen Jr JV, Kirwan GM, Collar N
(2020b) Rufous-breasted Leaftosser (_Sclerurus scansor_), version 1.0. In: Billerman SM, Keeney BK, Rodewald PG, Schulenberg TS (eds) Birds of the world. Cornell Lab of Ornithology, Ithaca
Google Scholar * d’Horta FM, Cabanne GS, Meyer D, Miyaki CY (2011) The genetic effects of Late Quaternary climatic changes over a tropical latitudinal gradient: diversification of an
Atlantic Forest passerine. Mol Ecol 20:1932–1935. https://doi.org/10.1111/j.1365-294X.2011.05063.x Article Google Scholar * Eaton DA, Overcast I (2020) ipyrad: Interactive assembly and
analysis of RADseq datasets. Bioinformatics 36:2592–2594. https://doi.org/10.1093/bioinformatics/btz966 Article CAS PubMed Google Scholar * Ellegren H, Galtier N (2016) Determinants of
genetic diversity. Nat Rev Genet 17:422–433. https://doi.org/10.1038/nrg.2016.58 Article CAS PubMed Google Scholar * Excoffier L, Foll M (2011) fastsimcoal: a continuous-time coalescent
simulator of genomic diversity under arbitrarily complex evolutionary scenarios. Bioinformatics 27:1332–1334. https://doi.org/10.1093/bioinformatics/btr124 Article CAS PubMed Google
Scholar * Excoffier L, Dupanloup I, Huerta-Sánchez E, Sousa VC, Foll M (2013) Robust demographic inference from genomic and SNP data. PLoS Genet 9:e1003905.
https://doi.org/10.1371/journal.pgen.1003905 Article CAS PubMed PubMed Central Google Scholar * Ferrier S, Manion G, Elith J, Richardson K (2007) Using generalized dissimilarity
modelling to analyse and predict patterns of beta diversity in regional biodiversity assessment. Divers Distrib 13:252–264. https://doi.org/10.1111/j.1472-4642.2007.00341.x Article Google
Scholar * Frichot E, François O (2015) LEA: an R package for landscape and ecological association studies. Methods Ecol Evol 6:925–929. https://doi.org/10.1111/2041-210X.12382 Article
Google Scholar * Frichot E, Mathieu F, Trouillon T, Bouchard G, François O (2014) Fast and efficient estimation of individual ancestry coefficients. Genetics 196:973–983.
https://doi.org/10.1534/genetics.113.160572 Article PubMed PubMed Central Google Scholar * Gehara M, Garda AA, Werneck FP, Oliveira EF, Fonseca EM, Camurugi F et al. (2017) Estimating
synchronous demographic changes across populations using hABC and its application for a herpetological community from northeastern Brazil. Mol Ecol 26:4756–4771.
https://doi.org/10.1111/mec.14239 Article PubMed Google Scholar * Greeney HF (2020) Hooded Gnateater (_Conopophaga roberti_), version 1.0. In: Schulenberg TS (ed) Birds of the world.
Cornell Lab of Ornithology, Ithaca Google Scholar * Heller H, Chikhi L, Siegismund HR (2013) The confounding effect of population structure on Bayesian skyline plot inferences of
demographic history. PLoS ONE 8:e62992. https://doi.org/10.1371/journal.pone.0062992 Article CAS PubMed PubMed Central ADS Google Scholar * Jacob J, Huang Y, Disnar JR, Sifeddine A,
Boussafir M, Albuquerque ALS et al. (2007) Paleohydrological changes during the last deglaciation in Northern Brazil. Quat Sci Rev 26:1004–1015.
https://doi.org/10.1016/j.quascirev.2006.12.004 Article ADS Google Scholar * Jenkins DG, Carey M, Czerniewska J, Fletcher J, Hether T, Jones A et al. (2010) A meta-analysis of isolation
by distance: relic or reference standard for landscape genetics? Ecography 33:315–320. https://doi.org/10.1111/j.1600-0587.2010.06285.x Article ADS Google Scholar * Jombart T (2008)
adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405. https://doi.org/10.1093/bioinformatics/btn129 Article CAS PubMed Google Scholar *
Karger DN, Conrad O, Böhner J, Kawohl T, Kreft H, Soria-Auza RW, Zimmermann NE, Linder P, Kessler M (2017) Climatologies at high resolution for the Earth land surface areas. Sci Data
4:170122. https://doi.org/10.1038/sdata.2017.122 Article PubMed PubMed Central Google Scholar * Köhler P, Knorr G, Buiron D, Lourantou A, Chappellaz J (2011) Abrupt rise in atmospheric
CO2 at the onset of the Bølling/Allerød: in-situ ice core data versus true atmospheric signals. Clim 7:473–486. https://doi.org/10.5194/cp-7-473-2011 Article ADS Google Scholar * Lanna
FM, Gehara M, Werneck FP, Fonseca EM, Colli GR, Sites JW, Rodrigues MT, Garda AA (2020) Dwarf geckos and giant rivers: the role of the São Francisco River in the evolution of _Lygodactylus
klugei_ (Squamata: Gekkonidae) in the semi-arid Caatinga of north-eastern Brazil. Biol J Linn Soc 129:88–98. https://doi.org/10.1093/biolinnean/blz170 Article Google Scholar * Ledo RMD,
Colli GR (2017) The historical connections between the Amazon and the Atlantic Forest revisited. J Biogeogr 44:2551–256. https://doi.org/10.1111/jbi.13049 Article Google Scholar * Ledru
M-P, Montade V, Blanchard G, Hély C (2016) Long-term spatial changes in the distribution of the Brazilian Atlantic forest. Biotropica 48:159–169. https://doi.org/10.1111/btp.12266 Article
Google Scholar * Lee C-R, Mitchell-Olds T (2011) Quantifying effects of environmental and geographical factors on patterns of genetic differentiation. Mol Ecol 20:4631–4642.
https://doi.org/10.1111/j.1365-294X.2011.05310.x Article PubMed PubMed Central Google Scholar * Liu X, Fu YX (2020) Stairway plot 2: demographic history inference with folded SNP
frequency spectra. Genome Biol 21:280. https://doi.org/10.1186/s13059-020-02196-9 Article PubMed PubMed Central Google Scholar * Lourenço A, Sequeira F, Buckley D, Velo-Antón G (2018)
Role of colonization history and species-specific traits on contemporary genetic variation of two salamander species in a Holocene island-mainland system. J Biogeogr 45:1054–1066.
https://doi.org/10.1111/jbi.13192 Article Google Scholar * Maldonado-Coelho M (2012) Climatic oscillations shape the phylogeographical structure of Atlantic Forest fire-eye antbirds (Aves:
Thamnophilidae). Biol J Linn Soc 105:900–924. https://doi.org/10.1111/j.1095-8312.2011.01823.x Article Google Scholar * Manel S, Schwartz MK, Luikart G, Taberlet P (2003) Landscape
genetics: combining landscape ecology and population genetics. Trends Ecol Evol 18:189–197. https://doi.org/10.1016/s0169-5347(03)00008-9 Article Google Scholar * Marini MA (2010) Bird
movement in a fragmented Atlantic Forest landscape. Stud Neotrop 45:1–10. https://doi.org/10.1080/01650521003656606 Article Google Scholar * McRae BH (2006) Isolation by resistance.
Evolution 60:1551–1561. https://doi.org/10.1554/05-321.1 Article PubMed Google Scholar * McRae BH, Dickson BG, Keitt TH, Shah VB (2008) Using circuit theory to model connectivity in
ecology and conservation. Ecology. 10:2712–2724. https://doi.org/10.1890/07-1861.1 Article Google Scholar * Muscarella R, Galante PJ, Soley-Guardia M, Boria RA, Kass JM, Uriarte M et al.
(2014) ENMeval: an R package for conducting spatially independent evaluations and estimating optimal model complexity for Maxent ecological niche models. Methods Ecol Evol 5:1198–1205.
https://doi.org/10.1111/2041-210X.12261 Article Google Scholar * Mazet O, Rodríguez W, Grusea S, Boitard S, Chikhi L (2016) On the importance of being structured: instantaneous coalescence
rates and human evolution—lessons for ancestral population size inference? Heredity 116:362–371. https://doi.org/10.1038/hdy.2015.104 Article CAS PubMed Google Scholar *
Nadachowska-Brzyska K, Li C, Smeds L, Zhang G, Ellegren H (2015) Temporal dynamics of avian populations during pleistocene revealed by whole-genome sequences. Curr Biol 25:1375–1380.
https://doi.org/10.1016/j.cub.2015.03.047 Article CAS PubMed PubMed Central Google Scholar * Nei M (1987) Molecular evolutionary genetics. Columbia Univ. Press, New York Google Scholar
* Nielsen R, Beaumont M (2009) Statistical inferences in phylogeography. Mol Ecol 18:1034–1047. https://doi.org/10.1111/j.1365-294X.2008.04059.x Article CAS PubMed Google Scholar *
Oliveira PE, Barreto AMF, Suguio K (1999) Late Pleistocene/Holocene climatic and vegetational history of the Brazilian caatinga: the fossil dunes of the middle São Francisco River.
Palaeogeogr Palaeoclimatol Palaeoecol 152:319–337. https://doi.org/10.1016/S0031-0182(99)00061-9 Article Google Scholar * Parchman TL, Gompert Z, Mudge J, Schilkey FD, Benkman CW, Buerkle
CA (2012) Genome-wide association genetics of an adaptive trait in lodgepole pine. Mol Ecol 21:2991–3005. https://doi.org/10.1111/j.1365-294X.2012.05513.x Article CAS PubMed Google
Scholar * Perez MF, Franco FF, Bombonato JR, Bonatelli IAS, Khan G, Romeiro-Brito M et al. (2018) Assessing population structure in the face of isolation by distance: are we neglecting the
problem. Divers Distrib 24:1883–1889. https://doi.org/10.1111/ddi.12816 Article Google Scholar * Pessenda LCR, Gouveia SEM, Ribeiro AS, De Oliveira PE, Aravena R (2010) Late Pleistocene
and Holocene vegetation changes in northeastern Brazil determined from carbon isotopes and charcoal records in soils. Palaeogeogr Palaeoclimatol Palaeoecol 297:597–608.
https://doi.org/10.1016/j.palaeo.2010.09.008 Article Google Scholar * Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE (2012) Double Digest RADseq: an inexpensive method for de novo
SNP discovery and genotyping in model and non-model species. PLoS ONE 7:e37135. https://doi.org/10.1371/journal.pone.0037135 Article CAS PubMed PubMed Central ADS Google Scholar *
Petkova D, Novembre J, Stephens M (2016) Visualizing spatial population structure with estimated effective migration surfaces. Nat Genet 48:94–100. https://doi.org/10.1038/ng.3464 Article
CAS PubMed Google Scholar * Pfeifer B, Wittelsbürger U, Ramos-Onsins SE, Lercher MJ (2014) PopGenome: an efficient Swiss Army Knife for population genomic analyses in R. Mol Biol Evol
31:1929–1936. https://doi.org/10.1093/molbev/msu136 Article CAS PubMed PubMed Central Google Scholar * Phillips SJ, Anderson RP, Schapire RE (2006) Maximum entropy modeling of species
geographic distributions. Ecol Model 190:231–259. https://doi.org/10.1016/j.ecolmodel.2005.03.026 Article Google Scholar * Piacsek P, Behling H, Ballalai JM, Nogueira J, Venancio IM,
Albuquerque ALS (2021) Reconstruction of vegetation and low latitude ocean-atmosphere dynamics of the past 130 kyr, based on South American montane pollen types. Glob Planet Change
201:103477. https://doi.org/10.1016/j.gloplacha.2021.103477 Article Google Scholar * Pinaya JLD, Cruz FW, Ceccantini GCT, Corrêa PLP, Pitman N, Vemado F et al. (2019) Brazilian montane
rainforest expansion induced by Heinrich Stadial 1 event. Sci Rep. 9:17912. https://doi.org/10.1038/s41598-019-53036-1 Article CAS PubMed PubMed Central ADS Google Scholar * R Core
Team (2018) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria, https://www.R-project.org Google Scholar * Rozas J, Ferrer-Mata
A, Sánchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE et al. (2017) DnaSP v6: DNA sequence polymorphism analysis of large datasets. Mol Biol Evol 34:3299–3302.
https://doi.org/10.1093/molbev/msx248 Article CAS PubMed Google Scholar * Sick H (1997) Ornitologia Brasileira. Nova Fronteira, Rio de Janeiro Google Scholar * Silveira MHB, Mascarenhas
R, Cardoso D, Batalha-Filho H (2019) Pleistocene climatic instability drove the historical distribution of forest islands in the northeastern Brazilian Atlantic Forest. Palaeogeogr
Palaeoclimatol Palaeoecol 527:67–76. https://doi.org/10.1016/j.palaeo.2019.04.028 Article Google Scholar * Storfer A, Murphy M, Evans J, Goldberg CS, Robinson S, Spear SF et al. (2007)
Putting the ‘landscape’ in landscape genetics. Heredity 98:128–142. https://doi.org/10.1038/sj.hdy.6800917 Article CAS PubMed Google Scholar * Tabarelli M, Santos AMM (2004) Uma breve
descrição sobre a história natural dos brejos nordestinos. In: Pôrto KC, Cabral JJP, Tabarelli M (eds) Brejos de altitude em Pernambuco e Paraíba: história natural, ecologia e conservação.
Ministério do Meio Ambiente Press, Brasília, pp. 17–24 Google Scholar * Tajima F (1993) Measurement of DNA polymorphism. In: Takahata N, Clark AG (eds) Mechanisms of molecular evolution.
Sinauer Associates. Inc, Sunderland, MA, pp. 37–59 Google Scholar * Thomé MTC, Carstens BC, Rodrigues MT, Alexandrino J, Haddad CFB (2021) Genomic data from the Brazilian sibilator frog
reveal contrasting Pleistocene dynamics and regionalism in two South American dry biomes. J Biogeogr 48:1112–1123. https://doi.org/10.1111/jbi.14064 Article Google Scholar * Violi B, Jong
MN, Frantzis A, Alexiadou P, Tardy C, Ody D et al. (2023) Genomics reveals the role of admixture in the evolution of structure among sperm whale populations within the Mediterranean Sea. Mol
Ecol 32:2715–2731. https://doi.org/10.1111/mec.16898 Article CAS PubMed Google Scholar * Wakeley J (1999) Nonequilibrium migration in human history. Genetics 153:1863–1871.
https://doi.org/10.1093/genetics/153.4.1863 Article CAS PubMed PubMed Central Google Scholar * Wang X, Auler AS, Edwards RL, Cheng H, Cristalli PS, Smart PL et al. (2004) Wet periods in
northeastern Brazil over the past 210 kyr linked to distant climate anomalies. Nature 432:740–743. https://doi.org/10.1038/nature03067 Article CAS PubMed ADS Google Scholar * Wang IJ,
Bradburd GS (2014) Isolation by environment. Mol Ecol 23:5649–5662. https://doi.org/10.1111/mec.12938 Article PubMed Google Scholar * Wang P, Burley JT, Liu Y, Chang J, Chen D, Lu Q et
al. (2021) Genomic consequences of long-term population decline in Brown Eared Pheasant. Mol Biol Evol 38:263–273. https://doi.org/10.1093/molbev/msaa213 Article CAS PubMed Google Scholar
* Wright S (1943) Isolation by distance. Genetics 28:139–156. https://doi.org/10.1093/genetics/28.2.114 Article CAS PubMed PubMed Central Google Scholar Download references
ACKNOWLEDGEMENTS The authors thank Caio Graco (UEFS) and Helder Araújo (UFPB) for providing some of the tissues used in this study, and Sidnei Santos, Rilquer Mascarenhas, and Lucas Passos
for their help during the fieldwork. The authors are grateful to Pedro Andrade for helping with the demultiplexing of Illumina runs. The authors also thank Rilquer Mascarenhas for help with
ecological niche modeling scripts; the editor and two anonymous reviewers for immensely improving this manuscript; and the National Laboratory for Scientific Computing (LNCC/MCTI, Brazil)
for providing HPC resources for the SDumont supercomputer. Instituto Brasileiro do Meio Ambiente e dos Recursos Naturais Renováveis and Instituto Chico Mendes de Conservação da
Biodiversidade (ICMBio license number 52235-1) and Secretaria do Meio Ambiente of the state of Bahia (SEMA/INEMA license number 11553) provided permits to collect the samples. This study was
funded by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq 443249/2014-8, 303713/2015-1,
406968/2021-7), Fundação de Amparo à Pesquisa do Estado da Bahia (FAPESB JCB0026/2016), and the joint National Science Foundation, Fundação de Amparo à Pesquisa do Estado de São Paulo and
NASA grant (Dimensions US-Biota-SP, FAPESP 2013/50297-0/NSF DOB 1343578). FS was supported by National Funds through FCT-Fundação para a Ciência e a Tecnologia under the project
UIDB/50027/2020. HB-F and CYM also acknowledge the CNPq Research Productivity Fellowships (310673/2021-6 and 306204/2019-3, respectively). MC was supported by a research contract from the
Fundação para a Ciência e Tecnologia (FCT; CEECINST/00014/2018/CP1512/CT0002). AUTHOR INFORMATION Author notes * Fernando Sequeira Present address: Departamento de Biologia, Faculdade de
Ciências, Universidade do Porto, Porto, Portugal AUTHORS AND AFFILIATIONS * National Institute of Science and Technology in Interdisciplinary and Transdisciplinary Studies in Ecology and
Evolution (INCT IN-TREE), Institute of Biology, Federal University of Bahia, Salvador, BA, Brazil Henrique Batalha-Filho, Silvia Britto Barreto & Mario Henrique Barros Silveira *
Departamento de Genética e Biologia Evolutiva, Instituto de Biociências, Universidade de São Paulo, São Paulo, Brazil Cristina Yumi Miyaki * CIBIO, Centro de Investigação em Biodiversidade e
Recursos Genéticos, InBIO Laboratório Associado, BIOPOLIS Program in Genomics, Biodiversity and Land Planning, Campus de Vairão, Universidade do Porto, 4485-661, Vairão, Portugal Sandra
Afonso, Nuno Ferrand, Miguel Carneiro & Fernando Sequeira * Departamento de Biologia, Faculdade de Ciências, Universidade do Porto, Porto, Portugal Nuno Ferrand Authors * Henrique
Batalha-Filho View author publications You can also search for this author inPubMed Google Scholar * Silvia Britto Barreto View author publications You can also search for this author
inPubMed Google Scholar * Mario Henrique Barros Silveira View author publications You can also search for this author inPubMed Google Scholar * Cristina Yumi Miyaki View author publications
You can also search for this author inPubMed Google Scholar * Sandra Afonso View author publications You can also search for this author inPubMed Google Scholar * Nuno Ferrand View author
publications You can also search for this author inPubMed Google Scholar * Miguel Carneiro View author publications You can also search for this author inPubMed Google Scholar * Fernando
Sequeira View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS HB-F conceived the original idea. HB-F, MC, and FS designed the research. HB-F and
MHBS conducted the fieldwork. SA performed the laboratory work. HB-F and SBB performed the analyses. HB-F wrote the manuscript with significant contributions from SBB and FS. All other
authors contributed to the revision of the final version of the manuscript. CORRESPONDING AUTHOR Correspondence to Henrique Batalha-Filho. ETHICS DECLARATIONS COMPETING INTERESTS The authors
declare no competing interests. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional
affiliations. Associate editor: Lounès Chikhi. SUPPLEMENTARY INFORMATION SUPPLEMENTAL MATERIAL RIGHTS AND PERMISSIONS Springer Nature or its licensor (e.g. a society or other partner) holds
exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely
governed by the terms of such publishing agreement and applicable law. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Batalha-Filho, H., Barreto, S.B., Silveira, M.H.B. _et
al._ Disentangling the contemporary and historical effects of landscape on the population genomic variation of two bird species restricted to the highland forest enclaves of northeastern
Brazil. _Heredity_ 132, 77–88 (2024). https://doi.org/10.1038/s41437-023-00662-1 Download citation * Received: 18 January 2023 * Revised: 02 November 2023 * Accepted: 02 November 2023 *
Published: 20 November 2023 * Issue Date: February 2024 * DOI: https://doi.org/10.1038/s41437-023-00662-1 SHARE THIS ARTICLE Anyone you share the following link with will be able to read
this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative