Play all audios:
ABSTRACT Climate change is rapidly affecting species distributions across the globe, particularly in the North Atlantic. For highly mobile and elusive cetaceans, the genetic data needed to
understand population dynamics are often scarce. Cold-water obligate species such as the white-beaked dolphin (_Lagenorhynchus albirostris_) face pressures from habitat shifts due to rising
sea surface temperatures in addition to other direct anthropogenic threats. Unravelling the genetic connectivity between white-beaked dolphins across their range is needed to understand the
extent to which climate change and anthropogenic pressures may impact species-wide genetic diversity and identify ways to protect remaining habitat. We address this by performing a
population genomic assessment of white-beaked dolphins using samples from much of their contemporary range. We show that the species displays significant population structure across the
North Atlantic at multiple scales. Analysis of contemporary migration rates suggests a remarkably high connectivity between populations in the western North Atlantic, Iceland and the Barents
Sea, while two regional populations in the North Sea and adjacent UK and Irish waters are highly differentiated from all other clades. Our results have important implications for the
conservation of white-beaked dolphins by providing guidance for the delineation of more appropriate management units and highlighting the risk that local extirpation may have on species-wide
genetic diversity. In a broader context, this study highlights the importance of understanding genetic structure of all species threatened with climate change-driven range shifts to assess
the risk of loss of species-wide genetic diversity. SIMILAR CONTENT BEING VIEWED BY OTHERS DISTRIBUTION OF GENETIC DIVERSITY REVEALS COLONIZATION PATTERNS AND PHILOPATRY OF THE LOGGERHEAD
SEA TURTLES ACROSS GEOGRAPHIC SCALES Article Open access 22 October 2020 GENETIC AND DEMOGRAPHIC HISTORY DEFINE A CONSERVATION STRATEGY FOR EARTH’S MOST ENDANGERED PINNIPED, THE
MEDITERRANEAN MONK SEAL _MONACHUS MONACHUS_ Article Open access 11 January 2021 GENOMIC INSIGHTS INTO THE HISTORICAL AND CONTEMPORARY DEMOGRAPHICS OF THE GREY REEF SHARK Article 16 March
2022 INTRODUCTION Understanding within-species connectivity and diversity is essential for informing conservation management and can help in assessing the impact of local extinctions for
species-wide genetic variation (Palsbøll et al. 2007; Pavlova et al. 2017). In the marine environment, limitations to dispersal are more subtle than in terrestrial systems due to the
scarcity of geophysical barriers. Nevertheless, marine species often display genetic structuring influenced by environmental conditions such as physiography, salinity, thermal niches, social
structure, movement patterns, and behavioural specialisation (Craig and Herman 1997; Foote et al. 2011; Hoelzel 2009). Disentangling these patterns can be challenging, yet is critical for
conservation management of species threatened with anthropogenic impacts and environmental shifts driven by global climate change (Palsbøll et al. 2007). The latter is particularly alarming
for species inhabiting cold temperatures, as their available habitat is likely to shift under global warming (Louis et al. 2020; Pauls et al. 2013). This can have strong impacts on
distribution, abundance, and species-wide genetic diversity. For example, when differentiated populations are present in areas subject to strong environmental change with limited
availability to new suitable habitat, extirpation may result in the loss of a significant proportion of species-wide genetic diversity (Razgour et al. 2013). This, in turn, can negatively
affect the ability of the species to adapt to future changes, as high genetic diversity is believed to be a major driver of positively selected mutations (Kardos et al. 2021). The risk of
local extirpation, either driven by climate change or by direct anthropogenic impact, can only be accurately assessed when sufficient information on range-wide population structure of a
species is available. As global recognition of the significance of genetic variation in biodiversity conservation grows and national and international bodies increasingly enforce commitments
to protect ocean habitats (CBD 2022; DeWoody et al. 2021; United Nations 2015), there has never been a more urgent time to explore the connectivity of cold-water marine species to achieve
appropriate conservation management strategies in response to challenges posed by global climate change. The white-beaked dolphin (_Lagenorhynchus albirostris_) is a cold-water obligate
cetacean inhabiting continental shelf, shelf edge and continental slope waters of the temperate and sub-polar North Atlantic (Galatius and Kinze 2016). The species is common in the Canadian
Atlantic, Greenland, Iceland, the Barents Sea, and parts of the North Sea and adjacent UK and Irish waters (Hammond et al. 2013; Hansen and Heide-Jørgensen 2013; Kinze et al. 2018; Lien et
al. 2001; Øien 1996; Pike et al. 2019; Fig. 1a). In past decades a considerable northward shift in its southern distribution has been detected suggesting that white-beaked dolphins in the
North Sea and adjacent UK waters avoid waters with higher sea surface temperatures (SSTs) (IJsseldijk et al. 2018; MacLeod et al. 2007; Waggitt et al. 2020). As SSTs in the North Sea are
projected to further increase, more frequently exceeding the suitable threshold for white-beaked dolphins, the species risks facing a considerable northward-shift in this region (Dieterich
et al. 2019; Evans and Waggitt 2020; Johns et al. 2003; Lambert et al. 2014). Additionally, the species faces numerous direct anthropogenic pressures, such as bycatch in commercial fisheries
(Reeves et al. 2013), local unregulated harvesting (Takekawa 2000; Piniarneq 2021), prey depletion (Jackson et al. 2001), anthropogenic noise and chemical contaminants (Stone and Tasker
2006; Galatius, Bossi et al. 2013; Williams et al. 2023). In order to understand the consequences of predicted habitat shifts and other threats, it is necessary to investigate how
white-beaked dolphins are connected across their range. As of now, morphometric studies have described differences in skull characteristics between the western North Atlantic and the North
Sea indicative of separate populations (Mikkelsen and Lund 1994), suggesting some level of population structure. This was confirmed by genetic studies, supporting a distinction between
northeast and northwest Atlantic populations, but also within the northeast Atlantic (Banguera-Hinestroza et al. 2010). Fernández et al. (2016) generated a panel of genome-wide SNPs, yet the
study lacks an assessment of within-species genetic structure. For the conservation management of the white-beaked dolphin, a comprehensive assessment of population structure is needed to
delineate more appropriate management units, as highlighted within the Agreement on the Conservation of Small Cetaceans of the Baltic, Northeast Atlantic, Irish and North Seas (ASCOBANS;
ASCOBANS 2019). Here, we explore species-wide population structure, genetic diversity and contemporary geneflow of the white-beaked dolphin across the North Atlantic with the aim of
providing guidance for improved conservation management of the species. METHODS SAMPLING AND DNA EXTRACTION A total of 169 tissue samples were obtained from preexisting archives and
consisted of stranded (_n_ = 133), by-caught (_n_ = 23) or biopsied (_n_ = 13) white-beaked dolphins sampled between 1992 and 2021. Prior to any further processing, the tissue samples were
stored either dry frozen or in a ≥80% ethanol solution at −80 °C. The geographical distribution of the samples ranged from eastern Canada in the western North Atlantic (_n_ = 3; biopsies),
Iceland in the central North Atlantic (_n_ = 23; bycatch) and the Barents Sea (_n_ = 10; biopsies), Scotland (_n_ = 81; strandings), England (_n_ = 24; strandings), Ireland (_n_ = 3;
strandings), Denmark (_n_ = 4; strandings), Germany (_n_ = 12; strandings), The Netherlands (_n_ = 8; strandings), and France (_n_ = 1; stranding) in the eastern North Atlantic or adjacent
waters (Figs. 1a and 2a). Genomic DNA was extracted using the Maxwell PureFood & GMO Authentication kit on a Maxwell RSC extraction robot. DNA was quantified using a Qubit fluorometer
and tested for high molecular weight DNA content on a 1% agarose gel. DNA concentrations were standardised across samples and subsequently, 166 samples and 18 duplicate samples were
submitted for DArTseq™ (Diversity Arrays Technology, Canberra, Australia). The DArTseq™ assay involves complexity reduction using a pair of restriction enzymes and amplification of the
fragments via PCR. The resulting library is then shotgun-sequenced on an Illumina HiSeq 2500, producing single-end sequenced reads with a length of 130 bp. Due to later acquisition, the
three samples from eastern Canada were submitted to Azenta Life Sciences (Chelmsford, Massachusetts, United States) for Short-Read Non-Human Whole Genome Sequencing (WGS) on an Illumina
NovaSeq to ensure coverage of the same markers retained by the DArTseq™ approach. The raw data produced by Azenta was paired-end shotgun-sequenced with a fragment size of 150 bp. READ
PROCESSING AND MAPPING Single-end DArTseq™ reads were quality-checked using FastQC and barcodes were trimmed using the _process_radtags_ function within STACKS v2.5.4 (Andrews 2010; Catchen
et al. 2013). Similarly, for the paired-end WGS reads FastQC was used for quality control and Trim Galore v0.6.6 was used to remove Illumina adapters. Both the DArTseq™ and WGS reads were
mapped to the chromosomal-level genome assembly of _Lagenorhynchus albirostris_ (Accession number: GCA_949774975.1) using the _bwa mem_ function in BWA v0.7.17 (Li 2013). The output was
assessed for mapping percentage and written to bam files using samtools v1.9 (Li et al. 2009). Following this, the mapped files were sorted (_SortSam_) and all reads were assigned to a read
group (_AddOrReplaceReadGroups_) using Picard Tools (Broad Institute 2019). Additionally, duplicates which can arise during library preparation were tagged and removed (_MarkDuplicates_) in
the WGS reads. The output was indexed, and depth of coverage was calculated using samtools. VARIANT CALLING Analysis of Next Generation Sequencing Data (ANGSD) software was used to detect
variants and calculate genotype likelihoods across the 169 samples (Korneliussen et al. 2014). An initial variant calling step was performed on all samples using base call and mapping
quality filters (-_minMapQ_ 30, -_minQ_ 30, -_SNP_pval_ 1e−6) and the output was written to PLINK format by specifying the _-doPlink_ flag, which translates the variants to called genotypes.
This initial dataset was inspected for levels of missing data and distribution of heterozygosity using the _--het_ and --_missing_ functions within PLINK v.1.09 (Purcell et al. 2007). Seven
samples showed missing data ≥ 30% and three samples displayed above-average heterozygosity suggesting cross-contamination issues. These samples were removed from the workflow. Furthermore,
pairwise relatedness between individuals was calculated by combining output from the PLINK _--genome_ function and output from the programme NGSrelate (Korneliussen and Moltke 2015). Two
pairs of samples showed a pairwise relatedness coefficient (_PI_HAT_) above 0.5 corresponding to first-degree relatedness (parent-offspring or full-siblings) and the sample with the lower
genotyping rate of each pair was removed from subsequent analyses (Supplementary Fig. S1). The dataset for investigation of population structure thus comprised 157 individuals and the
genotype likelihood calculation in ANGSD was repeated with additional filters on read depth (-_setMinDepth 785_, -_setMaxDepth 3140_) corresponding to a minimum depth of coverage of 5X and a
maximum depth of coverage of 20X per locus per individual to avoid potential bases arising from sequencing errors following recommendations by O’Leary et al. (2018). Furthermore, we
identified variants that were located in an interspersed repeat region using the programme RepeatMasker and excluded those from the variant calling by specifying the remaining sites using
the _-sites_ flag in ANGSD. Further filtering of the multilocus genotypes was conducted in PLINK using a minor allele count of 2 to remove variants generated through uncertainties in base
calling during sequencing. We examined the patterns of linkage disequilibrium decay in our data and observed a relatively steep decline in linkage disequilibrium in the initial portion of
your linkage disequilibrium decay graph drawn by the programme ngsLD (Fox et al. 2019). This suggests stronger linkage patterns among nearby SNPs and therefore, we used the --_indep_
function in PLINK to prune loci affected by linkage disequilibrium with a window size of 50 kb, a step size of 5 and a variant inflation factor of 2. The final dataset comprised 1092 Single
Nucleotide Polymorphisms (SNPs) for all downstream population genetic analyses. POPULATION STRUCTURE We investigated population structure using a number of different approaches. First, we
performed a Principal Components Analysis (PCA) in PCAngsd (Meisner and Albrechtsen 2018). PCA is a dimensionality reduction approach, summarising genetic variation into Principal Components
(PCs), which can be projected into axes to visualise genetic clustering. This approach is not influenced by geographic information. Eigenvalues of the first 20 PCs were inferred from the
covariance matrix generated by PCAngsd. In R, population structure was visualised by plotting PCs one and two and PCs two and three (R Core Team 2022). To investigate patterns of fine-scale
and sex-mediated population structure, the PCA was also performed with putative population assignments based on sampling site and for each sex separately, respectively. Second, _K_-means
clustering was conducted to estimate the number of ancestral populations in the dataset using a maximum likelihood approach in NGSadmix (Skotte et al. 2013) and a Bayesian approach in
Structure both with and without a-priori population assignments. A-priori assignments were informed by the clustering retained from the PCA. We investigated the most likely number of genetic
clusters present in the dataset by calculating both DeltaK and Log Likelihood from the NGSadmix output and DeltaK using the Evanno method from the Structure output (Evanno et al. 2005;
Supplementary Figs. S2 and S3). We examined the fit of the admixture proportions derived from the NGSadmix algorithm to its model assumptions by correlating the residual differences between
called and predicted genotypes with the EvalAdmix software (Garcia-Erill and Albrechtsen 2020; Supplementary Fig. S4). The individual admixture proportions for each _K_ and the correlation
of residuals were plotted in R. Finally, in order to investigate the significance of genetic population structure in the dataset, we grouped the samples into the populations informed by the
approaches above and calculated the Weir and Cockerham pairwise fixation index (_F__ST_) with 10,000 bootstraps using functions embedded in the _DartR_ package (Weir and Cockerham 1984;
Mijangos et al. 2022). Additionally, we grouped the samples by sampling sites (i.e., by country) and calculated pairwise _F__ST_ to investigate patterns of fine-scale population structure.
CONTEMPORARY GENE FLOW We estimated the proportion and direction of contemporary geneflow between genetic populations and between sampling sites using the BA3-SNPS extension of the software
BayesAss, which enables computation of large genomic datasets (Mussmann et al. 2019; Wilson and Rannala 2003). In the first instance, initial runs were performed using the BA3-SNPs-autotune
function to determine the optimal combination of the mixing parameters _deltaM_ (mixing parameter for migration rates), _deltaA_ (mixing parameter for allele frequencies) and _deltaF_
(mixing parameter for inbreeding coefficients). These parameters were set to _deltaM_ = 0.1563, _deltaA_ = 0.3250 and _deltaF_ = 0.0500. Five separate runs of BA3-SNPS were performed on
different seeds with 10,000,000 MCMC iterations and 1,000,000 burn-ins on sampling intervals of 1000. Chain convergence of the runs was assessed in R and significance of the retained
migration rates was assessed by a 95% confidence interval calculated as mean migration rate ±1.96 x mean standard deviation (Supplementary Fig. S5). The proportion and directionality of
geneflow between populations was visualised in R. ISOLATION BY DISTANCE We tested the correlation of geographic and genetic distance by performing redundancy analysis and an ANOVA test on
Euclidean distance matrices of geographic distances (_km_) and pairwise fixation indices (_F__ST_) calculated between sampling sites using the _vegan_ package in R. To achieve this, we
calculated the minimum marine distance between sampling sites using a workflow described in Assis et al. (2013). Genetic distances were transformed to a continuous scale as
\({GD}=\frac{{F}_{{ST}}}{(1-{F}_{{ST}})}\) to allow for correlation with geographic distances. The correlation of geographic distances and corresponding fixation indices was subsequently
visualised in R. MULTILOCUS HETEROZYGOSITY To investigate variation in genetic diversity across populations, we calculated the multilocus heterozygosity (_MLH_) across the 1092 SNPs as
described in Stoffel et al. (2016) per population using the package _InbreedR_. Based on the detected population structure in the dataset, we grouped the individuals into their corresponding
populations. Additionally, we visualised _MLH_ distribution across the entire sample set. INVESTIGATION OF A CONTACT ZONE Upon initial inspection of the observed structure, each analysis
was repeated within a more localised approach in the North Sea and adjacent UK and Irish waters, to investigate finer-scale structure and the putative existence of a region of strong
admixture in east Scotland in greater detail. For calculation of fixation indices, estimation of migration rates and heterozygosity, this was achieved by removing the admixed individuals
from east Scotland to retain unbiased estimates. RESULTS DATA QUALITY The percentage of reads that mapped to the reference genome was 100% in almost all the samples. The mean coverage of all
covered regions in the genome across all DArTseq™ samples was 12.75X and across the three WGS samples it was 8–10X across the entire genome. The initial number of variants detected in the
unfiltered dataset was 542,232 SNPs across 169 individuals, which was reduced to 1092 highly informative SNPs across 157 individuals. Comparison of the 18 duplicate pairs ensured no
genotyping errors were present our analyses. POPULATION STRUCTURE We investigated the population structure present in the dataset using complexity reduction, maximum likelihood and Bayesian
approaches combined with estimation of pairwise fixation indices and isolation-by-distance analysis. Combining results from all analyses, we observed both significant broad-scale and
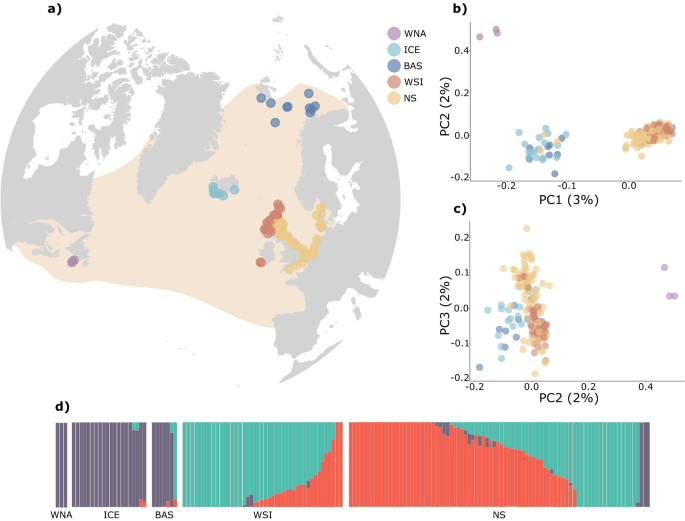
fine-scale population structure across the range of the white-beaked dolphin. Mapping genetic origin against sampling location shows a clear differentiation of geographically isolated
populations into four genetic clusters (Fig. 1b, c). Samples collected in both Iceland and the Barents Sea were assigned to the same genetic clade with a clear separation from the British
Isles and the North Sea along the first PC axis. Similarly, the three individuals sampled in eastern Canada (WNA) were separated further along PC1, forming a separate cluster (Fig. 1b).
Interestingly, NGSadmix and STRUCTURE analysis did not identify the WNA samples as a separate genetic cluster and grouped them together with the Icelandic and Barents Sea samples (Fig. 1d,
Supplementary Figs. S6 and S7). Finer structure could be identified with separation of white-beaked dolphins sampled around west Scotland and Ireland and the coastlines of the North Sea
along the third PC axis (Fig. 1c). This was further corroborated by NGSadmix and Structure analyses (Fig. 1d, Supplementary Figs. S6 and S7). A subsequent assessment of finer-scale and
sex-mediated structure by a separate PCA confirms the overall structure detected by the previous approaches, demonstrating no clear difference in population structure between male and female
white-beaked dolphins, implying the absence of sex-mediated dispersal (Supplementary Fig. S8). Based on these results, naming conventions for the genetic clusters are introduced as the
following regions: Western North Atlantic (WNA), Iceland and Barents Sea (ICE&BAS), west Scotland and Ireland (WSI) and North Sea (NS). Additionally, based on the pattern observed
between NS and WSI, subsequent analyses were also performed with the eastern Scottish (E_SCOT) individuals as a separate group to investigate the influence a potential contact zone in this
region may have on the estimation of population genetic parameters (Fig. 2a). We find that the majority of individuals previously grouped with NS but clustering with WSI were indeed from
eastern Scotland (Fig. 2b). Admixture proportions for the three regions separately further visualise that eastern Scotland seems to be a contact zone between individuals of the southern and
central North Sea and individuals sampled in Ireland and the west coast of Scotland (Fig. 2c). Pairwise fixation indices between the four populations retained by the PCA confirm that the
samples from the western North Atlantic were significantly differentiated from the North Sea and western Scotland and Ireland samples (_F__ST_WNAvs.NS_ = 0.05943055, _p_ = 0.000;
_F__ST_WNAvsWSI_ = 0.078774106, _p_ = 0.000), but displayed a lower, yet significant, differentiation to the Iceland and Barents Sea samples (_F__ST_WNAvs.ICE&BAS_ = 0.01886975, _p_ =
0.002). Similarly, the animals sampled in Iceland and the Barents Sea were significantly differentiated from the North Sea (_F__ST_ICE&BASvsNS_ = 0.03946870, _p_ = 0.000) and western
Scotland and Ireland (_F__ST_ICE&BASvs.WSI_ = 0.05496347, _p_ = 0.000), while the latter two regions displayed a weak, but statistically significant differentiation (_F__ST_NSvs.WSI_ =
0.006011101, _p_ = 0.000). When excluding eastern Scottish samples, the fixation index between the North Sea and western Scotland and Ireland clades increases (_F__ST_NSvs.WSI_ =
0.008685244, _p_ = 0.000), indicative of a large proportion of admixed individuals from both populations in this region. The pairwise _F__ST_ values calculated between sampling sites are
visualised in Fig. 3a and exact values can be obtained from Supplementary Table S2. CONTEMPORARY GENE FLOW Estimation of the proportion and direction of geneflow between populations
performed in BayesAss suggested little introgression from the western North Atlantic population into any of the other populations (Fig. 3c). Likewise, the Iceland and Barents Sea population
and the two populations of the North Sea and western Scotland and Ireland showed very little evidence of gene flow in either direction. However, a higher proportion of gene flow was detected
between Iceland and the Barents Sea and western North Atlantic populations in the direction of the western North Atlantic, specifically from Iceland to western North Atlantic with an
estimated 0.166 migrants per generation (see Supplementary Table S1 for details). Similarly, there is a strong signal of unidirectional geneflow from Iceland into the Barents Sea (_m_ =
0.222). Furthermore, a high level of introgression was detected between the two geographically neighbouring North Sea and western Scotland and Ireland populations with the majority of
geneflow being facilitated by the North Sea population (_m_ = 0.306). Interestingly, although the estimated migration rate remained similar when excluding eastern Scottish samples (_m_ =
0.3265), the direction of geneflow reversed to a unidirectional influx from western Scotland and Ireland to the North Sea. All other migration rates did not exceed 0.06 migrants per
generation and were therefore considered low. ISOLATION BY DISTANCE We investigated patterns of isolation by distance (IBD) by correlating geographic distance with pairwise fixation indices
(genetic distance) between all sampling sites. The redundancy analysis (RDA) showed a moderate but statistically significant correlation between geographic and genetic distance (_r__2_ =
0.3989931, _p_ = 0.001). The IBD curve confirms that most datapoints fit within the confidence interval, but some points outside the general trend suggest deviations from IBD in both
directions that is, stronger connectivity than expected under pure IBD (below curve) and stronger differentiation than expected under pure IBD (above curve; Fig. 3b). GENETIC DIVERSITY To
assess genetic variation across the dataset, we calculated individual multilocus heterozygosity (_MLH_). We found _MLH_ was normally distributed with a mean of 0.147 across all samples (min
= 0.079, max = 0.173, Supplementary Fig. S9). When comparing _MLH_ between genetic populations, we found little difference in heterozygosity between all sampled populations, but the western
North Atlantic population displays slightly higher yet non-significant _MLH_ compared to the other populations (Fig. 3d). The admixed eastern Scottish individuals did not inflate the
heterozygosity estimates of the North Sea population as assessed by excluding these in a separate estimate (_MLH_ = 0.1444 with E_SCOT vs. _MLH_ = 0.1447 with E_SCOT removed). DISCUSSION
Exploring the extent of genetic connectivity and differentiation across the range of the white-beaked dolphin is essential for conservation management, particularly given the numerous
anthropogenic impacts on dolphin populations such as bycatch, accumulation of chemical contaminants and traditional hunts, as well as the putatively rapidly progressing effects of increasing
SSTs threatening cold-water obligate species with habitat shifts. Using a combination of reduced representation and whole-genome sequencing, we investigated population structure, gene flow
and genetic diversity in white-beaked dolphins from ten different sampling locations. We detect both broad-scale structure across the North Atlantic and fine-scale structure in the eastern
North Atlantic and adjacent waters. The results of this study allow for a more informed delineation of management units for conservation and highlight the importance of population genomics
in biodiversity conservation of species facing changes in their habitat amid global climate change. Principal Components Analysis of 157 white-beaked dolphins detected a pattern of four
differentiated clusters. Three of the four populations were also detected using _K_-means clustering approaches, but western North Atlantic samples were continuously grouped with Icelandic
and Barents Sea individuals. This is likely due to limitations of the programmes Structure and NGSadmix to detect structure when gene flow is high and sample sizes are small (Waples and
Gaggiotti 2006). The evaluation of the fit of our data to the admixture algorithm confirms that this method may not be able to disentangle the full ancestral history, likely due to gaps in
sampling coverage across the species’ range and associated assumptions the algorithm makes based on the provided data set. Statistical evaluation of panmixia using Weir and Cockerham’s
pairwise fixation indices reject the null hypothesis of continuous genetic connectivity between Iceland and Barents Sea and western North Atlantic, supporting the existence of four
differentiated populations as shown in the PCA. We therefore conclude that _K_ = 4 comprising of the western North Atlantic, Iceland and Barents Sea, North Sea, and west Scotland and
Ireland, is the most likely number of populations in our dataset. Pairwise _F__ST_ values were highest between western North Atlantic and west Scotland and Ireland and North Sea populations.
Indeed, migration between these regions was very low in our data and a significant correlation of geographic and genetic distance suggests isolation by distance contributing largely to the
observed differentiation. This is in agreement with previous genetic studies using mtDNA and microsatellite loci and a morphometric study describing distinct differences in skull
characteristics between dolphins from the North Sea and the western North Atlantic (Banguera-Hinestroza et al. 2010; Mikkelsen and Lund 1994). In direct contrast to the differentiation
between the western North Atlantic and the North Sea, western Scotland and Ireland is the relatively strong connectivity between western North Atlantic and Iceland and Barents Sea
populations, despite an apparent hiatus in distribution between the two latter areas (Pike et al. 2019). Remarkably, a complete homogeneity of genotypes from Iceland and the Barents Sea was
found and is indicative of frequent (0.222 migrants per generation) long-distance individual migration events between the two regions as corroborated by our analysis on contemporary gene
flow and in line with observed movement capabilities of the species (Rasmussen et al. 2013). Similarly, a relatively high migration rate (0.166 migrants per generation) was found between
Iceland and the western North Atlantic, yet genetic distinction is persistent but weak. Abundance estimates from Iceland and the Barents Sea suggest large population sizes (Byrd et al. 2020;
Øien 1996; Pike et al. 2019) which could be obscuring the presence of differentiation between two otherwise demographically separate clades (Waples 1998). Regular geneflow between
populations and large population sizes could also contribute to the higher heterozygosity values in the western North Atlantic, though this could also be an artifact of different sequencing
techniques in this population and the possibility of introducing batch effects in the estimates (Lou and Therkildsen 2022). Within the eastern North Atlantic and adjacent waters, we detected
a clear separation of Icelandic and Barents Sea white-beaked dolphins from individuals sampled around western Scotland and Ireland and the North Sea, as well as a regional separation of
individuals sampled off the coast of the North Sea and those sampled off western Scotland and Ireland with a region of strong admixture in eastern Scotland. This result was in part
anticipated and consistent with results from a previous study by Banguera-Hinestroza et al. (2010) who compared Barents Sea samples to the British Isles and North Sea. The introduction of
samples from Iceland in our study gives a new dimension to the overall pattern of structure found in this species, as the minimal marine distance between sampling sites in Iceland and, for
example, the Netherlands is comparable to the distance between Iceland and the Barents Sea (~2200 km) yet genetic distances and migration rates are in stark contrast. Hence, there is a
strong implication that ecological factors could influence population structure in the eastern North Atlantic and adjacent waters. This is further supported by the consistent pattern of
regional structure found between the North Sea and west Scotland and Ireland. The region of strong admixture between white-beaked dolphins of the North Sea and of western Scotland and
Ireland, located at the eastern Scottish coast, brings up interesting questions about the factors driving this pattern. Ecological differences may in part be responsible for the detected
differentiation between the two neighbouring clades, and the occurrence of a contact zone could reflect a response to environmental change and resulting change in behaviour suggesting the
contact zone is a recent phenomenon. An alternative explanation to the pattern could be a retrieval of a separate refugium population to the southern North Sea during the most recent LGM,
which has been argued as a potential driver for regional structure in marine species of the North Atlantic (Hewitt 2000; Hoarau et al. 2007). The limited understanding of white-beaked
dolphin ecology, life history and habitat use hampers the interpretation of drivers of the observed population structure. Notably, seasonal migration from higher latitudes in the winter to
lower latitudes in the summer have been observed in various regions (Canning et al. 2008; Fall and Skern-Mauritzen 2014; Pike et al. 2019). This could be influenced by numerous factors such
as responses to migratory prey, site fidelity to certain areas during mating season, competition from other species or predator avoidance. Regarding diet, white-beaked dolphins have been
reported to target higher level trophic gadoid fish with some regional variation across their range based on stomach content analyses (Dong et al. 1996; Jansen et al. 2010; Fall and
Skern-Mauritzen 2014; Schick et al. 2020; Samarra et al. 2022). However, studies using stable isotopes show a clear preference for pelagic squids in the western North Atlantic versus a
preference for higher trophic level fish in the eastern North Atlantic and Iceland (Samarra et al. 2022; Plint et al. 2023; Kiszka and Caputo, unpublished data). This difference may in part
explain the elevated genetic distance that we found in our IBD analysis between the two sites of the North Atlantic. Within the eastern North Atlantic, seasonal occurrence during summer
months has been argued to possibly result from site fidelity in both the North Sea and west Scotland and Ireland, potentially driving the fine-scale structure as observed in our study
(Reeves et al. 1999; Canning et al. 2008; Brereton et al. 2013; Galatius, Jansen et al. 2013). Contrastingly, long-term photo ID monitoring of Icelandic dolphins suggest no strong signal of
site fidelity (Bertulli et al. 2015). Altogether, further studies on white-beaked dolphin movement, diet, behaviour, and ecology of different populations are needed to explore potential
drivers of the observed population structure and inform a more targeted management approach. IMPLICATIONS FOR CONSERVATION MANAGEMENT Our findings could have significant implications for
conservation management at both regional and North Atlantic basin-wide scales by providing new evidence on fine-scale population structure of white-beaked dolphins. Currently, the species
receives varying degrees of management; in the western North Atlantic, white-beaked dolphins are considered a single stock across their western range (Byrd et al. 2020). In Icelandic and
Norwegian waters, the species receives no targeted management, while in its southern distribution it is managed as a single management unit (MU) comprising of the North Sea and the waters
extending beyond the western coast of Ireland and the UK (IAMMWG 2015). Evans and Teilmann (2009) compiled all available information on the species and recommended four MUs comprising of the
Labrador shelf, Icelandic waters, the Barents Sea and the North Sea and adjacent waters. Our results largely support this delineation, but based on our genetic analysis, our recommendations
differ slightly. The Labrador shelf population is only represented by three samples in our study from eastern Canada. These samples represent a differentiated clade in our analyses,
generally supportive of the distinction of this region as a separate MU. However, a more in-depth assessment of structure is needed in this region, covering larger areas, and increasing
sample size. An important aim for future studies will be the introduction of samples from Greenland, especially in the light of increasing rates of removals through traditional hunts and the
uncertainty on the sustainability of these hunts (Piniarneq 2021). Our findings also reveal strong connectivity between white-beaked dolphins sampled in Iceland and the Barents Sea, and
strong differentiation between the Iceland and Barents Sea and the North Sea and west Scotland and Ireland. Populations in higher latitudes such as the Iceland and Barents Sea population are
unlikely to experience habitat loss due to increasing SSTs and may in fact find more available habitat as sea ice retreats (Stafford et al. 2022). However, it may still be useful to assess
white-beaked dolphins in these regions regarding their distribution, habitat use and behaviour as well as impact of anthropogenic activities to investigate their responses to potential
environmental change and learn more about this populations’ ecology. It is recommended that the genetic connectivity between Iceland and the Barents Sea should be considered in future
assessments of this population. Most strikingly, our findings on fine-scale structure between the North Sea and western Scotland and Ireland warrant reconsideration of current local
management (IAMMWG 2015). In this part of their range, white-beaked dolphins appear to strongly associate with SSTs below 12–13 °C (MacLeod et al. 2007) and therefore are likely to be
especially vulnerable to increasing SSTs (Evans and Waggitt 2020). Furthermore, this region has been identified as a high- risk area for strong anthropogenic impact from climate change,
pollution, and fishing (Davidson et al. 2012). Populations of white-beaked dolphins on the edge of their southern distribution are therefore likely to be impacted by climate-change
associated habitat shifts (Lambert et al. 2014), in addition to numerous direct threats (Stone and Tasker 2006; Bearzi et al. 2006; Reeves et al. 2013; Galatius, Bossi et al. 2013; Williams
et al. 2023). A recent northward-shift in their distribution based on strandings data (IJsseldijk et al. 2018; Williamson et al. 2021) and predictive habitat modelling (Lambert et al. 2014)
is indicative of an ongoing contraction of suitable habitat around the British Isles and North Sea. The responses of the two local populations are difficult to predict. Possible scenarios
range from a retreat to small pockets of suitable habitat, leading to small vulnerable populations, the total extirpation of the species in the area or a northward-shift and subsequently
increased connectivity into waters currently occupied by dolphins of the genetically differentiated population around Iceland and the Barents Sea. Our assessment of local structure in this
region indicates that eastern Scotland may currently be a contact zone for dolphins from the two southern clades, suggesting that further admixture could weaken the observed structure over
time. Future genetic monitoring of these regions could help to predict how those populations may interact and what the genetic consequences could be. The potential risk of local extirpation
of these two southern populations, and consequently the loss of a significant proportion of species-wide genetic diversity, should be emphasised in future management plans. Furthermore,
formal assessment of the impact of factors that may cause additional mortality such as bycatch, pollution, and marine development should be a priority in future conservation efforts. In a
wider context, our study provides an example of the importance of assessing population genomics in marine species facing pressures from climate change and human impact. As the relevance of
genetic diversity as a pillar of biodiversity conservation for long-term species survival gains acknowledgement from international and national policymakers (United Nations 2015; CBD 2022),
detailed knowledge on the population structure and genetic variability is urgently needed. Using these data to understand the dynamics of these species can help in identifying vulnerable
populations and assess the risk for the loss of species-wide genetic diversity by local depletion and continued human impact. DATA AVAILABILITY Raw sequence reads are available at the
European Nucleotide archive (ENA) under the accession number PRJEB71584. CODE AVAILABILITY All code for analyses and plotting can be accessed at https://github.com/MarcGose/WBD_PopGen.
CHANGE HISTORY * _ 29 JULY 2024 A Correction to this paper has been published: https://doi.org/10.1038/s41437-024-00699-w _ REFERENCES * Andrews S (2010) Babraham Bioinformatics - FastQC A
Quality Control tool for High Throughput Sequence Data * ASCOBANS (2019) Report of the 25th meeting of the ASCOBANS Advisory Committee * Assis J, Coelho NC, Alberto F, Valero M, Raimondi P,
Reed D et al. (2013) High and distinct range-edge genetic diversity despite local bottlenecks. PLoS One 8:e68646 CAS PubMed PubMed Central Google Scholar * Banguera-Hinestroza E, Bjørge
A, Reid RJ, Jepson P, Hoelzel AR (2010) The influence of glacial epochs and habitat dependence on the diversity and phylogeography of a coastal dolphin species: lagenorhynchus albirostris.
Conserv Genet 11:1823–1836 Google Scholar * Bearzi G, Politi E, Agazzi S, Azzellino A (2006) Prey depletion caused by overfishing and the decline of marine megafauna in eastern Ionian Sea
coastal waters (central Mediterranean). Biol Conserv 127:373–382 Google Scholar * Bertulli CG, Tetley MJ, Magnusdottir EE, Rasmussen MH (2015) Observations of movement and site fidelity of
white-beaked dolphins (Lagenorhynchus albirostris) in Icelandic coastal waters using photo-identification. J Cetacea Res Manag 15:27–34 Google Scholar * Brereton T, Lewis K, MacLeod CD
(2013) A recently discovered hotspot for white-beaked dolphins in the English Channel in Proceedings of the ECS / ASCOBANS / WDC Workshop Towards a Conservation Strategy for White-beaked
Dolphins in the Northeast Atlantic. Warsaw, Poland * Broad Institute (2019) Picard Toolkit. Available at http://broadinstitute.github.io/picard * Byrd BL, Chavez-Rosales S, Cole TVN,
Garrison LP, Hatch J, Henry A et al. (2020) U.S. Atlantic and Gulf of Mexico marine mammal stock assessments - 2019 (SA Hayes, E Josephson, K Maze-Foley, and PE Rosel, Eds.). Northeast
Fisheries Science Center (U.S.) NOAA technical memorandum NMFS-NE; 264 * Canning SJ, Santos MB, Reid RJ, Evans PGH, Sabin RC, Bailey N et al. (2008) Seasonal distribution of white-beaked
dolphins (Lagenorhynchus albirostris) in UK waters with new information on diet and habitat use. J Mar Biol Assoc U K 88:1159–1166 Google Scholar * Catchen J, Hohenlohe PA, Bassham S,
Amores A, Cresko WA (2013) Stacks: an analysis tool set for population genomics. Mol Ecol 22:3124–3140 PubMed PubMed Central Google Scholar * CBD (2022) Report of the subsidiary body on
scientific, technical and technological advice on its twenty-fourth meeting CBD/SBSTTA/24/12 * Craig AS, Herman LM (1997) Sex differences in site fidelity and migration of humpback whales
(Megaptera novaeangliae) to the Hawaiian Islands. Can J Zool 75:1923–1933 Google Scholar * Davidson AD, Boyer AG, Kim H, Pompa-Mansilla S, Hamilton MJ, Costa DP et al. (2012) Drivers and
hotspots of extinction risk in marine mammals. Proc Natl Acad Sci 109:3395–3400 CAS PubMed PubMed Central Google Scholar * DeWoody JA, Harder AM, Mathur S, Willoughby JR (2021) The
long-standing significance of genetic diversity in conservation. Mol Ecol 30:4147–4154 PubMed Google Scholar * Dieterich C, Wang S, Schimanke S, Gröger M, Klein B, Hordoir R et al. (2019)
Surface heat budget over the north sea in climate change simulations. Atmosphere 10:272 Google Scholar * Dong JH, Lien J, Nelson D, Curren K (1996) A contribution to the biology of the
White-beaked Dolphin, Lagenorhynchus albirostris, in waters off Newfoundland. Can Field Nat 110:278–287 Google Scholar * Evanno G, Regnaut S, Goudet J (2005) Detecting the number of
clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620 CAS PubMed Google Scholar * Evans, PGH and Waggitt, JJ (2020) Impacts of climate change on
marine mammals, relevantto the coastal and marine environment around the UK. MCCIP Sci Rev 2020:420–454 * Evans PGH, Teilmann J (2009) Report of the ASCOBANS/HELCOM small cetacean population
structure workshops. ASCOBANS/HELCOM, Bonn, Germany Google Scholar * Fall J, Skern-Mauritzen M (2014) White-beaked dolphin distribution and association with prey in the Barents Sea. Mar
Biol Res 10:957–971 Google Scholar * Fernández R, Schubert M, Vargas‐Velázquez AM, Brownlow A, Víkingsson GA, Siebert U et al. (2016) A genomewide catalogue of single nucleotide
polymorphisms in white-beaked and Atlantic white-sided dolphins. Mol Ecol Resour 16:266–276 PubMed Google Scholar * Foote AD, Vilstrup JT, De Stephanis R, Verborgh P, Abel Nielsen SC,
Deaville R et al. (2011) Genetic differentiation among North Atlantic killer whale populations. Mol Ecol 20:629–641 PubMed Google Scholar * Fox EA, Wright AE, Fumagalli M, Vieira FG (2019)
ngsLD: evaluating linkage disequilibrium using genotype likelihoods. Bioinformatics 35:3855–3856 CAS PubMed Google Scholar * Galatius A, Bossi R, Sonne C, Rigét FF, Kinze CC, Lockyer C
et al. (2013) PFAS profiles in three North Sea top predators: metabolic differences among species? Environ Sci Pollut Res Int 20:8013–8020 CAS PubMed Google Scholar * Galatius A, Jansen
OE, Kinze CC (2013) Parameters of growth and reproduction of white-beaked dolphins (Lagenorhynchus albirostris) from the North Sea. Mar Mammal Sci 29:348–355 Google Scholar * Galatius A,
Kinze CC (2016) Lagenorhynchus albirostris (Cetacea: Delphinidae). Mamm Species 48:35–47 Google Scholar * Garcia-Erill G, Albrechtsen A (2020) Evaluation of model fit of inferred admixture
proportions. Mol Ecol Resour 20:936–949 CAS PubMed Google Scholar * Hammond PS, Macleod K, Berggren P, Borchers DL, Burt L, Cañadas A et al. (2013) Cetacean abundance and distribution in
European Atlantic shelf waters to inform conservation and management. Biol Conserv 164:107–122 Google Scholar * Hansen RG, Heide-Jørgensen MP (2013) Spatial trends in abundance of
long-finned pilot whales, white-beaked dolphins and harbour porpoises in West Greenland. Mar Biol 160:2929–2941 Google Scholar * Hewitt G (2000) The genetic legacy of the Quaternary ice
ages. Nature 405:907–913 CAS PubMed Google Scholar * Hoarau G, Coyer JA, Veldsink JH, Stam WT, Olsen JL (2007) Glacial refugia and recolonization pathways in the brown seaweed Fucus
serratus. Mol Ecol 16:3606–3616 CAS PubMed Google Scholar * Hoelzel AR (2009) Evolution of population genetic structure in marine mammal species. In: Population Genetics for Animal
Conservation, Cambridge University Press * IAMMWG (2015) Management Units for cetaceans in UK waters (January 2015). IAMMWG, Peterborough Google Scholar * IJsseldijk LL, Brownlow A, Davison
N, Deaville R, Haelters J, Keijl G et al. (2018) Spatiotemporal analysis in white-beaked dolphin strandings along the North Sea coast from 1991–2017. Lutra 61:153–163 Google Scholar *
Jackson JBC, Kirby MX, Berger WH, Bjorndal KA, Botsford LW, Bourque BJ et al. (2001) Historical overfishing and the recent collapse of coastal ecosystems. Science 293:629–637 CAS PubMed
Google Scholar * Jansen OE, Leopold MF, Meesters EHWG, Smeenk C (2010) Are white-beaked dolphins Lagenorhynchus albirostris food specialists? Their diet in the southern North Sea. J Mar
Biol Assoc U K 90:1501–1508 Google Scholar * Johns TC, Gregory JM, Ingram WJ, Johnson CE, Jones A, Lowe JA et al. (2003) Anthropogenic climate change for 1860 to 2100 simulated with the
HadCM3 model under updated emissions scenarios. Clim Dyn 20:583–612 Google Scholar * Kardos M, Armstrong EE, Fitzpatrick SW, Hauser S, Hedrick PW, Miller JM et al. (2021) The crucial role
of genome-wide genetic variation in conservation. Proc Natl Acad Sci 118:e2104642118 CAS PubMed PubMed Central Google Scholar * Kinze CC, Thostesen CB, Olsen MT (2018) Cetacean stranding
records along the Danish coastline: records for the period 2008-2017 and a -comparative review. Lutra 61(1):87–105 Google Scholar * Korneliussen TS, Albrechtsen A, Nielsen R (2014) ANGSD:
analysis of next generation sequencing data. BMC Bioinforma 15:356 Google Scholar * Korneliussen TS, Moltke I (2015) NgsRelate: a software tool for estimating pairwise relatedness from
next-generation sequencing data. Bioinformatics 31:4009–4011 CAS PubMed PubMed Central Google Scholar * Lambert E, Pierce GJ, Hall K, Brereton T, Dunn TE, Wall D et al. (2014) Cetacean
range and climate in the eastern North Atlantic: future predictions and implications for conservation. Glob Change Biol 20:1782–1793 Google Scholar * Li H (2013) Aligning sequence reads,
clone sequences and assembly contigs with BWA-MEM. Preprint at arXiv https://arxiv.org/abs/1303.3997 * Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N et al. (2009) The sequence
alignment/map format and SAMtools. Bioinformatics 25:2078–2079 PubMed PubMed Central Google Scholar * Lien J, Nelson D, Hai DJ (2001) Status of the White-beaked Dolphin, Lagenorhynchus
albirostris, in Canada. Can Field Nat 115:118–126 Google Scholar * Lou RN, Therkildsen NO (2022) Batch effects in population genomic studies with low-coverage whole genome sequencing data:
causes, detection and mitigation. Mol Ecol Resour 22:1678–1692 CAS PubMed Google Scholar * Louis M, Skovrind M, Samaniego Castruita JA, Garilao C, Kaschner K, Gopalakrishnan S et al.
(2020) Influence of past climate change on phylogeography and demographic history of narwhals, Monodon monoceros. Proc R Soc B Biol Sci 287:20192964 CAS Google Scholar * MacLeod CD, Weir
CR, Pierpoint C, Harland EJ (2007) The habitat preferences of marine mammals west of Scotland (UK). J Mar Biol Assoc U K 87:157–164 Google Scholar * Meisner J, Albrechtsen A (2018)
Inferring population structure and admixture proportions in low-depth NGS data. Genetics 210:719–731 PubMed PubMed Central Google Scholar * Mijangos JL, Gruber B, Berry O, Pacioni C,
Georges A (2022) dartR v2: an accessible genetic analysis platform for conservation, ecology and agriculture. Methods Ecol Evol 13:2150–2158 Google Scholar * Mikkelsen AMH, Lund A (1994)
Intraspecific variation in the dolphins Lagenorhynchus albirostris and L. acutus (Mammalia: Cetacea) in metrical and non-metrical skull characters, with remarks on occurrence. J Zool
234:289–299 Google Scholar * Mussmann SM, Douglas MR, Chafin TK, Douglas ME (2019) BA3-SNPs: contemporary migration reconfigured in BayesAss for next-generation sequence data. Methods Ecol
Evol 10:1808–1813 Google Scholar * Øien N (1996) Lagenorhynchus species in Norwegian waters as revealed from incidental observations and recent sighting surveys. Paper SC/48/SM15 to the IWC
Scientific Committee, Aberdeen * O’Leary SJ, Puritz JB, Willis SC, Hollenbeck CM, Portnoy DS (2018) These aren’t the loci you’e looking for: principles of effective SNP filtering for
molecular ecologists. Mol Ecol 27:3193–3206 Google Scholar * Palsbøll PJ, Bérubé M, Allendorf FW (2007) Identification of management units using population genetic data. Trends Ecol Evol
22:11–16 PubMed Google Scholar * Pauls SU, Nowak C, Bálint M, Pfenninger M (2013) The impact of global climate change on genetic diversity within populations and species. Mol Ecol
22:925–946 PubMed Google Scholar * Pavlova A, Beheregaray LB, Coleman R, Gilligan D, Harrisson KA, Ingram BA et al. (2017) Severe consequences of habitat fragmentation on genetic diversity
of an endangered Australian freshwater fish: a call for assisted gene flow. Evol Appl 10:531–550 PubMed PubMed Central Google Scholar * Pike D, Gunnlaugsson T, Sigurjonsson J, Vikingsson
G (2019) Distribution and abundance of cetaceans in icelandic waters over 30 years of aerial surveys. NAMMCO Sci Publ 11:1–22 Google Scholar * Piniarneq (2021) Jagtinformation og
Fangstregistrering. Government of Greenland, Nuuk, Greenland Google Scholar * Plint T, ten Doeschate MTI, Brownlow AC, Davison NJ, Hantke G, Kitchener AC et al. (2023) Stable isotope
ecology and interspecific dietary overlap among dolphins in the Northeast Atlantic. Front Mar Sci 10:1111295 Google Scholar * Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR,
Bender D et al. (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81:559–575 CAS PubMed PubMed Central Google Scholar * R Core
Team (2022). R: A language and environment for statistical computing. R Found Stat Comput * Rasmussen MH, Akamatsu T, Teilmann J, Vikingsson G, Miller LA (2013) Biosonar, diving and
movements of two tagged white-beaked dolphin in Icelandic waters. Deep Sea Res Part II Top Stud Oceanogr 88–89:97–105 Google Scholar * Razgour O, Juste J, Ibáñez C, Kiefer A, Rebelo H,
Puechmaille SJ et al. (2013) The shaping of genetic variation in edge-of-range populations under past and future climate change. Ecol Lett 16:1258–1266 PubMed PubMed Central Google Scholar
* Reeves RR, McClellan K, Werner TB (2013) Marine mammal bycatch in gillnet and other entangling net fisheries, 1990 to 2011. Endanger Species Res 20:71–97 Google Scholar * Reeves RR,
Smeenk C, Kinze CC, Brownell RL, Lien J (1999) White-beaked dolphin Lagenorhynchus albirostris Gray, 1846. In: Handbook of marine mammals, volume 6: the second book of dolphins and the
porpoises, Academic Press: San Diego, California, pp 1–29 * Samarra FIP, Borrell A, Selbmann A, Halldórson SD, Pampoulie C, Chosson V et al. (2022) Insights into the trophic ecology of
white-beaked dolphins Lagenorhynchus albirostris and harbour porpoises Phocoena phocoena in Iceland. Mar Ecol Prog Ser 702:139–152 CAS Google Scholar * Schick L, IJsseldijk LL, Grilo ML,
Lakemeyer J, Lehnert K, Wohlsein P et al. (2020) Pathological findings in white-beaked Dolphins (Lagenorhynchus albirostris) and atlantic white-sided dolphins (Lagenorhynchus acutus) from
the South-Eastern North Sea. Front Vet Sci 7:262 PubMed PubMed Central Google Scholar * Skotte L, Korneliussen TS, Albrechtsen A (2013) Estimating individual admixture proportions from
next generation sequencing data. Genetics 195:693–702 CAS PubMed PubMed Central Google Scholar * Stafford KM, Farley EV, Ferguson M, Kuletz KJ, Levine R (2022) Northward range expansion
of subarctic upper trophic level animals into the pacific arctic region. Oceanography 35:158–166 Google Scholar * Stoffel MA, Esser M, Kardos M, Humble E, Nichols H, David P et al (2016)
inbreedR: an R package for the analysis of inbreeding based on genetic markers. Methods Ecol Evol 7:1331–1339 Google Scholar * Stone AJ, Tasker ML (2006) The effects of seismic airguns on
cetaceans in UK waters. J. Cetacean Res. Manag. 8:255–263 Google Scholar * Takekawa D (2000) Hunting method and the ecological knowledge of dolphins among the Fanalei villagers of Malaita,
Solomon Islands. SPC Traditional Marine Resource Management and Knowledge Information Bulletin 12:3–11 Google Scholar * United Nations (2015) UN General Assembly Transforming our World: The
2030 Agenda for Sustainable Development A/RES/70/1 * Waggitt JJ, Evans PGH, Andrade J, Banks AN, Boisseau O, Bolton M et al. (2020) Distribution maps of cetacean and seabird populations in
the North-East Atlantic. J Appl Ecol 57:253–269 Google Scholar * Waples R (1998) Separating the wheat from the chaff: patterns of genetic differentiation in high gene flow species. J Hered
89:438–450 Google Scholar * Waples RS, Gaggiotti O (2006) INVITED REVIEW: What is a population? An empirical evaluation of some genetic methods for identifying the number of gene pools and
their degree of connectivity. Mol Ecol 15:1419–1439 CAS PubMed Google Scholar * Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution
38:1358–1370 CAS PubMed Google Scholar * Williams RS, Brownlow A, Baillie A, Barber JL, Barnett J, Davison NJ et al. (2023) Spatiotemporal trends spanning three decades show toxic levels
of chemical contaminants in marine mammals. Environ Sci Technol 57:20736–20749 CAS PubMed PubMed Central Google Scholar * Williamson MJ, ten Doeschate MTI, Deaville R, Brownlow AC,
Taylor NL (2021) Cetaceans as sentinels for informing climate change policy in UK waters. Mar Policy 131:104634 Google Scholar * Wilson GA, Rannala B (2003) Bayesian inference of recent
migration rates using multilocus genotypes. Genetics 163:1177–1191 PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS Samples were collected, stored, and curated by
many organisations involved in this study. We thank all volunteers and staff of the Scottish Marine Animal Stranding Scheme (SMASS) and of the Cetacean Strandings and Investigation
Programme (CSIP) at Zoological Society of London for their effort in collecting samples from Scotland, England, and Wales. We wish to thank all individuals in Germany who helped to collect
carcasses, perform necropsies, and conduct further investigations. The study was partly funded by the German Federal Ministry for Environment, Nature Conservation and Nuclear Safety, the
German Federal Ministry for Research and Education and Ministry of Energy Transition, Agriculture, Environment, Nature and Digitalisation of Schleswig-Holstein (MELUND) and samples from
Germany were transferred under CITES permits DE 207-02 and GB 034. Post-mortem investigations and tissue sampling in the Netherlands is conducted at the Faculty of Veterinary Medicine of
Utrecht University, commissioned by the Ministry of Agriculture, Nature and Food Quality. We are thankful for the help of stranding network volunteers in reporting and retrieving the animals
for post-mortem investigation, and staff and students of Utrecht University for assisting the examinations. Dutch samples were transferred under CITES permit 610516/01. Samples from eastern
Canada (Saint Pierre et Miquelon) were collected under permit #431 from Préfecture de Saint Pierre et Miquelon through a grant from the Office Français de la Biodiversité (COPEMAM) to
Université de La Rochelle and Florida International University and transferred under CITES permit 615900/01. This is contribution #1674 of the Institute of Environment at Florida
International University. In Ireland, data were reported through the Irish Whale and Dolphin group stranding scheme and collection and storage of samples was facilitated by the Irish
Cetacean Genetic Tissue Bank at the National Museum of Ireland and was funded in part by the Heritage Council and the Ireland-Wales Interreg programme. We wish to thank staff and volunteers
of the Fisheries and Maritime Museum for providing samples from Denmark. Samples from Denmark, Ireland, Iceland, and Norway were transferred under CITES permits DK 014 and GB 034. Many
thanks to Ruth Fernandez and Kyle Ewart for their contribution with communication to sample providers and sample transfer logistics. We thank Gísli Víkingsson for organising access to
Icelandic dolphin samples. Gísli passed away in the summer of 2022. We want to thank the editor, Morten Tange Olsen, and two anonymous reviewers for their insightful comments on the
manuscript. For the purpose of open access, the author has applied for a CC-BY public copyright licence to any author accepted manuscript version arising from this submission. FUNDING
Funding was provided through a Ph.D. studentship from the Royal (Dick) School of Veterinary Studies and the Roslin Institute, University of Edinburgh. AUTHOR INFORMATION AUTHORS AND
AFFILIATIONS * Royal (Dick) School of Veterinary Studies and the Roslin Institute, University of Edinburgh, Edinburgh, UK Marc-Alexander Gose, Emily Humble & Rob Ogden * Scottish Marine
Animal Stranding Scheme, School of Biodiversity, One Health and Veterinary Medicine, College of Medical, Veterinary and Life Science, University of Glasgow, Glasgow, UK Andrew Brownlow,
Mariel ten Doeschate & Nicholas J. Davison * Irish Whale and Dolphin Group (IWDG), Kilrush, Ireland Dave Wall * School of Biological, Earth & Environmental Sciences, University
College Cork, Cork, Ireland Emer Rogan * Marine and Freshwater Research Institute, Hafnarfjörður, Iceland Guðjón Már Sigurðsson * Institute of Environment, Department of Biological Sciences,
Florida International University, North Miami, FL, USA Jeremy J. Kiszka * Fisheries and Maritime Museum, Esbjerg, Denmark Charlotte Bie Thøstesen * Division of Pathology, Department of
Biomolecular Health Sciences, Faculty of Veterinary Medicine, Utrecht University, Utrecht, the Netherlands Lonneke L. IJsseldijk * Institute of Marine Research (IMR), Bergen, Norway Nils
Øien * Institute of Zoology, Zoological Society of London, London, UK Rob Deaville * Institute for Terrestrial and Aquatic Wildlife Research, University of Veterinary Medicine Hannover
Foundation, Hannover, Germany Ursula Siebert Authors * Marc-Alexander Gose View author publications You can also search for this author inPubMed Google Scholar * Emily Humble View author
publications You can also search for this author inPubMed Google Scholar * Andrew Brownlow View author publications You can also search for this author inPubMed Google Scholar * Dave Wall
View author publications You can also search for this author inPubMed Google Scholar * Emer Rogan View author publications You can also search for this author inPubMed Google Scholar *
Guðjón Már Sigurðsson View author publications You can also search for this author inPubMed Google Scholar * Jeremy J. Kiszka View author publications You can also search for this author
inPubMed Google Scholar * Charlotte Bie Thøstesen View author publications You can also search for this author inPubMed Google Scholar * Lonneke L. IJsseldijk View author publications You
can also search for this author inPubMed Google Scholar * Mariel ten Doeschate View author publications You can also search for this author inPubMed Google Scholar * Nicholas J. Davison View
author publications You can also search for this author inPubMed Google Scholar * Nils Øien View author publications You can also search for this author inPubMed Google Scholar * Rob
Deaville View author publications You can also search for this author inPubMed Google Scholar * Ursula Siebert View author publications You can also search for this author inPubMed Google
Scholar * Rob Ogden View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS MAG, RO, AB and EH designed the study. AB, DW, ER, GMS, JJK, CBT, LLI,
MtD, NJD, NØ, RD and US provided samples. MAG conducted all laboratory work and formal analysis, with substantial input from EH. MAG, RO and EH interpreted the results. MAG wrote the main
text with all authors providing feedback. RO and AB acquired funding for the project. CORRESPONDING AUTHOR Correspondence to Marc-Alexander Gose. ETHICS DECLARATIONS COMPETING INTERESTS The
authors declare no competing interests. ETHICS APPROVAL AND CONSENT TO PARTICIPATE The study was performed in concordance with the Animals Scientific Procedures Act (1986) and has been
approved by the Animal Welfare and Ethical Review Board of the University of Edinburgh under the reference number OS013-23. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains
neutral with regard to jurisdictional claims in published maps and institutional affiliations. Associate editor: Paul Sunnucks. SUPPLEMENTARY INFORMATION SUPPLEMENTARY MATERIAL RIGHTS AND
PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any
medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The
images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not
included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly
from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Gose, MA., Humble,
E., Brownlow, A. _et al._ Population genomics of the white-beaked dolphin (_Lagenorhynchus albirostris_): Implications for conservation amid climate-driven range shifts. _Heredity_ 132,
192–201 (2024). https://doi.org/10.1038/s41437-024-00672-7 Download citation * Received: 05 October 2023 * Revised: 18 January 2024 * Accepted: 18 January 2024 * Published: 01 February 2024
* Issue Date: April 2024 * DOI: https://doi.org/10.1038/s41437-024-00672-7 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link
Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative