Play all audios:
ABSTRACT Asparagine synthetase (ASNS) catalyses the ATP-dependent conversion of aspartate to asparagine. However, both the regulation and biological functions of asparagine in tumour cells
remain largely unknown. Here, we report that p53 suppresses asparagine synthesis through the transcriptional downregulation of ASNS expression and disrupts asparagine-aspartate homeostasis,
leading to lymphoma and colon tumour growth inhibition in vivo and in vitro. Moreover, the removal of asparagine from culture medium or the inhibition of ASNS impairs cell proliferation and
induces p53/p21-dependent senescence and cell cycle arrest. Mechanistically, asparagine and aspartate regulate AMPK-mediated p53 activation by physically binding to LKB1 and oppositely
modulating LKB1 activity. Thus, we found that p53 regulates asparagine metabolism and dictates cell survival by generating an auto-amplification loop via asparagine-aspartate-mediated
LKB1-AMPK signalling. Our findings highlight a role for LKB1 in sensing asparagine and aspartate and connect asparagine metabolism to the cellular signalling transduction network that
modulates cell survival. SIMILAR CONTENT BEING VIEWED BY OTHERS ASS1 METABOLICALLY CONTRIBUTES TO THE NUCLEAR AND CYTOSOLIC P53-MEDIATED DNA DAMAGE RESPONSE Article Open access 10 June 2024
TRANSKETOLASE REGULATES SENSITIVITY TO APR-246 IN P53-NULL CELLS INDEPENDENTLY OF OXIDATIVE STRESS MODULATION Article Open access 24 February 2021 MUTANT P53 SUSTAINS SERINE-GLYCINE
SYNTHESIS AND ESSENTIAL AMINO ACIDS INTAKE PROMOTING BREAST CANCER GROWTH Article Open access 25 October 2023 INTRODUCTION The dysregulation of asparagine synthetase (ASNS) expression in
childhood acute lymphoblastic leukaemia (ALL) cells is considered to increase cell susceptibility to the toxicity of l-asparaginase (ASNase), a first-line therapy for ALL that breaks down
asparagine1,2,3. ASNS inhibition also renders some types of tumour cells more susceptible to glutamine withdrawal-induced apoptosis, and asparagine addition sufficiently reverses this effect
independent of TCA cycle anaplerosis4. ASNS and asparagine may be crucial for tumour cell proliferation, as the depletion of either can arrest cell proliferation and/or induce apoptosis in
some types of tumour cells2,4,5. However, the mechanisms underlying these observations are poorly understood. The tumour suppressor p53 is the most frequently mutated gene in human cancers6.
Consistent with this, p53-knockout mice are highly prone to the spontaneous development of different tumours7,8. However, ~70% of spontaneous tumours arising in p53-deficient mice are
lymphomas9,10, with the underlying mechanisms unknown. The activation of p53 is able to induce a range of antiproliferative responses, including cell apoptosis, senescence and
differentiation, and metabolic regulation appears to be central to the tumour-suppressive function of p5311,12. However, in addition to the induction of permanent proliferation arrest or
cell death, under some mild metabolic stresses, such as transient nutrient starvation, p53 activation confers adaptation to stress and helps cells survive12,13,14,15. This
survival-supporting ability of p53 is mostly implemented by a successful p21-mediated pause in cell cycle progression, which allows cells to repair DNA lesions and/or maintain metabolic
homeostasis12,16. Here, we report that p53 plays a role in regulating asparagine metabolism by repressing the expression of ASNS. High levels of ASNS or asparagine maintain cell survival and
promote tumour cell proliferation via stifling AMPK-mediated p53 activation. Also, to generalise how asparagine may affect tumours in a broader context, we extended our study into various
human cell lines besides mouse lymphoma. Moreover, by studying these factors, we found that LKB1 is a natural sensor of cellular asparagine-aspartate homeostasis, uncovering a role for
asparagine as a signalling molecule in tumour growth. RESULTS P53 DEFICIENCY SUPPORTS CELL PROLIFERATION THOUGH ASN p53-null mice predominantly develop and succumb to lymphomas9,10,17. We
thus hypothesised that there might be some advantage(s) provided by p53 deficiency that can facilitate tumourigenesis in plasma. To test this hypothesis, we transplanted luciferase- and
GFP-labelled p53-wildtype murine lymphoma EL4 cells into mice via the tail vein, and tumour cell proliferation was then monitored by whole-body imaging for luciferase activity and flow
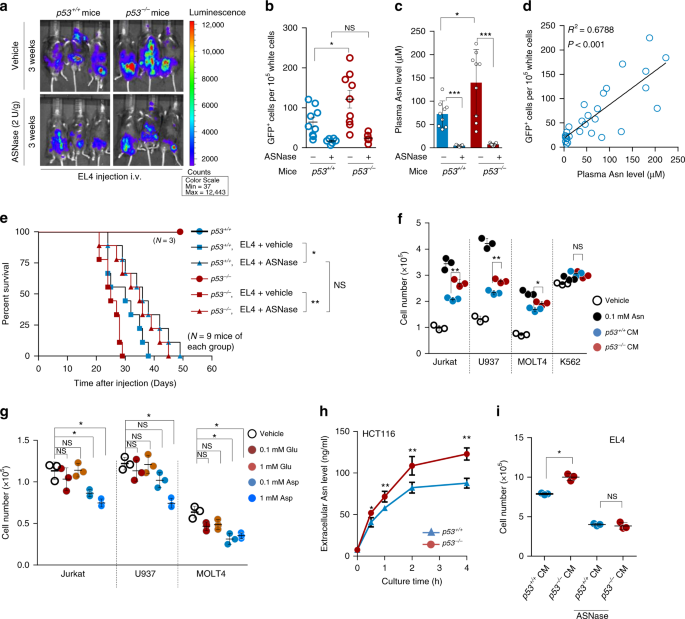
cytometry analysis for GFP expression. Strikingly, higher EL4 cell proliferation was observed in _p53__−/−_ mice than in _p53__+/+_ mice (Fig. 1a (top panels), b; and Supplementary Figs. 1a
and 11a), suggesting that _p53__−/−_ mouse plasma may provide signals that promote EL4 cell proliferation. To determine what those signals are, we found that the levels of asparagine (Asn),
not glutamate (Glu), were significantly higher in _p53__−/−_ mouse serum than in _p53__+/+_ mouse serum (Fig. 1c and Supplementary Fig. 1b, c). To explore whether asparagine mediates the
enhancement of lymphoma cell proliferation, we intraperitoneally injected mice with ASNase to remove plasma asparagine, which consequentially led to the accumulation of aspartate (Fig. 1c
and Supplementary Fig. 1d). Notably, the removal of plasma asparagine suppressed EL4 cell proliferation in vivo (Fig. 1a (bottom panels), b; and Supplementary Fig. 1a). Consistently, plasma
asparagine levels positively correlated with the EL4 cell proliferative rate (Fig. 1d). Moreover, the transplantation of EL4 cells substantially reduced the lifespan of mice, particularly
_p53__−/−_ mice (Fig. 1e). Treatment with ASNase reversed this effect and minimised the difference between _p53__+/+_ and _p53__−/−_ mice (Fig. 1e). The effect of ASNase on lymphoma
suppression may not be due to its toxicity, as no changes in body weight were observed (Supplementary Fig. 1e). These findings were further confirmed by subcutaneous cell transplantation
assays. _p53__−/−_ mice had obviously larger tumours than did _p53__+/+_ mice, and ASNase supplementation reduced tumour growth and abolished the difference in both tumour sizes and plasma
asparagine levels between _p53__+/+_ and _p53__−/−_ mice (Supplementary Fig. 1f–h). Furthermore, a positive correlation between plasma asparagine levels and tumour sizes was found
(Supplementary Fig. 1i), in agreement with the intravenous injection data (Fig. 1d). Together, increased lymphoma cell proliferation in _p53__−/−_ mice may be due to elevated plasma
asparagine. Next, we extended these findings by culturing lymphoma cells in vitro. Notably, asparagine addition promoted the proliferation of multiple types of lymphoma cells (Jurkat, U937
and MOLT4 cells) (Fig. 1f). Likewise, tumour-conditioned medium enhanced the proliferation of these cells, with _p53__−/−_ cell-conditioned medium having a more profound effect (Fig. 1f).
Consistently, asparagine or tumour-conditioned medium maintained cell survival, and _p53__−/−_ tumour-conditioned medium had a stronger effect (Supplementary Fig. 2a). To assess the
generalisability of these findings, we used tumour-conditioned medium from U2OS cells expressing p53 shRNA or control shRNA to culture lymphoma cells and similar results were obtained
(Supplementary Fig. 2b). In accordance with these findings, cells treated with asparagine at levels that were found in _p53__−/−_ mouse plasma (0.13 mM) proliferated faster and survived
better than those cultured in medium containing asparagine at 0.075 mM, as found in _p53__+/+_ mouse plasma (Fig. 1c and Supplementary Fig. 2c, d). Moreover, physiological levels of
asparagine sufficiently enhanced lymphoma growth in soft agar (Supplementary Fig. 2e, f). Asparagine is derived from glutamine or aspartate. In contrast to asparagine, aspartate or glutamate
did not promote cell proliferation, whereas aspartate visibly suppressed it (Fig. 1g and Supplementary Fig. 2g). We noticed that K562 cells, which are used as control cells, exhibited
resistance to treatment with either asparagine or tumour-conditioned medium (Fig. 1f and Supplementary Fig. 2c), which may be because these cells can de novo produce sufficient asparagine by
expressing high levels of ASNS (Supplementary Fig. 2h). Collectively, these data suggest that tumour cells, particularly p53-depleted cells, can fuel the proliferation of surrounding cells
by secreting asparagine. To further confirm this, we directly measured asparagine levels in cultured medium. Indeed, asparagine levels elevated rapidly, and p53-depleted cell medium had
higher levels of asparagine than did medium from p53-wildtype cells (Fig. 1h and Supplementary Fig. 2i). Consistent with the findings in vivo (Fig. 1a–d), EL4 cells cultured with _p53__−/−_
tumour-conditioned medium proliferated faster than those cultured with _p53__+/+_ tumour-conditioned medium. Conversely, ASNase treatment reduced cell proliferation and minimised the
differences between cells cultured with _p53__−/−_ and _p53__+/+_ tumour-conditioned medium (Fig. 1i and Supplementary Fig. 2j). Coculture experiments also revealed that _p53__−/−_ cells
markedly increased the proliferation of EL4 cells (Supplementary Fig. 2k), and the further addition of ASNase resulted in cell proliferation inhibition (Supplementary Fig. 2l). Taken
together, these findings indicate that tumour cells produce asparagine to promote cell proliferation, which is enhanced by p53 loss. IDENTIFICATION OF ASNS AS A TARGET OF P53 We next
investigated how p53 affects asparagine production. ASNS is critical for maintaining the physiological equilibrium between asparagine and aspartate (Fig. 2a). To study whether ASNS is a
physiological target for p53, and also due to the limited transfection efficiency of lymphomas, we examined the effect of p53 on ASNS expression in lymphomas and various human tumour cell
lines through different approaches. By comparing the gene expression of _p53__+/+_ and _p53__−/−_ HCT116 cells, we found that ASNS expression was significantly augmented in _p53__−/−_ cells
(Fig. 2b, c). Similar findings were observed in U2OS cells (Fig. 2b, c). Conversely, the forced expression of p53 reduced ASNS mRNA levels (Supplementary Fig. 3a). The pharmacological
activation of p53 by nutlin-3 decreased ASNS expression (Fig. 2d and Supplementary Fig. 3b, c), and the suppression of p53 by using PFT-α elevated ASNS expression (Supplementary Fig. 3b).
This p53-dependent repression was illustrated by the abrogation of ASNS expression following nutlin-3 treatment in p53-null cells (Fig. 2d and Supplementary Fig. 3c). DNA damage signals such
as etoposide (ETO) and doxorubicin (DOX) can stabilise p53 protein. Treatment with ETO decreased ASNS expression in _p53__+/+_ cells but not in _p53__−/−_ cells (Supplementary Fig. 3d).
Similarly, supplying EL4 cells with Nutlin-3, ETO or DOX resulted in a reduction in ASNS expression, whereas PFT-α treatment increased it (Fig. 2e). To both elucidate whether the
p53-mediated regulation of ASNS is cell type specific and further confirm the specificity of the effect observed with p53 depletion at the basal level, we knocked down p53 in a variety of
cell lines expressing endogenous wildtype p53 or mutant p53. The silencing of p53 increased ASNS expression in wildtype cell lines but not in mutant cells (Supplementary Fig. 3e, f). In
keeping with this, nutlin-3 treatment failed to alter the expression of ASNS in p53-mutated cell lines (Supplementary Fig. 3g). p53A138V is a tumour-associated, temperature-sensitive mutant
p53 (p53-ts) that exhibits wildtype p53 activity at 32 °C but induces transformation at 37 °C18. The expression of p53A138V inhibited _ASNS_ transcription at the permissive temperature of 32
°C but not at 37 °C (Supplementary Fig. 3h). Taken together, the results show that p53 mutant tumour cells lack the ability to suppress ASNS expression. ASNS is expressed in various mouse
tissues (Supplementary Fig. 3i). Consistent with the observations that plasma from _p53__−/−_ mice had higher levels of asparagine (Fig. 1c), multiple tissues from _p53__−/−_ mice had higher
levels of ASNS expression than did those from _p53__+/+_ mice (Fig. 2f and Supplementary Fig. 3j). Intriguingly, no significant enhancement in ASNS mRNA levels was observed in bone marrow
from _p53__−/−_ mice, despite a significant increase in ASNS protein (Fig. 2f and Supplementary Fig. 3j), indicating the existence of tissue-specific and transcription-independent
mechanism(s) for the regulation of ASNS expression by p53. Nevertheless, these data suggest that ASNS is a physiological target for p53, and its expression is suppressed by p53 at both the
genotoxic stress and basal levels. We next investigated the mechanism for the regulation of ASNS expression by p53. By analysing the _ASNS_ gene sequence for potential p53 protein response
elements, which share the consensus sequence of 5′-RRRCWWGYYY-(0-13 base pair (bp) spacer)-RRRCWWG YYY-3′ (where R is a purine, Y a pyrimidine, and W an A or T; ref. 19), we identified three
putative p53 response elements in the _ASNS_ gene (Supplementary Fig. 3k). Chromatin immunoprecipitation (ChIP) assays revealed that p53 bound to all these response element regions (Fig.
2g). Moreover, p53 repressed the expression of a luciferase gene driven by a genomic fragment containing these response elements (_ASNS_-RE1, _ASNS_-RE2, or _ASNS_-RE3, Supplementary Fig.
3l). Together, these results suggest that p53 binds to the _ASNS_ gene and suppresses ASNS expression. P53 REGULATES ASN-ASP HOMEOSTASIS Next, we investigated whether p53 regulates
asparagine metabolism by ASNS (Fig. 2a). Notably, the lack of p53 resulted in increased ASNS activities (Fig. 2h). Moreover, intracellular asparagine levels were significantly augmented in
p53-depleted cells (Fig. 2i), consistent with the observations that p53 loss enhances extracellular asparagine levels (Fig. 1h and Supplementary Figs. 2i and 4a). In line with the expression
data (Fig. 2e), pharmacological treatment of EL4 cells with p53 activators led to decreased intracellular asparagine (Fig. 2j). However, we found that extracellular asparagine levels
varied, which could be attributed to the potential side effects of these compounds on asparagine transportation (Supplementary Fig. 4b). Nevertheless, cells treated with PFT-α displayed
enhanced asparagine production (Fig. 2j and Supplementary Fig. 4b). p53 loss increased intracellular and extracellular asparagine (Figs. 1h and 2i and Supplementary Figs. 2i and 4a). In
contrast, aspartate both within and outside cells declined significantly when p53 was absent (Fig. 2k). Next, we directly assessed the effect of p53 on natural asparagine synthesis derived
from aspartate. Three cell lines (EL4, HCT116 and U2OS) were cultured in medium containing 15N-aspartate, and a portion of intracellular 15N-aspartate and 15N-asparagine was found,
suggesting that aspartate could be taken up and converted to asparagine by these cells (Fig. 2l and Supplementary Fig. 4c, d). Remarkably, isotope tracing using 15N-aspartate showed that p53
deficiency enhanced asparagine synthesis from aspartate (Fig. 2m), further confirming the data that a lack of p53 enhances ASNS expression and activity (Fig. 2b–h and Supplementary Fig.
3a–f). Similar findings were obtained in cells cultured with 13C-labelled glutamine ([U-13C5]Gln), which could support Asn synthesis by providing carbons through the TCA cycle. While p53
depletion caused an overall decline in aspartate levels (Fig. 2k), cellular [U-13C5]Gln-derived aspartate increased when p53 was absent (Fig. 2n). Nevertheless, a significant increase in
13C-asparagine was found in p53-deficient cells (Fig. 2o). To verify the cell culture findings in animals, we compared the asparagine levels in tissues from _p53__+/+_ mice and _p53__−/−_
mice. Consistent with the plasma data (Fig. 1c, Supplementary Fig. 1c and h), higher levels of asparagine were found in liver and pancreas tissues from _p53__−/−_ mice than in those from
_p53__+/+_ mice (Fig. 2p). These data together demonstrate that p53 regulates ASNS-mediated asparagine-aspartate homeostasis. ASNS PROMOTES TUMOUR CELL PROLIFERATION THROUGH ASN The
expression of ASNS is ultimately associated with the resistance of leukaemia cells and leukaemic lymphoblasts to asparagine depletion. However, the physiological effect of ASNS on other
somatic tumour cells remains largely unknown. As shown in Supplementary Fig. 5a–c, HCT116 cell proliferation was blocked when ASNS was knocked down. In addition, ASNS depletion exerted a
more profound effect on _p53__−/−_ cells than on wildtype control cells. Likewise, the depletion of ASNS impeded the growth of tumours derived from _p53__+/+_ and _p53__−/−_ HCT116 cells
(Fig. 3a, b), suggesting that ASNS is important for tumour growth. Similarly, the removal of asparagine from the cell medium by adding ASNase remarkably reduced the proliferation of both p53
knockdown HCT116 cells and their control counterparts (Fig. 3c). Analogously, siRNA-mediated silencing of ASNS led to proliferation arrest (Fig. 3d). The underlying mechanism seems to be
mediated by asparagine because the readdition of asparagine restored cell proliferation (Fig. 3d, e). Similar results were obtained using l-albizziine (l-Alb), a competitive inhibitor of
ASNS. Treatment with l-Alb led to a dose-dependent inhibition of the proliferation of _p53__+/+_ and _p53__−/−_ cells. Asparagine addition was sufficient to restore the proliferation of
_p53__+/+_ cells and partially restore that of _p53__−/−_ cells (Fig. 3f). Moreover, the silencing of ASNS reduced anchorage-independent tumour cell growth, while supplementation with
asparagine promoted overall tumour cell growth and almost restored the numbers of _p53__+/+_ colonies, with some restoration of _p53__−/−_ colonies (Supplementary Fig. 5d). The failure of
exogenous asparagine to restore the proliferation of ASNS-depleted _p53__−/−_ cells does not seem to be attributed to the low capability of _p53__−/−_ cells to take in environmental
asparagine because supplementation with 0.1 mM asparagine sufficiently restored intracellular asparagine in both _p53__+/+_ cells and _p53__−/−_ cells with ASNS knockdown (Supplementary Fig.
5e). Furthermore, when cells were cultured with 15N-asparagine, ASNS silencing resulted in higher levels of intracellular 15N-asparagine in _p53__−/−_ cells than in _p53__+/+_ cells, and
correspondingly, much lower levels of 15N-asparagine outside _p53__−/−_ cells were found (Supplementary Fig. 5f). Therefore, the lack of ASNS suppresses _p53__−_/_−_ cell proliferation
through both asparagine-dependent and asparagine-independent mechanisms. Notably, when ASNS was present, _p53__−/−_ cells had lower levels of 15N-asparagine than did _p53__+/+_ cells
(Supplementary Fig. 5f), suggesting that, under this condition, _p53__−/−_ cells are more dependent on the de novo synthesis of asparagine, correlating with the findings that p53 depletion
elevated ASNS expression (Fig. 2b–e and Supplementary Fig. 3). Taken together, these findings suggest that ASNS supports cell proliferation in a p53 context-dependent manner through
asparagine. ASNS MODULATES P53-DEPENDENT CELL SENESCENCE VIA ASN p53 is a critical regulator of senescence. We noticed that ASNS silencing increased the number of cells expressing
senescence-associated β-galactosidase in _p53__+/+_ cells but not _p53__−/−_ cells (Fig. 3g and Supplementary Fig. 5g). Notably, this ASNS knockdown-induced senescence could be reversed by
asparagine treatment (Fig. 3h and Supplementary Fig. 5h). Similar results were obtained in the primary human diploid WI-38 cell line. ASNS knockdown caused a profound increase in the
percentages of senescent cells in control siRNA-treated WI-38 cells but had little effect on cells without p53 (Fig. 3i). Again, asparagine addition reduced cell senescence (Fig. 3i). We
next further examined the effect of asparagine on other scenarios of p53-mediated senescence. Interestingly, asparagine addition inhibited the p53-dependent senescence induced by glutamine
starvation (Supplementary Fig. 5i). In WI-38 cells, ASNS expression declined as senescence progressed, especially at the late stage (Fig. 3j). To test whether the decline in ASNS contributes
to replicative senescence, we examined the replicative capacity of WI-38 cells treated with or without ASNS siRNA and/or asparagine. Compared with control cells, which could be cultured for
extended passages, ASNS-depleted cells exhibited a greatly accelerated onset of senescence (Fig. 3k). Strikingly, the addition of asparagine was sufficient to delay senescence and could
almost completely reverse the acceleration of senescence induced by ASNS depletion (Fig. 3k). To elucidate the senescence-associated function of asparagine in the context of oncogenic
signals, we investigated the effect of asparagine on HrasV12-induced premature senescence20. Although p53 depletion largely abolished HrasV12-induced senescence in U2OS cells (Supplementary
Fig. 5j, k), asparagine did not (Supplementary Fig. 5j, l). Taken together, these results indicate that ASNS regulates p53-dependent, but not oncogene-induced, senescence through asparagine.
ASNS AND ASN MAINTAIN TUMOUR CELL SURVIVAL To gain further insight into the functions of asparagine in cell survival, we studied the effect of asparagine and ASNS on cell survival. ASNase
treatment reduced the survival of both _p53__+/+_ and _p53__−/−_ HCT116 cells (Fig. 4a). These effects were also found in other tumour cell lines (Supplementary Fig. 6a–c). Similarly,
l-Alb-mediated inhibition of ASNS diminished the survival of these cells (Supplementary Fig. 6b, c). Consistent with these observations in tumour cells, when primary MEFs were treated with
ASNS siRNA, cell survival also declined, reflecting a physiological relevant effect of asparagine on cell survival (Fig. 4b). We noticed that in the HCT116 cell line, ASNase treatment
decreased the survival of sip53 cells compared to the control cells (Fig. 4a). In contrast, in the other cell lines examined, p53 depletion correlated with better cell survival in response
to ASNase or ASNS siRNA treatment (Fig. 4b and Supplementary Fig. 6a, b). This discrepancy might be due to the higher expression of p21 in HCT116 cells (Supplementary Fig. 6d), as p21
depletion further decreased the survival of HCT116 cells, not MEFs and HepG2 cells (Fig. 4c, d and Supplementary Fig. 6e). Taken together, these findings demonstrate that asparagine
maintains cell survival. ASNS IMPAIRS P53-DEPENDENT CELL CYCLE ARREST AND APOPTOSIS Failure to activate the cell cycle checkpoint impairs senescence and induces abnormal proliferation or
apoptosis under certain stress conditions12,13,21. The above findings that the alteration of asparagine metabolism influences various p53-dependent cell physiological events led us to
ascertain the effect of ASNS/asparagine on cell cycle progression. As shown in Fig. 4e, silencing ASNS resulted in p53 activation, as indicated by increased p53 phosphorylation (ser-15) and
p21 expression. Similar results were obtained in U2OS cells (Supplementary Fig. 7a). Likewise, treatment with ASNase remarkably activated p53 (Fig. 4f and Supplementary Fig. 7b). The
induction of p21 expression was p53 dependent, as this effect was abrogated when p53 was absent (Fig. 4e, f and Supplementary Fig. 7a, b). To investigate whether these effects were mediated
by asparagine, we added increasing amounts of asparagine to the medium of ASNS-KD cells. Notably, asparagine addition reduced the levels of p53 phosphorylation and p21 expression (Fig. 4g).
Moreover, l-Alb treatment triggered p53 activation, and asparagine supplementation almost completely reversed it (Supplementary Fig. 7c). Additionally, asparagine addition could also reverse
the enhancement of p53 phosphorylation and p21 expression induced by glutamine starvation, a metabolic stress that can induce a p53-dependent checkpoint for cell survival15 (Supplementary
Fig. 7d). Together, these data demonstrate a physiological function of asparagine in suppressing p53. We next investigated whether a p53/p21-dependent cell cycle checkpoint is initiated upon
asparagine depletion. Indeed, in HCT116 cells, ASNase treatment increased the numbers of p53 wildtype cells in the G1 phase. In contrast, the number of p53-depleted cells in this phase was
almost unaffected by ASNase treatment; instead, more sip53 HCT116 cells were in sub-G1 phase (apoptosis) than were their wildtype counterparts (Fig. 4h and Supplementary Fig. 11b). Likewise,
in p53-expressing U2OS cells and HepG2 cells, ASNase supplementation provoked G1 arrest, whereas the depletion of p53 totally abrogated this effect and induced increased the number of cells
in sub-G1 phase (Supplementary Figs. 7e, f and 11b). This effect of ASNase on apoptosis was further confirmed by flow cytometry (FACS) using Annexin V-FITC/PI staining. After 48 h of ASNase
treatment, ~20% of p53-depleted cells and ~6% of control cells underwent apoptosis (Supplementary Figs. 7g and 11c). Consistent with the ASNase data (Fig. 4h and Supplementary Fig. 7e, f),
the knockdown of ASNS in multiple tumour cell lines substantially increased G1 arrest in wildtype p53-expressing cells but not in p53-depleted cells. Instead, higher percentages of
p53-depleted cells in sub-G1 were found (Fig. 4i and Supplementary Fig. 8a–c). Intriguingly, in MEFs, silencing ASNS increased G1 arrest, yet no significant increase in the sub-G1 population
of sip53 MEFs was observed (Supplementary Fig. 8d), indicating that p53 loss renders tumour cells more susceptible to ASNS depletion. Nevertheless, asparagine addition abolished G1 arrest
in _p53__+/+_ cells and reduced the percentage of _p53__−/−_ cells in sub-G1 phase (Fig. 4i). In accordance with the findings that asparagine removal induced p53/p21-dependent cell cycle
arrest, ASNS knockdown failed to induce G1 arrest but increased the percentages of cells in sub-G1 when p21 was depleted (Fig. 4j). Moreover, similar to p53 deficiency, silencing p21
increased the apoptosis induced by ASNS siRNA (Supplementary Fig. 8e). Together, these data demonstrate that ASNS/asparagine depletion induces p53-dependent cell cycle arrest to protect
cells from apoptosis. ASNS AND ASN REGULATE P53 ACTIVITY VIA AMPK We next examined the mechanism by which asparagine regulates p53. AMP-activated protein kinase (AMPK) is an intracellular
energy-sensing kinase that can activate p53 through phosphorylation14,22. Interestingly, ASNase treatment elevated AMPK phosphorylation in both _p53__+/+_ and _p53__−/−_ HCT116 cells (Fig.
4f) and U2OS cells expressing p53 or control shRNA (Supplementary Fig. 7b). Similarly, when ASNS was knocked down, AMPK was activated, as evidenced by the increase in phosphorylation of AMPK
and its substrate ACC1 (Figs. 4e and 5a and Supplementary Figs. 7a and 9a). Moreover, the expression of RNAi-resistant ASNS almost completely abolished AMPK activation in ASNS-knockdown
cells (Fig. 5b). Based on these findings, we tested whether AMPK is required for ASNS silencing-triggered p53 activation by knocking down AMPK in HCT116 cells and by comparing AMPK null and
wildtype MEFs. In both situations, the loss of AMPK expression prevented ASNS depletion from activating p53 (Fig. 5c, d). Similarly, AMPK silencing attenuated ASNase-induced p53
phosphorylation (Fig. 5e). To directly assess whether asparagine is involved in ASNS depletion-mediated AMPK activation, we cultured l-Alb-treated EL4 cells in medium containing asparagine
or aspartate as a control. Interestingly, supplying cells with asparagine lessened the AMPK phosphorylation induced by l-Alb treatment (Supplementary Fig. 9b). Analogously, asparagine
addition led to the decreased phosphorylation of AMPK and p53 in ASNS-depleted cells (Fig. 5a, f, g and Supplementary Fig. 9d), and this phenomenon could be observed shortly after asparagine
treatment (Fig. 5g). Consistent with these findings, ASNase supplementation increased the level of AMPK-bound phosphorylated p53, whereas supplying cells with asparagine reversed this
effect (Fig. 5h), suggesting that asparagine suppresses AMPK activity towards p53. Aspartate supplementation increased overall cellular aspartate levels, particularly in ASNS-deprived cells
(Supplementary Fig. 9c). Surprisingly, aspartate treatment did not reduce AMPK and p53 activation but instead, to some extent, activated AMPK (Fig. 5f and Supplementary Fig. 9d). Similar
findings were obtained when cells were cultured in glutamine-free medium (Supplementary Fig. 9e). Consistently, asparagine addition restored cell survival in siASNS cells, while aspartate
supplementation did not (Fig. 5i). Taken together, the results show that ASNS and asparagine regulate p53 through AMPK. ASN-ASP HOMEOSTASIS DICTATES AMPK ACTIVITY Cellular aspartate is in
equilibrium with asparagine. We therefore investigated the effect of the dynamic asparagine-aspartate ratio on AMPK and p53. Targeting ASNS by siRNA resulted in a decrease in the levels of
cellular asparagine and a strong accumulation of aspartate, leading to a steep decline in the ratio of asparagine-aspartate (Fig. 6a, b). Consistent with this, AMPK and p53 were activated
(Fig. 6a, b), which negatively correlated with the decreased asparagine-aspartate ratio. Similarly, lowering the asparagine-aspartate ratio by l-Alb correlated with increased AMPK and p53
activation (Supplementary Fig. 9f). In contrast, the forced expression of ASNS induced asparagine synthesis, and intriguingly, aspartate levels did not decline under these conditions (Fig.
6c, d). The failure of ASNS overexpression to affect aspartate amounts might be due the compensation of other pathways (such as alanine metabolism and the TCA cycle) that can produce
aspartate. Nevertheless, the ratio of asparagine and aspartate remained significantly increased in a dose-dependent manner. Consistently, we found a dose-dependent reduction in the
activation of AMPK and p53 under these conditions (Fig. 6c, d). Together with the findings that p53 suppresses ASNS, our findings may reveal a positive feedback loop between p53 and
asparagine metabolism: the activation of p53 lessens ASNS expression, reduces asparagine-aspartate ratio and turns on AMPK, consequently leading to even higher p53 activation. Asparagine
addition was sufficient to block AMPK activation (Fig. 5a, f, g and Supplementary Fig. 9b, d). Next, we wanted to determine the effect of aspartate on AMPK-p53 signalling. In contrast to
asparagine, the addition of aspartate stimulated AMPK signalling (Supplementary Fig. 9g, h). This effect was largely abolished when AMPK was absent (Supplementary Fig. 9h). In accordance
with these observations, aspartate supplementation reduced cell proliferation (Supplementary Fig. 9i). Intriguingly, aspartate treatment elevated total AMPK levels in MEFs (Supplementary
Fig. 9g, h), which is unlikely due to aspartate-mediated AMPK phosphorylation by LKB1 because AMPK phosphorylation rarely correlates with its protein stabilisation. Next, we extended our
studies to animals. Mice receiving asparagine through either intraperitoneal injection or oral consumption via drinking water had increased sizes of tumours derived from EL4 cells (Fig. 6e).
In line with this observation, AMPK and p53 phosphorylation declined in these tumours (Fig. 6f). In contrast, aspartate administration impeded tumour growth (Fig. 6e) and activated AMPK and
p53 (Fig. 6f). These results were strengthened by the findings that body weight remained unchanged (Supplementary Fig. 9j), and plasma asparagine and aspartate correspondingly increased
following injection or oral administration (Supplementary Fig. 9k). Taken together, our findings reveal that LKB1-AMPK signalling may be the predominant mechanism by which asparagine and
aspartate regulate p53. ASN AND ASP DIRECTLY BIND TO LKB1 AND MODULATE ITS ACTIVITY Next, we investigated the mechanism(s) by which asparagine and aspartate regulate AMPK. AMPK is regulated
by LKB1-mediated phosphorylation22,23. Interestingly, ASNS depletion-induced AMPK activation (indicated by the increased phosphorylation of its substrates ACC1, ULK1, TSC2 and p53) was
abrogated when LKB1 was knocked down (Fig. 7a and Supplementary Fig. 10a). Similar results were observed in LKB1-knockout cells (Fig. 7b). Analogously, the knockdown of ASNS failed to
activate AMPK in LKB1-deficient A549 cells (Fig. 7c). Collectively, these findings suggest that LKB1 mediates the regulation of AMPK by ASNS. We next investigated whether asparagine and/or
aspartate act(s) directly on LKB1. When incubated with HEK293 cell-purified LKB1, asparagine dose-dependently abrogated LKB1 activity, while aspartate considerably augmented it (Fig. 7d).
Similar results were obtained using Myelin Basic Protein (MBP), another LKB1 substrate (Supplementary Fig. 10b). In contrast, when LKB1 was absent, asparagine and aspartate did not affect
AMPK and MBP phosphorylation (Supplementary Fig. 10c). To further verify these findings, we performed a sequential stimulation assay (Fig. 7e, left panel). HEK293 cell-purified LKB1 protein
was immobilised on agarose beads and stimulated by incubation with asparagine or aspartate (Asn/Asp). After washing the beads to eliminate unbound Asn/Asp, the Asn/Asp-stimulated LKB1 (on
beads) was further incubated with AMPK protein for the AMPK phosphorylation assay. Again, asparagine stimulation reduced LKB1 activity, whereas aspartate increased it (Fig. 7e). We next used
insect SF21 cell-purified proteins to further confirm these findings. Likewise, asparagine reduced AMPK phosphorylation in the presence of LKB1, while aspartate enhanced LKB1-mediated AMPK
phosphorylation, suggesting that asparagine and aspartate directly regulate LKB1 activity (Fig. 7f and Supplementary Fig. 10d). The binding of AMPK to AMP causes conformational changes that
promote the LKB1-mediated phosphorylation of AMPK at Thr17214,22,24. However, the addition of either asparagine or aspartate showed no effect on the LKB1-AMPK interaction in vitro
(Supplementary Fig. 10e). Similarly, changing the cellular asparagine-aspartate ratio by knocking down ASNS only slightly enhanced the LKB1-AMPK interaction (Supplementary Fig. 10f, g).
These data indicate that asparagine and aspartate may directly act on LKB1. Indeed, a real-time binding assay using surface plasmon resonance (SPR) (BIAcore) showed that both asparagine and
aspartate were able to directly bind to LKB1 with a dissociation constant (KD) of 45.5 μM and 64.6 μM, respectively (Fig. 7g and Supplementary Fig. 10h). Moreover, an equilibrium binding
assay25 using radioactive [3H]Asn revealed that [3H]Asn specifically bound to LKB1 but not AMPK, AKT1 or MBP. The binding between [3H]Asn and LKB1 proteins could be fully competed by excess
nonradiolabelled Asn (Fig. 7h). Likewise, Asp bound to LKB1 but not to AMPK, AKT1 or MBP (Fig. 7h). In contrast to Asn and Asp, other amino acids used as controls, such as methionine (Met),
leucine (Leu), glycine (Gly), alanine (Ala) and glutamate (Glu), did not bind to LKB1 (Supplementary Fig. 10i). Importantly, while ASNS silencing to some extent increased cellular aspartate
levels, cellular asparagine concentrations decreased significantly upon ASNS siRNA treatment, decreasing from above the dissociation constant of LKB1 for asparagine to below it (Fig. 7i and
Supplementary Fig. 10j), indicating that asparagine is a physiological modulator of LKB1 activity. Taken together, our findings suggest that asparagine and aspartate directly bind to LKB1 to
oppositely modulate its activity. DISCUSSION In this work, we found that some tumour cells, in particular p53-null cells, have active ASNS expression and asparagine synthesis. Increased
asparagine production helps cells proliferate and protects from senescence. Interestingly, ASNase treatment or ASNS knockdown reciprocally activates p53 to induce cell cycle arrest and
protect cells from apoptosis, suggesting a therapeutic approach (e.g. use of ASNase) for the treatment of p53-null tumours by interfering with asparagine synthesis. In addition to changes in
AMP or ADP, several metabolic stresses, such as the accumulation of reactive oxygen species (ROS) and the lack of fructose-1,6-bisphosphate (FBP) or ribulose-5-phosphate (Ru-5-P), have been
proposed to be involved in AMPK activation by different means26,27,28. In this work, we found an alternative mechanism for the regulation of AMPK and identified a role for LKB1 in sensing
cellular asparagine-aspartate homeostasis: LKB1 directly binds to asparagine and aspartate, and its activity is strongly suppressed by asparagine but enhanced by aspartate under certain
condition (Fig. 8). Although we found asparagine maintains both lymphoma and human cell survival, the mechanistic study was carried out mainly in human tumour cells due to the limited
transfection efficiency of mouse lymphoma tested. Thus, the molecular basis for the regulation and functions of asparagine in lymphoma may need further investigation. In some types of tumour
cells, aspartate can be directed to nucleotide biosynthesis to support proliferation under certain conditions29,30,31. Thus, it appears that both aspartate and asparagine promote
proliferation, and their role in tumour growth could be context dependent. In p53 wildtype cells, the disruption of ASNS expression or activity decreases asparagine generation and
accumulates aspartate, leading to the activation of LKB1-AMPK signalling and p53-dependent cell cycle arrest. When p53 is absent, the cell cycle continues, and abnormal proliferation and/or
apoptosis are triggered. Nevertheless, increased asparagine synthesis in p53-deficient tumour cells implies that, compared to aspartate, asparagine may provide more advantages to these
tumour cells. In addition, a long-standing mystery surrounding p53 is why p53-deficient mice predominantly develop lymphomas (~70% of all tumour types)9,10. Our findings presented here may
provide an important clue to the understanding of this phenomenon. Specifically, the elevation in ASNS expression and asparagine production may directly contribute to lymphomagenesis in
p53-null mice. METHODS ANTIBODIES AND REAGENTS The antibodies against the following proteins/epitopes were purchased from the indicated sources: ASNS (Proteintech, 14681-1-AP), p21 (BD
Biosciences, 556431), p53 (DO-1)-HRP (Santa Cruz, sc126-HRP), p53 (Pab 1801) (Santa Cruz, sc-98), Phosphorylated-p53(Ser15) (Cell signaling technology, 9284S), MDM2(SMP14) (Santa Cruz,
sc965), AMPK (Cell signaling technology, 23A3), phospho-AMPK(Thr 172) (Cell signaling technology, 40H9), Acetyl-CoA Carboxylase (Cell signaling technology, C83B10), Phospho-Acetyl-CoA
Carboxylase(Ser79) (Cell signaling technology, 3661), LKB1 (Cell signaling technology, 27D10), Phospho-(Ser/Thr) Phe (Cell signaling technology, 9631), HA(HA-7) (Sigma H3663), FLAG(M2)
(Sigma F3165), Actin (Proteintech, 66009-1-LG), Goat anti-rabbit IgG-HRP (Santa Cruz, sc-2004) and Goat anti-mouse IgG-HRP (Santa Cruz, sc-2302), ULK1 antibody [EPR4885(2)] (Abcam,
ab128859), Phospho-ULK1 (Ser555) (D1H4) (Cell signaling technology, 5869), Tuberin/TSC2 (Cell signaling technology, 3612), Phospho-Tuberin/TSC2 (Ser1387) (Cell signaling technology, 5584),
ATM [2C1 (1A1)] (Abcam, ab78), phospho-ATM (S1981) [EP1890Y] (Abcam, ab81292), Chk1 (Abcam, ab47574), Phospho-Chk1 (Ser345) (Cell signaling technology, 2341), p70 S6 Kinase (49D7) (Cell
signaling technology, 2708), Phospho-p70 S6 Kinase (Thr389) (108D2) (Cell signaling technology, 9234). The reagents were purchased from the following sources respectively: Nutlin-3α (Sigma,
SML0580),Doxorubicin (Sigma, D1515), Etopside (Sigma, E1383), l-Asparagine monohydrate (Sigma, A8381), l-Aspartic acid (Sigma, A9256), l-Glutamic acid (Sigma, G1251),
l-asparagine-(_amide_-15N) monohydrate (Sigma, 485896).ANTI-FLAG M2 affinity gel (Sigma, A2220), FLAG peptide (Sigma, F3290), HA peptide (Sino Biological, PP100028-1), Propidium iodide
(Sigma, P4170), Anti-HA magnetic beads (Pierce, 88837), Protein A/G agarose (Pierce, 20421), D-Luciferin monosodium salt (Pierce, 88291), l-Asparaginase (ProSpec, ENZ-287), l-Albizziine
(Goldbio, A-230-250), 0.4% Trypan blue solution (Amresco, K940). Lipofectamine 2000 (ThermoFisher Scientific, 12566014), RNAiMAX transfection agent (ThermoFisher Scientific, 13778075).
Crystal violet (Solarbio, C8470-25). PLASMIDS The coding sequences corresponding to the full-length human _ASNS_, _p53_, _AMPKα_, _LKB1_ and _MBP_ were amplified by polymerase chain reaction
(PCR) from cDNA library of 293T cells and then cloned into pRK5 empty vector tagged with Flag or HA epitope as indicated. The cloning sequences are as follows: Human _ASNS_: F:
5′-ACGCGTCGACATGTGTGGCATTTGGGC-3′; R: 5′-AACTGCAGCTAAGCTTTGACAGCTGACTTGTAGTGGG-3′. Human _p53_: F: 5′-GCTCTAGAATGGAGGAGCCGCAGTCA-3′; R: 5′ CCGCTCGAGTCAGTCTGAGTCAGGCCC-3′. Human _AMPKα_: F:
5′- GCTCTAGAATGCGCAGACTCAGTTCCTG-3′; R: 5′-CCGCTCGAGTTATTGTGCAAGAATTTTAATTAGA-3′. Human _LKB1_: F: 5′-ACGCGTCGACATGGAGGTGGTGGACCCG-3′; R: 5′-CCCAAGCTTTCACTGCTGCTTGCAGGC-3′. Human _MBP_: F:
5′- ACGCGTCGACATGGCGTCACAGAAGAGACC-3′; R: 5′- CCCAAGCTTTCAGCGTCTAGCCATGGGTG-3′. The full-length luciferase was amplified by PCR from pGL3-Basic vector (Promega, E1751) using the sequences as
follows: F: 5′-CCGCTCGAGATGGAAGACGCCAAAAACAT-3′; R: 5′-CGACGCGTTTACACGGCGATCTTTCCG-3′. The PCR product was then cloned into MSCV-IRES-EGFP vector (Addgene plasmid 20672) to construct
MSCV-Luciferase-IRES-GFP plasmid. All amplifications were made by PCR and confirmed by DNA sequencing. CELL CULTURE AND GENE KNOCKDOWN WITH SHRNA AND SIRNA All cells were cultured in a 5%
CO2 humidified incubator (ThermoFisher Scientific, USA) at 37 °C. 293T, HCT116, U2OS, A549, DU145, A431, MEF, L5178Y, EL4 and NCM460 cell lines were routinely maintained in standard
dulbecco’s modified eagle’s medium (DMEM) (ThermoFisher Scientific, C11995500BT) with 10% fetal bovine serum (FBS) (ThermoFisher Scientific, 16000044). L1210 cells were cultured in DMEM
(ThermoFisher Scientific, C11995500BT) with 10% horse serum (HS) (ThermoFisher Scientific, 16050122). HEK293, HepG2 and WI-38 cells were cultured in minimum essential medium (MEM) (Corning,
10-010-CVR) with 10% FBS (ThermoFisher Scientific, 16000044). Jurkat, U937, MOLT4, K652, H1299 and LNCaP cells were cultured in standard RPMI-1640 medium (ThermoFisher Scientific, 11875093)
with 10% FBS (ThermoFisher Scientific, 16000044), unless indicated otherwise. All cells were cultured without the addition of penicillin-streptomycin and examined for mycoplasma
contamination and cultured for no more than 2 consecutive months. Their morphologies were confirmed periodically to avoid cross-contamination or misuse of cell lines. None of the cell lines
used in this study was listed in the ICLAC database. shRNA-mediated knockdown of ASNS was performed using a specific targeting sequence shASNS#6
(5′-CCGGGCTCTGTTACAATGGTGAAATCTCGAGATTTCACCATTGTAACAGAGCTTTTTG-3′) or shASNS#10 (5′-CCGGGCTGTATGTTCAGAAGCTAAACTCGAGTTTAGCTTCTGAACATACAGCTTTTTG-3′). A non-specific ‘scrambled’ shRNA sequence
(5′-CCGGCAACAAGATGAAGAGCACCAACTCGAGTTGGTGCTCTTCATCTTGTTGTTTTT-3′) was used as control. These sequences were individually cloned into the pLKO.1-puro vector (Sigma, SHC201), which was then
co-transfected with the expression vectors containing gag/pol, rev and vsvg genes into 293T cells. The lentivirus was harvested 48 h after transfection and enriched with lentivirus
concentration solution (GeneCopoeia, LPR-LCS-01) for 48 h at 4 °C, followed by being added to nearly confluent HCT116 cells with 4 µg/ml polybrene and cultured for 16-24 h to complete
infection. The truly stable transfected cells were selected under the pressure of 2 µg/ml puromycin for 1–2 weeks. siRNA-mediated knockdown was performed with Lipofectamine RNAiMAX
transfection agent (ThermoFisher Scientific, 13778075) and siRNAs targeting human _TP53_, _ASNS_, _AMPKα_, _LKB1_ and mouse _Asns_, individually or combined for the purpose of experiment.
The targeting sequences were as follows: Human _TP53_: 5′-GACTCCAGTGGTAATCTAC-3′, Human _ASNS_ 5′-TGTATGTTCAGAAGCTAAA-3′. Human _AMPKα_ 5′-ACCAUGAUUGAUGAUGAAGCCUUAA-3′, Human _LKB1_ 5′-
GGACUGACGUGUAGAACAATT-3′, Human _p21_ 5′- CUUCGACUUUGUCACCGAG-3′, human _ATF4_ 5′-CAAGCACTTCAAACCTCAT-3′5, Human _p73_ 5′-GAGCUCGGGAGG GACUUCAACGAAG-3′, Human _p63_
5′-GCACACAAUUGAAACGUACAGGCAA-3′, Human _ATM_ 5′-CGTGTCTTAATGAGACTACAA-3′, and Mouse _asns_ 5′-GGCUUACUUAGGCAUGAAATT-3′. The siRNA sequence (5′-CGUACGCGGAAUACUUCGATT-3′) targeting luciferase
was used as control throughout this study. All siRNAs were used at a concentration of 20 nM. For co-transfection experiments, the total siRNA concentration was equalised under all conditions
by control siRNA. The siRNA transfection procedures were performed according to manufacturer’s instructions. SEMI-QUANTITATIVE RT-PCR AND QUANTITATIVE RT-PCR Briefly, total RNA was isolated
from triplicate wells in each condition using Total RNA purification Kit (GeneMark, TR01) and 2 µg RNA of each sample was complementarily reversed to cDNA by First-strand cDNA Synthesis
System (Thermo scientific, K1621). 0.04 µg cDNA product of each sample was used as template to conduct semi-quantitative or quantitative PCR. The primer pairs used in this study include:
Human _ASNS_: F: 5′-GGAAGACAGCCCCGATTTACT-3; R: 5′-AGCACGAACTGTTGTAATGTCA-3′. Mouse _Asns_: F: 5′-GCAGTGTCTGAGTGCGATGAA-3′; R: 5′-TCTTATCGGCTGCATTCCAAAC-3′. Human _MDM2_: F:
5′-ATGGTGAGGAGCAGGC-3′; R: 5′-CTAGATGAGGTAGATGGTC-3′. Human _TP53_: F: 5′-ATGGAGGAGCCGCAGTCAGA-3′; R: 5′-GGCATTCTGGGAGCTTCATC-3′. Human _ACTB_: F: 5′-GACCTGACTGACTACCTCATGAAGAT-3′; R:
5′-GTCACACTTCATGATGGAGTTGAAGG-3′. Mouse _Actb_: F: 5′-ACTACATTCAATTCCATC-3′; R: 5′-CTAGAAGCACTTGCGGTG-3′. Human _p21_: F: 5′- CCGGCGAGGCCGGGATGAG-3′; R: 5′-CTTCCTCTTGGAGAAGATC-3′. Mouse
_p21_: F: 5′-AACTTCGTCTGGGAGCGC-3′; R: 5′-TCAGGGTTTTCTCTTGCAGA-3′. Human _ABHD4_: F: 5′-TCACCCACTCTGTCCTTTCC-3′; R: 5′-GTGCAATCCCTTCACATCCT-3′. Human _PML_: F: 5′-CGCCCTGGATAACGTCTTTTT-3′;
R: 5′-TCCACAATCTGCCGGTACAC-3′. Human _SIDT2_: F: 5′-GCCAAATTGCTGCTTTCTTC-3′; R: 5′-TCCCTTCCATCCTTCCTCTT-3′. Human H-_RAS_: F: 5′-GACGTGCCTGTTGGACATC-3′; R: 5′-CTTCACCCGTTTGATCTGCTC-3′.
Semi-quantitative PCR was performed with Taq PCR StarMix (GeneStar, A112-100) in a thermal cycler (Bio-Rad, T100) according to a standard protocol as follows: 1 cycle at 95 °C for 3 min; 30
cycles at 94 °C for 45 sec, annealing for 45 sec, and 72 °C for 1 min; a final extension at 72 °C for 10 min; and holding at 4 °C. 10 μl PCR products were analysed by electrophoresis through
2% agarose gels with Gel-Red staining (Beyotime, D0139) and visualised by gel imaging analysis system (LIUYI, 130-1310). Quantitative RT-PCR was performed using CFX96 Real-Time PCR System
(Bio-Rad, USA) and the amplifications were conducted using the SYBR Green qPCR Master Mix (Biotool, B21202) according to manufacturer′s instructions. The thermal cycling conditions were set
as follows: 50 °C for 2 min followed by an initial de-naturation step at 95 °C for 10 min, 45 cycles at 95 °C for 15 s, 60 °C for 1 min, and a dissociation curve at 95 °C for 15 s and 60 °C
for 15 s. All experiments were performed in triplicate. The fold changes of gene expression were calculated after being normalised to _ACTB_. WESTERN BLOT ANALYSIS Cells were lysed by using
modified RIPA buffer containing 10 mM Tris-HCl at pH 7.5, 5 mM EDTA, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.025% SDS and proteinase inhibitors on ice for 10–20 min. Protein samples
were quantified using BCA protein assay kit (Macgene, MPK002), boiled in 5 × loading buffer and resolved by SDS-PAGE and transferred onto nitrocellulose membrane. In all, 5% skimmed milk
(BD Difco, 232100) in TBS supplemented with 0.1% Tween (TBST) was used to block the membrane before probing with indicated antibodies in TBST at 4 °C overnight. Membranes were washed with
TBST and then incubated with HRP-conjugated anti-rabbit or anti-mouse secondary antibodies at room temperature for 1 h and developed with ECL Western Blotting Detection Reagent (ThermoFisher
Scientific, 32132). Blot bands were quantified using ImageJ software. Uncropped scans of all the blots in this manuscript are shown in the Supplementary Information. ANIMALS Animal
experiments were performed with male C57BL/6J mice (Jackson Laboratory, Jax 664), _p53__−/−_ C57BL/6J mice (BIOCYTOGEN, BCG-DIS-0001) and BALB/c nude mice (Vital River Laboratory Animal
Technology, 401). All mice were maintained under specific pathogen-free conditions, and used in accordance with protocols approved by the Institutional Animal Care and Use Committees of
Tsinghua University for animal welfare. Mice were initially randomised by age and weight, and were 6–8 weeks of age at the time of injections. The maximum tumour size approved by IACUC
protocol was 2 cm in diameter and this was not exceeded in all experiments. ESTABLISHMENT OF EL4-LUC-GFP EL4 CELL LINE Retroviruses carrying MSCV-Luciferase-IRES-GFP plasmid were packaged
with the Plate-E system. For retroviral infection, EL4 cells were spin-infected with viral solution at 1500 × _g_ in the presence of 4 µg per ml polybrene for 2 h at 32 °C. Infected EL4
cells were expanded and GFP positive cells were sorted out by FACSArial II (BD Biosciences, USA) to obtain EL4-Luc-GFP cells. The purity of EL4-Luc-GFP cells was confirmed by a second
sorting 1 week after cultural expansion. FACS gating strategies for the identification of EL4-Luc-GFP cells (GFP+) were described as in Supplementary Fig. 11a. XENOGRAFT TUMOUR MODELS For
xenograft model established through i.v., viable EL4-Luc-GFP cells were washed twice with phosphate-buffered saline (PBS) and resuspended in PBS with the density of 5 × 106 cells/ml, which
were subsequently injected into the lateral tail vein in a volume of 0.1 ml. Mice were then treated i.p. with vehicle or ASNase (2 U per g of body weight) every 3 days for 3 weeks. During
these days, mice observed with paralysis in hind legs were transferred into new cage and fed with jellylike fodder until the end of the experiment. In total, 3 weeks after xenograft, nearly
100 μl blood samples were collected from the tail vein. In total, 20 μl volume of blood samples were added into PBS with 10% FBS and anticoagulant, after the procedure of red blood cell
lysis, the residual white cells were resuspended in PBS with 10% FBS and subjected to FACS analysis using LSR II cytometer (BD Biosciences, USA) to identify GFP positive cells. The rest
equal volume of blood samples were centrifuged at 5600 × _g_ for 5 min at 4 °C to obtain serum, and store at −80 °C for further analysis. After blood sampling, mice were anaesthetised and
injected i.p. with d-Luciferin monosodium salt (6 mg per mouse) and imaged for luciferase activity by IVIS Lumina II multispectral imaging system (Caliper, USA). Mice were further fed to
document survive status. For xenograft model established through s.c., viable EL4-Luc-GFP cells were resuspended in PBS with the density of 1 × 107 cells per ml, which were subsequently
injected into the flank of hind legs in a volume of 0.1 ml on both sides. In total, 1 week later, the tumour volume was measured by caliper, then mice were anaesthetised and injected i.p.
with d-Luciferin monosodium salt (6 mg per mouse) and imaged for luciferase activity by IVIS Lumina II multispectral imaging system. Next, mice received treatment i.p. with vehicle or ASNase
(2 U per g of body weight) every 3 days for 3 weeks. The tumour volume measurement and whole-mouse bioluminescence imaging were conducted every week until the end of the experiment. In
total, 3 weeks after xenograft, blood samples were collected and serum were obtained as mentioned before. After blood sampling, mice were killed and tumours were resected and weighted. For
another test using s.c. xenograft model to examine the effects of amino acid on tumour growth, viable EL4-Luc-GFP cells were resuspended in PBS with the density of 5 × 106 cells per ml,
which were subsequently injected into the flank of hind legs in a volume of 0.1 ml on both sides. One day later, mice were fed with drinking water containing with or without 10 mM Asn or
Asp, or intraperitoneally injected with or without 5 or 10 mM Asn or Asp at the end of the experiment. At 10, 14 and 17 days post-xenograft, tumour volume was measured by caliper, and then
mice were anaesthetised and imaged in vivo as mentioned before. After imaging, blood serum were obtained and the levels of Asn and Asp were measured by LC-MS as mentioned before. For
xenograft model established in BALB/c nude mice, similarly, viable HCT116 cells transfected with control siRNA or ASNS siRNA were resuspended in PBS with the density of 1 × 107 cells per ml,
followed by injection s.c. into the flank of hind legs in a volume of 0.1 ml at single side. In total, 3 weeks after implantation, mice were killed and tumours were resected, weighted and
pictured. METABOLIC FLUX AND LC-MS ANALYSIS HCT116 cells were seeded in 6-cm dishes and cultured overnight. At next day, cells were washed twice with PBS and cultured in medium containing 4
mM 15N-aspartate for 48 h. Polar metabolites were then extracted from cells using appropriate volume of 100% acetonitrile. Then, the fully vortexed cell lysates were centrifuged twice at
14,000×_g_ for 10 min to purify metabolites, and the soluble fractions were analysed by Multi-reaction monitoring (MRM) mode of UPLC-QQQ-MS/MS (Agilent 1290/6460 tandem mass spectrum,
Agilent USA). An ACQUITY UPLC® BEH HILIC, 2.1 mm × 100 mm, 1.7 μm (Waters) was used for LC separation, using gradient elution with 0.1% formic acid acetonitrile as solvent A and 0.1% formic
acid water as solvent B. The gradient programme is as follows: 0–1 min 80% A, 1–5 min 80% A to 50% A, 5–7 min 50% A, 7–7.1 min 50% A to 80% A, 7.1–10 min 80% A. The flow rate was set at 0.4
mL per min, and the injection volume was 10 μL. The total run time was 10 min for each sample. Using a Jet Stream electrospray ESI ion source in positive ion mode was used to detect 5 kinds
of amino acids, the nitrogen generator (PEAK Shanghai) was used for solvent removal and atomisation, and high purity nitrogen as colliding gas. Sheath Gas Temp is 350 °C, Sheath Gas Flow
flows at 10 L per min, Gas Temp is 325 °C and Gas Flow is 8 L per min. Capillary voltage is 4000 V, Nebulizing Gas is 45 psi, Nozzle Voltage is 500 V. Compound Precurosor ion (m/z) Product
ion (m/z) Fragmentor CE Asn 133 74 60 10 Asp 134 74 60 10 CHROMATIN IMMUNOPRECIPITATION The Genomatix Promoter Inspector software (http://www.genomatix.de) was used to search for potential
_TP53_ response elements in _ASNS_ gene with the consensus sequence 5′-RRRCWWGYYY-(0-13-base pair spacer)-RRRCWWGYYY-3′, where R is a purine, Y a pyrimidine and W either A or T. The
sequences for the putative _TP53_ response elements in _ASNS_ genes are: ASNS-RE1, 5′- ATCATCTTGTGGAGGCAAGTTGACA-3′; ASNS-RE2, 5′-ATGCACTGAAACTGCCATGTCCAGT-3′; ASNS-RE3,
5′-AAGTTCATGTTTAGACTTGGGTCTC-3′. For ChIP assay, cells were washed with PBS and crosslinked with 1% formaldehyde for 15 min at room temperature. The crosslinking reaction was stopped by the
addition of glycine to 125 mM final concentration. Cell lysates were sonicated to generate DNA fragments with the average size below 1000 base pairs and followed by immunoprecipitation with
indicated antibodies. Bounded DNA fragments were eluted and amplified by PCR. The used primer pairs were: ASNS-RE1, 5′-CCGCTCGAGATTTCTCAATTTATTTCGG-3′ and 5′-CCCAAGCTTCAAAATACATCAGTGGTC-3′;
ASNS-RE2, 5′-CCGCTCGAGACCTGTCTGTAGTTGGTTA-3′ and 5′-CCCAAGCTTACTGTTCTTCCTACTCCAAC-3′; ASNS-RE3, 5′-CCCTTTGCTTTCTGATGGTTCCATGTATGC-3′ and 5′-GATTGAGTATCCCTTATCTGAAATGTTTGGAACCAG-3′; p21-RE,
5′-CCGCTCGAGGTGGCTCTGATTGGCTTTCTG-3′ and 5′-CCCAAGCTTCTGAAAACAGGCAGCCCAAG-3′; ACTB-RE, 5′-CTAGGCGGACTATGAC-3′ and 5′-GACTTGGGAGAGGACT-3′. LUCIFERASE REPORTER ASSAY Briefly, the DNA fragment
containing the potential p53-binding region was amplified by PCR with primers used in ChIP assay and was cloned into a pGL3-promoter vector (Promega). In all, 293T cells were plated 18 h
before transfection in 24-well plates and transiently transfected with 500 ng of the reporter plasmid and 500 ng of the p53 expressing plasmid or vehicle using Lipofectamine 2000 reagent
(ThermoFisher Scientific, 12566014). The luciferase activity was determined according to the manufacturer’s instructions (Promega). Transfection efficiency was normalised on the basis of the
Renilla luciferase activity. SOFT AGAR AND COLONY FORMATION ASSAY HCT116 cells were transfected with control siRNA or _ASNS_ siRNA for 24 h and then suspended in 1 ml of DMEM medium
supplemented with or without 0.1 mM asparagine plus 20% FBS containing a 0.3% agarose and plated on a firm 0.6% agarose base in 12-well plates (5,00 cells per well). For L1210 and EL4 cells,
cells were suspended in 1 ml of DMEM medium supplemented with or without 50 or 150 µM asparagine plus 10% FBS containing a 0.3% agarose and plated on a firm 0.6% agarose base in 12-well
plates (800 cells per well). Cells were then cultured in a 5% CO2 incubator at 37 °C for 2 weeks. Colonies were fixed with 25% formaldehyde and stained with 0.0125% crystal violet till
colonies turned into blue. Colonies were then quantified by counting and images were obtained. For colony formation assay, 3000 viable cells were seeded in 6-well plate in triplicate with 2
ml culture medium in each well. Every 2 days, the medium were replaced with fresh medium until the end of the experiment. The experiment was ceased when the colonies were clearly visible
even without microscopic observation. At last, cellular colonies were fixed with 4% paraformaldehyde solution and stained with 0.05% crystal violet till colonies turned into blue. The ImageJ
software was used to analyse the area covered by colonies in each well. ANALYSIS OF CELL-CYCLE DISRUPTIONS Cells were washed twice with PBS and fixed in 75% ethanol overnight at 4 °C. Cells
were then incubated in the solution containing 0.1% Triton X-100, 100 µg/ml RNase A and 50 µg/ml propidium iodide for 30 min at 37 °C in the dark. Cell cycle distribution was analysed using
a LSR II cytometer (BD Biosciences, USA). The data were analysed using FlowJo software (TreeStar). FACS gating strategies for analysing PI-stained cells in cell cycle distribution analysis
were described as in Supplementary Fig. 11b. SENESCENCE-ASSOCIATED SA-Β-GAL ACTIVITY The SA-β-gal activity in cultured cells was determined using a Senescence Detection Kit (BioVision,
K320-250) according to the manufacturer’s instructions. After staining, cells were imaged and recorded under an inverted microscope (Olympus, IX70) in bright-field. Percentages of cells that
stained positive were calculated by counting 1000 cells in random fields per cell line. APOPTOSIS DETECTION The TUNEL staining was performed using the DeadEnd Fluorometric TUNEL system
according to the manufacturer’s instructions (Promega, G3250). After staining, cells were then observed under a confocal laser scanning microscope LSM710 (Zeiss, Germany), and a nucleus
displaying bright green fluorescence was recorded as a TUNEL-positive cell. For annexin V/PI double staining, cells were harvested and washed twice and resuspended in PBS. Apoptotic cells
were identified by double-stained with FITC-conjugated annexin-V monoclonal antibody and PI dye for 30 min by using the Annexin V-FITC Apoptosis Detection kit (BD Biosciences, 556547). After
staining and washing, cells were analysed by LSR II cytometer. Apoptotic cells were defined as annexin-V+PI− cells. FACS gating strategies were described as in Supplementary Fig. 11c. CELL
PROLIFERATION AND VIABILITY For leukaemia cell lines, 1 × 105 cells were plated in 12-well plate with 1.5 ml culture medium as indicated and supplemented with or without compound treatment.
In total, 2 days later, cells were washed with PBS and stained with 0.4% trypan blue for 2 min and counted by TC20 Automated Cell Counter (Bio-Rad, USA). Viable cells and dead cells were
recorded. In co-culture assay, 1 × 106 HCT116 cells were plated in 6-well plate with 2 ml culture medium, 12 hours later, culture medium were replaced with fresh medium containing 1 × 105
leukaemia cells, and further co-cultured for 2–3 days. The viable cells and dead cells were determined as mentioned above. For adherent cell lines, 2 × 106 cells were plated in 6-well plate
with 2 ml culture medium with or without treatment as indicated, Culture medium were replaced with fresh medium every 2 days, if needed. At the end of experiment, the viable cells and dead
cells were determined. IN VITRO MEASUREMENT OF ASNS ENZYME ACTIVITY Cells were collected in 1.5 ml tubes and 1/15 of them were prepared for protein quantification by BCA assay, and the rest
were washed three times with PBS and placed on ice. Each sample was resuspended in 400 μl sample buffer (50 mM Tris HCl, pH8.0, 0.5 mM EDTA, 1 mM EGTA, 1 mM DTT and 1 mM PMSF), frozen in
liquid nitrogen and dissolved in room temperature for 4 times. Cell extracts were centrifuged at 12,000 × _g_ for 30 min at 4 °C, and the supernatants were applied to analyse the enzymatic
activity of ASNS by reacting with a substrate mixture (85 mM Tris HCl, pH 8.0, 50 mM NaCl, 8.33 mM MgC12, 5 mM ATP, 10 mM aspartic acid and 5 mM glutamine). In each reaction, 40 μl of enzyme
solution were added to 20 μl substrate mixture and incubated with slight oscillation at 37 °C for 60 min. Each reaction was done in triplicate. The reaction system was placed on ice and
each was mixed with three times the volume of acetonitrile, followed by centrifuging twice at 14,000 × _g_ for 10 min to extract metabolites. Asn and Glu were identified and quantified by a
Triple Quadrupole LC/MS System (Agilent, 1290/6460) according to calibration curve. ASNS activity was obtained by calculating the production of Asn by cell extracts with determined amount of
protein in certain period. And the unit for ASNS activity is μg of Asn per minute per μg total protein (μg min−1 μg per protein). LC-MS ANALYSIS OF METABOLITES For fluid samples,
metabolites in mouse serum or 200 μl supernatants collected from culture medium were extracted by 100% acetonitrile by the ratio of 1:3. For cell samples, adhesive cells were harvested and
washed twice with PBS, the viable number of cells was recorded prior to metabolite extraction. The minimum 1 × 105 cells were required for LC/MS analysis. Metabolites in cells were extracted
by appropriate volume of 100% acetonitrile. For tissue samples, ultrasonication was utilised to obtain tissue extractions, which were centrifuged at 14,000 × _g_ for 20 min at 4 °C. The
supernatants were collected and quantified for protein level by BCA assay. Metabolites in these supernatants were extracted by 100% acetonitrile by the ratio of 1:3. Then, the mixtures were
centrifuged twice at 14,000 × _g_ for 10 min to extract metabolites, and the soluble fractions were analysed at original concentration or diluted in 80% acetonitrile by a Triple Quadrupole
LC/MS System (Agilent, 1290/6460). PROTEIN EXPRESSION AND PURIFICATION pRK5 plasmids tagged with Flag or HA epitope coding the full-length human AMPKα, LBK1 and MBP were transfected into
HEK-293T cells respectively. In total, 36 h after transfection, cells were harvested and lysed by sonification in lysis buffer (50 mM Tris HCl, pH 7.4, with 150 mM NaCl, 1 mM EDTA, and 1%
Triton X-100), and then centrifuged at 13,000 × _g_ for 10 min at 4 °C. FLAG M2 or HA beads were used to immunoprecipitate tagged proteins in supernatants according to the manufacturer’s
standard procedures. Beads were washed 3–5 times with wash buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 1% TritonX-100) at 4 °C on a rotator, followed by competitive elution with appropriate
synthetic FLAG or HA Peptide in TBS or PBS solution depending on experimental purpose. Purified proteins were used immediately or stored at −80 °C. MEASUREMENT OF LKB1 KINASE ACTIVITY Active
Flag-tagged LKB1 was immunoprecipitated from cell extracts of 293T cells using the FLAG M2 beads. Beads were washed three times in lysis buffer and twice in kinase buffer (50 mM Tris, pH
7.5, 10 mM MgCl2, 1 mM DTT, 100 µM ATP), and then resuspended in kinase buffer containing increasing concentrations of Asn or Asp for nearly 1 hour at 4 °C on a rotator. HA-MBP or HA-AMPKα
bound on HA beads were equally allocated into each reaction in 0.2 ml 8-Strip Tubes. These HA beads were resuspended by kinase buffer containing Flag-tagged LKB1 and kinase reaction was
performed for 30 min at 30 °C on a rotator. Samples were subjected to 8% SDS-PAGE, followed by Western blot. IN VITRO PULL-DOWN ASSAY For in vitro AMPKα and LKB1 interaction, Flag-tagged
LKB1 and HA-tagged AMPKα proteins were expressed in 293 T cells. Cells were lysed and proteins were immunoprecipitated using the FLAG M2 or HA beads according to the manufacturer’s standard
procedures. The Flag-tagged LKB1 proteins were purified and dissolved in the binding buffer (20 mM Tris pH 7.5, 150 mM NaCl) with or without different concentrations of Asn or Asp for nearly
1 h at 4 °C on a rotator. The HA beads bound with HA-tagged AMPKα were equally allocated into 0.2 ml 8-Strip Tubes and washed with the wash buffer. These HA beads were then resuspended with
binding buffer containing Flag-tagged LKB1 proteins and incubated for 4 h at room temperature, washed by the wash buffer for five times on a rotator, and subjected to 8% SDS-PAGE, followed
by Western blot. SURFACE PLASMON RESONANCE ANALYSIS SPR analysis was conducted at 25 °C with a Biacore T200 instrument (GE, USA) according to the manufacturer’s instructions. Purified LKB1
was immobilised on the surface of a Series S Sensor chip CM7 (GE, 28-9538-28) in 10 mM sodium acetate buffer (pH 4.5) and resulted in nearly 8000 response units. A reference surface was used
as a blank to correct for instrumental and buffer effects without protein injection. The amount of protein bound to the sensor chip was monitored by the change in refractive index. Asn or
Asp was dissolved in PBS and diluted with gradient concentrations and run across each sensor surface at eight different concentrations in a running buffer of PBS at a flow rate of 30 µl per
min for 90 s (contact phase), followed by 120 s of buffer flow (dissociation phase). Dissociation constants from eight serial dilutions of each compound and other kinetic parameters were
calculated using the Biacore T200 Evaluation software Version 1.0. CRISP/CAS9-MEDIATED KNOCKOUT OF LKB1 To generate a _LKB1_-knockout HCT116 cell line, a lentiviral CRISPR/Cas9 plasmid
targeting _LKB1_ was constructed by cloning the annealed sgRNA into pLenti-CRISPRv2 vector as previously described19. The sgRNAs were designed by CRISPER Design tool (crispr.mit.cn), and the
sequences are: 5′-CACCGAGCTTGGCCCGCTTGCGGCG-3′ and 5′-AAACCGCCGCAAGCGGGCCAAGCTC-3′. In all, 293T cells were cotranfected with pLenti-CRISPRv2, pVSVg and psPAX2 to produce lentiviruses,
which were used to infect HCT116 cells for 24 h, and then _LKB1_-knockout cell line were obtained by culturing in medium containing 2 μg/ml puromycin for a week. RADIOLABELED AMINO ACID
BINDING ASSAY Around five million 293T cells were seeded in a 10-cm plate, and the next day, cells were transfected with 10 µg pRK5-FLAG-based cDNA expression plasmids individually as
indicated in the experiment via Lipofectamine 2000. Forty-eight hours after transfection, cells were rinsed one time with ice-cold PBS and immediately lysed for 20 min with Triton lysis
buffer (1% Triton, 10 mM β-glycerol phosphate, 10 mM pyrophosphate, 40 mM Hepes pH 7.4, 2.5 mM MgCl2 and proteinase inhibitors). The cell lysates were cleared by centrifugation at 16,200 ×
_g_ at 4 °C in a microcentrifuge for 10 min. For anti-FLAG-immunoprecipitation, the FLAG-M2 affinity beads were blocked by rotating in 1 µg per µl bovine serum albumin (BSA) for 1 h at 4 °C
and subsequently washed for three times with lysis buffer. In total, 30 µl of a 50/50 slurry of the affinity beads were then added to clarified cell lysates and incubated with rotation for
overnight at 4 °C. Following immunoprecipitation, the beads were washed one time with lysis buffer and three times with lysis buffer containing 500 mM NaCl. If multiple samples of the same
type were represented in the experiment, the beads were equally distributed amongst the relevant tubes to ensure equal protein amounts across samples of the same type. For the binding
assays, the beads was incubated for 1 h on ice in cytosolic buffer (0.1% Triton, 40 mM HEPES pH 7.4, 10 mM NaCl, 150 mM KCl, 2.5 mM MgCl2) with 3 µCi [3H]-labelled amino acids with or
without 10 mM corresponding cold amino acids. During incubation, tubes were flicked every five minutes. At the end of one hour, the beads were briefly spun down, aspirated dry, and rapidly
washed three times with binding wash buffer (0.1% Triton, 40 mM HEPES pH 7.4, 150 mM NaCl). The beads were aspirated dry again and resuspended in 50 µl of binding wash buffer. With a cut
tip, each sample was mixed well and three 10 µl aliquots were separately added into each well of Isoplate-96 and quantified using a MicroBETA2 scintillation counter (PerkinElmer). This
process was repeated in pairs for each sample, to ensure similar incubation and wash times for all samples analysed across different experiments. After reading, beads were denatured by the
addition of 50 µl of sample buffer and boiling for 5 min, resolved by 10% SDS-PAGE, and analysed by Coomassie blue staining to detect the immunoprecipitated proteins. STATISTICAL ANALYSIS
All the data are presented as mean ± standard deviation of the mean (s.d.) unless indicated otherwise. An unpaired two-tailed Student’s _t_-test was used to calculate the _P_ values, except
specified otherwise. _P_ < 0.05 was considered significant. *_p_ < 0.05, **_p_ < 0.01, ***_p_ < 0.001, NS not significant. REPORTING SUMMARY Further information on research
design is available in the Nature Research Reporting Summary linked to this article. DATA AVAILABILITY The source data underlying Figs. 1b, c, e–i, 2b, f, h–p, 3a, c, d, f–k, 4a–d, h–j, 5i,
6a–e and 7i, g, h, and Supplementary Figs. 1b–e, g, h, 2a–g, i–l, 3i, l, 4a–c, 5a, c–f, i, j, 6a, b, e, 7e–g, 8b–e, 9c, f, i–k and 10i, j are provided as a Source Data file. All other data
are within the paper and its Supplementary Information files. REFERENCES * Richards, N. G. & Kilberg, M. S. Asparagine synthetase chemotherapy. _Annu. Rev. Biochem._ 75, 629–654 (2006).
Article CAS Google Scholar * Cantor, J. R. & Sabatini, D. M. Cancer cell metabolism: one hallmark, many faces. _Cancer Discov._ 2, 881–898 (2012). Article CAS Google Scholar * Li,
B. S. et al. The downregulation of asparagine synthetase expression can increase the sensitivity of cells resistant to l-asparaginase. _Leukemia_ 20, 2199–2201 (2006). Article CAS Google
Scholar * Zhang, J. et al. Asparagine plays a critical role in regulating cellular adaptation to glutamine depletion. _Mol. Cell_ 56, 205–218 (2014). Article CAS Google Scholar * Krall,
A. S., Xu, S., Graeber, T. G., Braas, D. & Christofk, H. R. Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. _Nat. Commun._ 7, 11457 (2016).
Article ADS CAS Google Scholar * Hollstein, M., Sidransky, D., Vogelstein, B. & Harris, C. C. p53 mutations in human cancers. _Science_ 253, 49–53 (1991). Article ADS CAS Google
Scholar * Donehower, L. A. et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. _Nature_ 356, 215–221 (1992). Article ADS CAS Google Scholar
* Lowe, S. W., Schmitt, E. M., Smith, S. W., Osborne, B. A. & Jacks, T. p53 is required for radiation-induced apoptosis in mouse thymocytes. _Nature_ 362, 847–849 (1993). Article ADS
CAS Google Scholar * Jacks, T. et al. Tumor spectrum analysis in p53-mutant mice. _Curr. Biol._ 4, 1–7 (1994). Article CAS Google Scholar * Harvey, M. et al. Spontaneous and
carcinogen-induced tumorigenesis in p53-deficient mice. _Nat. Genet._ 5, 225–229 (1993). Article CAS Google Scholar * Li, T. et al. Tumor suppression in the absence of p53-mediated
cell-cycle arrest, apoptosis, and senescence. _Cell_ 149, 1269–1283 (2012). Article CAS Google Scholar * Kruiswijk, F., Labuschagne, C. F. & Vousden, K. H. p53 in survival, death and
metabolic health: a lifeguard with a licence to kill. _Nat. Rev. Mol. Cell Biol._ 16, 393–405 (2015). Article CAS Google Scholar * Maddocks, O. D. et al. Serine starvation induces stress
and p53-dependent metabolic remodelling in cancer cells. _Nature_ 493, 542–546 (2013). Article ADS CAS Google Scholar * Jones, R. G. et al. AMP-activated protein kinase induces a
p53-dependent metabolic checkpoint. _Mol. Cell_ 18, 283–293 (2005). Article CAS Google Scholar * Reid, M. A. et al. The B55alpha subunit of PP2A drives a p53-dependent metabolic
adaptation to glutamine deprivation. _Mol. Cell_ 50, 200–211 (2013). Article CAS Google Scholar * Vousden, K. H. & Ryan, K. M. p53 and metabolism. _Nat. Rev. Cancer_ 9, 691–700
(2009). Article CAS Google Scholar * Liu, G. et al. Chromosome stability, in the absence of apoptosis, is critical for suppression of tumorigenesis in Trp53 mutant mice. _Nat. Genet._ 36,
63–68 (2004). Article CAS Google Scholar * Michalovitz, D., Halevy, O. & Oren, M. Conditional inhibition of transformation and of cell proliferation by a temperature-sensitive mutant
of p53. _Cell_ 62, 671–680 (1990). Article CAS Google Scholar * Sanjana, N. E., Shalem, O. & Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. _Nat. Methods_
11, 783–784 (2014). Article CAS Google Scholar * Serrano, M., Lin, A. W., McCurrach, M. E., Beach, D. & Lowe, S. W. Oncogenic ras provokes premature cell senescence associated with
accumulation of p53 and p16INK4a. _Cell_ 88, 593–602 (1997). Article CAS Google Scholar * Rufini, A., Tucci, P., Celardo, I. & Melino, G. Senescence and aging: the critical roles of
p53. _Oncogene_ 32, 5129–5143 (2013). Article CAS Google Scholar * Shaw, R. J. et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in
response to energy stress. _Proc. Natl Acad. Sci. USA_ 101, 3329–3335 (2004). Article ADS CAS Google Scholar * Garcia, D. & Shaw, R. J. AMPK: mechanisms of cellular energy sensing
and restoration of metabolic balance. _Mol. Cell_ 66, 789–800 (2017). Article CAS Google Scholar * Hardie, D. G. AMPK: positive and negative regulation, and its role in whole-body energy
homeostasis. _Curr. Opin. Cell Biol._ 33, 1–7 (2015). Article CAS Google Scholar * Wolfson, R. L. et al. Sestrin2 is a leucine sensor for the mTORC1 pathway. _Science_ 351, 43–48 (2016).
Article ADS CAS Google Scholar * Lin, R. et al. 6-Phosphogluconate dehydrogenase links oxidative PPP, lipogenesis and tumour growth by inhibiting LKB1-AMPK signalling. _Nat. Cell Biol._
17, 1484–1496 (2015). Article CAS Google Scholar * Zhang, C. S. et al. Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK. _Nature_ 548, 112–116 (2017). Article ADS
CAS Google Scholar * Blattler, S. M., Rencurel, F., Kaufmann, M. R. & Meyer, U. A. In the regulation of cytochrome P450 genes, phenobarbital targets LKB1 for necessary activation of
AMP-activated protein kinase. _Proc. Natl Acad. Sci. USA_ 104, 1045–1050 (2007). Article ADS Google Scholar * Birsoy, K. et al. An essential role of the mitochondrial electron transport
chain in cell proliferation is to enable aspartate synthesis. _Cell_ 162, 540–551 (2015). Article CAS Google Scholar * Sullivan, L. B. et al. Supporting aspartate biosynthesis is an
essential function of respiration in proliferating cells. _Cell_ 162, 552–563 (2015). Article CAS Google Scholar * Rabinovich, S. et al. Diversion of aspartate in ASS1-deficient tumours
fosters de novo pyrimidine synthesis. _Nature_ 527, 379–383 (2015). Article ADS CAS Google Scholar Download references ACKNOWLEDGEMENTS We thank all Jiang laboratory members for
discussion. We thank Yi Ding, Yu Tian, and Weihua Wang, Xueying Wang, Lina Xu, Xiaohui Liu, Huanhuan Pang and Dr. Zeping Hu for helping with the LC-MS/MS experiments. We would like to thank
Dr. De. Li for her assistance in the radiolabeling experiments. This research was supported by the 1000 Talents Program for Young Scholars, Tsinghua-Peking Center for Life Sciences, the
Tsinghua University Initiative Scientific Research Program, and National Natural Science Foundation of China (81930082; 31571470; 81722035) to P.J. AUTHOR INFORMATION AUTHORS AND
AFFILIATIONS * Tsinghua-Peking Joint Center for Life Sciences, Tsinghua University, 100084, Beijing, China Longfei Deng, Pengbo Yao, Le Li, Fansen Ji, Shuang Zhao, Chang Xu, Xun Lan &
Peng Jiang * School of Life Sciences, Tsinghua University, 100084, Beijing, China Longfei Deng, Pengbo Yao, Le Li, Shuang Zhao, Chang Xu & Peng Jiang * School of Medicine, Tsinghua
University, 100084, Beijing, China Fansen Ji & Xun Lan Authors * Longfei Deng View author publications You can also search for this author inPubMed Google Scholar * Pengbo Yao View
author publications You can also search for this author inPubMed Google Scholar * Le Li View author publications You can also search for this author inPubMed Google Scholar * Fansen Ji View
author publications You can also search for this author inPubMed Google Scholar * Shuang Zhao View author publications You can also search for this author inPubMed Google Scholar * Chang Xu
View author publications You can also search for this author inPubMed Google Scholar * Xun Lan View author publications You can also search for this author inPubMed Google Scholar * Peng
Jiang View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS L.D. performed the experiments during initiate submission. L.L. and P.Y. performed
the experiments raised during revisions. S.Z. helped with some of the FACS analysis experiments. C.X. and L.L. helped with metabolic and some of the animal experiments. C.X. generated SF21
expression system and purified the LKB-STRAD-MO25 complex and AMPK proteins. P.J. conceived and designed the research. F.J. and X.L. helped with the metabolomics data analysis. P.J.
organised and interpreted the data, and wrote the manuscript. CORRESPONDING AUTHOR Correspondence to Peng Jiang. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing
interests. ADDITIONAL INFORMATION PEER REVIEW INFORMATION _Nature Communications_ thanks the anonymous reviewer(s) for their contribution to the peer review of this work. PUBLISHER’S NOTE
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION REPORTING SUMMARY
SOURCE DATA SOURCE DATA RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation,
distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and
indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to
the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will
need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE
CITE THIS ARTICLE Deng, L., Yao, P., Li, L. _et al._ p53-mediated control of aspartate-asparagine homeostasis dictates LKB1 activity and modulates cell survival. _Nat Commun_ 11, 1755
(2020). https://doi.org/10.1038/s41467-020-15573-6 Download citation * Received: 28 August 2019 * Accepted: 17 March 2020 * Published: 09 April 2020 * DOI:
https://doi.org/10.1038/s41467-020-15573-6 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative