Play all audios:
ABSTRACT The natural antibiotic teixobactin kills pathogenic bacteria without detectable resistance. The difficult synthesis and unfavourable solubility of teixobactin require modifications,
yet insufficient knowledge on its binding mode impedes the hunt for superior analogues. Thus far, teixobactins are assumed to kill bacteria by binding to cognate cell wall precursors (Lipid
II and III). Here we present the binding mode of teixobactins in cellular membranes using solid-state NMR, microscopy, and affinity assays. We solve the structure of the complex formed by
an improved teixobactin-analogue and Lipid II and reveal how teixobactins recognize a broad spectrum of targets. Unexpectedly, we find that teixobactins only weakly bind to Lipid II in
cellular membranes, implying the direct interaction with cell wall precursors is not the sole killing mechanism. Our data suggest an additional mechanism affords the excellent activity of
teixobactins, which can block the cell wall biosynthesis by capturing precursors in massive clusters on membranes. SIMILAR CONTENT BEING VIEWED BY OTHERS TEIXOBACTIN KILLS BACTERIA BY A
TWO-PRONGED ATTACK ON THE CELL ENVELOPE Article Open access 03 August 2022 HOST DEFENCE PEPTIDE PLECTASIN TARGETS BACTERIAL CELL WALL PRECURSOR LIPID II BY A CALCIUM-SENSITIVE SUPRAMOLECULAR
MECHANISM Article Open access 23 May 2024 ENVIRONMENTAL AND DYNAMIC EFFECTS EXPLAIN HOW NISIN CAPTURES MEMBRANE-BOUND LIPID II Article Open access 01 June 2020 INTRODUCTION Teixobactin, the
first of a new class of antibiotics, kills a broad spectrum of clinically relevant multi-drug resistant pathogens, such as methicillin-resistant _Staphylococcus aureus_ and _Mycobacterium
tuberculosis_ with excellent activity1. Teixobactin is a natural undecapeptide from _Eleftheria terrae_ that comprises several uncommon residues including d-amino-acids and
l-_allo_-enduracididine, with the last four residues forming a ring motif. Given that it inhibits bacterial cell wall synthesis by targeting conserved non-proteinogenic molecules (Lipid II
and III) in the plasma-membrane (Fig. 1a)1, development of resistance against teixobactin is difficult2,3,4. Notwithstanding the high therapeutic potential of teixobactin, progress towards
its realization requires access to a large library of analogues due to high attrition rates in drug development5 and shortcomings, such as difficult and prohibitively expensive synthesis or
insufficient solubility. Several research groups have addressed these shortcomings6,7,8,9,10. However, due to a fundamental lack of knowledge on the binding mode of teixobactin, the search
for improved analogues has been largely based on coincidental discoveries. Structural insights on its binding mode are scarce11,12 and reported data were collected in water or micelles using
truncated Lipid II mimics, i.e., in non-physiological conditions. The lack of data in natural membrane settings is especially problematic because the binding mode and activity of Lipid
II-binding antibiotics can be highly sensitive to the cellular environment13. Here we use solid-state NMR (ssNMR) spectroscopy, fluorescence microscopy, and affinity studies to determine at
atomic resolution the native mode of action of the improved analogues14 [R4L10]-teixobactin and [L10]-teixobactin (Fig. 1b and Supplementary Fig. 1) in bacterial cellular membranes. We
disclose that teixobactins trap Lipid II in μm-sized clusters on membrane surfaces as an alternative killing mechanism. We solve the structure of the complex between [R4L10]-teixobactin and
Lipid II in these clusters, which provides detailed insights into the pharmacophore and explains how teixobactins mitigate antimicrobial resistance development. Unexpectedly, we find that
the binding affinity of teixobactins for Lipid II is strongly attenuated if membranes are anionic, which is commonly the case for bacterial plasma-membranes. Together, our data raise
astounding questions on the mode of action of teixobactins, which is more complex than previously thought. RESULTS TEIXOBACTINS FORM LARGE CLUSTERS ON MEMBRANE SURFACES We made
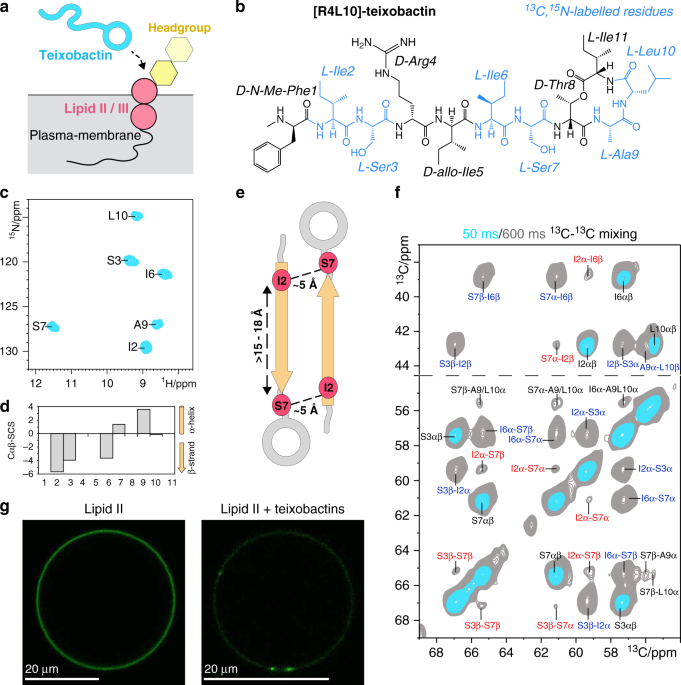
[R4L10]-teixobactin amenable to ssNMR by synthetically14 13C,15N-isotope labelling six residues (Ile2, Ser3, Ile6, Ser7, Ala9, Leu10) that cover the whole molecule. Co-assembly of
[R4L10]-teixobactin and Lipid II in DOPC membranes resulted in well-resolved ssNMR correlation spectra that demonstrate the formation of a well-defined complex (Fig. 1c and Supplementary
Fig. 2). We assigned the chemical shifts of [R4L10]-teixobactin using 2D 13C13C and 3D CαNH and CONH experiments. The chemical shifts report on the secondary structure15 and show that the
N-terminus of Lipid II-bound [R4L10]-teixobactin adopts a β-strand conformation (Fig. 1d), which matches with its high rigidity (Supplementary Fig. 2). β-structuring was also observed in a
teixobactin–sulfate complex in water11 and in micelles with a truncated Lipid II variant12. This implies that β-structuring is a natural part of the binding mode that neither requires a
membrane nor a complete receptor. We hypothesized that β-structuring is caused by the oligomerization of Lipid II-bound teixobactins, in line with previously observed peptide aggregation
behaviour in micelles12. We first examined the atomic-scale intermolecular arrangement of [R4L10]-teixobactins by a 2D 13C13C ssNMR spectrum with a long magnetization transfer time that
probes distances between 13C nuclei with a threshold of ~8 Å. We observed Cα–Cα contacts between residues Ile2–Ser7, Ile2–Ile6, and Ser3–Ser7 (Fig. 1e, f) that must stem from intermolecular
magnetization transfer between antiparallel β-strands, given that the Cαi–Cαi+5 distance within the same β-strand is 15–18 Å. We confirmed the oligomerization with pyrene excimer
fluorescence, which demonstrated that Lipid II molecules are within 1–2 nm of each other in the bound state (Supplementary Fig. 3)16. The oligomerization of Lipid II occurs virtually
immediately after addition of [R4L10]-teixobactin, which suggests that antibiotic molecules need to come together to efficiently bind Lipid II, in agreement with the loss of activity of
N-terminally truncated teixobactins that cannot oligomerise17. We used fluorescence microscopy and 7-nitrobenz-2-oxa-1,3-diazol-4-yl-(NBD)-tagged Lipid II in giant unilamellar vesicles
(GUVs) to visualize the macroscopic state of bound teixobactins (Fig. 1g and Supplementary Fig. 4)18. To our surprise, we observed that Lipid II segregated into μm-sized clusters upon
addition of [R4L10]-teixobactin, while it was initially homogeneously distributed over the GUV surface. We validated with [L10]-teixobactin, which is a close analogue of the natural
antibiotic1, that the segregation of Lipid II into clusters is a common property of teixobactins (Supplementary Fig. 4). While oligomerization is fast, cluster formation is a slower
subordinate process that occurs on a timescale of about 2–4 h, which is the same timescale on which bacterial killing by teixobactins is observed1,9,19. Together, our data suggest that the
capturing in large clusters makes Lipid II molecules unavailable for the peptidoglycan biosynthesis, which is an alternative killing mechanism similar to lantibiotics18. COMPLEX INTERFACE
AND TOPOLOGY Teixobactins target Lipid II, a complex lipid with a pyrophosphate (PPi) and a headgroup composed of two sugars (MurNAc and GlcNAc) and a pentapeptide (Fig. 2c). We used ssNMR
to determine the [R4L10]-teixobactin–Lipid II complex interface and topology in membranes. We first examined how the antibiotic targets PPi. 31P ssNMR data in membranes show stark changes of
the Lipid II PPi signals upon binding of [R4L10]-teixobactin, suggesting a direct interaction (Fig. 2a). Indeed, 2D 1H31P ssNMR data established that backbone amino protons of
[R4L10]-teixobactin directly coordinate the pyrophosphate (Fig. 2b). The interfacial correlations match with the amino protons of Ala9 and Leu10, showing that the ring motif of
[R4L10]-teixobactin coordinates PPi. Conversely, we observed no correlation for Ser7, which means that Ser7 and the β-structured N-terminus are not part of the PPi-binding interface. We next
show how the Lipid II sugar-pentapeptide headgroup participates in the complex. For this, we developed an approach to 13C,15N-label Lipid II, which has high potential to characterize the
binding modes of other promising antibiotics18,20,21. We used 2D 13C13C spectra to pinpoint the interface between 13C,15N-[R4L10]-teixobactin and 13C,15N-Lipid II (Fig. 2c, d and
Supplementary Fig. 5). While we observed intense signals for the Lipid II sugars, those of the pentapeptide were faint or absent. Since we used dipolar-based ssNMR experiments in which only
rigid residues give signals, our data imply that the pentapeptide is mobile and hence likely not part of the interface. This was confirmed with a scalar-based ssNMR experiment that enables
detection of mobile residues, showing the missing pentapeptide signals (Supplementary Fig. 6). For the sugars, we obtained many interfacial contacts that all related to Ala9 and Leu10,
confirming the interaction of the ring motif of [R4L10]-teixobactin with Lipid II (Fig. 2c). MurNAc, which is covalently attached to Lipid II PPi, shows a large number of contacts, implying
its direct presence at the interface (Fig. 2e), whereas we obtained only one low-intensity unambiguous correlation with GlcNAc, suggesting that the second sugar is only remotely involved in
the interface. We determined the membrane-topology of Lipid II-bound [R4L10]-teixobactin using a mobility-edited ssNMR experiment22 in which magnetization from mobile water or lipids is
transferred to the rigid antibiotic (Supplementary Fig. 7). We found that [R4L10]-teixobactin localizes at the water–membrane interface with the non-polar residues Ile2 and Ile6 partitioned
in the bilayer. Together, our data provide a detailed description of the complex in membranes (Fig. 2f). HIGH-RESOLUTION STRUCTURE OF THE COMPLEX We next calculated using HADDOCK23 a complex
of four [R4L10]-teixobactin and Lipid II molecules as minimal binding motif. The structure calculations were based on interfacial distance restraints between [R4L10]-teixobactin and Lipid
II, distance restraints between [R4L10]-teixobactin monomers, and dihedral angle restraints. We note that our structural insights are limited by the lack of isotope-labels for 5 out of 11
residues in [R4L10]-teixobactin. The obtained structures superimposed well (1.77 ± 0.49 Å RMSD for [R4L10]-teixobactin) (Fig. 3a and Supplementary Fig. 8) and reveal antiparallel
[R4L10]-teixobactin fibre-like β-sheets. Pairs of β-strands are shifted with respect to each other, which leaves the ring motif free to bind Lipid II. The complex shows that the critical
sequence of d- and l-amino acids in teixobactins10 enables separation of hydrophilic and hydrophobic residues (Fig. 3c), i.e., sidechains of sequential pairs of d- and l-residues (e.g.,
d-N-Me-Phe1 and l-Ile2/d-allo-Ile5 and l-Ile6) are located on the same side of the β-sheet. The long hydrophobic sidechains of N-Me-Phe1, Ile2, Ile5, and Ile6 align and act as
membrane-anchors (Fig. 3d), in agreement with computational data24, explaining why substitutions by polar8,25 or short26 sidechains or inversion from d to l configuration10 lead to complete
loss of activity. The only long hydrophobic sidechain that did not point towards the membrane-side is Ile11 (Supplementary Fig. 9), whose substitution by alanine is tolerated26. The
hydrophilic Ser3, Arg4, and Ser7 are water-exposed and form hydrogen bonds that, enabled by the long Arg4 sidechain, connect three [R4L10]-teixobactin molecules (Supplementary Fig. 9). The
ring amino protons (Thr8-Ile11) and the cationic N-terminus of a neighbouring teixobactin molecule coordinate the pyrophosphate of Lipid II. This explains why the presence of a positively
charged N-terminus is critical for the activity27 and shows that dimer formation of teixobactins is required for efficient Lipid II binding (Fig. 3b). The MurNAc sugar also directly contacts
the ring motif, while GlcNAc is distal from the drug and the mobile pentapeptide adopts random orientations. Non-polar groups of MurNAc face the hydrophobic Leu10 sidechain, which possibly
contributes to the good activity of teixobactins with branched-chain residues (Leu, Ile, Val) at position 1014. Diverse hydrophobic and cationic residues are however tolerated at positions 9
and 10 of the ring motif10,17,26, which shows that interactions with MurNAc and GlcNAc are of low specificity. Analogously, our ssNMR data, in line with MD simulations24, exclude specific
interactions with the pentapeptide. We validated that teixobactins are able to bind a broad spectrum of cognate cell wall precursors1 by comparing (i) Lys-Lipid II, (ii) mDAP-Lipid II, and
(iii) Lys-Lipid I. While Lipid I lacks the GlcNAc sugar, mDAP-Lipid II has a different pentapeptide. While Gram-positive bacteria have a cationic Lysine (Lys-Lipid II) in the pentapeptide,
it is replaced by a zwitterionic _meso_‐diaminopimelic acid (mDAP) in Gram-negative (mDAP-Lipid II) (Fig. 2c)2,3. As expected, [R4L10]-teixobactin bound to Lys-Lipid II or mDAP-Lipid II gave
the same ssNMR spectra, in line with high mobility of the pentapeptide and the absence of interfacial contacts with the pentapeptide. [R4L10]-teixobactin was also able to bind Lys-Lipid I
and exhibited clear signal changes in the ring motif (Fig. 4a and Supplementary Fig. 10), showing that GlcNAc modulates, presumably allosterically, drug binding. In summary, a picture of a
balanced binding mode emerges in which teixobactins use the conserved pyrophosphate as firm, specific anchor, whereas the interaction with the sugar-pentapeptide headgroup is more loosely
defined28,29. This behaviour enables teixobactins to recognize different cognate cell-wall building blocks and wall teichoic acids precursors, such as Lipid I, Lipid II, Lipid III, and
undecaprenyl pyrophosphate that all have the PPi moiety in common but feature different headgroups1. The capacity to recognize several conserved precursor molecules both maximizes the number
of targets and decreases the likelihood of the development of efficient resistance mechanisms against teixobactins. Taken together, a molecular rationale emerges for the broad activity
spectrum of teixobactins and for their ability to bind, unlike vancomycin, Lipid II versions with different pentapeptides30. CELLULAR CONDITIONS CHANGE THE BINDING MODE Eventually, we
examined the impact of the membrane composition that can be critical for the native binding mode and is unknown for teixobactins. We probed the impact of the membrane charge with comparative
ssNMR measurements in neutral DOPC and anionic DOPG liposomes (Fig. 4a, b). In anionic membranes, we observed widespread signal changes for residues Ile2, Ser7, Ala9, and Leu10, showing
impact on both the N- and C-termini which teixobactins use to coordinate Lipid II. Unexpectedly, ssNMR signal intensity dropped by a factor of six in anionic membranes (Supplementary Fig.
11). Since ssNMR sensitivity may inversely correlate with molecular mobility, we hypothesized that the dynamics of the complex was augmented in anionic membranes. Indeed, ssNMR relaxation
data confirmed a stark increase in mobility of Lipid II-bound [R4L10]-teixobactin with increasing anionic membrane charge (Fig. 4c). This mobility increase is of global nature and similar
for the entire peptide and the well-resolved residue Ser7. We validated with a second analogue, [L10]-teixobactin, that reduced ssNMR sensitivity and enhanced dynamics are common in anionic
membranes (Supplementary Fig. 11). The drop in sensitivity and the increase in dynamics likely point to a decreased binding affinity to Lipid II in anionic membranes. We compared binding
affinities between [R4L10]-teixobactin and Lipid II in neutral and anionic membranes with isothermal titration calorimetry (ITC)31. We found a much weaker binding affinity in membranes
containing anionic lipids, with an equilibrium dissociation constant _K_d that increased from 0.17 μM in neutral membranes to 21.9 μM in membranes containing 25% (mol/mol) of anionic lipids
(Fig. 4d, e). We hypothesized that the stark impact of the membrane composition should modulate the cellular binding mode of teixobactins, which is eventually decisive for drug design.
SSNMR32,33,34,35 enables high-resolution drug–receptor studies in crowded cellular conditions13, but requires highly sensitive methods such as 1H-detection13,36 and dynamic nuclear
polarization (DNP)33,35,37,38 due to the minute native Lipid II concentration39, which is maximally 0.5–1 % of the membrane lipids40. Here, we investigated the [R4L10]-teixobactin–Lipid II
complex directly in natural membranes isolated from the Gram-positive bacterium _M. flavus_. We first show that the oligomerization of teixobactins and the complex structure are conserved in
natural membranes, which is visible from the good match of DNP-ssNMR 2D 13C13C spectra in cellular and in vitro (DOPC) conditions (Supplementary Fig. 12). Next, we sought to reveal an
impact of the cellular environment with a well-resolved 2D NH spectrum (Fig. 4a, right panel). Intriguingly, the cellular 2D NH spectrum of bound [R4L10]-teixobactin strongly resembled the
one in anionic membranes, in line with a high content of anionic lipids (up to 90%) in _Micrococcal_ membranes41. Since the binding affinity to Lipid II is much lower in anionic membranes,
our data imply that teixobactins bind Lipid II with considerably reduced affinity under cellular conditions given that bacterial plasma-membranes are negatively charged41. A molecular
rationale for the observed effect could be the interaction of anionic lipids with the cationic N-terminal charge and the ring motif. DISCUSSION In this study, we have presented an in-depth
analysis of the binding mode of teixobactins in cellular membranes. We have presented a high-resolution structure of a teixobactin-analogue in complex with Lipid II in lipid membranes (Fig.
3). The structure provides a wealth of information on the pharmacophore and demonstrates that teixobactins balance specific and fuzzy binding to recognize a maximal number of targets and to
minimize the likelihood of resistance development. Furthermore, our structure explains the critical sequence of d- and l-amino acids that enables a clear-cut separation of hydrophilic and
hydrophobic sidechains and thereby also provides a molecular rationale why certain positions do not tolerate substitutions of hydrophobic residues by hydrophilic residues and vice
versa8,10,25,26,42,43. Besides the structure, our study presents two other major findings, which are that teixobactins form μm-sized clusters on membrane surfaces and that their binding
affinity to Lipid II is markedly reduced in anionic membranes. The impact of membrane lipids on teixobactins was previously not considered. The modest affinity for Lipid II in membranes that
contain high amounts of anionic lipids is particularly surprising because most bacterial membranes are strongly anionic and teixobactins show high activity against a broad bacterial
spectrum1,14. The direct binding of cell wall precursors is hence unlikely solely responsible for bacterial killing by teixobactins. Our data suggest that the subsequent slower formation of
μm-sized clusters, that separates precursors from the cell wall biosynthesis and occurs on the timescale on which teixobactins kill bacteria, is an additional mode of action, and potentially
even the more relevant one in physiological conditions. The relevance of cluster formation for activity is also congruent with studies of fluorescent teixobactins that showed pronounced
staining of cell wall synthesis sites in Gram-positive bacteria44. Cell wall precursors presumably get effectively entangled in the massive clusters despite a relatively weak binding
affinity to teixobactins in anionic cellular conditions. Indeed, we validated with fluorescence microscopy that teixobactins are able to efficiently capture Lipid II in clusters in anionic
membranes (Supplementary Fig. 13). While cluster formation was previously only observed for lantibiotics18, it is hence a more common mode of action of antibiotics that target cell wall
precursors. Future studies on how membrane lipids and cluster formation modulate the activity of teixobactins are urgently required to understand the mode of action of teixobactins. Our data
also provide compelling evidence that such studies require physiologically relevant media and, ideally, cellular conditions13. In this regard, concerted optimization of cluster formation
and of the binding affinity to different cell wall building blocks in relevant membrane conditions may lead to further improvements in the design of teixobactins. These insights may
critically advance our capacity to develop potent antibiotics against multi-drug resistant bacteria. METHODS MATERIALS Phospholipids 1,2-dioleoyl-sn-glycero-3-phosphocholine (C18:1, DOPC)
and 1,2-dioleoyl-sn-glycero-3-phosphoglycerol (C18:1, DOPG) were purchased from Avanti Polar Lipids, Inc. ISOTHERMAL TITRATION CALORIMETRY Preparation of liposomes: For ITC measurements
large unilamellar vesicles (LUVs) containing Lys-Lipid II were prepared by incorporating 2% (mol/mol) of Lys-Lipid II in DOPC from the stock solution. The lipids were dried by nitrogen
stream and hydrated with 10 mM HEPES, 150 mM NaCl, pH 7.5 buffer to a lipid-phosphate concentration of ~20 mM. LUVs were obtained after 10 rounds of extrusion through 200 nm membrane filters
(Whatman Nuclepore, Track-Etch Membranes). The liposomes were dialysed to citrate phosphate buffer (100 mM NaCl, 50 mM citric acid, 100 mM Na2HPO4, pH ~6). Anionic LUVs were prepared by
mixing 25% (mol/mol) DOPG and 75% (mol/mol) DOPC and then incorporating 2% (mol/mol) Lys-Lipid II. All other steps were the same as for zwitterionic LUVs. ITC was performed with the Low
Volume NanoITC (Waters LLC, New Castle, DE, USA) to determine interaction between LUVs and [R4L10]-teixobactin. [R4L10]-teixobactin was diluted in a citrate phosphate buffer (100 mM NaCl, 50
mM citric acid, 100 mM Na2HPO4, pH ~6) to a final concentration of 65 μM. Samples were degassed before use. The chamber was filled with 164 μL of [R4L10]-teixobactin, and the LUVs were
titrated into the chamber at a rate of 1.96 μL/300 s with a stirring rate of 300 rpm. Experiments were performed at 37 °C and analysed using the Nano Analyse Software (Waters LLC). All
experiments were performed in triplicate for each system. Control experiments were performed with Lipid II-free LUVs. FLUORESCENCE MICROSCOPY GUVs were prepared using the electroformation
method. The GUV cell is a self-assembled system which consists of a 5 mm-thick Teflon plate which is aligned with two titanium electrodes in a closed Teflon chamber. 1.5 μL of 0.5 mM DOPC
doped with NBD-labelled Lipid II (0.1 mol%) were brushed on the electrodes. Next, the chamber was filled with 350 μL 0.1 M sucrose solution; the electrodes dipped in and then connected to a
power supply which generated a sine wave of voltage 2.5 V at a frequency of 10 Hz for 90 min. Each microscopy slide (μ-slide 8 well, Ibidi) was covered with 350 μL BSA solution (1 mg/mL). To
detach the GUVs from the electrodes the power supply was changed to square wave of voltage 2 V at a frequency of 2 Hz for 15 min. The slides were immersed in 300 μL 0.1 mM glucose solution
to which 80 μL of GUVs were added. These were incubated for 4 h with 1 μM [R4L10]-teixobactin and observed by confocal microscopy. Anionic GUVs were prepared using a homemade
electroformation setup with ITO coverslips (18 × 18 mm, 15–30 Ω, SPI supplies). On both slips, 3 μL of a chloroform–methanol (1:1, vol/vol) solution containing 1 mM of a 3:1 mixture of
DOPC:DOPG (mol/mol) and 0.1 mol% NBD-labelled Lipid II was deposited and dried in a vacuum desiccator. With these slips, the electroformation chamber was assembled and filled with 0.1 M
sucrose solution. The GUV cell was connected to the power supply and an alternating current applied (1 V, 10 Hz). GUV formation was carried out overnight. The GUVs were then incubated with 1
μM [L10]-teixobactin for 4 h in a 0.1 M glucose solution. Microscopy: GUVs were imaged using Zeiss LSM 880 with ×63/1.2NA glycerol objective lens. The NBD label appeared green upon
excitation by the 488 nm laser. The emission range for detection was 530–545 nm (_λ_em peak = 539 nm). The brightfield images were used for detection and location of the GUVs without a
fluorescent signal and also as a control to observe their shape. FIJI software was used for the image analysis45. FLUORESCENCE SPECTROSCOPY The oligomerisation of fluorescent pyrene-labelled
Lipid II was observed upon binding of [R4L10]-teixobactin. DOPC LUVs containing 0.5 (mol%) of pyrene-labelled Lipid II in buffer (10 mM Tris–Cl, 100 mM NaCl, pH 8.0) were prepared as
described above. All fluorescence experiments were performed with a Cary Eclipse (FL0904M005) fluorometer46. All samples (1.0 mL) were continuously stirred in a 10 × 4-mm quartz cuvette and
kept at 20 °C. [R4L10]-teixobactin was titrated to the LUVs. Pyrene fluorescence was followed with spectral recordings between 360 and 550 nm (_λ_ex 350 nm, bandwidth 5 nm). The emission at
380 and 495 nm was recorded and averaged over 50 s, to obtain the values for the monomer and excimer intensity, respectively, to determine the excimer over monomer ratio for all conditions.
SOLID-STATE NMR SPECTROSCOPY 1D, 2D, and 3D 1H-detected experiments were performed at 60 kHz MAS frequency using magnetic fields of 16.4, 18.8, and 22.2 T (700, 800, and 950 MHz
1H-frequency) at temperatures of 305 K. All 1H-detected experiments were dipolar-based. PISSARRO low-power decoupling47 was used during detection periods. 2D 13C13C spin diffusion
experiments were performed with PARISxy48 recoupling at 950 MHz and 15.5, 17, or 18 kHz MAS frequency at 270 K temperature using mixing times of 25–1000 ms. SPINAL6449 was used in detection
periods. The 2D scalar 13C13C TOBSY50 experiment was acquired at 950 MHz with 8 kHz MAS at 295 K temperature with 6 ms 13C–13C magnetization transfer time. Chemical shift assignments were
performed using standard 1H-detected 3D CANH and CONH experiments36,51 in combination with 2D 13C13C PARISxy48 experiments with different magnetization transfer times (see Supplementary
Tables 1 and 2). DNP-enhanced 2D 13C13C spin diffusion (20 ms mixing time) ssNMR experiments52 were carried out using a 263 GHz microwave frequency, 400 MHz 1H frequency setup (Bruker
Biospin) at 100 K temperature and 8.5 kHz MAS. Topological studies: The mobility-edited 2D 1H(1H)13C experiment22 was performed at 700 MHz with 16.5 kHz MAS at 300 K temperature using a T2
relaxation filter of 2.5 ms. After dephasing of the rigid signals, magnetization from mobile lipid and water molecules was transferred to the rigid antibiotic with 5 ms 1H–1H mixing, and
subsequently transferred to 13C nuclei of [R4L10]-teixobactin with a short (200 μs) cross-polarization step. Structural studies: To probe intermolecular contacts between 13C,15N-labelled
[R4L10]-teixobactin molecules bound to unlabelled (12C,14N) Lipid II, we ran 2D 13C13C PARIS experiments with magnetization transfer times of 50, 300, 600, and 1000 ms at 950 MHz and 15.5
kHz MAS. To probe interfacial contacts between 13C,15N-labelled [R4L10]-teixobactin and 13C,15N-labelled Lipid II, we ran 2D 13C13C PARIS experiments with magnetization transfer times of 25,
50, 600, and 1000 ms at 950 MHz and 18 kHz MAS. 1D 31P CP experiments were acquired at 500 MHz with 12 kHz MAS. The 2D 1H31P experiment was acquired at 800 MHz magnetic field and 60 kHz MAS
using 1.75 ms CP transfer from 1H to 31P. Studies of the impact of membrane and receptor composition: 2D NH experiments were conducted with different numbers of scans due to strongly
decreased spectral sensitivity with increasing anionic membrane charge. Relaxation studies: 1H-detected 15N T1rho relaxation experiments were carried out at 950 and 700 MHz magnetic field,
respectively, using 60 kHz MAS51. The 15N transverse magnetization decay was probed with a 15N spinlock field of 17.5 kHz. For the bulk, we measured the intensity of the most intense signal
at ~8.80 1H ppm. T1rho trajectories were fit to single exponentials. NMR STRUCTURE CALCULATION Parameterization of [R4L10]-teixobactin: An initial linear [R4L10]-teixobactin topology was
generated in CNS 1.253. d-amino acids were generated by inverting relevant dihedral and improper torsion angles. d-Me-Phe N-methyl group parameters were based on monomethyl lysine (as
accommodated by HADDOCK). The rings were defined by manually removing Thr8 hydroxyl proton and Ile11 carboxyl –OH from the topology, and introducing the relevant bond lengths, bond angles,
dihedral angles, and improper torsion angles in the topology. Ester bond geometric parameters were based on the crystal structure of a teixobactin analogue (PDB 6E00)11; partial charges were
based on protonated glutamic acid sidechain (as defined in the OPLS54 force field used in HADDOCK). A monomeric [R4L10]-teixobactin starting model for HADDOCK structure calculation was then
generated in CNS55, using only dihedral angle restraints for residues Ile2-Ser7 initially obtained from an X-ray structure11 and validated by our chemical shift analysis (see Supplementary
Information). Parameters for Lipid II were taken from ref. 56. NMR restraints: NMR structure calculations were performed with HADDOCK version 2.423. SSNMR restraints are described in detail
in the Supplementary Information. In total, we used 23 intermolecular [R4L10]-teixobactin–[R4L10]-teixobactin NMR distance restraints to define the fibre-like β-sheet formed by oligomerised
teixobactins; 22 intermolecular [R4L10]-teixobactin–Lipid II NMR distance restraints were used to define the antibiotic–receptor interface. The structure of [R4L10]-teixobactin was further
defined by 12 dihedral restraints and 7 intramolecular NMR distance restraints. The head-group structure of Lipid II was further defined by 21 NMR distance restraints. See Supplementary
Table 3 for a summary of the restraints. Structure calculation protocol: HADDOCK version 2.423 was used for the structure calculations. An eight-body docking (4 Lipid II and 4
[R4L10]-teixobactin molecules) was performed using the distance and dihedral restraints described above. 7000 models were generated in the rigid body docking stage of HADDOCK, of which the
best scoring 500 were subjected to the flexible refinement protocol of HADDOCK. The resulting models were finally refined in explicit solvent. Default HADDOCK settings were used except for
doubling the weighting of the distance restraints during all stages of the structure calculation. The final models were further filtered based on the topological requirements (i.e., the
hydrophobic tails of all four Lipid II molecules must point in the same direction as the membrane-anchoring residues Ile2, Ile5, and Ile6). This resulted in a final ensemble of 26
structures. Analysis of calculated structures: Structural and violation statistics of the final 26 structures are discussed in detail in the Supporting Information. The average backbone RMSD
(from the average structure) of the 26 [R4L10]-teixobactin molecules in the complex was 1.77 ± 0.49 Å. SAMPLE PREPARATIONS Lipid II was synthesized based on enzymatic lipid reconstitution
using the Lipid II precursors UDP-GlcNAc, UDP-MurNAc-pentapeptide, and polyisoprenolphosphate as substrates16. Lysine-form UDP-MurNAc-pentapeptide was extracted from _Staphylococcus
simulans_ 22, whereas the mDAP-form was obtained from _Bacillus subtills_ 168. 13C,15N-labelled UDP-GlcNAc and UDP-MurNAc-pentapeptide (lysine form) were extracted from _S. simulans_ 22
grown in [13C,15N]-labelled rich medium (Silantes) and supplemented with [U-13C]-d-glucose and [15N]-NH4Cl (Supplementary Fig. 14a). Polyisoprenophosphate was synthesized via phosphorylation
of polyisoprenol obtained from _Laurus nobilis_57. The head-group precursors were extracted from bacteria and polyisoprenol was extracted from leaves as described58,59. After synthesis,
Lipid II was extracted with 2:1 BuOH:(Pyr/Acetate, 6 M) and purified with a DEAE cellulose resin (acetate-form, BiopHoretics) using a salt gradient (Supplementary Fig. 14b, c). Fractions
containing pure Lipid II were pooled, dried, and dissolved in 2:1 chloroform/methanol. Lipid II concentration was estimated through an inorganic phosphate determination60. The labelled Lipid
II yield was ~1.0 μmol/L. Lipid I was produced in the same manner, but without adding UDP-GlcNAc. Lipid I or II-doped multi-laminar vesicle preparations: DOPC or DOPG lipids were mixed with
Lipid I or II in 1:1 MeOH:CHCl3 at the final Lipid I or II molar ratio (0–4 mol%/mol). The mixtures were dried and the lipid films were hydrated by vortexing with 2 mL of a teixobactin
solution in 40 mM phosphate, 25 mM NaCl, pH 7.0. SSNMR samples: MLVs were collected by centrifugation (20,000×_g_) and loaded into ssNMR rotors. For 3.2 mm rotors, we used 800 nmol of
antibiotic with unlabelled Lipid II or I, while we used 400 nmol with labelled Lipid II. For 1.3 mm rotors, samples contained 200 nmol of antibiotic for unlabelled Lipid II or I, while we
used 100 nmol for labelled Lipid II. For all ssNMR samples, we used 4 mol%/mol Lipid II or I in the MLVs. Native membrane vesicle preparations were obtained from _Micrococcus flavus_ DSM
179013,16. The amount of antibiotic was ~7.5 and 30 nmol for the 1.3 and 3.2 mm rotors, respectively. Prior to the DNP measurements, the DNP samples were suspended in 60% glycerol-d8, 35%
buffer solution (25 mM NaCl, 15 mM Tris–HCl pH 7.0 final concentration), and 5% 15 mM AMUPol (final concentration) in D2O. SYNTHESIS OF [R4L10]-TEIXOBACTIN Labelled teixobactins were
synthesized using adapted protocols, described in detail in Supplementary Figs. 15, 16, and 1710. For simplicity, in this paper d-Arg4-Leu10-teixobactin is represented as [R4L10]-teixobactin
and Leu10-teixobactin as [L10]-teixobactin. Steps are illustrated in Supplementary Fig. 15: (step a) 2-Chlorotrityl chloride resin (manufacturer’s loading = 1.6 mmol/g, 150 mg resin) was
swelled in DCM in a reactor. To this resin was added 4 eq. Fmoc-Ala-OH/8 eq. DIPEA in DCM and the reactor was shaken for 3 h. The loading determined by UV absorption of the
piperidine-dibenzofulvene adduct was calculated to be 0.56 mmol/g, (150 mg resin, 0.084 mmol). Any unreacted resin was capped with MeOH:DIPEA:DCM = 1:2:7 by shaking for 1 h. (step b) The
Fmoc protecting group was deprotected using 20% piperdine in DMF by shaking for 3 min, followed by draining and shaking again with 20% piperidine in DMF for 10 min. AllocHN-D-Thr-OH was then
coupled to the resin by adding 3 eq. of the AA, 3 eq. HATU, and 6 eq. DIPEA in DMF and shaking for 1.5 h at room temperature. (step c) Esterification was performed using 10 eq. of
Fmoc-Ile-OH, 10 eq. DIC, and 5 mol% DMAP in DCM and shaking the reaction for 2 h. This was followed by capping the unreacted alcohol using 10% Ac2O/DIPEA in DMF shaking for 30 min and Fmoc
was removed using protocol described earlier in step (b). (step d) Fmoc-Leu-OH was coupled using 4 eq. of AA, 4 eq. HATU, and 8 eq. DIPEA in DMF and shaking for 1 h followed by Fmoc
deprotection using 20% piperidine in DMF as described earlier. (step e) The N terminus of Leu was protected using 10 eq. Trt-Cl and 15% Et3N in DCM and shaking for 1 h. Protection was
verified by Ninhydrin colour test. (step f) The Alloc protecting group of D-Thr was removed using 0.2 eq. [Pd(PPh3)]0 and 24 eq. PhSiH3 in dry DCM under argon for 20 min. This procedure was
repeated and the resin was washed thoroughly with DCM and DMF to remove any excess Pd stuck to the resin (step g). All amino acids were coupled using 4 eq. amino acid, 4 eq. HATU, and 8 eq.
DIPEA for 1 h. (step h) The peptide was cleaved from the resin without cleaving off the protecting groups of the amino acid sidechains using TFA:TIS:DCM = 2:5:93 and shaking for 1 h. (step
i) The solvent was evaporated and the peptide was dissolved in DMF to which 1 eq. HATU and 10 eq. DIPEA were added and the reaction was stirred for 30 min to perform the cyclisation. (step
j) The sidechain protecting groups were then cleaved off using TFA:TIS:H2O = 95:2.5:2.5 by stirring for 1 h. The peptide was precipitated using cold Et2O (−20 °C) and centrifuging at 7000
rpm to obtain a white solid. This solid was further purified using RP-HPLC using the protocols described in Supplementary Fig. 16c, d10. The teixobactin analogues (1–2) were identified by MS
in positive mode (Supplementary Table 4 and Supplementary Fig. 16e, f). REPORTING SUMMARY Further information on research design is available in the Nature Research Reporting Summary linked
to this article. DATA AVAILABILITY Data supporting the findings of this manuscript are available from the corresponding author upon reasonable request. A reporting summary for this Article
is available as a Supplementary Information file. The solid-state NMR assignments of [R4L10]-teixobactin and Lysine-Lipid II have been deposited in the BMRB (accession number 50202). The
coordinates of the [R4L10]-teixobactin–Lipid II complex have been deposited in the PDB database PDB 6YFY. The source data underlying Figs. 1d, 2e, 4c, and Supplementary Figs. 2b, 3a–c, 10a,
11b, c are provided as a Source Data file. REFERENCES * Ling, L. L. et al. A new antibiotic kills pathogens without detectable resistance. _Nature_ 517, 455–459 (2015). Article ADS CAS
PubMed PubMed Central Google Scholar * Breukink, E. & de Kruijff, B. Lipid II as a target for antibiotics. _Nat. Rev. Drug Discov._ 5, 321–332 (2006). Article CAS PubMed Google
Scholar * Muller, A., Klockner, A. & Schneider, T. Targeting a cell wall biosynthesis hot spot. _Nat. Prod. Rep._ 34, 909–932 (2017). Article PubMed Google Scholar * Medeiros-Silva,
J., Jekhmane, S., Breukink, E. & Weingarth, M. Towards the native binding modes of antibiotics that target lipid II. _Chembiochem_ 20, 1731–1738 (2019). CAS PubMed PubMed Central
Google Scholar * Shih, H. P., Zhang, X. & Aronov, A. M. Drug discovery effectiveness from the standpoint of therapeutic mechanisms and indications. _Nat. Rev. Drug Discov._ 17, 19–33
(2018). Article CAS PubMed Google Scholar * Jad, Y. E. et al. Synthesis and biological evaluation of a teixobactin analogue. _Org. Lett._ 17, 6182–6185 (2015). Article CAS PubMed
Google Scholar * Jin, K. et al. Total synthesis of teixobactin. _Nat. Commun_ 7, https://doi.org/10.1038/ncomms12394 (2016). * Zong, Y. et al. Developing equipotent teixobactin analogues
against drug-resistant bacteria and discovering a hydrophobic interaction between lipid II and teixobactin. _J. Med. Chem._ 61, 3409–3421 (2018). Article CAS PubMed Google Scholar *
Zong, Y. et al. Gram-scale total synthesis of teixobactin promoting binding mode study and discovery of more potent antibiotics. _Nat. Commun._ 10, 3268 (2019). Article ADS PubMed PubMed
Central CAS Google Scholar * Parmar, A. et al. Teixobactin analogues reveal enduracididine to be non-essential for highly potent antibacterial activity and lipid II binding. _Chem. Sci._
8, 8183–8192 (2017). Article CAS PubMed PubMed Central Google Scholar * Yang, H., Wierzbicki, M., Du Bois, D. R. & Nowick, J. S. X-ray crystallographic structure of a teixobactin
derivative reveals amyloid-like assembly. _J. Am. Chem. Soc._ 140, 14028–14032 (2018). Article CAS PubMed PubMed Central Google Scholar * Oster, C. et al. Structural studies suggest
aggregation as one of the modes of action for teixobactin. _Chem. Sci._ 9, 8850–8859 (2018). Article CAS PubMed PubMed Central Google Scholar * Medeiros-Silva, J. et al. High-resolution
NMR studies of antibiotics in cellular membranes. _Nat. Commun_ 9, 3963 (2018). Article ADS PubMed PubMed Central CAS Google Scholar * Parmar, A. et al. Design and syntheses of highly
potent teixobactin analogues against _Staphylococcus aureus_, methicillin-resistant _Staphylococcus aureus_ (MRSA), and vancomycin-resistant enterococci (VRE) in vitro and in vivo. _J. Med.
Chem._ 61, 2009–2017 (2018). Article CAS PubMed Google Scholar * Wang, Y. J. & Jardetzky, O. Probability-based protein secondary structure identification using combined NMR
chemical-shift data. _Protein Sci._ 11, 852–861 (2002). Article CAS PubMed PubMed Central Google Scholar * Breukink, E. et al. Lipid II is an intrinsic component of the pore induced by
nisin in bacterial membranes. _J. Biol. Chem._ 278, 19898–19903 (2003). Article CAS PubMed Google Scholar * Yang, H., Chen, K. H. & Nowick, J. S. Elucidation of the teixobactin
pharmacophore. _ACS Chem. Biol._ 11, 1823–1826 (2016). Article CAS PubMed PubMed Central Google Scholar * Hasper, H. E. et al. An alternative bactericidal mechanism of action for
lantibiotic peptides that target lipid II. _Science_ 313, 1636–1637 (2006). Article ADS CAS PubMed Google Scholar * Ramchuran, E. J. et al. In vitro antibacterial activity of
teixobactin derivatives on clinically relevant bacterial isolates. _Front. Microbiol._ 9, 1535 (2018). Article PubMed PubMed Central Google Scholar * Hover, B. M. et al.
Culture-independent discovery of the malacidins as calcium-dependent antibiotics with activity against multidrug-resistant Gram-positive pathogens. _Nat. Microbiol._ 3, 415–422 (2018).
Article CAS PubMed PubMed Central Google Scholar * Schneider, T. et al. Plectasin, a fungal defensin, targets the bacterial cell wall precursor lipid II. _Science_ 328, 1168–1172
(2010). Article ADS CAS PubMed Google Scholar * Doherty, T. & Hong, M. 2D 1H-31P solid-state NMR studies of the dependence of inter-bilayer water dynamics on lipid headgroup
structure and membrane peptides. _J. Magn. Reson._ 196, 39–47 (2009). Article ADS CAS PubMed Google Scholar * van Zundert, G. C. P. et al. The HADDOCK2.2 web server: user-friendly
integrative modeling of biomolecular complexes. _J. Mol. Biol._ 428, 720–725 (2016). Article PubMed CAS Google Scholar * Wen, P. C., Vanegas, J. M., Rempe, S. B. & Tajkhorshid, E.
Probing key elements of teixobactin-lipid II interactions in membranes. _Chem. Sci._ 9, 6997–7008 (2018). Article CAS PubMed PubMed Central Google Scholar * Abdel Monaim, S. A. H. et
al. Lysine scanning of Arg10-teixobactin: deciphering the role of hydrophobic and hydrophilic residues. _ACS Omega_ 1, 1262–1265 (2016). Article CAS PubMed PubMed Central Google Scholar
* Chen, K. H., Le, S. P., Han, X., Frias, J. M. & Nowick, J. S. Alanine scan reveals modifiable residues in teixobactin. _Chem. Commun._ 53, 11357–11359 (2017). Article CAS Google
Scholar * Monaim, S. et al. Investigation of the N-terminus amino function of Arg10-teixobactin. _Molecules_ 22, 1632 (2017). Article PubMed Central CAS Google Scholar * Sharma, R.,
Raduly, Z., Miskei, M. & Fuxreiter, M. Fuzzy complexes: specific binding without complete folding. _FEBS Lett._ 589, 2533–2542 (2015). Article CAS PubMed Google Scholar * Majewski,
M., Ruiz-Carmona, S. & Barril, X. An investigation of structural stability in protein–ligand complexes reveals the balance between order and disorder. _Commun. Chem_. 2, (2019). * Munch,
D. & Sahl, H. G. Structural variations of the cell wall precursor lipid II in Gram-positive bacteria—impact on binding and efficacy of antimicrobial peptides. _BBA-Biomembranes_ 1848,
3062–3071 (2015). Article PubMed CAS Google Scholar * Chiorean, S. et al. Dissecting the binding interactions of teixobactin with the bacterial cell wall precursor lipid II.
_Chembiochem_, https://doi.org/10.1002/cbic.201900504 (2019). * Theillet, F. X. et al. Structural disorder of monomeric alpha-synuclein persists in mammalian cells. _Nature_ 530, 45–50
(2016). Article ADS CAS PubMed Google Scholar * Kaplan, M. et al. EGFR dynamics change during activation in native membranes as revealed by NMR. _Cell_ 167, 1241–1251 (2016). Article
CAS PubMed Google Scholar * Banci, L. et al. Atomic-resolution monitoring of protein maturation in live human cells by NMR. _Nat. Chem. Biol._ 9, 297–299 (2013). Article CAS PubMed
PubMed Central Google Scholar * Frederick, K. K. et al. Sensitivity-enhanced NMR reveals alterations in protein structure by cellular milieus. _Cell_ 163, 620–628 (2015). Article CAS
PubMed PubMed Central Google Scholar * Medeiros-Silva, J. et al. 1H-detected solid-state NMR studies of water-inaccessible proteins in vitro and in situ. _Angew. Chem. Int. Ed. Engl._ 55,
13606–13610 (2016). Article CAS PubMed PubMed Central Google Scholar * Joedicke, L. et al. The molecular basis of subtype selectivity of human kinin G-protein-coupled receptors. _Nat.
Chem. Biol._ 14, 284–290 (2018). Article CAS PubMed PubMed Central Google Scholar * Narasimhan, S. et al. DNP-supported solid-state NMR spectroscopy of proteins inside mammalian cells.
_Angew. Chem. Int. Ed. Engl._ 58, 12969–12973 (2019). Article CAS PubMed PubMed Central Google Scholar * Plitzko, J. M., Schuler, B. & Selenko, P. Structural biology outside the
box-inside the cell. _Curr. Opin. Struct. Biol._ 46, 110–121 (2017). Article CAS PubMed Google Scholar * Kramer, N. E. et al. Resistance of Gram-positive bacteria to nisin is not
determined by Lipid II levels. _FEMS Microbiol. Lett._ 239, 157–161 (2004). Article CAS PubMed Google Scholar * O’Leary, W. M. & Wilkinson, S. G. _Gram-positive Bacteria_, vol. 117
(Academic Press, 1988). * Parmar, A. et al. Defining the molecular structure of teixobactin analogues and understanding their role in antibacterial activities. _Chem. Commun._ 53, 2016–2019
(2017). Article CAS Google Scholar * Karas, J. A. et al. Synthesis and structure–activity relationships of teixobactin. _Ann. N. Y. Acad. Sci._ 1459, 86–105 (2020). Article CAS PubMed
ADS Google Scholar * Morris, M. A. et al. A fluorescent teixobactin analogue. _ACS Chem. Biol._ https://doi.org/10.1021/acschembio.9b00908 (2020). * Schindelin, J. et al. Fiji: an
open-source platform for biological-image analysis. _Nat. Methods_ 9, 676–682 (2012). Article CAS PubMed Google Scholar * Hasper, H. E., de Kruijff, B. & Breukink, E. Assembly and
stability of nisin-Lipid II pores. _Biochem.-Us_ 43, 11567–11575 (2004). Article CAS Google Scholar * Weingarth, M., Bodenhausen, G. & Tekely, P. Low-power decoupling at high spinning
frequencies in high static fields. _J. Magn. Reson._ 199, 238–241 (2009). Article ADS CAS PubMed Google Scholar * Weingarth, M., Bodenhausen, G. & Tekely, P. Broadband
magnetization transfer using moderate radio-frequency fields for NMR with very high static fields and spinning speeds. _Chem. Phys. Lett._ 488, 10–16 (2010). Article ADS CAS Google
Scholar * Fung, B. M., Khitrin, A. K. & Ermolaev, K. An improved broadband decoupling sequence for liquid crystals and solids. _J. Magn. Reson._ 142, 97–101 (2000). Article ADS CAS
PubMed Google Scholar * Baldus, M. & Meier, B. H. Total correlation spectroscopy in the solid state. The use of scalar couplings to determine the through-bond connectivity. _J. Magn.
Reson. Ser. A_ 121, 65–69 (1996). Article ADS CAS Google Scholar * Jekhmane, S. et al. Shifts in the selectivity filter dynamics cause modal gating in K+ channels. _Nat. Commun._ 10, 12
(2019). Article ADS CAS Google Scholar * Maly, T. et al. Dynamic nuclear polarization at high magnetic fields. _J. Chem. Phys._ 128, 052211 (2008). Article ADS PubMed CAS Google
Scholar * Brunger, A. T. Version 1.2 of the crystallography and NMR system. _Nat. Protoc._ 2, 2728–2733 (2007). Article CAS PubMed Google Scholar * Jorgensen, W. L. & Tirado-Rives,
J. The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. _J. Am. Chem. Soc._ 110,
1657–1666 (1988). Article CAS PubMed Google Scholar * Nederveen, A. J. et al. RECOORD: a recalculated coordinate database of 500+proteins from the PDB using restraints from the
BioMagResBank. _Proteins_ 59, 662–672 (2005). Article CAS PubMed Google Scholar * Hsu, S. T. D. et al. The nisin-lipid II complex reveals a pyrophosphate cage that provides a blueprint
for novel antibiotics. _Nat. Struct. Mol. Biol._ 11, 963–967 (2004). Article CAS PubMed Google Scholar * Danilov, L. L., Druzhinina, T. N., Kalinchuk, N. A., Maltsev, S. D. &
Shibaev, V. N. Polyprenyl phosphates—synthesis and structure–activity relationship for a biosynthetic system of Salmonella-Anatum O-specific polysaccharide. _Chem. Phys. Lipids_ 51, 191–203
(1989). Article CAS PubMed Google Scholar * Kohlrausch, U. & Holtje, J. V. One-step purification procedure for Udp-N-acetylmuramyl-peptide murein precursors from _Bacillus-cereus_.
_FEMS Microbiol. Lett._ 78, 253–258 (1991). Article CAS Google Scholar * Swiezewska, E. et al. The search for plant polyprenols. _Acta Biochim. Pol._ 41, 221–260 (1994). Article CAS
PubMed Google Scholar * Rouser, G., Fleischer, S. & Yamamoto, A. 2 Dimensional thin layer chromatographic separation of polar lipids and determination of phospholipids by phosphorus
analysis of spots. _Lipids_ 5, 494–496 (1970). Article CAS PubMed Google Scholar Download references ACKNOWLEDGEMENTS This work is part of the research programmes ECHO, TOP, TOP-PUNT,
VICI, and VIDI with project numbers 723.014.003, 711.018.001, 700.26.121, 700.10.443, and 718.015.00, which are financed by the Dutch Research Council (NWO). I.S. acknowledges the Innovate
UK and Department of Health and Social Care (DHSC), UK and Rosetrees Trust for their kind support (SBRI grant 106368-623146 and Rosetrees Trust grant JS15/M783). The views expressed in this
publication are those of the authors and not necessarily those of Innovate UK or DHSC, UK. Experiments at the 950 MHz instrument were supported by uNMR-NL, an NWO-funded Roadmap NMR Facility
(no. 184.032.207). NMR measurements at the 800 MHz instrument at the University of Florence were supported by iNEXT (project number 653706), a Horizon 2020 programme of the European Union.
The HADDOCK software development is supported by the European Union Horizon 2020 BioExcel project numbers 675728 and 823830. AUTHOR INFORMATION Author notes * Anish Parmar, Sanjit Das &
Ishwar Singh Present address: Antimicrobial Pharmacodynamics and Therapeutics, Department of Molecular and Clinical Pharmacology, University of Liverpool, Sherrington Building, L69 3GA,
Liverpool, UK * Anish Parmar, Sanjit Das & Ishwar Singh Present address: Department of Chemistry, The Robert Robinson Laboratories, University of Liverpool, L69 3BX, Liverpool, UK *
These authors contributed equally: Rhythm Shukla, João Medeiros-Silva. AUTHORS AND AFFILIATIONS * NMR Spectroscopy, Bijvoet Centre for Biomolecular Research, Department of Chemistry, Faculty
of Science, Utrecht University, Padualaan 8, 3584 CH, Utrecht, The Netherlands Rhythm Shukla, João Medeiros-Silva, Bram J. A. Vermeulen, Alessandra Lucini Paioni, Shehrazade Jekhmane,
Alexandre M. J. J. Bonvin, Marc Baldus & Markus Weingarth * Membrane Biochemistry and Biophysics, Bijvoet Centre for Biomolecular Research, Department of Chemistry, Utrecht University,
Padualaan 8, 3584 CH, Utrecht, The Netherlands Rhythm Shukla, Joseph Lorent & Eefjan Breukink * School of Pharmacy, JBL Building, University of Lincoln, Beevor St, Lincoln, UK Anish
Parmar, Sanjit Das & Ishwar Singh * Department of Chemistry ‘Ugo Schiff’, University of Florence, Via della Lastruccia 3, 50019, Sesto Fiorentino (FI), Italy Moreno Lelli * Section
Molecular Host Defence, Division Infectious Diseases & Immunology, Department Biomolecular Health Sciences, Faculty of Veterinary Medicine, Utrecht University, Yalelaan 1, 3584, CL
Utrecht, The Netherlands Edwin J. A. Veldhuizen Authors * Rhythm Shukla View author publications You can also search for this author inPubMed Google Scholar * João Medeiros-Silva View author
publications You can also search for this author inPubMed Google Scholar * Anish Parmar View author publications You can also search for this author inPubMed Google Scholar * Bram J. A.
Vermeulen View author publications You can also search for this author inPubMed Google Scholar * Sanjit Das View author publications You can also search for this author inPubMed Google
Scholar * Alessandra Lucini Paioni View author publications You can also search for this author inPubMed Google Scholar * Shehrazade Jekhmane View author publications You can also search for
this author inPubMed Google Scholar * Joseph Lorent View author publications You can also search for this author inPubMed Google Scholar * Alexandre M. J. J. Bonvin View author publications
You can also search for this author inPubMed Google Scholar * Marc Baldus View author publications You can also search for this author inPubMed Google Scholar * Moreno Lelli View author
publications You can also search for this author inPubMed Google Scholar * Edwin J. A. Veldhuizen View author publications You can also search for this author inPubMed Google Scholar *
Eefjan Breukink View author publications You can also search for this author inPubMed Google Scholar * Ishwar Singh View author publications You can also search for this author inPubMed
Google Scholar * Markus Weingarth View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS R.S., J.M.-S., A.L.P., B.J.A.V., M.B., M.L., and M.W. did
and analysed NMR experiments; J.M.-S., R.S., and E.B. prepared ssNMR samples; I.S. conceived the design and synthesis of teixobactins; A.P., S.D., and I.S. synthesized teixobactins; R.S.,
J.L., and E.B. did fluorescence measurements; J.M.-S. and E.B. prepared Lipid II; B.J.A.V., A.M.J.J.B., and M.W. did structure calculations; R.S., S.J., E.J.A.V., E.B., and M.W. did and
analysed ITC experiments. All authors wrote and edited the manuscript. CORRESPONDING AUTHOR Correspondence to Markus Weingarth. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no
competing interests. ADDITIONAL INFORMATION PEER REVIEW INFORMATION _Nature Communications_ thanks James Nowick, Tuo Wang, and the other, anonymous, reviewer(s) for their contribution to
the peer review of this work. Peer reviewer reports are available. PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional
affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION PEER REVIEW FILE REPORTING SUMMARY SOURCE DATA SOURCE DATA RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under
a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article
are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and
your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this
license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Shukla, R., Medeiros-Silva, J., Parmar, A. _et al._ Mode of action
of teixobactins in cellular membranes. _Nat Commun_ 11, 2848 (2020). https://doi.org/10.1038/s41467-020-16600-2 Download citation * Received: 14 February 2020 * Accepted: 12 May 2020 *
Published: 05 June 2020 * DOI: https://doi.org/10.1038/s41467-020-16600-2 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link
Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative