Play all audios:
ABSTRACT BRCA1 mutation carriers have a higher risk of developing triple-negative breast cancer (TNBC), which is a refractory disease due to its non-responsiveness to current clinical
targeted therapies. Using the Sleeping Beauty transposon system in Brca1-deficient mice, we identified 169 putative cancer drivers, among which _Notch1_ is a top candidate for accelerating
TNBC by promoting the epithelial-mesenchymal transition (EMT) and regulating the cell cycle. Activation of NOTCH1 suppresses mitotic catastrophe caused by BRCA1 deficiency by restoring S/G2
and G2/M cell cycle checkpoints, which may through activation of ATR-CHK1 signalling pathway. Consistently, analysis of human breast cancer tissue demonstrates NOTCH1 is highly expressed in
TNBCs, and the activated form of NOTCH1 correlates positively with increased phosphorylation of ATR. Additionally, we demonstrate that inhibition of the NOTCH1-ATR-CHK1 cascade together with
cisplatin synergistically kills TNBC by targeting the cell cycle checkpoint, DNA damage and EMT, providing a potent clinical option for this fatal disease. SIMILAR CONTENT BEING VIEWED BY
OTHERS LOSS OF FUNCTION OF BRCA1 PROMOTES EMT IN MAMMARY TUMORS THROUGH ACTIVATION OF TGFΒR2 SIGNALING PATHWAY Article Open access 02 March 2022 _BRCA1_ HAPLOINSUFFICIENCY PROMOTES EARLY
TUMOR ONSET AND EPIGENETIC ALTERATIONS IN A MOUSE MODEL OF HEREDITARY BREAST CANCER Article 11 November 2024 PATHOGENIC _BRCA1_ VARIANTS DISRUPT PLK1-REGULATION OF MITOTIC SPINDLE
ORIENTATION Article Open access 22 April 2022 INTRODUCTION Breast cancer gene 1 (BRCA1), is the first identified breast cancer susceptibility gene, and is responsible for ~20–25% of
hereditary breast cancers and 5–10% of total breast cancers1. Low levels of BRCA1 expression are detected in ~30% of sporadic breast cancers, possibly due to hypermethylation of the promoter
or other transcriptional regulatory mechanisms2,3. Inherited mutations in the BRCA1 gene predispose carriers to early-onset tumourigenesis and an up to 87% cumulative lifetime risk of
developing breast cancer and/or ovarian cancer4. BRCA1-defective breast cancers are usually high grade and have poor prognoses. Moreover, ~48–66% of BRCA1 mutation carriers develop
triple-negative breast cancer (TNBC), a rate that is much higher than that of non-carriers (~20%)5,6,7. BRCA1 is crucial for multiple biological processes, including DNA damage repair,
cell-cycle checkpoints, ubiquitination and transcriptional regulation8. Studies have demonstrated that loss of BRCA1 results in defective DNA damage repair, abnormal centrosome duplication,
G2-M cell-cycle checkpoint defects, growth retardation, increased apoptosis, genetic instability and tumourigenesis9,10,11. Using mouse models, we and others have demonstrated that complete
knockout of Brca1 in the whole body (_Brca1__−/−_) causes lethality at embryonic day 7–8 (E7-8)12,13; in contrast, mammary-specific deletion of exon 11 of Brca1 (_Brca1__Co/Co__;MMTV-Cre_)
results in mammary tumour formation accompanied by massive genomic alterations and cellular lethality14. These findings prompted us to hypothesise that tumourigenesis triggered by Brca1
deficiency must encounter a lethal block that retards tumour progression, at least in early stages. Therefore, Brca1 deficiency is a double-edged sword, i.e., genome instability dysregulates
massive tumour suppressor and oncogenic factors to promote tumourigenesis, but too much DNA damage initiates a lethal block by inducing apoptosis to retard tumour formation. A genetic
approach by directly breeding mutant mice carrying various _Brca1__Δ11/Δ11_ mutations has indicated that loss of function of _Trp53, Atm, Chk1_ or _Chk2_ suppresses the embryonic lethality
caused by _Brca1_ deficiency and accelerats tumourigenesis to varying degrees15,16,17. Although many oncogenic drivers for tumourigenesis remain elusive, because the absence of _Brca1_
initially triggers the lethal block by inducing mitotic catastrophe and apoptosis, researchers have hypothesised the existence of secondary “hits” to modifying some cancer drivers (oncogenes
or tumour suppressors) to attenuate this block, allowing Brca1-mutant cells to overcome mitotic catastrophe and ultimately resulting in mammary tumourigenesis18. Identifying these second
“hits” may benefit clinical treatment by rebooting mitotic catastrophe after modifying the target gene or pathway. The Sleeping Beauty (SB) DNA transposon system has been used to identify
genes involved in multiple types of cancers19,20,21. This system consists of a conditionally expressed transposase and mutagenic transposon allele flanked by inverted/direct repeats. The SB
transposon can be adapted to initiate mutagenesis through random insertions to cause loss or gain of gene function while tagging potential cancer driver genes. SB transposition can also be
controlled to induce mutations in a specific target tissue by restricting conditional expression of the SB transposase via tissue-specific expression of _Cre_ to avoid potential side
effects. Together with high-throughput sequencing, these methods can rapidly identify cancer driver genes and related pathways, providing insight into human cancers through the use of mouse
models. In our efforts to identify these cancer drivers, we have conducted a functional-based driver gene screen by introducing the SB system (_SB_+_;T2Onc3_+)22 into the Brca1
mammary-specific knockout mouse model and identified 169 candidate genes correlating with tumourigenesis. Further analysis identifies Notch1 as a top putative oncogene that overcomes
apoptosis caused by Brca1 deficiency and promotes TNBC formation by activating the epithelial–mesenchymal transition (EMT) signalling pathway. RESULTS SB MUTAGENESIS ENHANCES _BRCA1_-MUTANT
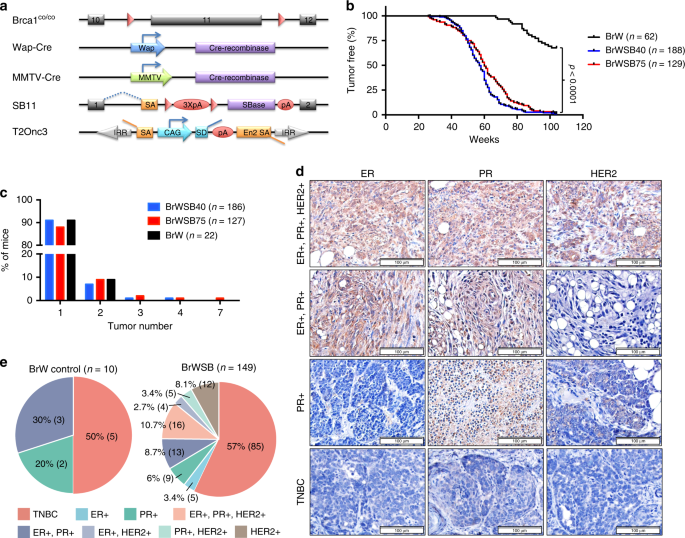
MAMMARY TUMOURIGENESIS We introduced the SB system into _Brca1__Co/Co__;WAP-Cre_ mice or _Brca1__Co/Co__;MMTV-Cre_ mice to generate four experimental cohorts of mice by interbreeding them
with two independent SB transposase-T2Onc3 transposon mouse lines: _SB;T2Onc3-12740_ and _SB;T2Onc3-12775_22 (Fig. 1a; Supplementary Fig. 1a). The mouse strains were
_Brca1__Co/Co__;WAP-Cre;SB;T2Onc3-12740_ (BrWSB40, _n_ = 188), _Brca1__Co/Co__;WAP-Cre;SB;T2Onc3-12775_ (BrWSB75, _n_ = 129), _Brca1__Co/Co__;MMTV-Cre;SB;T2Onc3-12740_ (BrMSB40, _n_ = 34),
and _Brca1__Co/Co__;MMTV-Cre;SB;T2Onc3-12775_ (BrMSB75, n = 36). _Brca1__Co/Co__;Wap-Cre_ (BrW, _n_ = 62) mice and _Brca1__Co/Co__;MMTV-Cre_ (BrM, _n_ = 56) mice without the SB transposase
or transposon were used as controls (Supplementary Data 1). The mice were monitored twice a week for tumourigenesis, and tumours were collected when they reached ~1–2 cm in diameter or the
mice were moribund. Complete necropsy was performed to assess primary and metastatic tumours. A total of 22/62 (35.5%) BrW mice developed mammary tumours in the study period of approximately
100 weeks. The SB tumourigenesis system markedly accelerated tumourigenesis and increased tumour incidence to almost 100% and tumour burden/mouse in both the 12740 and 12775 strains (Fig.
1b, c). Similarly, 26/56 (46.4%) BrM mice and all (_n_ = 70) BrMSB mice developed mammary tumours during the same period of time, with a faster acceleration of tumourigenesis in BrMSB75 than
in BrMSB40 strains (Supplementary Fig. 1b, c; Supplementary Data 1). The data showing that mammary tumourigenesis was accelerated in both _Brca1__Co/Co__;WAP-Cre_ and
_Brca1__Co/Co__;MMTV-Cre_ mice by two independent mammary-specific SB transposition strains underscore the idea that secondary “hits” are required to attenuate the lethal block in
Brca1-deficient mammary epithelial cells. Next, we conducted molecular subtyping for tumours from these mice using Hematoxylin and eosin (H&E) and immunohistochemistry (IHC) staining for
TNBC markers (Fig. 1d). Tumours from both the BrWSB and BrW groups demonstrated comparable diverse histology18 (Supplementary Fig. 1d). The tumours of both groups also had a similar
incidence of TNBC, i.e., 57% (85/149) and 50% (5/10), respectively (Fig. 1e), which is also comparable to the incidence of TNBC in human BRCA1-related breast cancers5. Furthermore we
analysed 24 tumours from BrM mice, and 45.8% (11/24) were TNBC (Supplementary Fig. 1e). These data indicate that SB-mediated insertional mutagenesis in Brca1 mammary gland-specific knockout
mice accelerates mammary tumour formation without changing the total tumour spectrum. IDENTIFICATION OF DRIVER GENES IN BRCA1-DEFICIENT TUMOURS To identify genes involved in promoting
mammary tumourigenesis, we performed high-throughput sequencing for transposon insertion sites of 306 SB tumours from 245 mice (Supplementary Data 1). Common insertion sites (CISs) were
identified using the TAPDANCE method, as previously described23, which led to the identification of over 17,626 non-redundant transposon insertion sites among the SB strains 12740 and 12775
(Supplementary Data 2). Analysis of CIS distribution patterns revealed no obvious bias between the different strains (i.e., 12740 or 12775 with MMTV-Cre or WAP-Cre) for all chromosomes,
including chromosomes 9 and 12, which are the original sites of the transposons in strains 12740 and 12775, respectively (Supplementary Fig. 2a–c). Therefore, all insertion sites were
further analysed. Using a cut-off point of their appearance in at least 5% of tumours in the 12740 and 12775 strains, we identified 119 putative driver genes (Supplementary Data 3) from the
BrWSB group and 90 genes from the BrMSB group (Supplementary Data 4). Combining the two groups together yielded 169 distinct genes with 40 overlapping genes (Fig. 2a), which appeared in
6–36% of all tumour samples (Fig. 2b). We hypothesised that other genes, in addition to the 40 overlapping genes, might be involved in accelerating tumourigenesis. Therefore, we conducted
functional enrichment analysis using the KEGG and GO database, as well as gene set enrichment analysis (GSEA), for all 169 candidate genes. Some functional items were enriched, including
ubiquitin-mediated proteolysis and cell adhesion-related functions, (Supplementary Data 5–7). We also conducted protein–protein interaction analysis for all 169 candidate genes. The results
highlighted several clusters, including the ubiquitination system, cytoskeleton and cell junction, gene expression regulation, and protein kinase/phosphatase modification-related functions
(Fig. 2c). ACTIVATED NOTCH1 IS AN ONCOGENIC DRIVER FOR TUMOURIGENESIS We divided the candidate drivers into oncogenes and tumour suppressors based on the insertion patterns of the T2Onc3
transposon (i.e., whether the insertions are clustered at hot-spot regions or widely distributed and in the same or opposite transcriptional direction of the host gene) (Fig. 3a,
Supplementary Data 8). Among all 169 potential cancer drivers, 33 genes, including _Notch1, Jup_ and _Met_, showed oncogenic features (Supplementary Fig. 3a), whereas the majority of genes,
including _Lipc, Cntn5, Hdac4, Nrxn3_ and _Cntnap5c_, exhibited tumour suppressor features (Supplementary Fig. 3b). Notch1 is a large transmembrane protein that is associated with several
human malignancies. Interaction of Notch1 with its ligands will trigger two sequential proteolytic cleavages of Notch1 to release ICN1 (intracellular domain of Notch1), allowing it to be
translocated to the nucleus to participate in transcriptional regulation24. Although the role of Notch1 in cancer formation has been well established, it is somewhat surprising that it can
act as an oncogene or a tumour suppressor in different contexts25,26. Thus, we decided to focus on Notch1 to illustrate the mechanism underlying its oncogenic function. Of 138 insertion
sites identified in 108 Notch1-trapped SB tumours, 136 (98.6%) were concentrated between exon 25 and exon 30 (Fig. 3b), and 73.5% (100/136) were in the same direction as the direction of
gene expression, indicating that the transposon specifically inserted to result in overexpression of ICN1. RNA-sequencing data also demonstrated that overexpression of Notch1 starts from
exons 25–27 (Fig. 3c), which is consistent with activated Notch1 protein expression analysis (Fig. 3d). This finding suggests an oncogenic role for Notch1 activation in Brca1-mutant
tumourigenesis (Supplementary Fig. 4a). In addition, we designed two shRNAs targeting endogenous Notch1 (the N terminus of Notch1, shRNA-N) or pan-Notch1, including both SB transposon-driven
Notch1 and endogenous Notch1 (the C terminus of Notch1, shRNA-ICN) (Supplementary Fig. 4a, b), and applied them to cell lines derived from Notch1-driven SB tumours (MK1370-3R) and cell
lines derived from non-Notch1-driven SB tumours (MK1097-5R) (Fig. 3e, Supplementary Fig. 4c, d). Knockdown of ICN1 blocked the growth of MK1370-3R tumours, whereas targeting endogenous
Notch1 did not have a significant effect (Fig. 3e, f). In contrast, targeting either the N terminus or C terminus of Notch1 in MK1097-5R tumours did not result in any obvious effects (Fig.
3e, g). These data confirm that transposon insertion-induced overexpression of ICN1 is the driver for MK1370-3R tumours but not for MK1097-5R tumour, which may be driven by other factors.
Consistently, we found that tumours with Notch1 activation exhibited faster progression than did non-Notch1-driven tumours (Fig. 3h). We previously showed that Brca1 deficiency results in
cell death that can be repressed by p53 loss15. We suspected that Notch1 activation might also affect cell lethality to attenuate the lethal block. To examine this possibility, we generated
a Brca1 conditional knockout ES cell line (_Brca1__Co/−__;Cre-ERT2_), in which Cre-mediated recombination is controlled by 4-hydratamoxifen (4-HT) from our Brca1fl/Exon11 ES cells27. These
cells enabled us to compare the impact of Notch1 activation or p53 loss on the viability of Brca1-deficient cells (Supplementary Fig. 4e, f). Acute knockout of Brca1 by 4-HT treatment
resulted in no _Brca1__−/−_ ES colonies (_n_ = 36); we observed 37.5% (18/48) of _Brca1__−/−_ ES colonies among p53-knockout ES cells (_Brca1__Co/−__;Cre-ERT2;p53__−/−_). In two
independently derived ES cell lines overexpressing ICN1 (_Brca1__Co/−__;Cre-ERT2;ICN1__OV_), acute knockout of Brca1 after administration of 4-HT yielded 16.7% (6/36) and 30.6% (11/36)
_Brca1__−/−_ ES colonies (Fig. 3i), indicating that activation of Notch1 suppressed, at least in part, the lethality caused by Brca1 deficiency. Overall, these data suggest that ICN serves
as an oncogene to promote the initiation of Brca1-associated mammary tumourigenesis, which may occur by suppressing the lethality effect caused by Brca1 deficiency. NOTCH1 SUPPRESSES BRCA1
DEFECTS CAUSED MITOTIC CATASTROPHE To explore how ICN1 suppresses the lethality effect under Brca1-deficient conditions, we used a tet-on system to induce ICN1 overexpression in MCF10A cells
by doxycycline (Dox) administration (Supplementary Fig. 5a–c). Acute knockdown of BRCA1 by using shRNA led to cell death, whereas Dox-induced ICN1 overexpression suppressed this lethal
effect (Fig. 4a, b). Our further analysis detected markedly increased γ-H2AX and 53BP1 levels upon acute knockdown of BRCA1, but overexpression of ICN1 blocked this effect (Fig. 4c–e,
Supplementary Fig. 5d, e), suggesting that activation of NOTCH1 abolishes the mitotic catastrophe caused by BRCA1 deficiency by decreasing DNA damage. The rescue effect was also observed
when we treated the cells with the NOTCH1 ligand Jagged1 (Supplementary Fig. 5f, g). Moreover, the cell growth rate was decreased after ICN1 overexpression before cell confluence at day 5
(Supplementary Fig. 5h); and the stall in growth was maintained after replating the cells (Supplementary Fig. 5i). By synchronising the cell cycle with thymidine, we found that
overexpression of ICN1 caused a G2/M-phase lag (Supplementary Fig. 5j, k), even with BRCA1 knockdown. We previously showed that BRCA1 deficiency impairs the G2/M cell-cycle checkpoint, which
causes severe genomic instability28. Thus, we investigated whether ICN1 is able to restore the G2/M checkpoint, which accounts for the decreased DNA damage. By assessing the mitotic index
(MI) with ICN1 overexpression and/or BRCA1 knockdown after gamma-irradiation (IR), we found that ICN1 decreased the mitotic population in both parental and BRCA1-knockdown cells after DNA
damage in both MCF10A (Fig. 4f, g), and T47D cells (Supplementary Fig. 5l, m). These data suggest a role for ICN in activation of the G2/M cell-cycle checkpoint upon irradiation, which is
sufficient to override the lethal block caused by BRCA1 deficiency. We then conducted a time course experiment after IR, and the data also indicated that ICN significantly reduced the
population of MI in BRCA1 WT and knockdown cells at multiple time points (Fig. 4h, Supplementary Fig. 5n). Next, we specifically evaluated S-G2 and S-M progression using quantitative
image-based cytometry combined with a pulse-chase assay (Fig. 4i). The fraction of EdU-positive cells that progressed to G2 phase (BrdU-negative) during the chase indicated that BRCA1
deficiency resulted in premature entry into mitosis and that hyperactivation of NOTCH1 restored cell-cycle regulation (Fig. 4j). Meanwhile, ICN1 also markedly delayed S-M progression (Fig.
4k). To provide further evidence, we performed western blotting for key proteins involved in cell-cycle checkpoints, and the results showed increased CHK1 phosphorylation at Ser317 and
Ser345 along with ICN1 induction in both parental and BRCA1-knockdown cells to varying degrees (Fig. 5a). To investigate the function of p-CHK1, we knocked down CHK1 with shRNA (Fig. 5b) and
found that it increased MI (Fig. 5c) and abolished the suppressive effect of ICN1 on the lethality caused by acute knockdown of BRCA1 (Fig. 5d). These data suggest that activated NOTCH1
functions through p-CHK1 to restore the S/G2 or G2/M checkpoint to suppress apoptosis caused by BRCA1 defects. The CHK1 kinase acts downstream of the ATR/ATM kinase, which is essential for
embryonic development and tumour suppression29,30. To determine whether CHK1 phosphorylation is mediated by ATR or ATM after ICN1 overexpression, we examined the expression levels of these
two kinases and found that ATR phosphorylation was moderately increased but that ATM phosphorylation was not (Fig. 5a). In addition, ATR knockdown blocked ICN1-triggered CHK1 phosphorylation
(Fig. 5e), increased MI (Fig. 5f), and, similar to CHK1 knockdown, abolished the ability of ICN1 to suppress the apoptosis caused by acute knockdown of BRCA1 (Fig. 5g). However, such
effects were not found when ATM was inhibited (Supplementary Fig. 6a). These observations indicate that ICN1 regulates CHK1 phosphorylation through ATR but not ATM. Immunofluorescent
staining results showed that ICN1 induction markedly increased p-ATR as well as p-CHK1 localisation to the nucleus (Supplementary Fig. 6d, e). Furthermore, immunoprecipitation analysis
demonstrated physical binding between ICN1 and ATR (Fig. 5h), indicating that NOTCH1 activation modulates p-ATR localisation to regulate the ATR–CHK1 pathway; these findings may need further
validation. Moreover, ATR–CHK1 activation by ICN1 was also detected in T47D cells, as well as in MCF10A cells treated with the NOTCH1 ligand Jagged1 (Supplementary Fig. 6b, c), suggesting a
general feature of NOTCH1 to regulate the cell cycle in breast cancer cells. To further confirm the correlation between ICN1 and p-ATR, we used two sets of human TNBC patient tissue
microarrays to conduct immunohistochemistry staining (Supplementary Fig. 6f). The results showed that the p-ATR protein level correlated significantly with the NOTCH1 activation level (Fig.
5i, j); conversely, other proteins did not show a consistent correlation with NOTCH1 (Supplementary Fig. 6g, h). Taken together, these data reveal that BRCA1 deficiency impairs the S/G2 and
G2/M cell-cycle checkpoints, which triggers the lethal block, but that ICN1 overexpression restores these checkpoints and suppresses the mitotic catastrophe caused by BRCA1 deficiency
through a non-canonical target: the ATR–CHK1 axis. NOTCH1 CORRELATES WITH INCREASED HUMAN TNBC FORMATION BRCA1 mutation increases the formation of malignant TNBC in humans5. To identify the
driver genes responsible for _BRCA1_-deficient TNBC formation, we used comparative coefficient analysis to select candidates correlating closely with TNBC incidence. First, we converted 169
candidate genes to human genes based on mouse-human orthologues. Then, we traced the expression data and corresponding pathology information for each gene from the human clinical databases
TCGA31 and METABRIC32 and separated the patients into defined equal cohorts based on the expression level of candidate genes. Finally, the TNBC percentage was calculated for each cohort to
determine the correlation between gene expression and TNBC incidence (Fig. 6a, Supplementary Fig. 7). Fifteen genes in our 169-gene pool showed good correlations with TNBC patients in both
TCGA and METABRIC databases (Fig. 6b). _NOTCH1, ARHGAP21, FMNL2_ and _TCF20_ correlated positively with TNBC incidence, i.e., patients with high expression of these genes tended to develop
TNBC. Expression pattern analysis of the _NOTCH1_ gene indicated that TNBC and basal-type tumours exhibited significantly increased levels of NOTCH1, regardless of whether BRCA1 was mutated
(Fig. 6c). TNBC and basal tumour incidence markedly increased with increasing NOTCH1 expression (Fig. 6d). In 54 BRCA1-deficient breast cancers in this cohort, increased expression of NOTCH1
also correlated with TNBC and the basal-type. Analysis of protein levels in BRCA1-deficient tumours indicated that a high protein expression level of NOTCH1 correlated with TNBC incidence
(Supplementary Fig. 8a, b). Moreover, under BRCA1-defective conditions, NOTCH1 tended to display an enhanced ability to promote TNBC and basal-type breast cancer (Supplementary Fig. 8c, d).
A Kaplan–Meier survival curve indicated high expression levels of NOTCH1 to be associated with worse outcomes in TNBC patients (Supplementary Fig. 8e, f). To investigate whether Notch1
activation in our SB tumours also contributes to TNBC formation, we conducted IHC staining for ER, PR and Her2, The data demonstrated that 74.24% (49/66) of ICN1-driven SB tumours were TNBC
(Supplementary Fig. 9a), which was much higher than the average percentage of BrWSB (57%) and BrW control (50%) tumours (Fig. 1e). Moreover, we observed that 62.5% (20/32) of ICN1-driven SB
tumours were the basal-type, compared with 40% (8/20) of randomly selected non-ICN1-driven SB tumours (Supplementary Fig. 9b), suggesting that ICN1 also enhances basal-type tumour formation.
In conclusion, our results indicate that NOTCH1 may be an oncogenic driver for BRCA1-related TNBC and basal-type tumours. NOTCH1 ACTIVATION STIMULATES EMT TO PROMOTE TNBC FORMATION To
further explore the related mechanism, we conducted gene expression analysis of Notch1-driven TNBC tumours compared with non-Notch1-driven TNBC tumours (Fig. 7a). Gene set enrichment
analysis (GSEA) demonstrated that genes upregulated in the EMT process were highly expressed in Notch1-driven tumours (Fig. 7b, Supplementary Data 9). In contrast, genes downregulated during
EMT revealed low levels of expression in Notch1-driven tumours (Fig. 7c, Supplementary Data 9). These results indicate that EMT-related genes correlated strictly with Notch1 activation in
TNBC tumours. It has been reported that Notch1 signalling regulates the EMT in human breast cancer33,34, and that expression of EMT-related markers in TNBCs can serve as a signature of a
certain subgroup of TNBC35,36. Therefore, we studied whether NOTCH1 promotes EMT transition, leading to TNBC formation in BRCA1-defective conditions. By using a tet-on system to induce ICN1
overexpression in different mammary cell lines, we found that activated NOTCH1 upregulated Fibronectin 1, Vimentin and Slug to varying degrees in MCF10A, T47D and MCF7 cells (Fig. 7d, e,
Supplementary Fig. 9c, d). Moreover, we induced Notch1 expression in ATR or CHK1 knockdown cells and then assessed their EMT status. As shown in the Supplementary Fig. 9e, f, Dox-mediated
Notch1 induction increased expression of Fibronectin; in contrast, knockdown of ATR or CHK1 did not affect expression. These data suggest that the function of Notch1 in EMT stimulation is
independent of ATR or CHK1. Human BRCA1-mutant breast cancer samples also showed high levels of mesenchymal markers (Fig. 7f). Furthermore, a meta-analysis of BRCA1-related cancer revealed
that all basal-like tumours belong to the TNBC subtype, much higher than the average level (Supplementary Fig. 8g, h). These data suggest the EMT might be the driver of BRCA1-mutant cell
transdifferentiation. Therefore, we conclude that NOTCH1 hyperactivation promotes EMT, which might induce the formation of TNBC and basal-like breast cancer. INHIBITION OF ATR–CHK1 ENHANCES
THE TUMOURICIDAL ACTIVITY Cisplatin preferentially kills BRCA1-deficient breast cancers compared to BRCA1-proficient cancers; however, the inhibition efficiency is always compromised when
cancers gain various additional alterations37,38. Furthermore, a high dosage of cisplatin can cause serious nephrotoxicity39,40. Our previous data indicate that cisplatin inhibits tumour
metastasis by blocking EMT, though prolonged treatment may result in drug resistance41. TNBC is the most difficult type of breast cancer to treat due to its unresponsiveness to current
clinical targeted therapies, high rate of recurrence and poor prognosis42. A high expression level of Notch1 correlates closely with TNBC incidence and activates ATR–CHK1 signalling to
compensate for the DNA damage repair deficiency and benefit tumourigenesis, and cisplatin causes DNA crosslinking, leading to DNA double strand breaks (DSBs). Accordingly, we hypothesised
that ATR or CHK1 inhibitor combined with cisplatin might have a synthetic lethal effect on TNBC samples. To investigate this, we first assessed the effect on the HCC1937 cell line, and the
results indicated that a low dosage of an ATR inhibitor (AZD6738 or VE821) combined with different concentrations of cisplatin effectively killed HCC1937 cells (Fig. 8a, b). Notably,
combined treatment with cisplatin and an ATM inhibitor (KU-60019) failed to show a synergistic effect in killing cancer cells, which is consistent with our previous finding that ICN1
regulates downstream effects through ATR but not ATM. Moreover, the CHK1 inhibitor CCT245737 or LY2603618 also increased sensitivity to cisplatin (Fig. 8a, b). Next, we tested four
additional BRCA1-mutant TNBC cell lines and found that the ATR or CHK1 inhibitor markedly enhanced killing but that the ATM inhibitor showed the opposite effect (Fig. 8c). These data verify
our model that ICN1 can, through the non-canonical target ATR or CHK1, regulate the cell cycle and promote TNBC formation. Furthermore, we conducted in vivo treatment of TM00091 PDX tumours,
which carry two BRCA1 point mutations (Q356R and C61G), with NOTCH1 hyperactivation (Supplementary Fig. 10a). Because a high dose of cisplatin markedly decreased mouse body weight
(Supplementary Fig. 10b) indicating a toxic effect, we administered cisplatin at a low dose. The results showed that AZD6738 single treatment did not affect tumour growth, even at high
doses. A low dosage of AZD6738 markedly increased the killing effect of cisplatin (Fig. 8d, Supplementary Fig. 10c). Notably, combined with ATRi, cisplatin at a low dose (1.5 mg/kg)
significantly inhibited tumour progression. According to IHC staining, double treatment induced massive apoptosis/death compared with high-dosage single treatment of ATRi or cisplatin (Fig.
8e, f). In conclusion, cisplatin-mediated inhibition of the NOTCH1 non-canonical target ATR–CHK1 cascade can synergistically kill BRCA1-related TNBC both in vitro and in vivo. DISCUSSION
Tumours initiate from normal cells through a long “evolution” process, encountering various blocks, including immune checkpoints, telomere limitation, contact inhibition and genome
instability-induced apoptosis, among others. Our previous studies indicated that the progression of Brca1-related tumourigenesis occurs through a long latency that is accompanied by initial
cellular lethality12,14, suggesting the existence of such blocks. In the present study, SB-mediated mutagenesis revealed several findings that advance our understanding of this situation. We
demonstrated that a defective G2/M cell-cycle checkpoint caused by Brca1 deficiency serves as one such block, as restoration of this checkpoint by Notch1 activation suppressed lethality and
allowed Brca1-mutant cells to grow. Further analysis revealed several previously unknown functions of Notch1 that may be necessary in accelerating Brca1-related TNBC tumourigenesis. The
Notch signalling pathway has an important role in several biological processes. Deregulated Notch signalling is associated with several human malignancies, such as carcinomas of the lung,
colon, head and neck, breast, pancreas and kidney, either as an oncogene or a tumour suppressor26,43,44. In the present study, our data clearly demonstrate that expression of ICN1,
representing Notch1 activation, promotes Brca1-associated mammary tumour formation. One of the major abnormalities induced by Brca1 deficiency is a defective G2/M cell-cycle checkpoint28,
which may serve as a double-edged sword that, on the one hand, causes genetic instability leading to cell death and, on the other hand, creates an environment for mutations leading to
tumourigenesis if p53 is mutated14. We further showed that loss of p53 enables Brca1-deficient cells to survive despite the presence of widespread genetic instability due to inactivation of
apoptosis mediated by p5315. Notably, we found that activation of Notch1 also enables Brca1-deficient cells to survive through a distinct mechanism, namely, partial restoration of the G2/M
cell-cycle checkpoint possibly through the ATR–CHK1 cascade, which is known to have an essential role in activating G2/M29. ATR also activates intra-S and S/G2 cell-cycle checkpoints45,46,
which prompted us to examine these checkpoints. Our data indicate that Notch1 activation does not have an obvious role in the intra-S checkpoint but that it does activate the S/G2 cell-cycle
checkpoint. Activation of these checkpoints delays cell-cycle progression, as reflected by reduced cell proliferation and attenuation of the lethal block caused by BRCA1 deficiency.
Clinical sample analysis demonstrated that NOTCH1 activation correlated positively with the phosphorylation level of ATR plus the capacity of direct binding between NOTCH1 and ATR;
therefore, we speculate that NOTCH1 rescues BRCA1-defective lethality through a non-canonical target, the ATR–CHK1 signalling pathway. Consequently, we conclude that NOTCH1 has an oncogenic
function in the presence of BRCA1 loss. Nonetheless, we should note that under some other conditions, e.g., in tumours with intact DNA damage repair systems or cell-cycle checkpoints,
activation of ATR–CHK1 signalling would inhibit cell-cycle progression and elicit tumour suppressor functions to block tumour development. This may help to explain the dual functions of
NOTCH1 as a tumour suppressor and an oncogene under different circumstances26. Obviously, further studies are needed to support this hypothesis. Previous studies have revealed that
activation of NOTCH1 correlates with cancer stem cell maintenance and expansion47 and that high NOTCH1 expression may serve as a prognostic marker in patients with TNBC48, yet the underlying
mechanism remains elusive. Our study demonstrates that activation of Notch1 promotes TNBC formation. BRCA1-deficient cancers originate from luminal cells, which are largely ERα positive
initially49. Our previous study analysing tumours at different stages also showed that Brca1_−_/_−_ mammary tumours are initially ERα positive and gradually become negative as they grow50;
nonetheless, it remains unclear how ERα is lost. EMT-related markers in TNBC might comprise a signature of a certain subgroup of TNBC35,36,51, and the EMT programme has an important role in
basal breast cancer52,53. Thus, in light of our finding that activation of Notch1 can enhance expression of EMT signature genes, including Fibronectin and Slug, we believe that Notch1 may
drive TNBC formation under BRCA1-defective conditions by inducing expression of EMT signature genes, thereby promoting the transdifferentiation of luminal cells into the basal-type and at
the same time strongly enhancing TNBC progression. This mechanism may also explain why basal-like tumours largely overlap with TNBC in both human and mouse BRCA1-related breast tumours.
Analysis of the human breast cancer database revealed a close correlation between high expression levels of NOTCH1 and TNBC incidence, especially BRCA1-related TNBC. Thus, it is important to
develop an effective therapy for this group of tumours. We recently found that cisplatin, which kills BRCA1-deficient cancers with high efficiency by causing DNA crosslinking54, also blocks
tumour metastasis through inhibition of EMT41. Given our finding that NOTCH1 promotes BRCA1-deficient tumour growth by activating ATR–CHK1 and inducing EMT, we inhibited the ATR/CHK1
pathway in combination with a low dose of cisplatin to effectively kill TNBC in our mouse model and PDX model55. This approach might avoid the non-specific cytotoxic side effects commonly
associated with high doses of cisplatin, thus providing a potent clinical option for TNBC treatment. PARP1 inhibitors are used for the treatment of BRCA1-deficient cancers, as inhibition of
PARP1 generates more single-strand DNA damage, which results in DSBs and consequently leads to a synthetic lethality with BRCA1 deficiency. Our strategy of blocking ATR–CHK1 is likely to
target the cell cycle, i.e., it targets different aspects of PARP1. Our data indicate that inhibition of ATR or CHK1 works with cisplatin, which causes DNA inter-strand crosslink, leading to
DSBs. As the effect of cisplatin shares some similarities with that of PARP1 inhibitors, we suspect that blocking ATR–CHK1 might also involve PARP1 function, which should be investigated in
the near future. Of note, alteration of many genes involved in some other systems and pathways can accelerate tumourigenesis, including the ubiquitination system, cytoskeleton and cell
junction, gene expression regulation, and protein kinase/phosphatase modification-related functions. As Brca1 is an E3 ligase56, enrichment of ubiquitin-related genes may have a compensatory
role in Brca1 deficiency. Cytoskeleton rearrangement is involved in the EMT and tumour metastasis, whereas tight junctions are a specialised structure of epithelial and endothelial cell
membranes and have an important role in maintaining most apical junctional complexes57. Indeed, components of all enriched pathways among our candidate genes are known to participate in cell
proliferation, transformation and metastasis suppression, though their specific role in Brca1-related tumourigenesis remains elusive. Regardless, these data demonstrate that these mutations
can activate multiple oncogenic pathways to overcome the lethal block of Brca1 deficiency in mammary epithelial cells to promote the initiation and progression of cancer formation. In
summary, our study revealed that activation of Notch1 has various important consequences, including cell-cycle checkpoint activation and promotion of transdifferentiation by inducing
expression of EMT signature genes. By stimulating ATR–CHK1 signalling, activation of the G2/M and S/G2 cell-cycle checkpoints, at least in part, overcomes the lethal block caused by Brca1
deficiency through suppression of the mitotic catastrophe and promotes tumour initiation (Fig. 8g). However, activation of Notch1 also promotes TNBC formation by activating the EMT
signalling pathway. The combination of these effects enhances tumourigenesis; conversely, treatment with cisplatin, which inhibits the EMT, and ATR–CHK1 inhibitors that target the cell-cycle
checkpoint display good synergy in inhibiting TNBC, thus providing a potent clinical option for this fatal disease. METHODS MICE All mouse experiments were performed under the ethical
guidelines of the University of Macau (animal protocol number: UMAEC-037-2015). Mice were housed in a Specific-pathogen-free (SPF) facility at 23–25 °C on a 12-h light/dark cycle. The
following mouse strains were used in this study. (1) Brca1 conditional knockout (_Brca1__Co/Co_) mice, in which deletion of exon 11 of Brca1 is controlled by two mammary tissue-specific
_Cre_ transgenes (_WAP-Cre_ or _MMTV-Cre_)14. (2) Two strains with a conditionally expressed SB11 transposase that includes a floxed transcriptional stop cassette to be activated by _Cre_.
(3) Independent transgenic lines of T2onc3 (12740 and 12775)22, located on chromosome 9 and chromosome 12, respectively. Because of the several transgenic strains used in this study, the
resulting cohorts of mice were of a mixed genetic background, including C57BL/6J, 129SVE and FVB. Mice without the SB transposase or transposon served as a negative control to compare the
function of the transposon on tumour formation under Brca1-defective conditions. Animals in all experimental groups were examined twice a week for tumourigenesis. Complete necropsies were
performed to assess the primary tumour and metastasis in other organs. Only female mice were used for the experiment, and all the mice were pregnant once at 2–4 months of age to activate
expression of _WAP-Cre_ or _MMTV-Cre_. We named the tumour based on the mouse eartag number and the tumour location information; for example, MK1370-3R means that the tumour was collected
from the third right mammary gland of mouse MK1370. HISTOLOGY AND IHC Routine haematoxylin and eosin staining was performed on 6-μm sections of formalin-fixed, paraffin-embedded (FFPE)
tissues. IHC for ERα (1:50, Santa Cruz, sc-542), PR (1:50, Santa Cruz, sc-538), HER2 (1:50, Santa Cruz, sc-284), cleaved caspase-3 (1:500, CST, 9664) and Ki67 (1:200, Abcam, ab16667) was
performed to characterise tumour histopathology using a Histostain-Plus IHC kit (Thermo, 859043, Lot: 1954379A) after antigen retrieval (pH 6.0), quenching of endogenous peroxidase and
overnight incubation with the primary antibody. CIS IDENTIFICATION AND ANNOTATION A sequence library of SB transposon insertion sites was constructed using Splinkerette-PCR58 followed by a
second round of PCR with SB Illumina adaptors (Supplementary Data 10). Samples were sequenced using the Illumina HiSeq X-Ten platform with paired-end 150-bp reads. The resulting reads were
filtered and trimmed using cutadapt version 1.16 for contaminant sequences, including Illumina adaptor sequences, splinkerette linker sequences and transposon sequences. Reads without a
valid adaptor and transposon sequence were excluded from further analysis. Reads <20 bp after trimming were discarded. Clean reads were aligned to the mouse reference genome mm10,
allowing 3 mismatches, using Bowtie2 version 2.3.4 followed by TAPDANCE analysis23. The aligned locations identified as CISs were automatically annotated based on mouse reference genes
(mm10.gtf). CISs located in the gene body plus 3 kb upstream with _p_-values < 0.05 were analysed further. CIS genes were then annotated with the criteria of gene location plus the
upstream 3 kb. Although local hopping of the SB transposon always increased within the chromosome where the transposon concatamer was located, the insertion patterns among the SB strains
12740 or 12775 did not show bias with regard to chromosome, regardless of which Cre (MMTV-Cre or WAP-Cre) was used (Supplementary Fig. 2a). Therefore, we assessed all insertion sites for
candidate gene identification, including those in Chr9 or Chr12, which are the original insertion sites for the T2Onc3 transposon in strains 12740 or 12775, respectively. However, to avoid
the bias of local hopping, we chose genes identified in more than 5% of the samples in both 12740 and 12775 as strict criteria to filter out fake genes. CIS genes were considered oncogenic
drivers if all insertions were located in a specific region and the CAG promoter of the transposon was in the same orientation as the gene’s ORF, which will drive target gene overexpression
from the insertion site. Conversely, genes were considered tumour suppressors in the event of an unbiased insertion site and no bias orientation of the CAG promoter, whereby the
polyadenylation signal of the transposon most likely disrupts the gene. TRANSCRIPTOME SEQUENCING AND ANALYSIS RNA-seq libraries were constructed using an Illumina TruSeq RNA library Prep Kit
and sequenced with the Illumina HiSeq X-Ten platform to generate a minimum of 25 million paired-end 150-bp reads. Sequencing reads were aligned to the mouse reference genome mm10 and
processed using HISAT2 version 2.1.059. Differential expression analysis was conducted using DESeq2 version 1.22.260. For exon-level expression, reads from each exon of the target gene were
counted followed by normalisation with total reads and length of individual exons to avoid bias due to different exon sizes. PAM50 subtype assignment was conducted using Genefu (2.18.1)61.
FUNCTIONAL ENRICHMENT ANALYSIS Gene function annotation enrichment analysis was performed with DAVID Bioinformatics v6.8 using the Gene Ontology and KEGG pathway data sets62,63. GSEA (GSEA
v3.0)64 was also used for gene expression difference analysis. Analysis of driver gene molecular interactions among CIS genes was conducted using STRING v11.0 online tools65. CROSS-SPECIES
TNBC CORRELATION ANALYSIS Gene expression data for human patient samples were downloaded from TCGA31 and METABRIC32 databases by using R studio (version 3.5.1) with cgdsr packages. Clinical
pathology information was also retrieved. We obtained 1898 and 403 patients from the METABRIC and TCGA databases, respectively. Among them, 37 BRCA1-mutant samples were obtained from the
former. For each gene, we ranked the samples based on expression levels. The samples were separated equally into 10 (for all patients) or 6 (for BRCA1-mutant patients) cohorts. Finally, we
counted the TNBC proportion of each single cohort to determine the correlation between the gene expression level and TNBC morbidity for each individual gene. WESTERN BLOTTING Cultured cells
were homogenised in RIPA buffer supplemented with phosphatase and protease inhibitor cocktails (Sigma). After measurement and normalisation of the protein concentration, whole-cell lysates
were loaded onto polyacrylamide gels for electrophoresis. Proteins were electrotransferred onto PVDF membranes (Bio-Rad), blocked with 5% BSA in TBS-T buffer for 1 h at room temperature and
incubated with the primary antibody overnight at 4 °C. The blots were incubated with the corresponding secondary antibody (Cell Signaling, CST), and immunoreactive bands were developed with
the ECL substrate (Millipore). The primary antibodies used in this study were as follows: BRCA1 (1:200, Santa Cruz, sc-642), NOTCH1 (1:1000, CST 4380), E-cadherin (1:1000, CST 3195),
β-catenin (1:1000, Abcam, ab32572), N-cadherin (1:1000, Abcam, ab76057), Vimentin (1:1000, CST 5741), Fibronectin (1:1000, Abcam, ab2431), Slug (1:1000, CST 9585), β-actin (1:4000, Sigma,
A5316), ATM (1:1000, Abcam ab78), pATM (1:1000, Abcam, ab81292), ATR (1:1000, CST 2790), p-ATR-1989 (1:1000, Abcam, ab227851), p-ATR-428 (1:1000, CST 2853), CHK1 (1:200, Santa Cruz,
sc-56288), p-CHK1-317 (1:1000,CST 12302) and p-CHK1-345 (1:1000, CST 2348). The secondary antibodies used in this study were as follows: Anti-rabbit IgG, HRP-linked Antibody (1:5000, CST
7074, Lot:28), Anti-mouse IgG and HRP-linked Antibody (1:5000, CST 7076, Lot:32). QUANTITATIVE RT-PCR Total RNA was extracted using TRIzol reagent (Thermo) by following their routine
procedure. cDNA was synthesised with the QuantiTect Reverse Transcription kit (Qiagen, 205313). Quantitative PCR was performed using FastStart Universal SYBR Green Master (Rox) mix (Sigma)
on a QuantStudio 7 Flex Real-Time PCR System (Thermo). The primer sequences used are listed in Supplementary Data 10. VECTOR CONSTRUCTION shRNA plasmids were constructed by inserting
annealed targeting oligos into the pLKO.1 backbone (Addgene, 8453) after digestion with _Age_I and _Eco_RI (NEB, R0552 and R0101). ICN1 cDNA was cloned from the Puro-iNotch1IC plasmid
(Addgene, 75338)66 for mice and EF.hICN1.CMV.GFP plasmid (Addgene, 17623)67 for humans and then inserted into the tet-on plasmid (Addgene, 80921)68. An HA tag was also added to the N
terminus of ICN1. Routine PCR and Sanger sequencing were performed to confirm successful insertion into the vector. The oligonucleotide sequences used for shRNA knockdown are listed in
Supplementary Data 10. CELL CULTURE Mouse primary mammary tumour cells were derived from SB tumours after dissociation with digestion buffer, which contained DMEM/F12 medium (Thermo), 5%
FBS, 10 ng/ml EGF (Thermo, 13247-051), 0.5 mg/ml hydrocortisone (Sigma, H0888), 20 ng/ml cholera toxin (Sigma, C-3012), 5 μg/ml insulin (Sigma, 350-020), 300 U/ml collagenase III
(Worthington, S4M7602S) and hyaluronidase (Sigma, H3506)10. MCF10A cells were cultured in DMEM/F12 medium containing 5% horse serum (Thermo), 20 ng/ml EGF, 0.5 mg/ml hydrocortisone, 100
ng/ml cholera toxin, 10 μg/ml insulin and pen/strep (Thermo). T47D, MCF7, MDA-MB-231, MDA-MB-436, MDA-MB-468, Sum149 and HCC1937 cells were cultured in DMEM supplemented with 10% FBS,
glutamine (Thermo), insulin and pen/strep. The human primary breast cancer cell line TM00091 was derived from a PDX model (Jax, TM00091)55, which carries a BRCA1 mutation with high
expression of NOTCH1, by using the same approach used for the mouse primary cells. All cells were routinely tested to exclude mycoplasma contamination. CELL-CYCLE SYNCHRONISATION The double
thymidine block approach was applied for cell-cycle synchronisation as previously reported69. Briefly, cells were blocked using 2 mM thymidine for 18 h when they reached 25–30% confluence.
After 9 h of culture in regular medium, 15 h of thymidine incubation was performed for the second round of blocking. The cells were then released and collected at different time points for
cell-cycle flow cytometry analysis. MI ANALYSIS Cells were cultured in 6-well plates. After irradiation, the cells were trypsinised and fixed in 70% cold ethanol for at least 1 h. The cells
were permeabilised with 0.25% Triton X-100 for 15 min at room temperature and incubated with 5% BSA/PBS for blocking. The cells were then incubated sequentially with an anti-p-H3 antibody
(1:500, Millipore, 06-570), Alexa 488-conjugated secondary antibody (1:1000, Thermo, A-11070, Lot:1431810) and PI. Fluorescence-activated cell sorting (FACS) was performed immediately to
analyse the MI28. FLUORESCENCE MICROSCOPY Cultured cells were fixed with 4% PFA/PBS for 10 min, permeabilised with 0.25% Triton X-100 for 15 min and blocked in 5% BSA/PBS for 1 h at room
temperature. For BrdU (1:200, Santa Cruz, sc-32323) staining, DNA was denatured with 2 N HCl for 30 min and neutralised with two round washes of PBS before the blocking step. For EdU
staining, the click-iT reaction was carried out after permeabilisation using a Clic-iT EdU Alexa Fluor imaging kit (Thermo, C10339). The primary antibody was incubated overnight at 4 °C.
After three washes with PBS, secondary antibodies and DAPI were incubated for 1 h at room temperature. For the quantitative image-based cytometry assay, images were captured and analysed
using an In Cell Analyzer 2000 (GE). The γ-H2AX (1:500, Millipore, 05-636, Lot: 3076468) and 53BP1 (1:200, Santa Cruz, sc-22760, Lot: I1813) immunofluorescence staining are same with MI
analysis. Secondary antibodies for this experiments are: F(ab′)2-Goat anti-Rabbit IgG (H + L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 (1:1000, Thermo, A-11070, Lot: 1494754),
F(ab′)2-Goat anti-Rabbit IgG (H + L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 (1:1000, Thermo, A11072, Lot: 1431810), F(ab′)2-Goat anti-Mouse IgG (H + L) Cross-Adsorbed Secondary
Antibody, Alexa Fluor 594 (1:1000, Thermo, A-11020, Lot: 1454439), F(ab′)2-Goat anti-Mouse IgG (H + L) Cross-Adsorbed Secondary Antibody and Alexa Fluor 488 (1:1000, Thermo, A-11017, Lot:
1557766) CELL VIABILITY ANALYSIS Cell viability was assessed using different approaches, including the MTT assay and the Alamar Blue assay. For the MTT assay, MTT dye
(3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide; thiazolyl blue, Sigma-Aldrich) was added (final concentration: 0.5 mg/ml) to cultured cells for 4 h. The formazan crystals were
solubilised with DMSO, and absorbance was measured at 570 nm using a plate spectrophotometer. The Alamar Blue assay was used for cell drug treatment analysis. Briefly, culture medium
containing 0.02% Alamar Blue was added to cultured cells, and absorbance at 590 nm under 560 nm excitation was measured 2 h of incubation at 37 °C. ALLOGRAFT/XENOGRAFT STUDIES Tumours or
cultured cells were dissociated into single cells and resuspended in 50% Matrigel (Corning, 356234) for inoculation. NSG or nude mice were anaesthetised with tribromoethanol, and a small
abdominal incision was made. Mammary fat pads were exposed gently by forceps, and 1 million cells were injected using a microlitre syringe with a 27-gauge needle. The mice were maintained in
an SPF facility for further drug administration. The mice began receiving drug treatment when the tumours reached ~200 mm3. Tumour volume was calculated as _V_ = (_W_2 × _L_)/270. AZD6738
was administered by oral gavage at 10 or 0.5 mg/kg every 3 days together with cisplatin, which was administered by intraperitoneal injection at 1.5 or 5 mg/kg. STATISTICS AND REPRODUCIBILITY
Microsoft Excel, GraphPad Prism (version 6) and R (version 3.5.1) software were used for statistical calculations. Specific statistical tests, sample number and other information are
indicated in the main text or figure legends. The _t_-test and two-sided statistical analysis approach was used to determine the significance of the difference between the different sets of
data if there was no specific indication. All experiments were conducted at least three times independently, and similar results were adopted for further analysis to guarantee
reproducibility. REPORTING SUMMARY Further information on research design is available in the Nature Research Reporting Summary linked to this article. DATA AVAILABILITY The RNA sequence
data have been deposited in the Sequence Read Archive (SRA) database under the accession code PRJNA529536. The data from TCGA and METABRIC referenced in the study are available in a public
repository from the cBioPortal website (https://www.cbioportal.org/). All the other data supporting the findings of this study can be found within the supplementary files. A reporting
summary for this article is available as a Supplementary Information file. Source data for all the plots are provided as a Source Data file. REFERENCES * Hall, M. J. et al. BRCA1 and BRCA2
mutations in women of different ethnicities undergoing testing for hereditary breast-ovarian cancer. _Cancer_ 115, 2222–2233 (2009). CAS PubMed Google Scholar * Esteller, M. et al.
Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. _J. Natl Cancer Inst._ 92, 564–569 (2000). CAS PubMed Google Scholar * Lambie, H. et al. Prognostic
significance of BRCA1 expression in sporadic breast carcinomas. _J. Pathol._ 200, 207–213 (2003). CAS PubMed Google Scholar * Ford, D., Easton, D. F., Bishop, D. T., Narod, S. A. &
Goldgar, D. E. Risks of cancer in BRCA1-mutation carriers. Breast Cancer Linkage Consortium. _Lancet_ 343, 692–695 (1994). CAS PubMed Google Scholar * Lee, E. et al. Characteristics of
triple-negative breast cancer in patients with a BRCA1 mutation: results from a population-based study of young women. _J. Clin. Oncol._ 29, 4373–4380 (2011). PubMed PubMed Central Google
Scholar * Spurdle, A. B. et al. Refined histopathological predictors of BRCA1 and BRCA2 mutation status: a large-scale analysis of breast cancer characteristics from the BCAC, CIMBA, and
ENIGMA consortia. _Breast Cancer Res_ 16, 3419 (2014). PubMed PubMed Central Google Scholar * Zhang, J. et al. Comprehensive analysis of BRCA1 and BRCA2 germline mutations in a large
cohort of 5931 Chinese women with breast cancer. _Breast Cancer Res. Treat._ 158, 455–462 (2016). CAS PubMed Google Scholar * Deng, C. X. & Scott, F. Role of the tumor suppressor gene
Brca1 in genetic stability and mammary gland tumor formation. _Oncogene_ 19, 1059–1064 (2000). CAS PubMed Google Scholar * Deng, C. X. BRCA1: cell cycle checkpoint, genetic instability,
DNA damage response and cancer evolution. _Nucleic Acids Res_ 34, 1416–1426 (2006). CAS PubMed PubMed Central Google Scholar * Xu, X. et al. BRCA1 represses DNA replication initiation
through antagonizing estrogen signaling and maintains genome stability in parallel with WEE1-MCM2 signaling during pregnancy. _Hum. Mol. Genet_. 28, 842–857 (2018). * Lahusen, T. J. et al.
BRCA1 function in the intra-S checkpoint is activated by acetylation via a pCAF/SIRT1 axis. _Oncogene_ 37, 2343–2350 (2018). CAS PubMed Google Scholar * Shen, S. X. et al. A targeted
disruption of the murine Brca1 gene causes gamma-irradiation hypersensitivity and genetic instability. _Oncogene_ 17, 3115–3124 (1998). CAS PubMed Google Scholar * Hakem, R. et al. The
tumor suppressor gene Brca1 is required for embryonic cellular proliferation in the mouse. _Cell_ 85, 1009–1023 (1996). CAS PubMed Google Scholar * Xu, X. et al. Conditional mutation of
Brca1 in mammary epithelial cells results in blunted ductal morphogenesis and tumour formation. _Nat. Genet._ 22, 37–43 (1999). CAS PubMed Google Scholar * Xu, X. L. et al. Genetic
interactions between tumor suppressors Brca1 and p53 in apoptosis, cell cycle and tumorigenesis. _Nat. Genet._ 28, 266–271 (2001). CAS PubMed Google Scholar * Cao, L. et al. ATM-Chk2-p53
activation prevents tumorigenesis at an expense of organ homeostasis upon Brca1 deficiency. _Embo J._ 25, 2167–2177 (2006). CAS PubMed PubMed Central Google Scholar * Cao, L., Li, W.,
Kim, S., Brodie, S. G. & Deng, C. X. Senescence, aging, and malignant transformation mediated by p53 in mice lacking the Brca1 full-length isoform. _Genes Dev._ 17, 201–213 (2003). CAS
PubMed PubMed Central Google Scholar * Brodie, S. G. et al. Multiple genetic changes are associated with mammary tumorigenesis in Brca1 conditional knockout mice. _Oncogene_ 20, 7514–7523
(2001). CAS PubMed Google Scholar * Bard-Chapeau, E. A. et al. Transposon mutagenesis identifies genes driving hepatocellular carcinoma in a chronic hepatitis B mouse model. _Nat.
Genet._ 46, 24–32 (2014). CAS PubMed Google Scholar * Mann, M. B. et al. Transposon mutagenesis identifies genetic drivers of Braf(V600E) melanoma. _Nat. Genet._ 47, 486–U86 (2015). CAS
PubMed PubMed Central Google Scholar * Chen, L. et al. Transposon insertional mutagenesis in mice identifies human breast cancer susceptibility genes and signatures for stratification.
_Proc. Natl Acad. Sci. USA_ 114, E2215–E2224 (2017). CAS PubMed PubMed Central Google Scholar * Dupuy, A. J. et al. A modified sleeping beauty transposon system that can be used to model
a wide variety of human cancers in mice. _Cancer Res._ 69, 8150–8156 (2009). CAS PubMed PubMed Central Google Scholar * Sarver, A. L., Erdman, J., Starr, T., Largaespada, D. A. &
Silverstein, K. A. TAPDANCE: an automated tool to identify and annotate transposon insertion CISs and associations between CISs from next generation sequence data. _BMC Bioinformatics_ 13,
154 (2012). PubMed PubMed Central Google Scholar * Iso, T., Kedes, L. & Hamamori, Y. HES and HERP families: multiple effectors of the Notch signaling pathway. _J. Cell. Physiol._ 194,
237–255 (2003). CAS PubMed Google Scholar * Lobry, C., Oh, P., Mansour, M. R., Look, A. T. & Aifantis, I. Notch signaling: switching an oncogene to a tumor suppressor. _Blood_ 123,
2451–2459 (2014). CAS PubMed PubMed Central Google Scholar * Radtke, F. & Raj, K. The role of Notch in tumorigenesis: oncogene or tumour suppressor? _Nat. Rev. Cancer_ 3, 756–767
(2003). CAS PubMed Google Scholar * Willis, N. A. et al. BRCA1 controls homologous recombination at Tus/Ter-stalled mammalian replication forks. _Nature_ 510, 556–559 (2014). ADS CAS
PubMed PubMed Central Google Scholar * Xu, X. et al. Centrosome amplification and a defective G2-M cell cycle checkpoint induce genetic instability in BRCA1 exon 11 isoform-deficient
cells. _Mol. Cell_ 3, 389–395 (1999). CAS PubMed Google Scholar * Liu, Q. H. et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint.
_Genes Dev._ 14, 1448–1459 (2000). CAS PubMed PubMed Central Google Scholar * Zhao, H. & Piwnica-Worms, H. ATR-mediated checkpoint pathways regulate phosphorylation and activation
of human Chk1. _Mol. Cell Biol._ 21, 4129–4139 (2001). CAS PubMed PubMed Central Google Scholar * Ciriello, G. et al. Comprehensive molecular portraits of invasive lobular breast cancer.
_Cell_ 163, 506–519 (2015). CAS PubMed PubMed Central Google Scholar * Pereira, B. et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic
landscapes. _Nat. Commun._ 7, 11479 (2016). ADS CAS PubMed PubMed Central Google Scholar * Shao, S. et al. Notch1 signaling regulates the epithelial-mesenchymal transition and invasion
of breast cancer in a Slug-dependent manner. _Mol. Cancer_ 14, 28 (2015). * Stylianou, S., Clarke, R. B. & Brennan, K. Aberrant activation of Notch signaling in human breast cancer.
_Cancer Res._ 66, 1517–1525 (2006). CAS PubMed Google Scholar * Cheung, S. Y. et al. Role of epithelial-mesenchymal transition markers in triple-negative breast cancer. _Breast Cancer
Res. Treat._ 152, 489–498 (2015). CAS PubMed Google Scholar * Jang, M. H., Kim, H. J., Kim, E. J., Chung, Y. R. & Park, S. Y. Expression of epithelial-mesenchymal transition-related
markers in triple-negative breast cancer: ZEB1 as a potential biomarker for poor clinical outcome. _Hum. Pathol._ 46, 1267–1274 (2015). CAS PubMed Google Scholar * Chisholm, C. L. et al.
Ammonium tetrathiomolybdate treatment targets the copper transporter ATP7A and enhances sensitivity of breast cancer to cisplatin. _Oncotarget_ 7, 84439–84452 (2016). PubMed PubMed Central
Google Scholar * Zheng, H. P., Shao, F. Y., Martin, S., Xu, X. L. & Deng, C. X. WEE1 inhibition targets cell cycle checkpoints for triple negative breast cancers to overcome cisplatin
resistance. _Sci. Rep._ 7, 43517 (2017). * Volarevic, V. et al. Molecular mechanisms of cisplatin-induced nephrotoxicity: a balance on the knife edge between renoprotection and tumor
toxicity. _J. Biomed. Sci._ 26, 25 (2019). * Miller, R. P., Tadagavadi, R. K., Ramesh, G. & Reeves, W. B. Mechanisms of cisplatin nephrotoxicity. _Toxins_ 2, 2490–2518 (2010). CAS
PubMed PubMed Central Google Scholar * Vassilopoulos, A. et al. Synergistic therapeutic effect of cisplatin and phosphatidylinositol 3-kinase (PI3K) inhibitors in cancer growth and
metastasis of Brca1 mutant tumors. _J. Biol. Chem._ 289, 24202–24214 (2014). CAS PubMed PubMed Central Google Scholar * Shao, F. Y., Sun, H. & Deng, C. X. Potential therapeutic
targets of triple-negative breast cancer based on its intrinsic subtype. _Oncotarget_ 8, 73329–73344 (2017). PubMed PubMed Central Google Scholar * Wang, Z. W., Li, Y. W. & Sarkar, F.
H. Notch signaling proteins: legitimate targets for cancer therapy. _Curr. Protein Pept. Sci._ 11, 398–408 (2010). CAS PubMed PubMed Central Google Scholar * Avila, J. L. & Kissil,
J. L. Notch signaling in pancreatic cancer: oncogene or tumor suppressor? _Trends Mol. Med._ 19, 320–327 (2013). CAS PubMed PubMed Central Google Scholar * Saldivar, J. C. et al. An
intrinsic S/G2 checkpoint enforced by ATR. _Science_ 361, 806–810 (2018). ADS CAS PubMed PubMed Central Google Scholar * Iyer, D. R. & Rhind, N. The intra-S checkpoint responses to
DNA damage. _Genes_ 8, 74 (2017). * Xie, X. et al. c-Jun N-terminal kinase promotes stem cell phenotype in triple-negative breast cancer through upregulation of Notch1 via activation of
c-Jun. _Oncogene_ 36, 2599–2608 (2017). CAS PubMed Google Scholar * Kim, D. et al. Notch1 in tumor microvascular endothelial cells and tumoral miR-34a as prognostic markers in locally
advanced triple-negative breast cancer. _J. Breast Cancer_ 22, 562–578 (2019). PubMed PubMed Central Google Scholar * Lim, E. et al. Aberrant luminal progenitors as the candidate target
population for basal tumor development in BRCA1 mutation carriers. _Nat. Med._ 15, 907–913 (2009). CAS PubMed Google Scholar * Li, W., Xiao, C., Vonderhaar, B. K. & Deng, C. X. A role
of estrogen/ERalpha signaling in BRCA1-associated tissue-specific tumor formation. _Oncogene_ 26, 7204–7212 (2007). CAS PubMed Google Scholar * Al Moustafa, A. E. Epithelial-mesenchymal
transition and its regulators are major targets of triple-negative breast cancer. _Cell Adhes. Migr._ 7, 424–425 (2013). Google Scholar * Sarrio, D. et al. Epithelial-mesenchymal transition
in breast cancer relates to the basal-like phenotype. _Cancer Res._ 68, 989–997 (2008). CAS PubMed Google Scholar * Guen, V. J. et al. EMT programs promote basal mammary stem cell and
tumor-initiating cell stemness by inducing primary ciliogenesis and Hedgehog signaling. _Proc. Natl Acad. Sci. USA_ 114, E10532–E10539 (2017). CAS PubMed PubMed Central Google Scholar *
Jamieson, E. R. & Lippard, S. J. Structure, recognition, and processing of cisplatin-DNA adducts. _Chem. Rev._ 99, 2467–2498 (1999). CAS PubMed Google Scholar * Conte, N. et al. PDX
finder: a portal for patient-derived tumor xenograft model discovery. _Nucleic Acids Res._ 47, D1073–D1079 (2019). CAS PubMed Google Scholar * Densham, R. M. et al. Human BRCA1-BARD1
ubiquitin ligase activity counteracts chromatin barriers to DNA resection. _Nat. Struct. Mol. Biol._ 23, 647–655 (2016). CAS PubMed PubMed Central Google Scholar * Runkle, E. A. &
Mu, D. Tight junction proteins: from barrier to tumorigenesis. _Cancer Lett._ 337, 41–48 (2013). CAS PubMed PubMed Central Google Scholar * Friedrich, M. J. et al. Genome-wide transposon
screening and quantitative insertion site sequencing for cancer gene discovery in mice. _Nat. Protoc._ 12, 289–309 (2017). CAS PubMed Google Scholar * Pertea, M., Kim, D., Pertea, G. M.,
Leek, J. T. & Salzberg, S. L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. _Nat. Protoc._ 11, 1650–1667 (2016). CAS PubMed PubMed
Central Google Scholar * Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. _Genome Biol._ 15, 550 (2014). * Gendoo,
D. M. A. et al. Genefu: an R/Bioconductor package for computation of gene expression-based signatures in breast cancer. _Bioinformatics_ 32, 1097–1099 (2016). CAS PubMed Google Scholar *
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. _Nat. Protoc._ 4, 44–57 (2009). CAS Google
Scholar * Huang, D. W., Sherman, B. T. & Lempicki, R. A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. _Nucleic Acids Res._
37, 1–13 (2009). Google Scholar * Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. _Proc. Natl Acad. Sci.
USA_ 102, 15545–15550 (2005). ADS CAS PubMed PubMed Central Google Scholar * Szklarczyk, D. et al. STRING v10: protein-protein interaction networks, integrated over the tree of life.
_Nucleic Acids Res._ 43, D447–D452 (2015). CAS PubMed Google Scholar * Zhu, Z. R. et al. Genome editing of lineage determinants in human pluripotent stem cells reveals mechanisms of
pancreatic development and diabetes. _Cell Stem Cell_ 18, 755–768 (2016). CAS PubMed PubMed Central Google Scholar * Yu, X. B. et al. HES1 inhibits cycling of hematopoietic progenitor
cells via DNA binding. _Stem Cells_ 24, 876–888 (2006). CAS PubMed Google Scholar * Barger, C. J., Branick, C., Chee, L. & Karpf, A. R. Pan-cancer analyses reveal genomic features of
FOXM1 overexpression in cancer. _Cancers_ 11, 251 (2019). * Whitfield, M. L. et al. Identification of genes periodically expressed in the human cell cycle and their expression in tumors.
_Mol. Biol. Cell_ 13, 1977–2000 (2002). CAS PubMed PubMed Central Google Scholar * Faustino-Rocha, A. et al. Estimation of rat mammary tumor volume using caliper and ultrasonography
measurements. _Lab Anim._ 42, 217–224 (2013). Google Scholar Download references ACKNOWLEDGEMENTS We thank members of the C. Deng and X. Xu laboratories for helpful advice and discussion,
the Animal Research Core for providing the animal housing, and the Information and Communication Technology Office (ICTO) for providing the HPC for data processing. This work was supported
by the Chair Professor Grant granted to C.D., multi-year research grant (MYRG) 2016-00139-FHS to C.D. and 2018-00186-FHS to K.M. by the University of Macau, Macau SAR, China; the Macao
Science and Technology Development Fund (FDCT) Grant 094/2015/A3, 0011/2019/AKP, 0034/2019/AGJ and 0048/2019/A1 to C.D.; 029/2017/A1 and 0101/2018/A3 to X.X., and 111/2017/A to K.M.; and the
National Natural Science Foundation of China grant 81672603 to Q.C. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Cancer Center, Faculty of Health Sciences, University of Macau, Macau, SAR,
China Kai Miao, Josh Haipeng Lei, Monica Vishnu Valecha, Aiping Zhang, Jun Xu, Lijian Wang, Xueying Lyu, Si Chen, Zhengqiang Miao, Xin Zhang, Sek Man Su, Fangyuan Shao, Barani Kumar
Rajendran, Jiaolin Bao, Jianming Zeng, Heng Sun, Ping Chen, Kaeling Tan, Qiang Chen, Koon Ho Wong, Xiaoling Xu & Chu-Xia Deng * Centre for Precision Medicine Research and Training,
Faculty of Health Sciences, University of Macau, Macau, SAR, China Kai Miao, Josh Haipeng Lei, Monica Vishnu Valecha, Aiping Zhang, Jun Xu, Lijian Wang, Xueying Lyu, Si Chen, Sek Man Su,
Fangyuan Shao, Barani Kumar Rajendran, Jiaolin Bao, Jianming Zeng, Heng Sun, Ping Chen, Qiang Chen, Xiaoling Xu & Chu-Xia Deng * Genomics & Bioinformatics Core, Faculty of Health
Sciences, University of Macau, Macau, SAR, China Zhengqiang Miao, Kaeling Tan & Koon Ho Wong * Transgenic and Knockout Core, Faculty of Health Sciences, University of Macau, Macau, SAR,
China Xin Zhang & Xiaoling Xu Authors * Kai Miao View author publications You can also search for this author inPubMed Google Scholar * Josh Haipeng Lei View author publications You can
also search for this author inPubMed Google Scholar * Monica Vishnu Valecha View author publications You can also search for this author inPubMed Google Scholar * Aiping Zhang View author
publications You can also search for this author inPubMed Google Scholar * Jun Xu View author publications You can also search for this author inPubMed Google Scholar * Lijian Wang View
author publications You can also search for this author inPubMed Google Scholar * Xueying Lyu View author publications You can also search for this author inPubMed Google Scholar * Si Chen
View author publications You can also search for this author inPubMed Google Scholar * Zhengqiang Miao View author publications You can also search for this author inPubMed Google Scholar *
Xin Zhang View author publications You can also search for this author inPubMed Google Scholar * Sek Man Su View author publications You can also search for this author inPubMed Google
Scholar * Fangyuan Shao View author publications You can also search for this author inPubMed Google Scholar * Barani Kumar Rajendran View author publications You can also search for this
author inPubMed Google Scholar * Jiaolin Bao View author publications You can also search for this author inPubMed Google Scholar * Jianming Zeng View author publications You can also search
for this author inPubMed Google Scholar * Heng Sun View author publications You can also search for this author inPubMed Google Scholar * Ping Chen View author publications You can also
search for this author inPubMed Google Scholar * Kaeling Tan View author publications You can also search for this author inPubMed Google Scholar * Qiang Chen View author publications You
can also search for this author inPubMed Google Scholar * Koon Ho Wong View author publications You can also search for this author inPubMed Google Scholar * Xiaoling Xu View author
publications You can also search for this author inPubMed Google Scholar * Chu-Xia Deng View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS
C.D. and K.M. designed this research; K.M., H.L., J.X., S.C., X.Z., S.M.S. and F.S. performed experiments; H.L., L.W. and S.M.S. performed the histopathological analysis and
immunohistochemical staining; J.X., J.B., H.S. and Q.C. assisted with the cell-cycle experiments and data analysis; M.V.V., A.Z., X.L., B.K.R. and J.Z. identified the CISs, analysed the
RNA-sequencing data and performed other bioinformatics analyses; K.M., F.S. and P.C. performed the drug treatment experiments; Z.M. and K.H.W. initiated the bioinformatics analysis for
identifying CISs; K.T. was responsible for sequencing the SB libraries; K.H.W. and K.M. supervised the bioinformatics analysis; C.D., X.X. and Q.C. supervised the experiments; and K.M. and
C.D. wrote the manuscript. CORRESPONDING AUTHOR Correspondence to Chu-Xia Deng. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION
PEER REVIEW INFORMATION _Nature Communications_ thanks Branden Moriarity, Marcel van Vugt and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer
reviewer reports are available. PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY
INFORMATION SUPPLEMENTARY INFORMATION PEER REVIEW FILE DESCRIPTION OF ADDITIONAL SUPPLEMENTARY FILES SUPPLEMENTARY DATA 1 SUPPLEMENTARY DATA 2 SUPPLEMENTARY DATA 3 SUPPLEMENTARY DATA 4
SUPPLEMENTARY DATA 5 SUPPLEMENTARY DATA 6 SUPPLEMENTARY DATA 7 SUPPLEMENTARY DATA 8 SUPPLEMENTARY DATA 9 SUPPLEMENTARY DATA 10 REPORTING SUMMARY SOURCE DATA SOURCE DATA RIGHTS AND
PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any
medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The
images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly
from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Miao, K., Lei,
J.H., Valecha, M.V. _et al._ NOTCH1 activation compensates BRCA1 deficiency and promotes triple-negative breast cancer formation. _Nat Commun_ 11, 3256 (2020).
https://doi.org/10.1038/s41467-020-16936-9 Download citation * Received: 11 June 2019 * Accepted: 02 June 2020 * Published: 26 June 2020 * DOI: https://doi.org/10.1038/s41467-020-16936-9
SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to
clipboard Provided by the Springer Nature SharedIt content-sharing initiative