Play all audios:
ABSTRACT The optical properties of chromophores can be efficiently tuned by electrostatic fields generated in their close environment, a phenomenon that plays a central role for the
optimization of complex functions _within_ living organisms where it is known as internal Stark effect (ISE). Here, we realised an ISE experiment at the lowest possible scale, by monitoring
the Stark shift generated by charges confined within a single chromophore on its emission energy. To this end, a scanning tunneling microscope (STM) functioning at cryogenic temperatures is
used to sequentially remove the two central protons of a free-base phthalocyanine chromophore deposited on a NaCl-covered Ag(111) surface. STM-induced fluorescence measurements reveal
spectral shifts that are associated to the electrostatic field generated by the internal charges remaining in the chromophores upon deprotonation. SIMILAR CONTENT BEING VIEWED BY OTHERS
CONTROLLED SINGLE-ELECTRON TRANSFER ENABLES TIME-RESOLVED EXCITED-STATE SPECTROSCOPY OF INDIVIDUAL MOLECULES Article Open access 26 September 2024 FLUORESCENCE FROM A SINGLE-MOLECULE PROBE
DIRECTLY ATTACHED TO A PLASMONIC STM TIP Article Open access 10 November 2024 AN OPTOFLUIDIC ANTENNA FOR ENHANCING THE SENSITIVITY OF SINGLE-EMITTER MEASUREMENTS Article Open access 21 March
2024 INTRODUCTION In many chemical and biological systems, the electric fields generated by embedded electrostatic charges regulate the absorption or emission energies of
chromophores.1,2,3. This phenomenon, known as internal Stark effect (ISE),3,4,5 contrasts with Stark shifts induced by external electric fields6,7. It is at play in natural light-harvesting
complexes where the sensitive optical properties of chlorophyll8,9 and carotenoid2,10 are adjusted by local charges in surrounding proteins and neighboring compounds to enable energy
funneling. Similarly, electrostatic interactions between retinal chromophores and neighboring charged groups are responsible for wavelength regulation of vision1. In these examples, ISE
occurs in complex landscapes composed of a large number of interacting organic systems. Scaling down these effects to single-molecules would be a step towards understanding the intimate
interaction between biological pigments and their electrostatic environment, but has been so far limited to the spectroscopy of molecules in frozen matrices11,12,13,14 and scanning-tunneling
microscopy (STM) experiments15,16,17,18 where _external_ electric fields were used to shift the molecular states. The effect of an electric field generated by an elementary charge located
within a chromophore on its optical properties remains up to now a Gedankenexperiment. Here, we use free-base phthalocyanine (H2Pc) molecules, deposited on a NaCl-covered silver sample, as a
model system to study the ISE induced by one or two charges localised at the center of the chromophore on its fluorescence properties. To do so, we successively remove the two central
protons of a H2Pc molecule. STM images, topographic time traces, and differential conductance spectra are used to identify the nature of the deprotonated species and their electronic
structure. STM-induced luminescence (STML) spectra recorded on the singly and doubly deprotonated compounds reveal a fluorescence emission blue-shifted compared to the original H2Pc
chromophore. Based on a comparison with time-dependent density functional theory (TD-DFT) simulations, we show that these shifts can be traced back to the radial electric field generated by
charges confined to the _σ_-orbitals of the deprotonated chromophores whereas their _π_-orbitals remain unchanged. As a consequence, the neutral and deprotonated compounds are
iso-electronic, in contrast with scanning probe experiments in which charging a molecule alters its _π_-orbitals19,20,21,22,23,24. The deprotonation procedure also affects the vibronic
emission of the molecule, inducing measurable frequency shifts for several modes, an effect that is discussed in terms of a vibrational Stark effect25 and mass changes similar to isotopic
shifts. Overall, our experiment constitutes an ultimate ISE experiment where the electric field is generated directly inside the probed chromophore, a model landmark for more complex
ISE-induced color-tuning phenomena occurring in biological systems, and a novel strategy to develop tunable optoelectronic devices relying on single molecules as active components. RESULTS
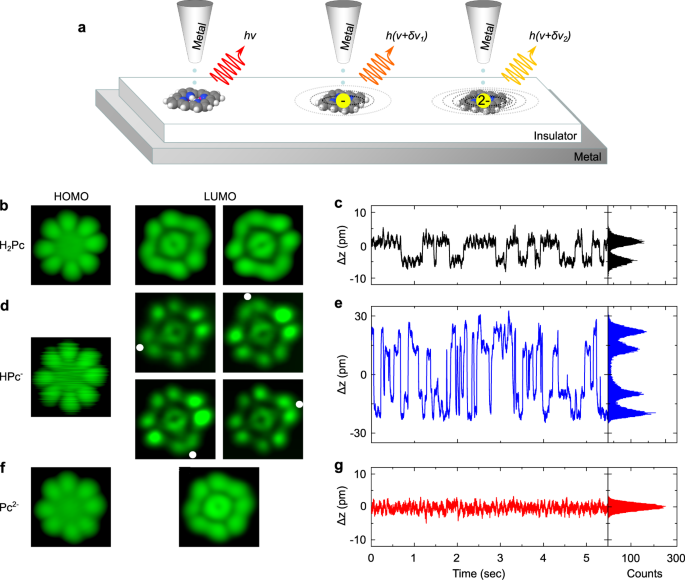
DEPROTONATION OF H2PC MOLECULES ON NACL/AG(111) Figure 1a shows a sketch of the STM-induced luminescence (STML) experiment used to probe the Stark effect generated by central charges on the
fluorescence of a phthalocyanine chromophore deposited on a NaCl-covered Ag(111) surface (see “Methods” for details). To realise this scheme, we worked with H2Pc molecules whose typical STM
images recorded at _V_ = −2.5 V and _V_ = 0.5 V are displayed in Fig. 1b. While the left image reveals the characteristic four-fold symmetry pattern of the highest occupied molecular orbital
(HOMO), the right one reflects the twofold symmetry pattern of the lowest unoccupied molecular orbital (LUMO) (see Supplementary Figs. 1–3 and Supplementary Table 1). This latter can adopt
two configurations rotated by 90∘ from each other in successive images26,27, a behavior that has been formerly assigned to tautomerization, i.e., the permutation of the central hydrogen
atoms between two equivalent sites of the molecule. This phenomenon can be tracked in the variation of the tip-sample distance (Δz) versus time (Fig. 1c) recorded at constant current where
it appears as two-level fluctuations24,27,28,29,30,31. As a next step of the experiment, we located the tip on top of the center of the H2Pc molecule and applied a positive voltage ramp at a
constant current of 10 pA while simultaneously recording the relative tip-sample distance “z”. In most cases, this procedure reveals a sudden “z” decrease for _V_ ≈ 3.2 V (see Supplementary
Note 1 for details), hinting towards a change of the molecular structure. STM images recorded at _V_ = −1.5 V (HOMO) after such an event are similar to the one measured before, but are
fuzzier (Fig. 1d). Δz time-traces now reveal four-level fluctuations with a high switching frequency (Fig. 1e). This explains the fuzzy appearance of the HOMO image and hints towards the
inner motion of a single proton in a HPc molecule24,29,31. This is confirmed by STM images acquired at _V_ = 1.43 V [LUMO, (Fig. 1d)] where each tautomer can be stabilized and identified.
The voltage is then ramped a second time until another sudden distance drop occurs. The typical four-fold symmetry STM images (Fig. 1f) observed for both HOMO and LUMO of the resulting
compound32, together with non-fluctuating Δz time traces (Fig. 1g) now suggest a doubly dehydrogenated (or deprotonated) phthalocyanine molecule (Pc). ELECTRONIC STRUCTURE OF THE
DEPROTONATED H2PC MOLECULES Single and double STM-induced removal of central hydrogens of porphyrin and phthalocyanine molecules were reported in several prior works24,29,31,32,33. In some
cases, it was assumed that only the proton was removed29,31,33 while some other works using partially decoupled molecules24,32 concluded on a full dehydrogenation (proton and electron). In
the latter case, the HPc molecule is in a neutral state with an unpaired electron in the _π_-orbital, which strongly affects its electronic properties. The former, which has not been
reported so far for decoupled molecules, should leave the molecule in a negatively charged state. Characterizing the charged state of the molecules may therefore unravel the exact nature of
the STM-induced chemical reaction: dehydrogenation or deprotonation. On 2 monolayer (ML) NaCl-covered (111) noble metal surfaces, charged atoms, and molecules were shown to scatter the
two-dimensional surface-state localised at the metal-salt interface (also called interface state) while neutral species do not34,35. In Fig. 2a, e, and i, we display differential conductance
images (i.e., constant current d_I_/d_V_ maps) recorded at a DC bias of 400 mV—some 200 meV above the onset of the interface state—for the same molecule prior to (a) and after the first (e)
and second (i) voltage ramp procedure (the topography is shown in the inset). These images reveal circular standing waves— which become even more apparent in difference images (see
Supplementary Fig. 4)—around the modified molecules that are absent for H2Pc. This indicates that, on NaCl, only the protons are removed from the H2Pc molecules ending up in singly (HPc−)
and doubly (Pc2−) negative charged species. Note that the molecule occupies the same adsorption site prior to and after the deprotonation procedures (see Supplementary Fig. 5). In Fig. 2b, f
and j, we first evaluate how these localised charges affect the electronic structure of the molecules by recording differential conductance (d_I_/d_V_) spectra for the three compounds.
These spectra reveal similar HOMO–LUMO gaps (≈2.7 eV), as well as rigid shifts of the frontier orbitals to higher energies (+0.94 eV for HPc− and +1.05 eV for Pc2−). For more information on
the interface states and scattering of charged species, as well as on the rigidity of the electronic gap of the molecules, please refer to Supplementary Figs. 4–7. These observations further
support the deprotonation mechanism, as a dehydrogenation would lead to a splitting of the HOMO into a singly occupied and a singly unoccupied molecular orbital24, or a shift of the
original HOMO above the Fermi level32. The d_I_/d_V_ spectra in Fig. 2b, f, j rather indicate that the _π_-orbitals are unaffected upon deprotonation, an observation that is backed-up by DFT
calculations of the electronic structure of the three species (Fig. 2c, g, k), see Methods for details. In contrast, these calculations reveal large modifications of some _σ_-orbitals,
originally involved in the N-H bond. For example, the orbital labeled _σ_-HOMO in Fig. 2g and k, located 1.7 eV below the _π_-HOMO in the calculation of H2Pc, appears 0.3 eV below and 0.4 eV
above the _π_-HOMO for HPc− and Pc2−, respectively. Altogether, these observations suggest that the excess negative charges do not localise in the frontier _π_-orbitals but in the
_σ_-orbitals originally involved in the N–H bonds. In fact, similar deprotonation effects were reported for porphycene compounds which were identified as _σ_-type anions and dianions36. To
better identify _where_ the excess charges are located on the HPc− and Pc2− chromophores, we represent in Fig. 2d, h, and l the total electrostatic potential generated jointly by the nuclei
and the distributed electron density of the singlet ground state of the molecule. The potential is displayed in a horizontal plane 0.21 nm above the molecule. The absence of clear contrast
in the calculated image of H2Pc confirms the neutral nature of the molecule. For HPc− and Pc2−, these calculated images reveal an overall negative potential on the molecules, that is of
maximum amplitude at the center of the chromophores where the protons have been removed. This indicates that the charges left over after deprotonation remain close to their original
positions, strongly affecting the _σ_-orbitals originally involved in the N–H bond but preserving the _π_-structure of the chromophore. The rigid shift of the frontier orbitals observed in
Fig. 2f, j can therefore be associated to a change in the molecular ionization energy due to the electric field generated by the excess inner charges. For more information about calculations
of the electronic structure and total potential, please refer to “Methods”. INTERNAL STARK-SHIFT OF THE MOLECULAR FLUORESCENCE LINES We now discuss the photonic properties of the three
chromophores and investigate the effect of one or two central charges on the fluorescence of the phthalocyanine molecule. Figure 3 displays the STM-induced fluorescence spectra of H2Pc, HPc−
and Pc2−. The spectrum of H2Pc, excited at a negative bias of _V_ = −2.5 V, is composed of two purely electronic contributions named Q_x_ and Q_y_ appearing respectively at ≈1.81 and ≈1.93
eV26,27,37,38. Q_x_ (Q_y_) is defined as the low (high) energy spectral contribution, which, for H2Pc, is associated to a dipole oriented along (perpendicular to) the axis formed by the two
inner hydrogen atoms. Q_y_ usually appears much weaker than the lower energy Q_x_ contribution, reflecting fast non-radiative decay channels between the excited states39. Peaks of low
intensities are also observed on the low energy side of the spectrum that reflect vibronic transitions characteristic of the H2Pc molecule27,40. Independently of the used polarity, HPc− does
not emit when directly excited by the STM tip, a behavior that will be discussed elsewhere but that we associate to the low absolute energies of both HOMO and LUMO that prevent the
excitation of the molecule by tunneling electrons41. However, the fluorescence of HPc− can be recovered through excitonic energy transfer37,42 from a higher energy gap molecule, here ZnPc,
positioned in direct contact to the “dark” HPc− (Supplementary Fig. 9). Hence, an energy-transfer mediated excitation enables probing the fluorescence of otherwise “dark” molecules, a
strategy that may be used with other “dark” chromophores in STML experiments. This spectrum exhibits two electronic contributions; the high energy one can be associated to the Q_Z__n_
emission line of the ZnPc donor40,43, whereas the Q_x_ fluorescence line of HPc− is at ≈1.86 eV, some 50 meV above the Q_x_ peak of H2Pc. Note that according to our TD-DFT calculations, the
low energy transition (Q_x_) of HPc− is oriented along the molecular axis that does not contain the hydrogen atom. The Q_y_ contribution of HPc− cannot be identified in the spectrum (Fig.
3b), probably because it is at a higher energy than the Q_Z__n_ contribution of the ZnPc donor. For Pc2−, Fig. 3c shows that it can only be excited at positive voltage (_V_ = 2.5 eV) with an
emission line at ≈1.88 eV. Similar to metal phthalocyanines37,40,43,44, only a single emission line is observed in this spectrum, reflecting the D4h symmetry of the doubly deprotonated
molecule and the associated degeneracy of the two first emission contributions. In summary, one notices that moving from H2Pc to Pc2−, the transition is blue-shifted by 50 meV upon the first
deprotonation, and by an additional 20 meV upon the second one. These shifts may originate from several physical effects. Due to the presence of the STM tip generating external static and
dynamic electrical fields, Stark and photonic Lamb effects may contribute to the observed energy shifts18,45,46,47,48. However, recent STML and photoluminescence works demonstrated that
these effects may at best lead to few meV shifts of the emission maxima18,48, and cannot explain the large blue shifts reported in Fig. 3b and c. The interaction between the indirectly
excited HPc− molecule with the donor molecule of ZnPc can also result in small energy shifts42 of the HPc− emission line, but which again are negligible compared to the shifts in Fig. 3.
Similarly, we theoretically ruled out the impact of the static screening of the NaCl substrate on the energy shifts by performing DFT and TD-DFT calculations44 (discussed in detail in
Supplementary Note 2). On the other hand, charged chromophores as _π_-type phthalocyanine anions and cations discussed in previous reports21,22,23,49 are systematically characterized by a
strongly red-shifted emission (≈400 meV) compared to neutral compounds, reflecting important modifications of the _π_-orbitals involved in the optical transition. Similarly, if one assumes
neutral dehydrogenated compounds, one is left with one unpaired electron in the HOMO (HPc) or with an empty HOMO (Pc)24,32. The fluorescence spectra of HPc and Pc should therefore reflect
those of a H2Pc _π_-type cation (H2Pc+) and a H2Pc _π_-type dication (H2Pc2+) with whom they share the same electronic structure. Fluorescence spectra of HPc and Pc should therefore display
emission lines red-shifted by roughly 400 meV with respect to the Q_x_ of H2Pc; this is inconsistent with the spectra of Fig. 3. In contrast, our DFT calculations of the deprotonated
compounds, HPc− and Pc2−, reveal an unchanged occupancy of the _π_-orbitals compared to H2Pc, explaining why the fluorescence characteristics of these three iso-electronic compounds do not
change drastically. The observed blue shifts are well reproduced by the TD-DFT simulations of HPc− and Pc2− (assuming a systematic shift of the theoretical data to account for the specific
environment of the chromophores, see Fig. 3 and “Methods” for details). These blue-shifts may either find their origin in tiny structural reorganisations of the molecule upon deprotonation,
or in the electric field generated by the excess _σ_-electrons. To address this issue, we considered the fully symmetric Pc2− molecule and progressively neutralized it by artificially adding
partial positive charges in its center, assuming an unchanged geometry. We then calculated the evolution of the optical gap as a function of the molecular charge (Fig. 3d). These
simulations show that the optical gaps of HPc− and H2Pc scale extremely well with those of Pc2− with one and two central positive charges, respectively, eventually demonstrating that the
observed shifts are due to the Stark effect generated by the internal charges. Assuming such an ISE, it may appear surprising to observe a much smaller relative shift (+20 meV) upon removal
of the second proton compared to the first deprotonation (+50 meV). This phenomenon is explained by the data of Fig. 3d, where a parabolic dependency of the optical gap on the central charge
is observed, and where the singly and doubly charged chromophores are at either side of the apex of the parabola, leading to very close optical gaps. In the usual case of a chromophore with
non-degenerated electronic states placed in a chiefly homogeneous external electrical field, the linear and quadratic Stark effects reflect the respective changes in the permanent dipole
moment, \({{\Delta }}\vec{\mu }\), and in the polarizability, Δ_α_, experienced by the ground and excited states of the chromophore25. As the H2Pc ground and excited states do not exhibit
permanent dipole moments (\({\vec{\mu }}_{{S}_{0}}\) = \({\vec{\mu }}_{{S}_{1}}\) = \(\vec{0}\)), the presence of a non vanishing linear term (see inset Fig. 3d) may be surprising. This can
be elucidated by accounting for the specific geometry of our system where central charges generate a strongly non-homogeneous electric field at the scale of the chromophore, eventually
resulting in a linear contribution to the Stark shift of the spectral line (see a detailed perturbation model in Supplementary Note 3). This behavior is therefore characteristic of the close
proximity between a chromophore and a point source of electric field. Clearly evidenced in our model system, this “local” effect is inherent to any ISE configuration, including the most
complex biological ones, and constitutes the main difference with the usual Stark effect generated by an external electric field. VIBRONIC SPECTRA OF THE DEPROTONATED MOLECULES The H2Pc and
Pc2− spectra also display several vibronic emission lines, similar to tip-enhanced Raman spectra, that can be used as accurate chemical fingerprints of the probed compounds, and which
provide detailed information regarding their chemical bond structures40. These spectra (Fig. 4a) reveal subtle changes of the intensity and energy of several vibronic peaks between H2Pc and
its doubly deprotonated counterpart. At this stage, however, these changes can indicate either a shift of the mode frequency or the appearance/disappearance of vibronic peaks. In Fig. 4b, we
show DFT calculations of the vibronic active modes for the two compounds, which reproduce the experimental frequencies and peak intensities remarkably well. We then use atomic-coordinate
displacements provided by DFT to identify the prominent modes of H2Pc and of Pc2− and indicate with black vertical lines in Fig. 4b those having a nearly perfect one-to-one correspondence.
Experimentally, while the modes below 900 cm−1 all shift to higher wavenumbers upon double deprotonation of H2Pc, those above 900 cm−1 display the opposite behavior. Numerically, only one of
the low wavenumber modes (_α_) clearly blue shifts, while the experimental trend in the high wavenumber modes is better accounted for. Two shifts that are well reproduced by the numerical
simulations, both in amplitude and sign, are those of the strongest peak close to 700 cm−1 (_α_ in Fig. 4) and the highest wavenumber mode around 1500 cm−1 (_β_ in Fig. 4). For both modes,
the four nitrogen atoms of the pyrrole cycles, whether protonated or not, stand nearly still as do the central protons when present (see Supplementary Fig. 10 for the real-space
representation of the _α_ and _β_ vibrational modes). Hence, their frequency shifts can hardly be explained by an isotopic-like effect associated to deprotonation. In contrast, the two modes
entail large motions of the nitrogen atoms bridging the isoindole units and of the carbon atoms they are bound to. The vibronic intensities of these modes are therefore linked to the
conjugation paths along the inner rings of _π_-orbitals in both species. Their shifts must therefore be attributed to a polarization of the _π_-electron system upon removal of the inner
proton. To conclude, these two modes may then be seen as undergoing pure vibrational ISE. Most of the other modes entail radial motions of the nitrogen of the isoindoles. Therefore, the
respective influence of the vibrational ISE and of isotopic effects induced by the removal of the central protons cannot be disentangled. DISCUSSION Charged states of single molecules have
been recently probed in a wide range of experimental schemes involving scanning probe approaches19,20,21,22,23,35,49. By simultaneously preserving the _π_-orbital structure of H2Pc and
leaving an excess _σ_-electron within the chromophore, the deprotonation procedure reported here provides a unique opportunity to study the Stark effect generated by an internal charge on
the fluorescence emission of an individual chromophore. The resulting _σ_-type anionic and dianionic molecules constitute model systems allowing us to identify the role played by the
proximity between a chromophore and a point-like electrostatic field source. This proximity is responsible for the combined linear and quadratic dependency of the emission energy, a behavior
that should occur in any biological systems subject to ISE. It also suggests that chromophores in STML experiments could be used as precise electrostatic sensors of their nanometer-scale
environment. Eventually, this work establishes a new biomimetic strategy, based on the control of the local electrostatic environment, to tune and optimise future artificial molecular
optoelectronic devices. METHODS EXPERIMENT The experiments are performed in ultra-high vacuum at low temperature (≈4.5 K) with an Omicron STM that is combined with an optical set-up adapted
to detect light emitted at the STM tip-sample junction. The emitted photons are collected with a lens located on the STM head and then redirected out of the chamber through optical
viewports. The light is then focused on an optical fiber coupled to a spectrograph itself connected to a low-noise liquid nitrogen-cooled CCD camera. The spectral resolution of the setup is
better than 1 nm. Further details regarding the optical detection setup can be found in the Supplementary Materials of ref. 50. The STM tips are prepared by electrochemical etching of a
tungsten wire in a NaOH solution. The tips are then sputtered with argon ions and annealed under UHV. To optimise their plasmonic response, the tips are eventually indented in clean Ag(111)
to cover them with silver. The Ag(111) substrate is cleaned by simultaneous argon-ion sputtering and annealing. After this cleaning procedure, NaCl has evaporated on the Ag(111) substrate
maintained at room temperature. Post-annealing at ≈370 K is performed to induce NaCl surface reorganization in bi- and tri-layers. Eventually, the 2–3ML NaCl/Ag(111) sample is introduced
into the STM chamber and cooled down to ≈4.5 K. H2Pc and ZnPc molecules are then sublimed in very small quantities on the cold sample from a powder located in a quartz crucible. ZnPc–HPc−
molecular dimers on NaCl are obtained by STM tip manipulation of ZnPc. To this end, the STM tip is first positioned at the edge of a ZnPc molecule at a bias _V_ = +2.5 V. In a second step,
the tip-molecule distance is slowly reduced until a jump of current occurs, indicating a motion of the molecule. The procedure is reproduced until the desired structure in obtained. The
d_I_/d_V_ maps of Fig. 2 in the main paper are recorded in constant current mode (closed feedback loop) with a voltage modulation of 50 mV at a DC voltage of 400 mV, whereas the d_I_/d_V_
spectra are recorded in constant height mode (open feedback loop) with a voltage modulation of 20 mV. A modulation frequency of 740 Hz is used for these measurements. DFT CALCULATION OF THE
GROUND-STATE ELECTRONIC PROPERTIES To analyze the molecular transport properties, the electronic structure of the molecule in a vacuum, and the distribution of the corresponding electron
charge density in the neutral and charged species, we perform ground-state DFT calculations using the OCTOPUS code51. In OCTOPUS the electron density is represented on a real-space grid
which does not constrain the localization of the density to predefined atomic orbitals and is therefore suitable to describe the ground-state electron densities of the charged molecules. The
electron-ion interaction is modeled in the framework of the pseudopotential approximation. We use the Perdew-Zunger52 parametrization of the local density approximation (LDA) correlation
and the Slater density functional for the LDA exchange functional53,54. We present further analysis of the molecular orbitals, electronic density, and comparison with experimental d_I_/d_V_
images of the molecules with theory in Supplementary Figs. 1–3 and Supplementary Table 1. NUMERICAL ANALYSIS OF THE GROUND-STATE ELECTRON DENSITIES We extract from OCTOPUS the ground-state
electron densities and calculate the total electrostatic potential _ϕ_tot generated by the molecular charges to visualize the localization of the excess charge of the deprotonized molecules
(as shown in Fig. 2). To that end we solve the Poisson equation: $${{\Delta }}{\phi }_{{{{{{{{\rm{tot}}}}}}}}}=\frac{{\rho }_{{{{{{{{\rm{e}}}}}}}}}({{{{{{{\bf{r}}}}}}}})}{{\varepsilon
}_{0}}+\frac{{\rho }_{{{{{{{{\rm{ion}}}}}}}}}({{{{{{{\bf{r}}}}}}}})}{{\varepsilon }_{0}},$$ (1) with _ε_0 being the vacuum permittivity. _ρ_e(R) is the electron charge density of the valence
electrons in the singlet ground state of the molecule and _ρ_ion(R) is the charge density of the positive nuclei screened by the core electrons of the respective atoms. We represent the
density of the screened nuclei as a sum of Gaussian charge distributions: $${\rho }_{{{{{{{{\rm{ion}}}}}}}}}({{{{{{{\bf{r}}}}}}}})=\mathop{\sum}\limits_{i}\frac{{Q}_{i}}{{\left(2\pi
\right)}^{3/2}{\sigma }_{{{{{{{{\rm{ion}}}}}}}}}^{3}}\exp\left( -\frac{{\left|{{{{{{{\bf{r}}}}}}}}-{{{{{{{{\bf{R}}}}}}}}}_{i}\right|}^{2}}{2{\sigma }_{{{{{{{{\rm{ion}}}}}}}}}^{2}}\right),$$
(2) where _Q__i_ is the total charge of the screened ion _i_ at position R_i_. The charge distribution has a width _σ_ion = 0.05 nm. The electron charge density is extracted from the
ground-state DFT calculations in the form of Gaussian cube files. The ground-state electron charge densities for H2Pc and HPc− are shown in Supplementary Fig. 3c, d, respectively, alongside
with the corresponding geometries of the molecules (H2Pc in Supplementary Fig. 3a and HPc− in Supplementary Fig. 3b). Finally, we solve the Poisson equation [Eq. (1)] numerically on a
homogeneously spaced grid by a Fourier-based method. TD-DFT CALCULATIONS OF THE EXCITED-STATE ELECTRONIC PROPERTIES To address the excited-state properties of the molecules we perform TD-DFT
calculations as implemented in the software Gaussian 09 Revision D.0155. The TD-DFT calculations (Fig. 3) are carried out with the B3LYP functional and the 6-311G(d,p) basis set. The
convergence of 64 roots is asked for when calculating the excited singlet states at the ground state equilibrium geometries of all three compounds which are optimized using the same
functional and basis set. The dependence on a central fractional charge of the transition energy to the first excited singlet state of the doubly deprotonated Pc dianion (Fig. 3d) is
calculated in the same way while keeping its equilibrium D4h geometry frozen. DFT CALCULATION OF THE GEOMETRY OF THE MOLECULES ON NACL The geometrical structure of the molecules on NaCl is
relaxed using Quantum Espresso56 which is a plane-wave pseudopotential DFT code suitable for the description of the extended substrate. The cubic supercell (_a_ = 53.02457485 Å) included
100 Na and 100 Cl substrate atoms and the calculation is performed at the Γ point. The exchange and correlation terms are described using the local density approximation under the approach
of Perdew and Zunger52. Projector augmented-wave pseudopotentials with core corrections are used to describe the electron-ion interaction57. The energy cutoff and density cutoff are set to
50 Ry and 500 Ry, respectively. A damped dynamics is used to perform the structural optimization of the system. The atoms are moved according to Newton’s equation by using a Verlet
algorithm58. The structural optimization is stopped when two subsequent total energy evaluations differed by <10−4 Ry and each force component is less than 10−3 Ry/bohr. DFT AND TD-DFT
CALCULATIONS OF THE MOLECULAR VIBRONIC PROPERTIES Finally, the vibronic intensities (Fig. 4b) are calculated as the square of the sum of the Franck and Condon and Herzberg-Teller amplitudes
for each relevant mode independently using Gaussian 09 Revision D.01. To this end, the results of the normal mode calculations are used to define equally spaced discrete distortions spanning
the range between the classical turning points of each mode. Repeated TD-DFT calculations of the vertical transition energy and dipole moment to the lowest excited singlet are used to
determine the mode displacements and the derivative of the transition dipole moments with respect to the dimensionless normal coordinate through a series of third order polynomial
regressions. Since the third order terms are found to be negligible and since the changes in frequencies (from second order terms) between ground and excited states of all the modes that are
identifiable in the experimental spectrum are all well below 5% and would not affect the Franck and Condon overlaps, Duschinsky rotations (mode mixing in the excited state) are ignored. The
theoretical vibronic spectra presented are then calculated using the TD-DFT determined intensities by scaling the normal mode frequencies obtained analytically at the DFT level by a factor
0.9659 and by broadening each line by a Lorentzian 20 cm−1 wide at half maximum. DATA AVAILABILITY The data supporting the findings of the present study can be found in Methods or
Supplementary Information. All the datasets are also available from the corresponding author upon request. Source data are provided with this paper. REFERENCES * Honig, B. et al. An external
point-charge model for wavelength regulation in visual pigments. _J. Am. Chem. Soc._ 101, 7084–7086 (1979). Article CAS Google Scholar * Gottfried, D., Steffen, M. & Boxer, S. Large
protein-induced dipoles for a symmetric carotenoid in a photosynthetic antenna complex. _Science_ 251, 662–665 (1991). Article ADS CAS PubMed Google Scholar * Lockhart, D. & Kim, P.
Internal Stark effect measurement of the electric field at the amino terminus of an alpha helix. _Science_ 257, 947–951 (1992). Article ADS CAS PubMed Google Scholar * Drobizhev, M.,
Tillo, S., Makarov, N. S., Hughes, T. E. & Rebane, A. Color hues in red fluorescent proteins are due to internal quadratic Stark effect. _J. Phys. Chem. B_ 113, 12860–12864 (2009).
Article CAS PubMed PubMed Central Google Scholar * Boxer, S. G. Stark realities. _J. Phys. Chem. B_ 113, 2972–2983 (2009). Article CAS PubMed Google Scholar * Wild, U. P., Güttler,
F., Pirotta, M. & Renn, A. Single molecule spectroscopy: Stark effect of pentacene in p-terphenyl. _Chem. Phys. Lett._ 193, 451–455 (1992). Article ADS CAS Google Scholar * Orrit,
M., Bernard, J., Zumbusch, A. & Personov, R. Stark effect on single molecules in a polymer matrix. _Chem. Phys. Lett._ 196, 595–600 (1992). Article ADS CAS Google Scholar * Schulte,
T. et al. Identification of a single peridinin sensing Chl-a excitation in reconstituted PCP by crystallography and spectroscopy. _Proc. Natl. Acad. Sci. USA_ 106, 20764–20769 (2009).
Article ADS CAS PubMed PubMed Central Google Scholar * Croce, R. & van Amerongen, H. Natural strategies for photosynthetic light harvesting. _Nat. Chem. Biol._ 10, 492 (2014).
Article CAS PubMed Google Scholar * Kakitani, T., Honig, B. & Crofts, A. Theoretical studies of the electrochromic response of carotenoids in photosynthetic membranes. _Biophys. J._
39, 57 – 63 (1982). Article PubMed PubMed Central Google Scholar * Kulzer, F., Matzke, R., Bräuchle, C. & Basché, T. Nonphotochemical hole burning investigated at the single-molecule
level: Stark effect measurements on the original and photoproduct state. _J. Phys. Chem. A_ 103, 2408–2411 (1999). Article CAS Google Scholar * Brunel, C., Tamarat, P., Lounis, B.,
Woehl, J. C. & Orrit, M. Stark effect on single molecules of dibenzanthanthrene in a naphthalene crystal and in a n-hexadecane Shpol’skii matrix. _J. Phys. Chem. A_ 103, 2429–2434
(1999). Article CAS Google Scholar * Karotke, S., Lieb, A. & Hecht, B. Stark-shift microscopy of single emitters. _Appl. Phys. Lett._ 89, 023106 (2006). Article ADS Google Scholar
* Moradi, A., Ristanović, Z., Orrit, M., Deperasińska, I. & Kozankiewicz, B. Matrix-induced linear Stark effect of single dibenzoterrylene molecules in 2,3-dibromonaphthalene crystal.
_Chem. Phys. Chem._ 20, 55–61 (2019). Article CAS PubMed Google Scholar * Limot, L., Maroutian, T., Johansson, P. & Berndt, R. Surface-state Stark shift in a scanning tunneling
microscope. _Phys. Rev. Lett._ 91, 196801 (2003). Article ADS CAS PubMed Google Scholar * Fernández-Torrente, I., Kreikemeyer-Lorenzo, D., Stróżecka, A., Franke, K. J. & Pascual, J.
I. Gating the charge state of single molecules by local electric fields. _Phys. Rev. Lett._ 108, 036801 (2012). Article ADS PubMed Google Scholar * Martínez-Blanco, J. et al. Gating a
single-molecule transistor with individual atoms. _Nat. Phys._ 11, 640–644 (2015). Article Google Scholar * Roslawska, A. et al. Mapping Lamb, Stark and Purcell effects at a
chromophore-picocavity junction with hyper-resolved fluorescence microscopy. _Phys. Rev. X_ 12, 011012 (2022). * Fatayer, S. et al. Reorganization energy upon charging a single molecule on
an insulator measured by atomic force microscopy. _Nat. Nanotechnol._ 13, 376–380 (2018). Article ADS CAS PubMed Google Scholar * Patera, L. L., Queck, F., Scheuerer, P. & Repp, J.
Mapping orbital changes upon electron transfer with tunnelling microscopy on insulators. _Nature_ 566, 245–248 (2019). Article ADS CAS PubMed Google Scholar * Doppagne, B. et al.
Electrofluorochromism at the single-molecule level. _Science_ 361, 251 (2018). Article ADS CAS PubMed Google Scholar * Rai, V. et al. Boosting light emission from single hydrogen
phthalocyanine molecules by charging. _Nano Lett._ 20, 7600–7605 (2020). Article ADS CAS PubMed Google Scholar * Doležal, J., Canola, S., Merino, P. & Švec, M. Exciton-trion
conversion dynamics in a single molecule. _ACS Nano_ 15, 7694–7699 (2021). Article PubMed Google Scholar * Reecht, G., Krane, N., Lotze, C. & Franke, K. J. _π_-radical formation by
pyrrolic H abstraction of phthalocyanine molecules on molybdenum disulfide. _ACS Nano_ 13, 7031–7035 (2019). Article CAS PubMed Google Scholar * Bishop, D. M. The vibrational stark
effect. _J. Chem. Phys._ 98, 3179–3184 (1993). Article ADS CAS Google Scholar * Imada, H. et al. Single-molecule investigation of energy dynamics in a coupled plasmon-exciton system.
_Phys. Rev. Lett._ 119, 013901 (2017). Article ADS PubMed Google Scholar * Doppagne, B. et al. Single-molecule tautomerization tracking through space- and time-resolved fluorescence
spectroscopy. _Nat. Nanotechnol._ 15, 207–211 (2020). Article ADS CAS PubMed Google Scholar * Liljeroth, P., Repp, J. & Meyer, G. Current-induced hydrogen tautomerization and
conductance switching of naphthalocyanine molecules. _Science_ 317, 1203 (2007). Article ADS CAS PubMed Google Scholar * Auwärter, W. et al. A surface-anchored molecular four-level
conductance switch based on single proton transfer. _Nat. Nanotechnol._ 7, 41–46 (2012). Article ADS Google Scholar * Kumagai, T. et al. Controlling intramolecular hydrogen transfer in a
porphycene molecule with single atoms or molecules located nearby. _Nat. Chem._ 6, 41–46 (2014). Article CAS PubMed Google Scholar * Kügel, J., Klein, L., Leisegang, M. & Bode, M.
Analyzing and tuning the energetic landscape of H2Pc tautomerization. _J. Phys. Chem. C_ 121, 28204–28210 (2017). Article Google Scholar * Néel, N., Lattelais, M., Bocquet, M.-L. &
Kröger, J. Depopulation of single-phthalocyanine molecular orbitals upon pyrrolic-hydrogen abstraction on graphene. _ACS Nano_ 10, 2010–2016 (2016). Article PubMed Google Scholar * Pham,
V. D. et al. Control of molecule-metal interaction by hydrogen manipulation in an organic molecule. _J. Phys. Chem. Lett._ 7, 1416–1421 (2016). Article CAS PubMed Google Scholar * Repp,
J., Meyer, G., Olsson, F. E. & Persson, M. Controlling the charge state of individual gold adatoms. _Science_ 305, 493–495 (2004). Article ADS CAS PubMed Google Scholar * Swart, I.,
Sonnleitner, T. & Repp, J. Charge state control of molecules reveals modification of the tunneling barrier with intramolecular contrast. _Nano Lett._ 11, 1580–1584 (2011). Article ADS
CAS PubMed Google Scholar * Rabbani, M. G. & Teraoka, J. Resonance Raman spectra of n-deprotonated _σ_-type dianion of porphycenes. _Spectrochim. Acta A_ 76, 207–212 (2010). Article
ADS Google Scholar * Imada, H. et al. Real-space investigation of energy transfer in heterogeneous molecular dimers. _Nature_ 538, 364–367 (2016). Article ADS CAS PubMed Google
Scholar * Chen, G. et al. Spin-triplet-mediated up-conversion and crossover behavior in single-molecule electroluminescence. _Phys. Rev. Lett._ 122, 177401 (2019). Article ADS CAS PubMed
Google Scholar * Murray, C. et al. Visible luminescence spectroscopy of free-base and zinc phthalocyanines isolated in cryogenic matrices. _Phys. Chem. Chem. Phys._ 13, 17543–17554
(2011). Article CAS PubMed Google Scholar * Doppagne, B. et al. Vibronic spectroscopy with submolecular resolution from stm-induced electroluminescence. _Phys. Rev. Lett._ 118, 127401
(2017). Article ADS PubMed Google Scholar * Kaiser, K., Gross, L. & Schulz, F. A single-molecule chemical reaction studied by high-resolution atomic force microscopy and scanning
tunneling microscopy induced light emission. _ACS Nano_ 13, 6947–6954 (2019). Article CAS PubMed PubMed Central Google Scholar * Cao, S. et al. Energy funneling within multi-chromophore
architectures monitored with sub-nanometer resolution. _Nat. Chem_. 13, 766–770 (2021). * Zhang, Y. et al. Visualizing coherent intermolecular dipole-dipole coupling in real space. _Nature_
531, 623 (2016). Article ADS CAS PubMed Google Scholar * Doležal, J. et al. Mechano-optical switching of a single molecule with doublet emission. _ACS Nano_ 14, 8931–8938 (2020).
Article PubMed PubMed Central Google Scholar * Zhang, Y. Sub-nanometre control of the coherent interaction between a single molecule and a plasmonic nanocavity. _Nat. Commun_. 8, 15225
(2017). * Kuhnke, K. et al. Pentacene excitons in strong electric fields. _Chem. Phys. Chem._ 19, 277–283 (2018). Article CAS PubMed Google Scholar * Yang, B. Sub-nanometre resolution in
single-molecule photoluminescence imaging. _Nat. Phot._ 14, 693–699 (2020). Article CAS Google Scholar * Imada, H. et al. Single-molecule laser nanospectroscopy with micro–electron volt
energy resolution. _Science_ 373, 95–98 (2021). Article ADS CAS PubMed Google Scholar * Farrukh, A. et al. Bias-polarity dependent electroluminescence from a single platinum
phthalocyanine molecule. _Chin. J. Chem. Phys._ 34, 87–94 (2021). Article CAS Google Scholar * Chong, M. C. et al. Narrow-line single-molecule transducer between electronic circuits and
surface plasmons. _Phys. Rev. Lett._ 116, 036802 (2016). Article ADS PubMed Google Scholar * Tancogne-Dejean, N. et al. Octopus, a computational framework for exploring light-driven
phenomena and quantum dynamics in extended and finite systems. _Phys. Chem. Chem. Phys._ 152, 124119 (2020). ADS CAS Google Scholar * Perdew, J. P. & Zunger, A. Self-interaction
correction to density-functional approximations for many-electron systems. _Phys. Rev. B_ 23, 5048 (1981). Article ADS CAS Google Scholar * Dirac, P. A. M. Note on exchange phenomena in
the Thomas atom. _Math. Proc. Camb. Philos. Soc._ 26, 376 (1930). Article ADS CAS MATH Google Scholar * Slater, J. A simplification of the Hartree-Fock method. _Phys. Rev._ 81, 385
(1951). Article ADS CAS MATH Google Scholar * Frisch, M. J. et al. Gaussian 09 Revision D.01Gaussian Inc. (Wallingford CT, 2009). * Giannozzi, P. et al. QUANTUM ESPRESSO: a modular and
open-source software project for quantum simulations of materials. _J. Phys.: Condens. Matter_ 21, 395502 (2009). Google Scholar * Blöchl, P. E. Projector augmented-wave method. _Phys. Rev.
B_ 50, 17953–17979 (1994). Article ADS Google Scholar * Verlet, L. Computer “experiments" on classical fluids. i. thermodynamical properties of lennard-jones molecules. _Phys. Rev._
159, 98–103 (1967). Article ADS CAS Google Scholar * NIST Computational Chemistry Comparison and Benchmark Database, NIST Standard Reference Database Number 101.
http://cccbdb.nist.gov/. Release 21, August 2020, Editor: Russell D. Johnson III. Download references ACKNOWLEDGEMENTS The authors thank Virginie Speisser and Michelangelo Romeo for
technical support. This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement No
771850 [G.S.]) and the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No 894434 [A.R.]. The Labex NIE (Contract No.
ANR-11-LABX-0058_NIE [G.S.]), and the International Center for Frontier Research in Chemistry (FRC) [G.S.] are acknowledged for financial support. This work was granted access to the HPC
resources of IDRIS under the allocation 2020-A0060907459 [H.B.] made by GENCI. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Université de Strasbourg, CNRS, IPCMS, UMR 7504, F-67000,
Strasbourg, France Kirill Vasilev, Benjamin Doppagne, Tomáš Neuman, Anna Rosławska, Hervé Bulou, Alex Boeglin, Fabrice Scheurer & Guillaume Schull Authors * Kirill Vasilev View author
publications You can also search for this author inPubMed Google Scholar * Benjamin Doppagne View author publications You can also search for this author inPubMed Google Scholar * Tomáš
Neuman View author publications You can also search for this author inPubMed Google Scholar * Anna Rosławska View author publications You can also search for this author inPubMed Google
Scholar * Hervé Bulou View author publications You can also search for this author inPubMed Google Scholar * Alex Boeglin View author publications You can also search for this author
inPubMed Google Scholar * Fabrice Scheurer View author publications You can also search for this author inPubMed Google Scholar * Guillaume Schull View author publications You can also
search for this author inPubMed Google Scholar CONTRIBUTIONS K.V., B.D., F.S., and G.S. conceived, designed, and performed the experiments. K.V., A.R., and G.S. analyzed the experimental
data. T.N., H.B., and A.B. performed the analytical and numerical calculations. All authors discussed the results and contributed to the redaction of the paper. CORRESPONDING AUTHOR
Correspondence to Guillaume Schull. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Nature Communications_ thanks the
anonymous reviewers for their contribution to the peer review of this work. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in
published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION SOURCE DATA SOURCE DATA RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a
Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit
to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are
included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and
your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this
license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Vasilev, K., Doppagne, B., Neuman, T. _et al._ Internal Stark effect
of single-molecule fluorescence. _Nat Commun_ 13, 677 (2022). https://doi.org/10.1038/s41467-022-28241-8 Download citation * Received: 08 August 2021 * Accepted: 07 January 2022 *
Published: 03 February 2022 * DOI: https://doi.org/10.1038/s41467-022-28241-8 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable
link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative