Play all audios:
ABSTRACT Thiamine (vitamin B1) functions as an essential coenzyme in cells. Humans and other mammals cannot synthesise this vitamin de novo and thus have to take it up from their diet.
Eventually, every cell needs to import thiamine across its plasma membrane, which is mainly mediated by the two specific thiamine transporters SLC19A2 and SLC19A3. Loss of function mutations
in either of these transporters lead to detrimental, life-threatening metabolic disorders. SLC19A3 is furthermore a major site of drug interactions. Many medications, including
antidepressants, antibiotics and chemotherapeutics are known to inhibit this transporter, with potentially fatal consequences for patients. Despite a thorough functional characterisation
over the past two decades, the structural basis of its transport mechanism and drug interactions has remained elusive. Here, we report seven cryo-electron microscopy (cryo-EM) structures of
the human thiamine transporter SLC19A3 in complex with various ligands. Conformation-specific nanobodies enable us to capture different states of SLC19A3’s transport cycle, revealing the
molecular details of thiamine recognition and transport. We identify seven previously unknown drug interactions of SLC19A3 and present structures of the transporter in complex with the
inhibitors fedratinib, amprolium and hydroxychloroquine. These data allow us to develop an understanding of the transport mechanism and ligand recognition of SLC19A3. SIMILAR CONTENT BEING
VIEWED BY OTHERS SUBSTRATE TRANSPORT AND DRUG INTERACTION OF HUMAN THIAMINE TRANSPORTERS SLC19A2/A3 Article Open access 30 December 2024 MOLECULAR BASIS OF SLC19A1-MEDIATED FOLATE AND CYCLIC
DINUCLEOTIDE TRANSPORT Article Open access 02 April 2025 SUBSTRATE BINDING PLASTICITY REVEALED BY CRYO-EM STRUCTURES OF SLC26A2 Article Open access 29 April 2024 INTRODUCTION Thiamine,
commonly known as vitamin B1, is crucial for the survival of cells and an essential micronutrient for humans and other mammals1. Inside the cell, thiamine is converted to thiamine
pyrophosphate (thiamine-pp) by the enzyme thiamine pyrophosphokinase 1 (TPK1)2,3. Thiamine-pp then acts as a coenzyme in central metabolic pathways, such as the citric acid cycle and the
pentose phosphate pathway4. Mammalian cells lack the ability to synthesise thiamine de novo. Instead, they import the vitamin over their plasma membrane through the transport activity of
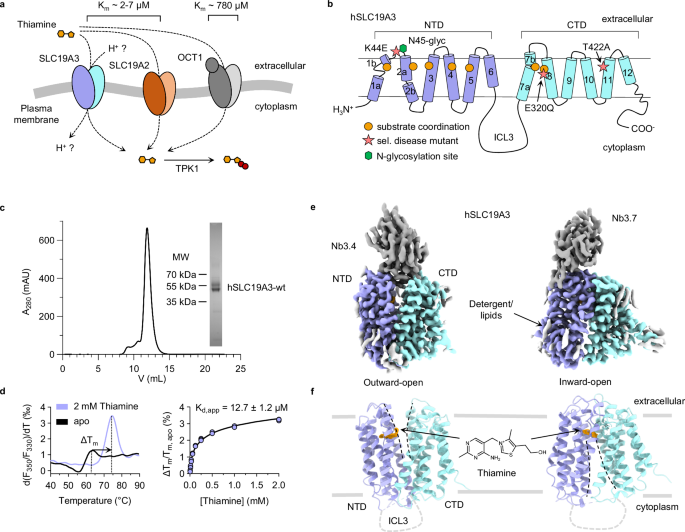
several integral membrane proteins of the solute carrier family (SLC, Fig. 1a)5. SLC19A2 and SLC19A3 are the main transporters for thiamine in humans and mediate the uptake of the vitamin
with high affinity and specificity (Km = 2–7 μM)6,7,8. They are closely related to the folate transporter SLC19A1 (35% sequence identity, 48% sequence similarity), with which they form the
SLC19A family of vitamin transporters5. In addition, the organic cation transporter 1 (OCT1, SLC22A1), which is known to accept a wide variety of substrates, can also mediate thiamine
uptake, though with lower affinity (Km~780 μM)9. Among the known thiamine transporters, SLC19A3 stands out in terms of its physiological and pharmacological importance, as it is crucial for
the transport of thiamine across the intestinal wall and the blood-brain barrier10,11. Deletion of SLC19A3 in mice leads to strongly decreased thiamine levels in the blood serum and brain
tissue under standard diet conditions11,12,13. Mutations of SLC19A3 in humans cause severe neurometabolic disorders in the form of Wernicke’s-like encephalopathy (WLE)14 and biotin- and
thiamine-responsive basal ganglia disease (BTBGD)15,16,17. In most cases, these diseases have an onset in infancy and can lead to life-long disabilities and early death15. Thiamine uptake
through SLC19A3 is further known to be inhibited by a broad spectrum of commonly prescribed drugs, including antidepressants, antibiotics and antineoplastic medications7,10. Being under
treatment with these thiamine uptake inhibitors (TUIs) can lead to drug-induced thiamine deficiencies on an organism-wide or tissue-specific level, with potentially fatal consequences11.
This is exemplified by the case of the Janus-kinase (JAK) inhibitor fedratinib. Treatment with this compound induced the development of Wernicke’s encephalopathy, a severe neurodegenerative
disorder characteristic for thiamine deficiency, in several patients18 (clinical trial ID: NCT01437787). This adverse event could eventually be linked to high-affinity inhibition of SLC19A3
by fedratinib19. The human SLC19A3 (hSLC19A3) transporter is a twelve-pass transmembrane protein of the Major Facilitator Superfamily (MFS)20,21,22. It is predicted to follow the canonical
MFS fold, which consists of two α-helical and symmetrically related domains, termed the N-terminal (NTD) and the C-terminal domain (CTD), as shown in Fig. 1b. In recent years, the transport
activity and drug-induced inhibition of SLC19A3 has been thoroughly studied on a cellular level7,8,10,23,24,25,26,27. The molecular basis of ligand recognition and the transport mechanism of
SLC19A3 were, however, for a long time not well understood. Here, we report seven cryo-EM structures of hSLC19A3. Structure determination is enabled by the generation of three
conformation-specific nanobodies against hSLC19A3. With these, we capture the solute carrier in its outward- and inward-open states, which provides detailed insights in its substrate
recognition and transport cycle. Furthermore, we use thermal shift assays in conjunction with cellular thiamine-uptake assays to screen for compounds that inhibit thiamine transport through
hSLC19A3. This brings about the discovery of seven previously unknown drug interactions of the transporter. We further present cryo-EM structures of the transporter in complex with the known
high-affinity inhibitors fedratinib, amprolium and hydroxychloroquine, which elucidates the structural basis of drug interactions of hSLC19A3. RESULTS CRYO-EM STRUCTURES OF HSLC19A3 IN
DIFFERENT CONFORMATIONS To study hSLC19A3 on a structural and biophysical level we made use of an expression system that has already been widely used for the functional characterisation of
the transporter in cell-based assays7,8,10,24. Full-length hSLC19A3 was expressed in HEK293-derived Expi293FTM cells and purified in a detergent solution using a mixture of lauryl maltose
neopentyl glycol (LMNG) and cholesterol hemisuccinate (CHS) (Fig. 1c). The recombinant transporter is strongly stabilised against heat denaturation in the presence of its known substrate
thiamine (ΔTm = 10.9 ± 0.3 °C, Fig. 1d). Concentration-dependent thermal shift assays revealed high-affinity binding of thiamine with an apparent dissociation constant (Kd,app) of 12.7 ± 1.2
μM (Fig. 1), which is in good agreement with previously reported transport affinities in the low micromolar range8,9. In contrast, phosphorylation of thiamine strongly reduced the
interaction between the vitamin and hSLC19A3 (Supplementary Fig. 1a). Since hSLC19A3 on its own proved to be too small for structure determination by cryo-EM (~55 kDa), we generated
hSLC19A3-specific nanobodies in a llama (Supplementary Fig. 2). For immunisation, a glycosylation-free mutant of hSLC19A3 (hSLC19A3-gf) was used in order to provide an increased accessible
surface area for antibody binding (Supplementary Fig. 3). To generate hSLC19A3-gf, the predicted N-glycosylation sites Asn45 and Asn166 were mutated to glutamine residues. Thermal stability
experiments did not show any interference of these mutations with thiamine binding (Supplementary Fig. 4). The immunisation and nanobody discovery were performed as described earlier28. In
total we identified three hSLC19A3-specific binders: Nb3.3, Nb3.4 and Nb3.7 with binding affinities in the range of 100-300 nM (Supplementary Fig. 2). While Nb3.3 and Nb3.4 bind stably to
wildtype hSLC19A3, Nb3.7 can sterically only interact with the glycosylation-free transporter (Supplementary Fig. 3b). All three nanobodies were used for structure determination of hSLC19A3
by single particle cryo-EM with map resolutions reaching between 2.9-3.8 Å on a global level and 2.4-3.2 Å in the substrate binding site (for an overview, see Supplementary table 1). Nb3.4
stabilises the outward-open state of the transporter, while Nb3.3 and Nb3.7 both bind to its inward-open state (Fig. 1e,f, Supplementary Fig. 6–10). Using this molecular toolset, we
determined the structures of hSLC19A3 in its outward-open and inward-open conformations, in both apo and thiamine-bound states. The experimental structures of hSLC19A3 confirm the MFS fold
of the transporter, with twelve transmembrane helices (TM) folding into two compact six-helix bundle domains (Fig. 1f, Supplementary Fig. 11). The domains are connected via a 76-residues
long (Lys195-Glu271), poorly conserved intracellular loop 3 (ICL3) (Supplementary Fig. 12). No density could be resolved for this loop, indicating that this part of the transporter is likely
disordered and flexible. Similarly, parts of the N-terminus (Met1-Ser10) and the C-terminus (Tyr459-Leu496) could not be resolved in any of the cryo-EM structures (Fig. 1e, f). Within the
well resolved helical domains, there are three discontinuous transmembrane helices in both the outward- and inward-open state: TM1, TM2, and TM7. In TM1, Ile13-Met27 form a stable α-helix
(TM1a). The helix then partially unwinds and is followed by a short helical segment (TM1b), which is flanked by two proline residues in position 33 and 42 (Supplementary Fig. 11a). TM2 is
interrupted close to its cytoplasmic end, between Val65 and Leu68. This discontinuity allows TM2 to bend around TM4 (Supplementary Fig. 11a). The helical structure of TM7 is also broken, in
this case close to the extracellular space, between Asn297 and Gln300 (Supplementary Fig. 11b). Our structural data further confirm that recombinant hSLC19A3 is N-glycosylated on Asn45,
whereas the second predicted glycosylation side, Asn166, appears to be glycosylation-free in the wildtype transporter (Supplementary Fig. 3). Additional non-protein density observed in the
EM-reconstructions likely originates from bound lipids or detergent molecules (Fig. 1e). THIAMINE BINDING SITE Comparison of the final cryo-EM reconstructions from data sets recorded in the
absence and presence of thiamine revealed the binding mode of the vitamin to hSLC19A3. In the thiamine-free samples, the central vestibule of the transporter retains some weak densities
(Supplementary Fig. 13). These originate most likely from small molecules of cellular origin, buffer components or coordinated water molecules. This observation is not uncommon in cryo-EM
maps29. In the thiamine-supplied samples, in contrast, extra densities appeared in the vestibule of hSLC19A3, which correspond well to the molecular shape of thiamine. The fitted ligand also
matches with the transporter structures in terms of coordinating interactions (Fig. 2, Supplementary Fig. 13). These structures highlight that the substrate binding site is positioned close
to the extracellular side of hSLC19A3 (Fig. 2a, b). Thus, the thiamine binding site in hSLC19A3 is similar to the reported substrate binding pocket of the closely related folate transporter
SLC19A130,31,32 (Supplementary Fig. 14b, d, f; the corresponding binding site residues are also conserved in hSLC19A2, Supplementary Fig. 15). This is, however, a rather unusual feature for
MFS transporters, which typically bind their substrates more centrally within the membrane plane33 (Supplementary Fig. 16). Since the discovered nanobodies allowed the determination of
different conformational states of hSLC19A3, a direct comparison of thiamine binding in the outward-open and the inward-open state was possible. In the outward-open state, thiamine is
exclusively coordinated by the N-terminal domain (NTD) of the transporter (Fig. 2a, c). Thiamine adapts a kinked conformation with regard to its two aromatic rings. Its aminopyrimidine
moiety inserts into a complementary pocket of the NTD, parallel to the membrane plane (Fig. 2a, c). Here, the aromatic ring interacts via π-π stacking with Tyr113 and to a lesser extend with
Trp59 (Fig. 2c, e). The methyl group of the aminopyrimidine ring extends into a hydrophobic pocket formed by the side chains of Thr93, Trp94 and Leu97 on TM3, and Met106 and Val109 on TM4
(Fig. 2e). Crucial polar contacts are formed with Glu110, which is within hydrogen bonding distance (≤ 3.0 Å)34 to both the ring-nitrogen (2.8 Å), as well as the primary amine of thiamine
(2.8 Å) (Fig. 2e). The latter ones constitute a hydrogen bond acceptor and donor, respectively. We hypothesise that Glu110 could be protonated in the thiamine-bound state to provide a
matching hydrogen bond donor-acceptor pair for the aminopyrimidine ring (Fig. 2e, Supplementary Fig. 1c, d). Structure-based pKa estimations using PROPKA35 rank the pKa of Glu110 in the
neutral range (pKa = 6.4–7.5), which would support the idea of at least partial protonation of this residue under physiological conditions. The specific hydrogen bonding between the
aminopyrimidine ring and Glu110 appears to be critical for the high-affinity binding of thiamine to hSLC19A3. In oxythiamine, a close derivative of thiamine, the primary amine of the
aminopyrimidine ring is substituted by a carbonyl oxygen. This subtle change already leads to a strongly decreased affinity and a lower thermostabilisation of the transporter (Supplementary
Fig. 1a, d). A similar effect is observed, when Glu110 is mutated to glutamine, resulting eventually in a drop of binding affinity from an apparent Kd of 12.7 ± 1.2 μM to 85 ± 13 μM. The
replacement of Glu110 by a positively charged lysine residue renders hSLC19A3 completely unresponsive to thiamine (Supplementary Fig. 5). Opposite of Glu110, Glu32 provides another polar
contact, which is coordinating the aminopyrimidine ring of thiamine and has been shown to be important for the transport activity of hSLC19A336,37 (Fig. 2e). In the outward-open state of
hSLC19A3, the thiazolium ring of thiamine is oriented approximately 90° relative to the aminopyrimidine ring and is primarily coordinated through π-π stacking with Phe56. The hydroxyethyl
group, the tail of thiamine, reaches even further up in the binding site, forming polar contacts with the backbone of Ile36 (Fig. 2c). When hSLC19A3 transitions from the outward- to the
inward-open state, thiamine undergoes a major rearrangement. It relaxes via its rotatable _C-C_ and _C-N_ bonds connecting the aminopyrimidine and thiazolium ring into an elongated
conformation (Fig. 2d, e). The aminopyrimidine ring stays coordinated in the same position in the NTD, with Phe56 closing on top of it. Together with Trp59 and Tyr113, Phe56 forms now a
hydrophobic pocket that we termed ‘aromatic clamp’ (Fig. 2e). Meanwhile, the positively charged thiazolium ring reorients and is accommodated between the electronegative backbone and side
chain carbonyl oxygen atoms of Leu296 and Asn297, respectively. Through the transition of hSLC19A3 to the inward-open conformation, the hydroxyethyl tail of thiamine detaches from the
backbone of Ile36 and reaches across to Glu320 on the CTD to form a hydrogen bond (2.7 Å). With this, thiamine tightly connects the two helical bundles of the transporter by non-covalent
interactions (Fig. 2d, e). Glu320 is further predicted to undergo strong pKa changes over the course of the transport cycle, which could potentially link thiamine transport to a proton
gradient over the membrane (see discussion). In order to obtain experimental insights into the interaction of individual residues with thiamine, we generated and analysed several single
point mutants of hSLC19A3 (Fig. 2f, Supplementary Fig. 4, 20). The mutation of residues of the aromatic clamp to alanine did not impair thiamine binding (Fig. 2f). We speculate that two out
of the three constituents of the aromatic clamp (Phe56, Trp59 and Tyr113) are sufficient to mediate stable π-π stacking of the aminopyrimidine ring of thiamine. In contrast, mutating Asn297
to alanine, and thereby removing a negative partial charge from the substrate binding site, led to a ~250-fold loss in binding affinity (Fig. 2f). In addition, we probed the role of Glu32 in
thiamine binding. The mutation of the equivalent residue Glu45 to lysine in the closely related folate transporter SLC19A1 leads to a loss of function and conveys methotrexate (MTX)
resistance38,39 (Supplementary Fig., 14). The analogous mutation (hSLC19A3-Glu32Lys) causes a 10-fold drop in the affinity for thiamine in hSLC19A3, from 12.7 ± 1.2 μM to 145 ± 27 μM. It is
likely that the placement of a positive charge in this position leads to an electrostatic or partially steric repulsion of the aminopyrimidine ring (Fig. 2e, f). The interaction between
Glu320 and the hydroxyethyl tail of thiamine is affected in a rare form of heritable Wernicke’s-like encephalopathy14. Our biophysical data illustrate that the disease-associated mutation
Glu320Gln leads to a 20-fold decrease in affinity for thiamine, from 12.7 ± 1.2 μM to 284 ± 45 μM (Fig. 2f), in agreement with recently published transport data36. SUBSTRATE BINDING SITES IN
OTHER HUMAN THIAMINE TRANSPORTERS The transport of thiamine can be mediated by different transporters, dependent on the tissue, cell type and subcellular compartment23,25,40,41 (Fig. 1a).
High-affinity transport of thiamine across the plasma membrane is shared between SLC19A3 and the closely related SLC19A26,26. In order to understand the structural relationship between these
two, we compared the experimentally determined structure of the inward-open hSLC19A3 with the AlphaFold2 (AF2) predicted structure of hSLC19A242 (Supplementary Figs. 14, 15). The global
fold of the two proteins is almost identical, with the only marked differences in the poorly conserved and disordered N- and C-terminal regions, as well as ICL3 (Supplementary Fig. 14b). The
substrate binding site appears to be structurally highly conserved. Only subtle differences in the form of two substitutions, (i) Phe56hSLC19A3 to Tyr74hSLC19A2 and (ii) Tyr151hSLC19A3 to
Phe169hSLC19A2 can be observed (Supplementary Fig. 14e). The first substitution introduces a minor change in the aromatic clamp. As shown in Fig. 2f, replacing Phe56hSLC19A3 with alanine in
hSLC19A3 did not impact the binding affinity for thiamine. Therefore, we assume that the even more similar substitution of Phe56 to tyrosine will not impact substrate binding. The effect of
the second substitution (ii) on thiamine binding was tested experimentally. When Tyr151hSLC19A3 was replaced with phenylalanine, no strong effect on the affinity for thiamine was observed
(Fig. 2f). This is in line with previous observations, which reported very similar transport affinities for the two thiamine transporters7,8. The situation is different, when comparing the
hSLC19A3 structures with the recently determined cryo-EM structure of OCT1 (SLC22A1)29 in complex with thiamine. The reported transport affinity of OCT1 for thiamine is much lower than for
the SLC19A transporters (Km ~780 μM compared to 2–7 μM)7,8,9. This difference could be explained by a looser coordination of thiamine in OCT1, compared to the tight binding observed in
hSLC19A329 (Supplementary Fig. 16). TRANSPORT CYCLE To obtain a better understanding of the conformational changes during the transport cycle we compared the cryo-EM structures of hSLC19A3
stabilised in the outward-open state by Nb3.4 with the inward-open state, bound by Nb3.3 or Nb3.7. An overlay of the individual N- and C-terminal domains of the different conformational
states revealed no major intradomain rearrangements (Cα RMSD of 0.52–1.94 Å). Only extracellular loop 4 (ECL4) and intracellular loop 5 (ICL5) reorient slightly (Supplementary Fig. 11).
Based on the available structures, we conclude that the conformational changes observed for hSLC19A3 follow a rocker-switch mechanism, in which the NTD and CTD move as rigid bodies to create
a moving barrier around the substrate binding site (Supplementary Movies 1–3) 33. This barrier consists of a cytoplasmic gate and an extracellular gate that open and close in an alternating
fashion. This provides alternating solvent accessibility to the substrate binding site from the extracellular and cytoplasmic space (Supplementary Fig. 17). The cytoplasmic gate of hSLC19A3
is formed by hydrophobic contacts of Tyr128 (TM4) and Ala395 (TM10) with Tyr403 (TM11). The gate is further stabilised by polar interactions of the side chains of Asp75 (TM2) and Gln137
(TM5) with the backbone of Ala404 and Leu405 (TM11) and Lys338 (ICL5), respectively (Supplementary Fig. 17a–c). These contacts are broken when the transporter transitions to the inward-open
state (Supplementary Fig. 17d–f). The long ICL3 of hSLC19A3 allows for a dilation of the cytoplasmic gate. Simultaneously, a barrier to the extracellular space is established by the closure
of the extracellular gate. The core of this gate is built by polar contacts of the side chains of Asn297 and Gln300 (TM7). They are within hydrogen bonding distance with the backbone of
Phe56 (TM2) and Pro33 (TM1a), respectively. These polar contacts are supported by hydrophobic interactions between Ile36 (TM1b) and Ile301 (TM7). With these in place, TM1b is coordinated as
a lid, sealing the extracellular gate (Supplementary Fig. 17d–f). MUTATION OF HSLC19A3 IN BIOTIN- AND THIAMINE RESPONSIVE BASAL GANGLIA DISEASE (BTBGD) Loss of function mutations of hSLC19A3
cause BTBGD in humans15,17,43,44,45,46,47 (Supplementary Fig. 18, 19). This disease is marked by severe neurological symptoms and is inherited genetically in an autosomal recessive
manner15. The median age of onset of symptoms varies between three months and four years after birth, dependent on the underlying mutations in the _SLC19A3_ gene. Under the currently advised
treatment - a combination therapy of high-dose thiamine and biotin - more than 30% of the patients still suffer from moderate to severe effects of the disease, while about 5% of those
affected die despite treatment15. To gain a better understanding of the molecular cause of this disorder, we investigated the effects of one of the most common BTBGD-causing point mutation
SLC19A3-Thr422Ala. Most cases are reported in Saudi Arabia, where the heterozygous carrier frequency is about 1 in 500 among the Saudi population17. The patients usually present with
symptoms including subacute encephalopathy with confusion, convulsions, dysarthria and dystonia48. On a cellular level, the SLC19A3-Thr422Ala mutant localises normally to the (apical) plasma
membrane in Caco-2 and MDCK cells25 and can also be expressed and purified in vitro (Supplementary Fig. 20a). However, its thiamine uptake activity is significantly impaired25. This agrees
with our data, highlighting that the Thr422Ala mutation leads to a more than 10-fold decrease of binding affinity for thiamine, from 12.7 ± 1.2 μM to 172 ± 35 μM (Fig. 2f, Supplementary Fig.
19d). Thr422 is located on TM11 and thus spatially in the outer region of the transporter. It is, however, physically connected to the core of hSLC19A3 by hydrogen bonding with the side
chain of Gln294 on TM7 (Supplementary Figs. 18b, c and 19a, d). This part of TM7 is involved in the formation of the substrate binding site and the extracellular gate through Asn297, Gln300
and Ile301 (Fig. 2c, d, Supplementary Fig. 17). Structural changes in this region might explain the loss of affinity and transport activity of the Thr422Ala mutant. The cryo-EM structures
determined in this study additionally provide hints on the disease-causing molecular effects of other BTBGD mutations. Trp59Arg, Trp94Arg, Tyr113His, and Glu320Lys would be directly
affecting thiamine binding by placing a positive charge in the substrate binding pocket of hSLC19A3 (Fig. 2e, Supplementary Fig. 18c). Binding data on the previously mentioned Glu32Lys
mutant illustrate that the insertion of a positive charge in this region can lead to a strong loss of affinity for thiamine (>10-fold, Fig. 2f, Supplementary Fig. 4). This would likely
render the transporter unreceptive for physiological thiamine concentrations, which is the possible cause for the pathogenicity of these mutations. DRUG INTERACTIONS OF SLC19A3 Previous
studies identified a broad spectrum of FDA-approved drugs that act as thiamine uptake inhibitors (TUIs) by blocking hSLC19A37,10,11. This has important clinical implications, as many of
these drugs are widely prescribed over extended periods of time. Among these drugs are antidepressants, such as sertraline (Zoloft® and others) and amitriptyline, the antiparasitic
hydroxychloroquine and the Janus-kinase (JAK) inhibitor fedratinib (Inrebic®). The inhibition of SLC19A3 can lead to organism-wide and tissue-specific thiamine deficiencies. In the latter
case, the deficiencies would not be reflected in serum thiamine levels and remain undetected in standard diagnostics. As BTBGD-causing mutations and the example of SLC19A3 knock-out mice
illustrate, this can lead to the decline of entire cell populations in vital organs, particularly in the brain11,49. Studying recombinantly purified hSLC19A3 in thermal shift assays, we
explored the interaction space of the transporter with six known TUIs and eleven pharmacologically related drugs (Fig. 3). The corresponding screen confirmed physical binding of known
inhibitors to hSLC19A3, and further led to the identification of nine previously unknown drug interactions, of which seven show inhibitor function (Fig. 3). To begin with, several drugs that
are structurally related to thiamine were probed. The known hSLC19A3 inhibitors amprolium and trimethoprim strongly stabilised the transporter against heat unfolding in a
concentration-dependent manner. This effect was exceeded by the antiparasitic pyrimethamine tested here (Fig. 3a). Even at a concentration of 20 μM, this compound led to a
thermostabilisation of 9.3 ± 0.5 °C and reduced thiamine-uptake through hSLC19A3 by about 50% in cell-based assays. Next, representative JAK inhibitors were analysed. Fedratinib, which is
known to be a potent inhibitor of hSLC19A3, also showed high-affinity binding to the transporter in the thermal shift assays (Kd,app = 1.0 ± 0.2 μM, Supplementary Fig. 21) and strongly
suppressed the thiamine transport activity of hSLC19A3 (Fig. 3b). The interaction of hSLC19A3 with three other JAK inhibitors, tofacitinib (Xeljanz®), baricitinib (Olumiant®) and momelotinib
(Ojjaara®) was tested as well. These compounds are functionally similar to, but structurally distinct from fedratinib. Tofacitinib is also stabilising hSLC19A3, but shows no inhibitory
effect on thiamine transport (Fig. 3a). Baricitinib and momelotinib did not stabilise hSLC19A3 and thus likely do not interact with the transporter. This agrees with previous cellular uptake
inhibition studies of JAK inhibitors7. Another important drug class that was tested were tyrosine kinase inhibitors, represented by imatinib (Gleevec®), gefitinib (Iressa®) and dasatinib
(Sprycel®). They strongly stabilized hSLC19A3 with apparent binding affinities of 4.4 ± 0.5 μM, 116 ± 36 nM and 7.5 ± 1.5 μM, respectively (Supplementary Fig. 21). All tested tyrosine kinase
inhibitors interfered with thiamine-uptake through hSLC19A3, with imatinib reaching an inhibitory effect comparable to the one of fedratinib (Fig. 3b). When comparing fedratinib and the
tyrosine kinase inhibitors on a chemical level, they share a central aminopyrimidine moiety (Supplementary Fig. 21). This moiety is also at the core of the interaction of thiamine within the
substrate binding site (Fig. 2e) and is thus potentially important for the interaction of the tyrosine kinase inhibitors with hSLC19A3, as already suggested by Giacomini and co-workers7.
All of the tested tricyclic antidepressants (TCA), including the known inhibitor amitriptyline and the ones assessed here, imipramine and doxepin, interact with hSLC19A3 and inhibit its
thiamine transport activity (Fig. 3). Interestingly, TCAs actually destabilize the transporter in a concentration-dependent manner, while compounds of other classes were generally
stabilising. In the group of selective serotonin reuptake inhibitors (SSRI), sertraline stabilises hSLC19A3 already at low micromolar concentrations (ΔTm = 2.5 ± 0.4 °C, Fig. 3), which is in
agreement with existing data on cellular thiamine uptake inhibition10. The compounds citalopram (Celexa® and others) and fluoxetine (Prozac® and others) tested here also interact with
hSLC19A3, but only citalopram interferes with thiamine uptake (Fig. 3). The last analysed compound was hydroxychloroquine (Fig. 3). This drug strongly stabilises hSLC19A3 and binds to the
transporter with high-affinity in the nanomolar range (Kd,app = 170 ± 55 nM, Supplementary Fig. 21). This agrees with the previously reported high-affinity inhibition of hSLC19A3 by
hydroxychloroquine10. In contrast to the known and the hSLC19A3 inhibitors identified here, biotin does not interact with the transporter (Fig. 3b), in agreement with our and previously
published cellular uptake data25 (Fig. 3b). STRUCTURAL BASIS FOR THIAMINE UPTAKE INHIBITION The known thiamine uptake inhibitors (TUIs), as well as the ones identified here, are structurally
diverse (Supplementary Fig. 21). Their chemical heterogeneity presents a challenge for the development of a universal ligand-based pharmacophore model7. To elucidate how these compounds,
bind to hSLC19A3, we determined cryo-EM structures of the transporter in complex with three known transport inhibitors: fedratinib, amprolium and hydroxychloroquine, (Fig. 4a–c). They
represent structurally and functionally different drug classes. Cryo-EM structures were solved using either Nb3.7 or Nb3.3 as structural fiducials, both of which stabilise the transporter in
the inward-open state. The cryo-EM maps of the transporter-drug complexes were resolved to 3.0-3.7 Å, reaching resolutions between 2.5 and 3.2 Å in the core of the protein (Supplementary
Figs. 22–24). All of the analysed inhibitors bind orthosterically in the substrate binding site of the transporter (Fig. 4). From a structural point of view, the binding of these compounds
to hSLC19A3 is determined by at least three key factors: (1) Intercalation of aromatic rings in the aromatic clamp: Our cryo-EM data show that the aromatic rings of all three structurally
resolved inhibitors form direct π-π stacking interactions with Tyr113 and to a lesser extent with Phe56 and Trp59 (Fig. 4a–c). This seems to be a universally important feature of the
interaction of TUIs with hSLC19A3, as most known and all ligands identified here comprise at least one aromatic ring. This interaction might be particularly important for the binding of
compounds that otherwise lack compatible polar contacts. This concerns specifically the tested antidepressants amitriptyline, imipramine, doxepin, sertraline and citalopram (Fig. 3a). (2)
Electrostatic compatibility with the polar contact points presented by the side chains of Glu32, Glu110 and Asn297: Fedratinib, amprolium and hydroxychloroquine provide hydrogen bond donors
and acceptors that pair spatially well with the polar residues of the substrate binding site (Fig. 4a–c). All three inhibitors are within hydrogen bonding distance to Glu110. As mentioned
above, this residue might be protonated, allowing Glu110 to act simultaneously as a hydrogen bond donor and acceptor. This would agree with the observed interactions of fedratinib and
amprolium with this residue (Fig. 4a). Another important polar residue of the substrate binding site is Asn297. Its side chain carbonyl oxygen contributes to the electrostatically negative
interaction surface of the transporter in general, and with 2.8 Å, it is within hydrogen bonding distance with the primary amine of amprolium (Fig. 4c). The coordination capability of Glu320
is essential for high-affinity binding of thiamine, as shown in Fig. 2f and Supplementary Fig. 19c. For inhibitor binding, however, this residue seems to play no major role, as no
interactions between its side chain and the inhibitors could be observed (Fig. 4a–c). Glu32, in contrast, is in clear hydrogen bond distance with one of the secondary amines of fedratinib
(2.8 Å, Fig. 4b). (3) Insertion of a lipophilic moiety in the hydrophobic pocket formed by Thr93, Trp94, Leu97, Met106 and Val109: The structurally resolved inhibitors all engage in this
type of interaction and protrude an apolar substituent into the hydrophobic pocket (Fig. 4a–c), namely, a methyl group (fedratinib), a propyl group (amprolium) and a chlorine
(hydroxychloroquine). This interaction is likely beneficial for high affinity binding to hSLC19A3. A structurally corresponding methyl group can, for example, also be found in the chemical
structures of the inhibitors imatinib and dasatinib, identified here (Supplementary Fig. 21). DISCUSSION Our structural and biophysical data reveal key aspects of the transport cycle and
drug interactions of hSLC19A3. The transporter moves in a rocker-switch motion over the course of its transport cycle, creating a moving barrier around its substrate (Fig. 2). Our data
support a model where thiamine is first recognised via the NTD in the outward-open hSLC19A3. Once the transporter changes to its inward-open conformation, thiamine rearranges to form a
molecular bridge between the NTD and the CTD, before it dissociates into the cytoplasm. The binding of thiamine follows a key-in-lock mechanism, with almost no structural differences between
apo and substrate-bound structures of the transporter (Cα RMSD of 0.30–0.62 Å; Supplementary Fig. 11). In our work, we identified the key residues mediating thiamine recognition in
hSLC19A3. Several of these are mutated in severe neurometabolic diseases (BTBGD and WLE, Supplementary Fig. 18). Regarding the transport mode of hSLC19A3, it is still not clear whether the
transporter operates in a uniporter fashion or requires co-substrates to drive thiamine uptake. Energetically, a uniporter mechanism is plausible. The membrane potential in combination with
the concentration gradient of thiamine might be sufficient for uptake of this organic cation across the plasma membrane. Our data highlight that thiamine is not able to bind to hSLC19A3 once
it is phosphorylated (Supplementary Fig. 1a,b). With this, the exit route for the vitamin via hSLC19A3 is blocked and shifts the transport equilibrium in favour of thiamine import (Fig.
1a). A coupling of the transport mechanism of hSLC19A3 to sodium ions was not observed in a previous study25. There are conflicting data about the role of protons for the activity of
hSLC19A3. While some authors find evidence for proton-dependent anti- or symport mechanisms8,50, others see no link between the uptake of thiamine and proton gradients24. We provide
structure-based predictions, suggesting that the binding site residue Glu320 might undergo a strong pKa shift (from neutral to acidic), as it forms a salt bridge with Lys380 upon the
transition of hSLC19A3 from the outward-open to the inward-open state. This could potentially connect the uptake of thiamine to the translocation of protons across the membrane. However,
functional studies, ideally in the controlled environment of liposome-based transport assays are needed to asses this hypothesis. hSLC19A3 interacts with many commonly used medical
compounds. This has implications for the adjustment and monitoring of existing treatment plans, as well as the development of new drugs10,11. In this work, we identified seven drugs that
inhibit thiamine uptake via hSLC19A3. Among the most notable ones are the tyrosine kinase inhibitors imatinib, gefitinib and dasatinib (Fig. 3). While these in vitro findings still need to
be validated in vivo, our study highlights the importance of screening drug interactions of vitamin transporters like hSLC19A3. One aspect we did not address in the present study is the mode
of inhibition of hSLC19A3 by the different drugs. For fedratinib and trimethoprim, it is known that they are xenobiotic substrates of the transporter7, and consequently inhibit thiamine
uptake by acting as competing substrates. In this context it seems plausible that hSLC19A3 might also represent a transport route for existing and prospective medical compounds. In
particular, it might be an interesting entry gate for drugs to cross the blood-brain barrier7,51. Our structural data provide a basis for the rational design of drugs to prevent the side
effects of thiamine uptake inhibition or to use hSLC19A3 for tissue-specific drug delivery51. The other high-affinity thiamine transporter, hSLC19A2, shares high sequence identity (48%)26
and, based on AF2 predictions42, high structural conservation with hSLC19A3 (Supplementary Fig. 14). We speculate that many of our findings also apply to hSLC19A2. It seems likely that this
transporter follows a similar transport mechanism. Since the substrate binding site is well conserved, most inhibitors of hSLC19A3 are expected to bind to its closest relative. During the
revision of our manuscript another study by Dang and co-workers36 was published, describing the structure of hSLC19A3 bound to thiamine and fedratinib in the inward-open state. Furthermore,
Qu and colleagues released a preprint on structural work of hSLC19A3 and hSLC19A237. Both studies are in line with the data and conclusions presented here and complement each other. Our
study presents a structural framework of thiamine transport and drug interactions of hSLC19A3. We provide structural snapshots of its transport cycle and detailed molecular insights into the
binding of its substrate and high-affinity inhibitors to the transporter. This work can serve as a basis for studies of the transport mechanism of human MFS transporters and can guide
future structure-based drug design approaches. METHODS CLONING, EXPRESSION AND PURIFICATION OF HSLC19A3 The full-length, wildtype hSLC19A3 (Uniprot accession number: Q9BZV2) was cloned from
cDNA (Source BioScience) into a modified pXLG vector52. In this vector, hSLC19A3 is fused with a C-terminal human rhinovirus 3 C (HRV-3C) protease cleavage site (LEVLFQGP), followed by a
short flexible linker (SSG) and a Twin-Strep-tag for affinity purification. All constructs, including mutants were generated using SLiCE cloning53. The corresponding construct and primer
sequences are listed in the Supplementary Data file. PCR reactions were performed using an in-house purified Phusion DNA polymerase, provided in a 2×Phusion reaction mix (40 mM Tris-HCl,
pH8.9, 4 mM MgCl2, 120 mM KCl, 20 mM (NH4)2SO4, 0.02 mM EDTA, 0.2 % TritonX-100, 8 % Glycerol, 0.005 % Xylene Cyanol FF, 0.05 % Orange G, 0.4 mM dNTPs, 0.04 U/µl Phusion Polymerase). The
transporter constructs were transiently transfected in Expi293FTM cells using PEI MAX® as a transfection agent (ratio 1:2 (w/w) of DNA to PEI MAX®) and grown for 96 h post-transfection in
FreeStyleTM medium, at 37 °C, 8% (v/v) CO2, and 270 rpm54. Cells were harvested at 3000×g, washed with 1×PBS (pH7.4) and subsequently frozen at -70°C. After thawing at room temperature, five
grams of cell pellet were resuspended in 25 mL of solubilisation buffer (1×PBS, pH7.4, 200 mM NaCl, 5% glycerol (v/v), 1% LMNG (w/v), 0.1% CHS (w/v), 1×EDTA-free protease inhibitors
(Roche), 0.5 mM TCEP, 20 U/mL DNase I, 2.5 U avidin) in a 50 mL tube and incubated on a shaking platform for 1 h at 4 °C. Cellular debris was removed via centrifugation for 30 min at
35,000×g at 4 °C. The supernatant was loaded onto 1 mL StrepTactin resin and incubated for 1 h at 4 °C. Afterwards, the resin was washed with 4×20 column volumes of StrepA buffer (20 mM
Tris-HCl, pH 7.4, 350 mM NaCl, 5% glycerol (v/v), 0.5 mM TCEP, 0.02% LMNG (w/v), 0.002% CHS (w/v)). The target proteins were eluted in 5 column volumes StrepB buffer (20 mM Tris-HCl, pH 7.4,
150 mM NaCl, 0.002% LMNG, 0.0002% CHS, 10 mM desthiobiotin). The eluted protein was concentrated to 0.5 mL and further purified by size-exclusion chromatography (SEC) in SEC buffer (20 mM
Tris-HCl, pH7.4, 150 mM NaCl, 0.002% LMNG (w/v), 0.0002% CHS (w/v)) on a Superdex S200 10/300 Increase column (Cytiva). Biotinylation of Avi-tagged protein for nanobody selection was
performed by adding 120 μg/mL of BirA, 5 mM ATP, 50 μM biotin, 0.5 mM TCEP and 40 μg/mL 3C protease to the concentrated and desalted (PD Miditrap G-25 (Cytiva), in SEC buffer) StrepB
elution. The mixture was then incubated at 4 °C overnight. Subsequently, the transporter was separated from the enzymes and the small molecule additives by SEC as described above. THERMAL
STABILITY MEASUREMENTS AND BINDING AFFINITY DETERMINATION The thermal stability of the hSLC19A3 constructs and their binding affinities for small molecules were determined by nanoDSF, using
a Prometheus NT.48 (NanoTemper Technologies). For sample preparation, the protein was diluted to a final concentration of 4 μM in SEC buffer and supplied with the respective compounds in a
concentration range from 2 μM to 2 mM. Before measurement, the samples were incubated for 30 min at room temperature. Melting scans were performed at a temperature range from 20 to 95 °C
with a temperature increment of 1 °C per minute. Thermal unfolding curves were recorded by measuring the ratio of the tryptophan-fluorescence at 350 nm (F350) and 330 nm (F330). The protein
melting temperature (Tm) of a given sample was determined as the inflection point of the thermal unfolding curves, or the local maximum of its first derivative. The measured curves were
analysed using the manufacturer’s software (PR.ThermControl, v 2.1.2, NanoTemper Technologies). Based on the determined Tm values, ligand induced thermal shifts (ΔTm) were determined as the
difference between the melting temperature of the ligand-bound state (Tm) and the apo state (Tmapo). For the determination of apparent binding affinities at 25 °C, the Tm values recorded
from dilution series of the respective ligands were used to fit the following equation: $$\frac{{\triangle T}_{m}}{{T}_{m}^{{apo}}}\left(L\right)=\frac{-R{T}_{{std}}}{{E}_{a1}} *
{{\mathrm{ln}}}\left(\frac{{K}_{d,{app}}}{{K}_{d,{app}}+L}\right)$$ (1) were L is the ligand concentration, R is the universal gas constant, Tstd is the chosen standard temperature of 298.15
K (25 °C), Ea1 is the activation energy for the unfolding of the apo state. As R, Tstd and L are known and the ΔTm and Tmapo are experimentally determined, non-linear curve fitting can be
used to estimate Ea1 and Kd,app. The fitting was performed using symfit (v. 0.5.3), as described in Kotov et al. 55, based on theoretical considerations formulated in Hall, 201956.
GENERATION OF NANOBODIES To generate nanobodies against the human thiamine transporter SLC19A3, the glycosylation-free mutant hSLC19A3-gf was injected into a llama (_Glama lama_) on a weekly
basis over the course of six weeks as described elsewhere28. Following mRNA extraction and cDNA library preparation from peripheral blood mononuclear cells of the immunized llamas,
VHH-fragments were cloned into the pMESy428 vector, which provides the insert with an N-terminal PelB signal peptide for periplasmic expression, and a C-terminal His6-tag, followed by a Myc
peptide. Subsequently, two rounds of phage display were performed to enrich for specific binders. From the enriched libraries, nanobody (VHH) families were identified by sequencing of 192
colonies. Eventually, three unique nanobodies, Nb3.3, Nb3.4 and Nb3.7 were identified and expressed. Binding to the transporters was confirmed using biolayer interferometry (Supplementary
Fig. 2). For the production of nanobodies, transformed _E. coli_ WK6 cells were grown in 1 L TB medium, containing 0.1% glucose, at 37 °C, and 165 rpm. Overexpression of the nanobodies was
induced with 1 mM IPTG at an OD of 0.7, where after the cells were grown for 18 h at 22 °C, and 165 rpm. After pelleting the cells by centrifugation for 15 min at 7000 rpm, periplasmic
extraction was performed by resuspending the pellet in 200 mL periplasmic extraction buffer (20% sucrose (w/v), 50 mM Tris-HCl, pH7.4, 0.5 mM EDTA, pH8.0, 1×EDTA-free protease inhibitors
(Roche), 0.5 µg/mL lysozyme), and stirring the suspension for 30 min at 4 °C. Subsequently, the periplasmic extract was incubated with 1 mL Ni-NTA resin for 1 hour at 4 °C. The resin was
then washed with 4×20 mL of wash buffer (20 mM Tris-HCl, pH7.4, 500 mM NaCl, 20 mM imidazole, pH7.4). The nanobodies were afterwards eluted from the resin with 5 mL of elution buffer (20 mM
Tris-HCl, pH7.4, 500 mM NaCl, 400 mM imidazole, pH7.4). The eluted protein was concentrated to 0.5 mL and further purified by SEC using a Superdex S75 10/300 Increase column (Cytiva) in TBS
buffer (20 mM Tris-HCl, pH7.4, 150 mM NaCl). BIOLAYER INTERFEROMETRY (BLI) Binding between hSLC19A3 and the nanobodies Nb3.3, Nb3.4 and Nb3.7 was measured by biolayer interferometry (BLI)
using an Octet RED96 system (FortéBio). The nanobodies were immobilised at 300 nM via their C-terminal 6×His-tag on Octet® HIS1K Biosensors, pre-equilibrated in BLI buffer (20 mM Tris-HCl,
pH7.4, 150 mM NaCl, 0.02% LMNG (w/v), 0.002% CHS (w/v), 0.1% BSA (w/v), 0.5 mM thiamine). After a baseline step of 60 s, the biosensors were dipped into BLI buffer containing hSLC19A3-gf at
concentrations ranging from 25-800 nM for 240 s to allow for the binding of the transporter to the immobilised nanobodies. Afterwards, the sensors were transferred into fresh BLI buffer and
the dissociation was followed over the course of 600 s. BLI experiments were performed at 22 °C. Data were reference-subtracted and aligned in the Octet Data Analysis software v10.0
(FortéBio). Binding affinities were determined using a 1:1 binding model and the maximum response values as readout. MASS SPECTROMETRY-BASED CELLULAR THIAMINE-UPTAKE ASSAYS The uptake of
deuterated thiamine (thiamine-d3) was measured in hSLC19A3-wt and mock transfected cells in using LC-MS/MS. For this assay, Expi293FTM cells were transfected as described above and incubated
for 48 h at 37 °C, 8% (v/v) CO2, and 270 rpm prior to the experiment. The cells were then pelleted for 5 min at 250×_g_ and resuspended in 1×PBS (pH 7.4) at a cell density of 2 million
cells per mL. The cell suspension was subsequently transferred to a 96-deep well plate in three technical replicates per condition (_n_ = 3, 1 mL per well, corresponding to 2 million cells
per condition). 200 μM of the selected compounds were added to the respective wells and the cells were incubated for 1 min on a shaking platform. Subsequently, 2 μM of thiamine-d3 were added
and the plate was incubated for another 5 min on a shaking platform. Afterwards, the cells were pelleted for 5 min at 500×_g_ and washed 3× with 1 mL 1×PBS (pH7.4), before transferring them
to a fresh 96-deep well plate. The cell pellets were stored at –70 °C. For the LC-MS/MS analysis, the samples were extracted by adding 500 µL of a mixture of H2O:MeOH:ACN (1:1:1, v/v),
containing 5% (v/v) formic acid. After vortexing and ultrasonication in a water bath for 5 min at 4 °C, samples were incubated for 20 min at –20 °C. Ultimately, samples were centrifuged at
15,000×_g_ and 4 °C for 10 min using a 5415 R microcentrifuge (Eppendorf, Hamburg, Germany). The supernatants were then transferred for LC-MS analysis, which was initiated within one hour
after sample preparation. LC-MS/MS analysis was performed on a Vanquish UHPLC system coupled to an Orbitrap Exploris 240 high-resolution mass spectrometer (Thermo Fisher Scientific, MA, USA)
in positive ESI (electrospray ionization) mode. Chromatographic separation was carried out on an Atlantis Premier BEH Z-HILIC column (Waters, MA, USA; 2.1 mm × 100 mm, 1.7 µm) at a flow
rate of 0.25 mL/min. The mobile phase consisted of water:acetonitrile (9:1, v/v; mobile phase phase A) and acetonitrile:water (9:1, v/v; mobile phase B), which were modified with a total
buffer concentration of 10 mM ammonium formate. The aqueous portion of each mobile phase was adjusted to pH 3.0 via addition of formic acid. The following gradient (8 min total run time
including re-equilibration) was applied (time [min]/%B): 0/90, 3/85, 3.5/60, 4/60, 5/90, 8/90. Column temperature was maintained at 40 °C, the autosampler was set to 4 °C and sample
injection volume was set to 3 µL. Analytes were recorded via a full scan with a mass resolving power of 120,000 over a mass range from 60 to 900 _m/z_ (scan time: 100 ms, RF lens: 70%).
MS/MS fragment spectra were recorded via targeted product ion scans for Thiamine ([M]+, _m/z_ = 265.1118) and Thiamine-d3 ([M]+, _m/z_ = 268.1306) at a resolving power of 15,000, stepped
collision energies [%]: 20/35/50, and total cycle time of 3 s. Ion source parameters were set to the following values: spray voltage: 3500 V, sheath gas: 30 psi, auxiliary gas: 5 psi, sweep
gas: 0 psi, ion transfer tube temperature: 350 °C, vaporizer temperature: 300 °C. All experimental samples were measured in a randomized manner. Pooled quality control (QC) samples were
prepared by mixing equal aliquots from each processed sample. Multiple QCs were injected at the beginning of the analysis in order to equilibrate the analytical system. A QC sample was
analyzed after every 6th experimental sample to monitor instrument performance throughout the sequence. For determination of background signals and subsequent background subtraction, an
additional processed blank sample was recorded. Data was processed using TraceFinder 5.1 and raw peak area data was exported for relative metabolite quantification. CRYO-EM SAMPLE
PREPARATION AND DATA ACQUISITION For cryo-EM, hSLC19A3 was purified in LMNG/CHS as described above. hSLC19A3-wt was prepared in complex with Nb3.3 and Nb3.4, while hSLC19A3-gf was used in
conjunction with Nb3.7. After concentrating the transporter to 60 μM, the sample was supplied with a 1.5× molar excess of nanobody and diluted to a total concentration of 30 μM of hSLC19A3
in SEC buffer. Compounds were added during the dilution step at the following concentrations: 500 μM thiamine, 250 μM fedratinib, 300 μM amprolium or 200 μM hydroxychloroquine,. The samples
were subsequently incubated for 4–18 h on ice. 3.6 μL of sample were applied to glow discharged holey carbon film grids (either Quantifoil 300 mesh, Au, R1.2/1.3, or R2/1, or C-flat, 200
mesh, Cu, 2/1) for 15 s at 10 °C and 100% humidity in a Vitrobot Mark IV (Thermo Fisher Scientific). Blotting was performed for 3.5 s at blot force -5 and with a drain time of 0.5 s. The
grids were plunged into liquid ethane/propane and transferred to liquid nitrogen for storage. Most data were collected in counting mode on a Titan Krios G3 (FEI) operating at 300 kV with a
BioQuantum imaging filter (Gatan) and a K3 direct detection camera (Gatan). Data were collected in fast acquisition mode by aberration-free image shift (AFIS) at 105,000× magnification, a
physical pixel size of 0.83 or 0.85 Å, a slit width of 20 eV and a total dose of 45-60 e-/Å2 (dose rate of ~15 e−/px/s, exposure time of ~2.4 s; for details, see Supplementary Table 1). The
nominal defocus was for most datasets was set to the range of -0.8 μM to -1.6 μm. The dataset for the hydroxychloroquine-bound hSLC19A3 was collected on a Titan Krios G4 equipped with a
cold-FEG, a Falcon 4i electron detector camera, and a SelectrisX imaging filter with its slit width set to 10 eV. The data were acquired at a nominal magnification of 215,000×, corresponding
to a pixel size of 0.572 Å. CRYO-EM DATA PROCESSING Processing of the acquired micrographs was performed using cryoSPARC (v. 4.4)57. Briefly, the collected movies were motion corrected
using patch motion correction. After patch CTF estimation, exposures with a CTF resolution <3.5 Å, a relative ice thickness <1.05 and a total frame motion distance <30 pixels were
selected for further processing. Particle picking was performed using template picker. The corresponding templates were generated from blob-picking (100 Å particle diameter) a small subset
of the data (200 - 600 micrographs), which was subjected to 2D classification, ab-initio reconstruction and non-uniform refinement (NU-refinement). After template-based particle picking of
the entire dataset, protein-containing and well-aligning particles were enriched using several rounds of 2D classification. The selected particles were then used in a sequence of ab-initio
reconstruction and NU-refinement to create a first higher resolution map, usually in the resolution range of 3.5-4.0 Å. This refined map was then, together with randomly generated volumes,
used for several rounds of heterogeneous refinement to retrieve more good particles and remove junk particles. The resulting map was then further refined using NU-refinement. The
corresponding particles were subsequently subjected to local motion correction and re-extracted with a larger box size of 384 pixels ( ~ 326 Å). After another round of NU-refinement, the
resulting density map was locally refined using a mask covering the transporter-nanobody complex. This was followed by local resolution estimation and local filtering to finalise the maps.
For more details, see Supplementary Fig. 6. Q-scores were determined using UCSF Chimera (v.1.18) and the MapQ plugin (v.1.9.14), developed by Grigore Pintilie58. MODEL BUILDING AND
REFINEMENT For starting models, AlphaFold2 predictions of hSLC19A3 and the nanobodies were used42. These were relaxed into the corresponding EM density maps using ISOLDE59. Ligands were
built and parametrised using the eLBOW package within PHENIX60 and integrated in the protein structures using Coot (v. 0.9.8.1)61. The resulting models were subsequently refined iteratively
in PHENIX and Coot. Figures were prepared using UCSF ChimeraX (v. 1.6.1)59. All reagents generated in this study are available from the Lead Contact with a completed Materials Transfer
Agreement. REPORTING SUMMARY Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article. DATA AVAILABILITY The EM data and fitted
models for hSLC19A3 generated in this study have been deposited in the Protein Data Bank (PDB) and the Electron Microscopy Data Bank (EMDB) under the following accession codes:
hSLC19A3-wt:Nb3.4-apo [https://doi.org/10.2210/pdb8S4U/pdb], [https://www.ebi.ac.uk/pdbe/entry/emdb/EMD-19716]; hSLC19A3-wt:Nb3.4:thiamine [https://doi.org/10.2210/pdb8S5U/pdb],
[https://www.ebi.ac.uk/pdbe/entry/emdb/EMD-19750]; hSLC19A3-wt:Nb3.3-apo [https://doi.org/10.2210/pdb9G5K/pdb], [https://www.ebi.ac.uk/pdbe/entry/emdb/EMD-51088]; hSLC19A3-gf:Nb3.7:thiamine
[https://doi.org/10.2210/pdb8S61/pdb], [https://www.ebi.ac.uk/pdbe/entry/emdb/EMD-19754]; hSLC19A3-gf:Nb3.7:Fedratinib [https://doi.org/10.2210/pdb8S5W/pdb],
[https://www.ebi.ac.uk/pdbe/entry/emdb/EMD-19752]; hSLC19A3-gf:Nb3.7:Amprolium [https://doi.org/10.2210/pdb8S62/pdb], [https://www.ebi.ac.uk/pdbe/entry/emdb/EMD-19755], hSLC19A3-gf:Nb3.7:
Hydroxychloroquine [https://doi.org/10.2210/pdb8S5Z/pdb], [https://www.ebi.ac.uk/pdbe/entry/emdb/EMD-19753]. Other data generated in this study are provided in the Supplementary Data 1 file
and Supplementary Movie 1, Supplementary Movie 2, and Supplementary Movie 3. Source Data are provided with this paper as source data file. Source data are provided with this paper.
REFERENCES * Bettendorff L. Thiamin. _Present Knowledge in Nutrition_. 261–279 (John Wiley & Sons, Inc., 2012). * Timm, D. E., Liu, J., Baker, L. J. & Harris, R. A. Crystal structure
of thiamin pyrophosphokinase. _J. Mol. Biol._ 310, 195–204 (2001). Article CAS PubMed Google Scholar * Baker, L. J., Dorocke, J. A., Harris, R. A. & Timm, D. E. The crystal
structure of yeast thiamin pyrophosphokinase. _Structure_ 9, 539–546 (2001). Article CAS PubMed Google Scholar * Tylicki, A., Lotowski, Z., Siemieniuk, M. & Ratkiewicz, A. Thiamine
and selected thiamine antivitamins — biological activity and methods of synthesis. _Biosci. Rep._ 38, 1–23 (2018). Article Google Scholar * Zhao, R. & Goldman, I. D. Folate and
thiamine transporters mediated by facilitative carriers (SLC19A1-3 and SLC46A1) and folate receptors. _Mol. Asp. Med._ 34, 373–385 (2013). Article Google Scholar * Dutta, B. et al. Cloning
of the human thiamine transporter, a member of the folate transporter family. _J. Biol. Chem._ 274, 31925–31929 (1999). Article CAS PubMed Google Scholar * Giacomini, M. M. et al.
Interaction of 2,4-diaminopyrimidine-containing drugs including fedratinib and trimethoprim with thiamine transporters. _Drug Metab. Dispos._ 45, 76–85 (2017). Article CAS PubMed Google
Scholar * Yamashiro, T., Yasujima, T., Said, H. M. & Yuasa, H. PH-dependent pyridoxine transport by SLC19A2 and SLC19A3: Implications for absorption in acidic microclimates. _J. Biol.
Chem._ 295, 16998–17008 (2020). Article CAS PubMed PubMed Central Google Scholar * Chen, L. et al. OCT1 is a high-capacity thiamine transporter that regulates hepatic steatosis and is a
target of metformin. _Proc. Natl Acad. Sci._ USA 111, 9983–9988 (2014). Article ADS CAS PubMed PubMed Central Google Scholar * Vora, B. et al. Drug-nutrient interactions: Discovering
prescription drug inhibitors of the thiamine transporter ThTR-2 (SLC19A3). _Am. J. Clin. Nutr._ 111, 110–121 (2020). Article PubMed Google Scholar * Wen, A. et al. The Impacts of Slc19a3
Deletion and Intestinal SLC19A3 Insertion on Thiamine Distribution and Brain Metabolism in the Mouse. _Metabolites_ 13, 885 (2023). Article CAS PubMed PubMed Central Google Scholar *
Reidling, J. C., Lambrecht, N., Kassir, M. & Said, H. M. Impaired Intestinal Vitamin B1 (Thiamin) Uptake in Thiamin Transporter-2-Deficient Mice. _Gastroenterology_ 138, 1802–1809
(2010). Article CAS PubMed Google Scholar * Kato, K. et al. Involvement of organic cation transporters in the clearance and milk secretion of thiamine in mice. _Pharm. Res._ 32,
2192–2204 (2015). Article CAS PubMed Google Scholar * Kono, S. et al. Mutations in a thiamine-transporter gene and wernicke’s-like encephalopathy. _N. Engl. J. Med._ 360, 1792–1794
(2009). Article CAS PubMed Google Scholar * Wang, J. et al. Report of the largest Chinese cohort with SLC19A3 gene defect and literature review. _Front. Genet._ 12, 1–9 (2021). Google
Scholar * Zeng, W. Q. et al. Biotin-responsive basal ganglia disease maps to 2q36.3 and is due to mutations in SLC19A3. _Am. J. Hum. Genet._ 77, 16–26 (2005). Article CAS PubMed PubMed
Central Google Scholar * Alfadhel, M. et al. Targeted SLC19A3 gene sequencing of 3000 Saudi newborn: a pilot study toward newborn screening. _Ann. Clin. Transl. Neurol._ 6, 2097–2103
(2019). Article CAS PubMed PubMed Central Google Scholar * Pardanani, A. et al. Safety and efficacy of fedratinib in patients with primary or secondary myelofibrosis: A randomized
clinical trial. _JAMA Oncol._ 1, 643–651 (2015). Article PubMed Google Scholar * Zhang, Q. et al. The Janus kinase 2 inhibitor fedratinib inhibits thiamine uptake: A putative mechanism
for the onset of Wernicke’s encephalopathy. _Drug Metab. Dispos._ 42, 1656–1662 (2014). Article PubMed Google Scholar * Quistgaard, E. M., Löw, C., Guettou, F. & Nordlund, P.
Understanding transport by the major facilitator superfamily (MFS): Structures pave the way. _Nat. Rev. Mol. Cell Biol._ 17, 123–132 (2016). Article CAS PubMed Google Scholar * Drew, D.,
North, R. A., Nagarathinam, K. & Tanabe, M. Structures and general transport mechanisms by the major facilitator superfamily (MFS). _Chem. Rev._ 121, 5289–5335 (2021). Article CAS
PubMed PubMed Central Google Scholar * Hou, Z. & Matherly, L. H. Biology of the major facilitative folate transporters SLC19A1 and SLC46A1. _Curr. Top. Membr._ 73, 175–204 (2014).
Article CAS PubMed PubMed Central Google Scholar * Said, H. M., Balamurugan, K., Subramanian, V. S. & Marchant, J. S. Expression and functional contribution of hTHTR-2 in thiamin
absorption in human intestine. _Am. J. Physiol. - Gastrointest. Liver Physiol._ 286, 491–498 (2004). Article Google Scholar * Liang, X. et al. Metformin is a substrate and inhibitor of the
human thiamine transporter, THTR-2 (SLC19A3). _Mol. Pharm._ 12, 4301–4310 (2015). Article CAS PubMed PubMed Central Google Scholar * Subramanian, V. S., Marchant, J. S. & Said, H.
M. Biotin-responsive basal ganglia disease-linked mutations inhibit thiamine transport via hTHTR2: Biotin is not a substrate for hTHTR2. _Am. J. Physiol. - Cell Physiol._ 291, 851–859
(2006). Article Google Scholar * Rajgopal, A., Edmondnson, A., Goldman, I. D. & Zhao, R. SLC19A3 encodes a second thiamine transporter ThTr2. _Biochim. Biophys. Acta - Mol. Basis Dis._
1537, 175–178 (2001). Article CAS Google Scholar * Miyake, K., Yasujima, T., Takahashi, S., Yamashiro, T. & Yuasa, H. Identification of the amino acid residues involved in the
species-dependent differences in the pyridoxine transport function of SLC19A3. _J. Biol. Chem._ 298, 102161 (2022). Article CAS PubMed PubMed Central Google Scholar * Pardon, E. et al.
A general protocol for the generation of Nanobodies for structural biology. _Nat. Protoc._ 9, 674–693 (2014). Article CAS PubMed PubMed Central Google Scholar * Zeng, Y. C. et al.
Structural basis of promiscuous substrate transport by Organic Cation Transporter https://doi.org/10.1038/s41467-023-42086-9 (2023). * Zhang, Q. et al. Recognition of cyclic dinucleotides
and folates by human SLC19A1. _Nature_ 612, 170–176 (2022). Article ADS CAS PubMed Google Scholar * Wright, N. J. et al. Methotrexate recognition by the human reduced folate carrier
SLC19A1. _Nature_ 609, 1056–1062 (2022). Article ADS CAS PubMed PubMed Central Google Scholar * Dang, Y. et al. Molecular mechanism of substrate recognition by folate transporter
SLC19A1. _Cell Discov._ 8, 141 (2022). Article CAS PubMed PubMed Central Google Scholar * Drew, D. & Boudker, O. Shared molecular mechanisms of membrane transporters. _Annu. Rev.
Biochem._ 85, 543–572 (2016). Article CAS PubMed Google Scholar * Harris, T. K. & Mildvan, A. S. High-precision measurement of hydrogen bond lengths in proteins by nuclear magnetic
resonance methods. _Proteins Struct. Funct. Genet._ 35, 275–282 (1999). Article CAS PubMed Google Scholar * Bas, D. C., Rogers, D. M. & Jensen, J. H. Very fast prediction and
rationalization of pKa values for protein-ligand complexes. _Proteins Struct. Funct. Genet._ 73, 765–783 (2008). Article CAS PubMed Google Scholar * Dang, Y. et al. Substrate and drug
recognition mechanisms of SLC19A3. _Cell Res._ 34, 458–461 (2024). Article PubMed Google Scholar * Qu, Q. et al. Preprint: Substrate transport and drug interaction of human thiamine
transporters SLC19A2 / A3. _Res. Sq._ 1, 25 (2024). Google Scholar * Zhao, R., Assaraf, Y. G. & Goldman, I. D. A mutated murine reduced folate carrier (RFC1) with increased affinity for
folic acid, decreased affinity for methotrexate, and an obligatory anion requirement for transport function. _J. Biol. Chem._ 273, 19065–19071 (1998). Article CAS PubMed Google Scholar
* Yee, S. W. et al. SLC19A1 pharmacogenomics summary. _Pharmacogenet Genomics_ 20, 708–715 (2010). Article CAS PubMed PubMed Central Google Scholar * Labay, V. et al. Mutations in
SLC19A2 cause thiamine-responsive megaloblastic anaemia associated with diabetes mellitus and deafness. _Nat. Genet._ 22, 300–304 (1999). Article CAS PubMed Google Scholar * Liberman, M.
C., Tartaglini, E., Fleming, J. C. & Neufeld, E. J. Deletion of SLC19A2, the high affinity thiamine transporter, causes selective inner hair cell loss and an auditory neuropathy
phenotype. _JARO - J. Assoc. Res. Otolaryngol._ 7, 211–217 (2006). Article CAS PubMed Google Scholar * Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold.
_Nature_ 596, 583–589 (2021). Article ADS CAS PubMed PubMed Central Google Scholar * Ozand, P. T. et al. Biotin-responsive basal ganglia disease: A novel entity. _Brain_ 121, 1267–1279
(1998). Article PubMed Google Scholar * Aburezq, M. et al. Biotin-thiamine responsive basal ganglia disease: a retrospective review of the clinical, radiological and molecular findings
of cases in Kuwait with novel variants. _Orphanet J. Rare Dis._ 18, 1–13 (2023). Article Google Scholar * Wesół-Kucharska, D. et al. Early treatment of biotin–thiamine–responsive basal
ganglia disease improves the prognosis. _Mol. Genet. Metab. Rep._ 29, 100801 (2021). Article PubMed PubMed Central Google Scholar * Kevelam, S. H. et al. Exome sequencing reveals mutated
SLC19A3 in patients with an early-infantile, lethal encephalopathy. _Brain_ 136, 1534–1543 (2013). Article PubMed Google Scholar * Rose-Sperling, D., Tran, M. A., Lauth, L. M., Goretzki,
B. & Hellmich, U. A. 19 F NMR as a versatile tool to study (membrane) protein structure and dynamics. _Biol. Chem._ 400, 1277–1288 (2019). Article CAS PubMed Google Scholar *
Aljabri, M. F., Kamal, N. M., Arif, M., AlQaedi, A. M. & Santali, E. Y. M. A case report of biotin–thiamine-responsive basal ganglia disease in a Saudi child. _Med. (Baltim.)_ 95, e4819
(2016). Article CAS Google Scholar * Schänzer, A. et al. Stress-induced upregulation of SLC19A3 is impaired in biotin-thiamine- responsive basal ganglia disease. _Brain Pathol._ 24,
270–279 (2014). Article PubMed PubMed Central Google Scholar * Dudeja, P. K., Tyagi, S., Kavilaveettil, R. J., Gill, R. & Said, H. M. Mechanism of thiamine uptake by human jejunal
brush-border membrane vesicles. _Am. J. Physiol. - Cell Physiol._ 281, 786–792 (2001). Article Google Scholar * Galetin, A. et al. Membrane transporters in drug development and as
determinants of precision medicine. _Nat. Rev. Drug Discov._ 23, 255–280 (2024). Article CAS PubMed Google Scholar * Backliwal, G. et al. Rational vector design and multi-pathway
modulation of HEK 293E cells yield recombinant antibody titers exceeding 1 g/l by transient transfection under serum-free conditions. _Nucleic Acids Res._ 36, e96 (2008). Article PubMed
PubMed Central Google Scholar * Zhang, Y., Uwe Werling, W. & Edelmann, W. Seamless ligation cloning extract (SLiCE) cloning method Yongwei. _Methods Mol. Biol._ 1116, 235–244 (2014).
Article CAS PubMed PubMed Central Google Scholar * Pieprzyk, J., Pazicky, S. & Löw, C. Transient expression of recombinant membrane-eGFP fusion proteins in HEK293 cells. _Methods
Mol. Biol._ 1850, 17–31 (2018). Article CAS PubMed Google Scholar * Kotov, V. et al. Plasticity of the binding pocket in peptide transporters underpins promiscuous substrate recognition.
_Cell Rep._ 42, 112831 (2023). Article CAS PubMed Google Scholar * Hall, J. A simple model for determining affinity from irreversible thermal shifts. _Protein Sci._ 28, 1880–1887
(2019). Article CAS PubMed PubMed Central Google Scholar * Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. CryoSPARC: Algorithms for rapid unsupervised cryo-EM
structure determination. _Nat. Methods_ 14, 290–296 (2017). Article CAS PubMed Google Scholar * Pintilie, G. et al. Measurement of atom resolvability in cryo-EM maps with Q-scores. _Nat.
Methods_ 17, 328–334 (2020). Article CAS PubMed PubMed Central Google Scholar * Pettersen, E. F. et al. UCSF ChimeraX: Structure visualization for researchers, educators, and
developers. _Protein Sci._ 30, 70–82 (2021). Article CAS PubMed Google Scholar * Afonine, P. V. et al. Real-space refinement in PHENIX for cryo-EM and crystallography. _Acta Crystallogr.
Sect. D. Struct. Biol._ 74, 531–544 (2018). Article ADS CAS Google Scholar * Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. _Acta Crystallogr.
Sect. D. Biol. Crystallogr._ 66, 486–501 (2010). Article ADS CAS Google Scholar Download references ACKNOWLEDGEMENTS We thank the Sample Preparation and Characterization facility at EMBL
(Hamburg, Germany) and the team of the cryo-EM Facility at CSSB for their support, technical assistance and advice. We want to acknowledge Tânia Custódio, Grzegorz Chojnowski and all group
members for fruitful discussions and continuous support and feedback on the project. Part of this work was performed at the CryoEM Facility at CSSB, supported by the UHH and DFG (grant
numbers INST 152/772-1 | 152/774-1 | 152/775-1 | 152/776-1 | 152/777-1 FUGG). We further acknowledge the access and services provided by the Imaging Centre at the European Molecular Biology
Laboratory (EMBL IC) in Heidelberg, generously supported by the Boehringer Ingelheim Foundation. In this context, we want to thank Joseph Bartho for his help with the cryo-EM data
acquisitions at the Imaging Centre. We want to acknowledge the support of the EMBL Metabolomics Core Facility (MCF), and in particular the help of Bernhard Drotleff, in the acquisition and
analysis of liquid chromatography-mass spectrometry data. Nanobody discovery was realised through the Nanobodies4Instruct centre and the support by Instruct-ERIC (PID: 23688), which is part
of the European Strategy Forum on Research Infrastructures (ESFRI). We want to thank in particular Alison Pirro Lundqvist and Eva Beke from the Steyaert Lab at VUB for their technical
assistance during nanobody discovery. The research-stay of F.G. at VUB was supported by an EMBO Scientific Exchange Grand (Nr. 10251). FUNDING Open Access funding enabled and organized by
Projekt DEAL. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Centre for Structural Systems Biology (CSSB), Notkestraße 85, 22607, Hamburg, Germany Florian Gabriel, Lea Spriestersbach, Antonia
Fuhrmann, Katharina E. J. Jungnickel, Siavash Mostafavi & Christian Löw * European Molecular Biology Laboratory (EMBL) Hamburg, Notkestraße 85, 22607, Hamburg, Germany Florian Gabriel,
Lea Spriestersbach, Antonia Fuhrmann, Katharina E. J. Jungnickel, Siavash Mostafavi & Christian Löw * Structural Biology Brussels, Vrije Universiteit Brussel (VUB), 1050, Brussels,
Belgium Els Pardon & Jan Steyaert * VIB-VUB Center for Structural Biology, VIB, 1050, Brussels, Belgium Els Pardon & Jan Steyaert * Bernhard Nocht Institute for Tropical Medicine,
20359, Hamburg, Germany Christian Löw Authors * Florian Gabriel View author publications You can also search for this author inPubMed Google Scholar * Lea Spriestersbach View author
publications You can also search for this author inPubMed Google Scholar * Antonia Fuhrmann View author publications You can also search for this author inPubMed Google Scholar * Katharina
E. J. Jungnickel View author publications You can also search for this author inPubMed Google Scholar * Siavash Mostafavi View author publications You can also search for this author
inPubMed Google Scholar * Els Pardon View author publications You can also search for this author inPubMed Google Scholar * Jan Steyaert View author publications You can also search for this
author inPubMed Google Scholar * Christian Löw View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS F.G. conceptualised the project and
experiments, cloned and expressed transporter constructs and nanobodies, performed the nanobody discovery, performed and analysed nanoDSF and BLI experiments, collected and processed all
cryo-EM data and prepared the manuscript. L.S. and A.F. cloned and expressed transporter constructs, as well as nanobodies, and collected biophysical data. K.E.J.J. and S.M. helped with
experiment design, as well as cryo-EM data processing, structure refinement and manuscript writing. J.S and E.P. immunised llamas and generated nanobody libraries for the discovery process.
C.L. was responsible for the overall project administration, experiment design, funding acquisition, and for reviewing and editing of the manuscript. CORRESPONDING AUTHOR Correspondence to
Christian Löw. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Nature Communications_ thanks Devanshu Kurre and the
other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral
with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION PEER REVIEW FILE DESCRIPTION OF ADDITIONAL
SUPPLEMENTARY FILES SUPPLEMENTARY MOVIE 1 SUPPLEMENTARY MOVIE 2 SUPPLEMENTARY MOVIE 3 SUPPLEMENTARY DATA 1 REPORTING SUMMARY SOURCE DATA SOURCE DATA RIGHTS AND PERMISSIONS OPEN ACCESS This
article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as
you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party
material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the
article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the
copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Gabriel, F.,
Spriestersbach, L., Fuhrmann, A. _et al._ Structural basis of thiamine transport and drug recognition by SLC19A3. _Nat Commun_ 15, 8542 (2024). https://doi.org/10.1038/s41467-024-52872-8
Download citation * Received: 02 April 2024 * Accepted: 20 September 2024 * Published: 02 October 2024 * DOI: https://doi.org/10.1038/s41467-024-52872-8 SHARE THIS ARTICLE Anyone you share
the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer
Nature SharedIt content-sharing initiative