Play all audios:
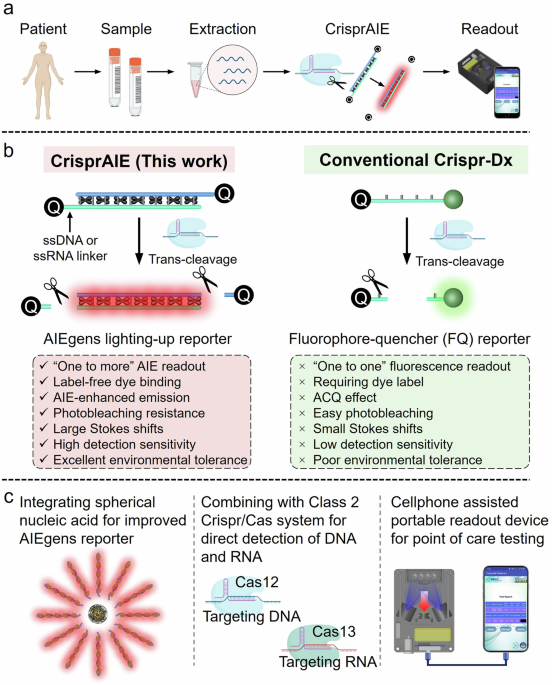
ABSTRACT CRISPR diagnostics are effective but suffer from low signal transduction efficiency, limited sensitivity, and poor stability due to their reliance on the trans-cleavage of
single-stranded nucleic acid fluorescent reporters. Here, we present CrisprAIE, which integrates CRISPR/Cas reactions with “one to more” aggregation-induced emission luminogen (AIEgen)
lighting-up fluorescence generated by the trans-cleavage of Cas proteins to AIEgen-incorporated double-stranded DNA labeled with single-stranded nucleic acid linkers and Black Hole Quencher
groups at both ends (Q-dsDNA/AIEgens-Q). CrisprAIE demonstrates superior performance in the clinical nucleic acid detection of norovirus and SARS-CoV-2 regardless of amplification. Moreover,
the diagnostic potential of CrisprAIE is further enhanced by integrating it with spherical nucleic acid-modified AIEgens (SNA/AIEgens) and a portable cellphone-based readout device. The
improved CrisprAIE system, utilizing Q-dsDNA/AIEgen-Q and SNA/AIEgen reporters, exhibits approximately 80- and 270-fold improvements in sensitivity, respectively, compared to conventional
CRISPR-based diagnostics. We believe CrisprAIE can be readily extended as a universal signal generation strategy to significantly enhance the detection efficiency of almost all existing
CRISPR-based diagnostics. SIMILAR CONTENT BEING VIEWED BY OTHERS ACCELERATED RNA DETECTION USING TANDEM CRISPR NUCLEASES Article 05 August 2021 DETECTION OF VIRAL RNAS AT AMBIENT TEMPERATURE
VIA REPORTER PROTEINS PRODUCED THROUGH THE TARGET-SPLINTED LIGATION OF DNA PROBES Article Open access 04 May 2023 ENGINEERED LWACAS13A WITH ENHANCED COLLATERAL ACTIVITY FOR NUCLEIC ACID
DETECTION Article 22 September 2022 INTRODUCTION The quick and accurate detection of nucleic acid biomarkers in complex biological systems is critical for the diagnosis and monitoring of
infectious diseases1,2. Real-time quantitative polymerase chain reaction (qPCR) and sequencing technologies have been clinically recognized as cutting-edge detection tools for nucleic acid
targets3,4. However, these techniques relying on target amplification are laborious, expensive, and time consuming and require complex multiple reagent reactions, trained technicians, and
sophisticated laboratory instrument. Despite the remarkable achievement in isothermal nucleic acid amplification, which effectively eliminates the thermal cycle, nonspecific amplification
remains a major challenge for these amplification-dependent techniques due to their low targeting specificity and high false-positive results5,6,7. As a natural adaptive immune system in
microorganisms, the clustered regularly interspaced short palindromic repeat (Crispr) and Crispr-associated (Cas) protein system has been skillfully integrated with various nucleic acid
amplification techniques to improve the sensitivity and specificity of traditional nucleic acid diagnostic techniques. It specifically targets nucleic acid sequences and can perform the
efficient nuclease-like collateral cleavage of ambient nontargeted single-stranded nucleic acids. With this design concept, several Crispr/Cas-based diagnostic methods (Crispr-Dx) combining
the advantages of Cas reactions and isothermal amplification have been developed as highly specific nucleic acid detection tools that can measure the collateral cleavage of single-stranded
nucleic acid probes after target binding8,9. The currently available Crispr-Dx assays mainly include Specific High-sensitivity Enzymatic Reporter unlocking (SHERLOCK)10,11, DNA
Endonuclease-Targeted Crispr Trans Reporter (DETECTOR)12,13, and one-Hour Low-Cost Multipurpose Highly Efficient System (HOLMES)14. These assays employ Cas12 or Cas13 effectors to trigger
single-stranded DNA (ssDNA)/RNA (ssRNA) cleavage and induce efficient signal readout for various nucleic acid targets, including Zika virus10, dengue virus10, human papillomavirus (HPV)12,
and single-nucleotide polymorphism14. Despite these achievements, the signal generation of these Crispr-Dx assays mainly rely on the trans-cleavage activity of Cas proteins on linear
single-stranded nucleic acid reporters and still has some problems, such as low signal transduction efficiency, limited detection sensitivity, and poor stability in complex biological
systems. An important strategy to improve the detection performance of Crispr-Dx systems is by evolving the signal-generation mechanism and modality of Cas reaction. Increasing attempts have
been directed to combine Cas reaction with a variety of enhanced signal readout alternatives, including electrochemistry15,16, electrochemiluminescence17, surface-enhanced Raman
scattering18,19, surface plasmon resonance20,21, and field-effect transistor22,23, to achieve efficient target detection. Nevertheless, fluorescence-based signal readouts are still
extensively investigated due to their integration and miniaturization potential. The majority of Crispr-Dx assays utilize the activated trans-cleavage activity to generate a detectable
fluorescent signal through the nonspecific cleavage of traditional synthetic fluorophore-quencher (FQ) pairs bound to the end of short ssDNA or ssRNA24,25,26. However, this signal
illuminating behavior usually suffers from insufficient sensitivity because one FQ reporter cleaved by activated Cas nuclease only can liberate one fluorescent signal molecule. Therefore,
breaking the limitations of existing fluorescent transduction systems will help promote the rapid development and application of Crispr-Dx. A preferable alternative to traditional FQ-based
reporter with “one to more” characteristic, that is, one trans-cleavage of FQ reporter can output many fluorescent signal molecules, is urgently needed to improve the existing Crispr-Dx.
Leveraging the amplification mechanism described above, we speculate that one promising fluorescent readout system for Crispr-Dx is aggregation-induced emission luminogens (AIEgens)27. As a
type of organic fluorescence dye, AIEgens exhibit many unique advantages over traditional aggregation-caused quenching probes, including enhanced aggregation emission, high signal-to-noise
ratio (S/N), excellent photostability, and large Stokes shift28,29. These characteristics make AIEgens a promising candidate for reducing the detection limit (LOD) and improving the
stability of Crispr-Dx. Owing to the positively charged AIEgens spontaneously bind to negatively charged nucleic acid chains through noncovalent interactions (e.g., van der Waals, π-π
stacking, and hydrogen bonding interactions) and electrostatic interactions, these AIEgens can light-up their fluorescence signals via the restriction of intramolecular motion (RIM)30,31. In
theory, a long nucleic acid chain can provide many negatively charged sites, endowing additional opportunities for AIEgen binding and generating intense fluorescence emission, which offer
the possibility of creating “one to more” signal reporters for amplifying Crispr/Cas detection. Here, we report the development of CrisprAIE, the combination of a Crispr/Cas reaction with a
“one to more” AIEgen lighting-up fluorescence readout (Fig. 1). The “one to more” AIEgen lighting-up readout is achieved using AIEgen-loaded double-stranded DNA (dsDNA) reporters, which are
synthesized by incorporating cationic AIEgens into the grooves of dsDNA. To enable the Cas protein to cleave the AIEgen-loaded dsDNA reporters and achieve the lighting-up fluorescence
readout, we introduce ssDNA or ssRNA linkers at both ends of dsDNA to bridge the Black Hole Quencher (BHQ) for producing AIEgen-enhanced CrisprAIE reporters (Q-dsDNA/AIEgens-Q). After
carefully exploring the factors affecting the fluorescence transduction efficiency of AIEgens, we find that the resulting CrisprAIE shows a LOD of femtomolar concentrations of at least 80
times more sensitive than the traditional FQ reporter-based Crispr-Dx without amplification. This LOD can be further reduced to attomolar levels through isothermal amplification. The
clinical diagnostic efficiency of our CrisprAIE is demonstrated by detecting norovirus and SARS-CoV-2 virus in their respective diagnostic samples. To further improve the sensitivity and
stability of our CrisprAIE, we develop AIEgen-enhanced spherical nucleic acid (SNA/AIEgen) reporters and demonstrate their potential in improving the nucleic acid diagnostic capabilities of
CrisprAIE in complex biological systems. We also integrate CrisprAIE assay with a portable cellphone-assisted fluorescence readout device for point-of-care (POC) diagnostic application and
evaluate its detection performance and consistency with commercial multimode fluorescence microplate readers. We believe that this AIEgen lighting-up reporting probe can serve as an
attractive alternative to traditional FQ reporter, and the developed CrisprAIE can be readily extended for improving the detection efficiency of almost all existing Crispr-Dx assays based on
single-stranded nucleic acid reporters. RESULTS CRISPR-DRIVEN AIEGEN LIGHTING-UP READOUT Cationic AIEgens can interact with negatively charged DNA strands through electrostatic or
noncovalent interactions, thereby enabling lighting-up fluorescence readout by activating the RIM. As a typical cationic AIEgen,
4ʹ,4ʹʹ,4ʹʹʹ-(((((1E,1ʹE)-benzo[c][1,2,5]thiadiazole-4,7-diylbis(ethene-2,1-diyl))bis(4,1-phenylene))bis(azanetriyl))tetrakis(benzene-4,1-diyl))tetrakis(1-methylpyridin-1-ium) iodide (TPBT)
with four positively charged pyridine groups is used for the label-free fluorescent detection of dsDNA and single-nucleotide polymorphisms30. The molecular structure of TPBT was confirmed by
1H NMR, 13C NMR, and high-resolution mass spectra (Supplementary Figs. 1–3), and its AIE feature was further characterized in Supplementary Fig. 4. Compared with FAM, a widely used
fluorophore, TPBT has a larger Stokes shift and better resistance to photobleaching (Fig. 2a, b) that are conducive to sensitive and accurate detection. Considering the preference of the
trans-cleavage activity of Cas nuclease for single-stranded oligonucleotides, we first designed two ssDNA-modified AIEgen reporters, namely, AIEgen-modified ssDNA (ssDNA/AIEgens) and
AIEgen-modified ssDNA labeled with a single-terminal quencher group BHQ by a five-base ssDNA linker (Q-ssDNA/AIEgens). Compared with the flexible ssDNA, the rigid dsDNA can more effectively
restrict the free intramolecular motion of the incorporated AIEgens, thereby facilitating enhanced fluorescent signal output. We then constructed two AIEgen-modified dsDNA reporters, namely,
AIEgen-modified dsDNA labeled with a single-terminal quencher group BHQ by a five-base ssDNA linker (Q-dsDNA/AIEgens) and AIEgen-modified dsDNA labeled with a double-terminal quencher group
BHQ by a five-base ssDNA linker (Q-dsDNA/AIEgens-Q). A traditional FQ reporter, denoted as Q-ssDNA-FAM, was also commercially synthesized for comparison. The related sequence is summarized
in Supplementary Table 1. S1 nuclease, a single-stranded specific endonuclease that hydrolyzes ssDNA into single nucleotides, was selected as an alternative of activated Cas nuclease to
study the feasibility of nuclease-driving AIEgen lighting-up readout. As shown in Fig. 2c, the presence of S1 nuclease led to the significant fluorescence recovery of the Q-ssDNA-FAM
reporter, accompanied by the departure of the quencher group (BHQ). This finding confirmed the validity of S1 nuclease against ssDNA degradation. The ssDNA/AIEgen probe showed significantly
increased fluorescence signals due to the activation of RIM when TPBT bound to ssDNA. The ssDNA part of the composite probe can be digested by S1 nuclease, thus decreasing its fluorescence
to achieve the “ON-OFF” signal output. By contrast, the Q-ssDNA/AIEgen reporter displayed fluorescent signal recovery in the presence of S1 nuclease, thus favoring the “OFF-ON” signal output
(Fig. 2c). When TPBT was incubated with dsDNA, the fluorescence signal of TPBT was markedly increased. Its fluorescence can be effectively quenched by labeling the BHQ quenching groups at
one or both ends of dsDNA (Fig. 2d). S1 nuclease can induce BHQ detachment by digesting the ssDNA linker, resulting in significant fluorescence enhancement to obtain the “OFF-ON” signal
output. These results demonstrated that the designed DNA-modified AIEgen probes are suitable as the fluorescent signal reporters for nuclease-based assays, including Crispr/Cas-based
detection. Encouraged by this finding, we reported the development of CrisprAIE by combining a Crispr/Cas-assisted nucleic acid cleavage reactions with DNA-modified AIEgen lighting-up
readout. The effect of the length and base type of DNA sequence, ionic strength, and AIEgen concentration on the fluorescent properties of the resulting ssDNA-modified AIEgen reporters was
studied to obtain the best binding between TPBT and DNA oligonucleotides and the enhanced fluorescence of ssDNA-modified AIEgen probes for the fabrication of optimal ssDNA/AIEgen and
Q-ssDNA/AIEgen reporters (Fig. 2e, f). As revealed in Supplementary Fig. 5, the ssDNA (A10 or T10) containing 10 repeating units of adenine (A) or thymine (T) obtained higher fluorescence
signal response than that containing cytosine (C) and guanine (G) bases in the presence of 100 mM NaCl and 2.5 µM TPBT. In particular, the enhanced fluorescence of the A10 was the highest
among them. One possible reason is that the stronger intermolecular interaction of TPBT with A base than with T, C, and G bases contributes to restricting the intramolecular motion of TPBT
and boosting the fluorescent emission. Further DNA sequence length analysis showed that ssDNA (A15) could produce suitable relative fluorescence intensity, highest fluorescence decay rate,
and short cleavage equilibrium time (Supplementary Figs. 6, 7). On the basis of the above results, the A15 and A-T base pairs were selected as the building blocks for fabricating
DNA-modified AIEgen reporters and achieving the lowest LOD of CrisprAIE. Under the developed conditions, we first fabricated the CrisprAIE assay using ssDNA/AIEgens and Q-ssDNA/AIEgens as
signal reporters (Fig. 2e, f). The LOD was determined as the mean plus three times the standard deviation of the measured concentration, derived from twenty repeated measurements of the
blank sample, following a previously reported method32. Figure 2i shows that the LOD of this ssDNA/AIEgens-based CrisprAIE was 12 pM, which was close to that of conventional Crispr-Dx (LOD:
15 pM) based on the Q-ssDNA-FAM reporter (Supplementary Fig. 8). Moreover, we observed that when Q-ssDNA/AIEgens were used as CrisprAIE signal reporters, the sensitivity was further reduced
with a LOD of 195 pM (Fig. 2j, Supplementary Fig. 9). The possible reason for the low sensitivity of these two CrisprAIE assays is that multiple independent nuclease cleavages are required
to fully release AIEgens from the composite ssDNA/AIEgen reporters to achieve efficient fluorescence transduction. In addition, the binding of AIEgens with ssDNA resulted in limited RIM and
reduced fluorescence enhancement, thus yielding low S/N ratio. Compared with ssDNA, dsDNA can better restrict the intramolecular movement of AIEgens by binding AIEgens to the major or minor
grooves of dsDNA, thus improving the sensitivity of DNA-lighting-up AIEgen probes. On this basis, two AIEgen-modified dsDNA probes, namely, Q-dsDNA/AIEgens and Q-dsDNA/AIEgens-Q, were
designed and proposed for enhancing the sensitivity of CrisprAIE. As revealed in Fig. 2g, h and Supplementary Fig. 10, Q-dsDNA/AIEgens consisted of a dsDNA containing multiple repeating
units of A and T for AIEgen loading, an ssDNA linker for Crispr/Cas cleavage, and a BHQ group for quenching the fluorescence of AIEgens bound to dsDNA. Meanwhile, Q-dsDNA/AIEgens-Q consisted
of a dsDNA containing multiple repeating A-T base pairs, two ssDNA linkers, and two BHQ groups at both ends. Q-dsDNA/AIEgen and Q-dsDNA/AIEgen-Q reporters were separated and purified by
agarose gel electrophoresis (Supplementary Figs. 12). To achieve the highest S/N ratio, we studied the effect of the composition and length of dsDNA and TPBT concentration on the groove
binding between dsDNA and TPBT. Supplementary Fig. 11 implies that the optimal combination for Q-dsDNA/AIEgens was A15T15 of dsDNA and 5 μM of TPBT, resulting in the highest S/N ratio due to
the high load efficiency of TPBT (the binding number was 11.8 TPBT molecules per A15T15 dsDNA) and the efficient quenching of the entire fluorescence of TPBT binding on dsDNA by BHQ. The
length of dsDNA was too long or too short, resulting in a relatively low S/N ratio due to the insufficient AIEgen loading and incomplete fluorescence quenching. Further molecular simulation
calculations between TPBT and dsDNA were performed to simulate the interaction. The docking results in Supplementary Fig. 12 show that compared with those in dsDNA with C-G repeating units,
the benzothiadiazole core and the two vinylene bridges on TPBT were perfectly inserted into the minor grooves of dsDNA containing A-T base pairs. This finding suggested that the dsDNA
consisting of repeating units of A and T can restrict the intramolecular motion of TPBT to enhance fluorescence emission by blocking the nonradiative decay. These observations are consistent
with the results from ssDNA, where the A and T bases were superior to the C and G bases in restricting the molecular motion of TPBT and enhancing the fluorescence. Considering that the
optimal dsDNA length for individual BHQ group quenching was 15 repeating A-T base pairs, we synthesized Q-dsDNA/AIEgens-Q by incorporating TPBT (5 μM) with A30T30 of dsDNA. Prior to the
Crispr detection, we monitored the double-stranded structure integrity of Q-dsDNA/AIEgens-Q in the presence of activated Cas12 protein using gel electrophoresis. The results in Supplementary
Fig. 13 demonstrated that the ssDNA component was completely cleaved by activated Cas12 protein, whereas the dsDNA segment remained intact even after trans-cleavage, which is crucial for
achieving enhanced AIE fluorescence signal output. We then measured the concentration of synthetic DNA by incorporating Q-dsDNA/AIEgens and Q-dsDNA/AIEgens-Q into Crispr/Cas12-based
reaction. With the optimized assay design, the LOD of CrisprAIE based on Q-dsDNA/AIEgen reporters was 0.37 pM of synthetic DNA (Fig. 2k). With the use of Q-dsDNA/AIEgen-Q reporters, this
value can be further reduced to 0.18 pM, which is about 80 folds lower than that of conventional Q-ssDNA-FAM-based Crispr-Dx (Fig. 2l). This dramatically reduced LOD is superior or
comparable with those of other reported amplification-free Crispr/Cas strategies involving electrochemical15 and nanozyme-catalyzed readout33. We speculated that the high sensitivity of
CrisprAIE may be due to the increased signal output capacity caused by loading additional AIEgens onto dsDNA, enhanced AIE fluorescence by dsDNA-caused efficient intramolecular motion
restriction, large Stokes shift, high extinction coefficient, “one to more” signal amplification, and superior S/N ratio (Fig. 2a and Supplementary Fig. 14). Subsequently, we evaluated the
long-stem storage stability and repeatability of our CrisprAIE over 21 consecutive days by freeze-drying the Q-dsDNA/AIEgen-Q reporter. Supplementary Fig. 15 indicates that the developed
AIEgen reporter maintains excellent stability and reproducibility under lyophilized conditions, showing compatibility with the current freeze-dried CRISPR reagent. Two other AIEgens with
excellent binding ability to DNA strands, namely, TPE-1OH and TPA-1OH31 (Supplementary Figs. 16–22), were introduced to replace TPBT for constructing CrisprAIE to broaden the optional
ability of AIEgen toolbox. Supplementary Figs. 23, 24 verify the versatility and feasibility of CrisprAIE for the detection of synthetic DNA targets with acceptable analytical performance.
This optimized Q-dsDNA/AIEgens-Q reporter was used in the following experiments unless otherwise indicated. RNA QUANTIFICATION USING CRISPRAIE COMBINED WITH RT-RPA The improvement in the
detection performance of CrisprAIE encouraged us to investigate its real-world application. For the sensitive detection and quantification of target RNA, we first combined the
Q-dsDNA/AIEgen-Q lighting-up fluorescence reporting strategy with Crispr/Cas12-based viral RNA detection. This assay design performs simultaneous reverse transcription and isothermal
amplification using recombinase polymerase amplification (RT-RPA) for the RNA extracted from actual samples, such as vomitus or nasopharyngeal swabs, in universal transport medium, followed
by the Cas12 detection of target RNA sequences. The cleavage of an AIEgen reporter verifies the presence of the virus. If the target RNA is present in the reaction system, then the
Cas12a/crRNA binary complex forms a ternary complex with the reverse-transcribed amplicons, which then trans-cleave nontargeted ssDNA linker in the Q-dsDNA/AIEgens-Q to light up AIEgen
fluorescence. With this design concept, we developed a single-tube isothermal CrisprAIE assay for the rapid and accurate detection of target RNA (Fig. 3a). As a proof of concept for
potential applications, we first applied this CrisprAIE assay for the detection of norovirus from clinical vomit RNA extracts. The primers targeting the ORF2 gene of norovirus and the crRNA
sequence were designed as summarized in Supplementary Table 2. To obtain the best analytical performance, we optimized the detection conditions of norovirus ORF2 gene by CrisprAIE assay,
including RT-RPA reaction at 37 °C for 15 min and Cas12 detection reaction at 37 °C for 15 min. Figure 3b displays that only the combination of target RNA and RPA can obtain fluorescence
signal response, confirming the validity and feasibility of this CrisprAIE system. A series of ORF2 gene templates with concentrations ranging from 10-1 to 104 aM were determined, and 1 aM
of the target gene was successfully detected (Fig. 3c). The same gene templates were simultaneously detected by the Q-ssDNA-FAM-based Cas detection and agarose gel electrophoresis. The LOD
values for both methods were 100 and 10 aM of the gene template (Fig. 3d and Supplementary Fig. 25), indicating that the developed CrisprAIE assay had better detection efficiency than
traditional Crispr-Dx and agarose gel electrophoresis. The clinical practicability and reliability of this CrisprAIE assay was further compared with that of a well-approved RT-qPCR method
for the detection of clinical vomit samples of patients infected with norovirus. Fifteen clinical vomitus samples collected from Jiangxi Provincial Center for Disease Control and Prevention
(China) were tested. As shown in Figs. 3e, 9 and 6 samples were detected as norovirus positive and negative, respectively, which is completely consistent with the detection results obtained
by RT-qPCR (Fig. 3f, Supplementary Fig. 26 and Supplementary Table 3). Neither of the two methods showed false-positive and false-negative results. Further analysis of the area under the
curve (AUC) of receiver operating characteristic (ROC) curve indicated that CrisprAIE obtained 100% (9 out of 9) positive predictive agreement and 100% (6 out of 6) negative predictive
agreement relative to the RT-qPCR results (Fig. 3g). CrisprAIE was then extended for the quantification of SARS-CoV-2 RNA to evaluate its versatility in detecting different target RNAs.
SARS-CoV-2 pseudovirus with the same virus structure but no infectivity was selected as a standard in this experiment. The primers targeting the N gene of SARS-CoV-2 and the corresponding
crRNA sequence are listed in Supplementary Table 2. Prior to CrisprAIE detection, the viral RNA was first extracted and amplified by RT-RPA, and its amplification products were verified by
agarose gel electrophoresis (Supplementary Fig. 27). We then combined RT-RPA with CrisprAIE assay to detect SARS-CoV-2 pseudovirus in a single-tube reaction system. Under the optimal
conditions, the LOD of the developed CrisprAIE detection method was as low as 1 copy/μL (Fig. 4a). To evaluate the specificity of the established CrisprAIE assay for SARS-CoV-2 RNA
detection, we performed synthetic DNA template detection for other four coronaviruses and human respiratory virus, namely, SARS-CoV, MERS-CoV, bat-SL-CoVZC45, and hCoV-229E, by using our
CrisprAIE system corresponding to N gene detection. The results showed that only the presence of SARS-CoV-2 DNA template could result in significantly increased fluorescence response. The
other interfering gene templates exhibited negligible signal response relative to the control group, suggesting the excellent specificity of the assay for all the tested coronavirus (Fig.
4b). The clinical detection performance of this CrisprAIE system was validated by conducting an assay of SARS-CoV-2 N gene in 75 clinical nasopharyngeal swab samples, which were
synchronously identified by the standard RT-qPCR. As recommended by the RT-qPCR kit and the results of RT-qPCR in Supplementary Fig. 28, when analyzing clinical unknown nasopharyngeal swab
samples, Ct values less than or equal to 38 are considered as SARS-CoV-2 positive, Ct values between 38 and 40 indicate suspiciously or weakly positive, and Ct values more than 40 are judged
as negative. The relative fluorescence intensities for N gene detection with the CrisprAIE assay are shown in Fig. 4c. The SARS-CoV-2-positive and -negative samples were distinguished
according to the cut-off value, that is, the mean plus threefold standard deviations of the nontemplate control. As indicated in Supplementary Fig. 29 and Supplementary Table 4, the Ct
obtained by RT-qPCR showed that 50 out of 75 tested samples tested SARS-CoV-2 positive, 10 samples were suspiciously positive, and 15 samples were negative. Compared with RT-qPCR, our
CrisprAIE accurately identified 30 samples with Ct values below 35, demonstrating 100% sensitivity (Fig. 4c and Supplementary Table 4). Significantly, out of 20 samples with Ct values
ranging from 35 to 38, our method successfully detected 18 SARS-CoV-2 positive samples, achieving 90% sensitivity (18 out of 20), showcasing its reliability in detecting clinical samples
with low viral loads. Furthermore, the proposed CrisprAIE achieved 96% (48 out of 50) and 88% (53 out of 60) sensitivity for samples with Ct values less than or equal to 38 and 40 for the
analysis of the N gene, respectively (Fig. 4d). Notably, among 10 suspected samples with Ct values between 38 and 40, 5 tested positive and 5 were negative according to our CrisprAIE. All 15
SARS-CoV-2 negative samples with Ct values more than 40 were accurately identified as negative by our method. These findings indicate that the developed CrisprAIE assay exhibits sensitivity
comparable to that of the standard RT-qPCR assay, which not only facilitates the detection of clinical samples with high viral loads but also reliably identifies low-concentration samples
with Ct values ranging from 35 to 40. Using a Ct value of 38 as the judgment criterion, the ROC curve of our CrisprAIE method showed an AUC of 0.969 for the SARS-CoV-2 N gene (Fig. 4e),
showcasing its great potential for RNA quantification in actual clinical samples. IMPROVED SENSITIVITY AND STABILITY OF CRISPRAIE USING AIEGENS-ASSISTED SNA REPORTERS Despite the clinical
success of our CrisprAIE, its signal generation still relies on the trans-cleavage activity on the ssDNA linker of the composite linear DNA Q-dsDNA/AIEgen-Q substrates. Compared with
traditional Q-ssDNA-FAM reporters, the constructed Q-dsDNA/AIEgen-Q probes had higher stability and better detection efficiency in complex biological samples. However, these probes still
carry the risk of nuclease degradation in the linear ssDNA linker in biological fluids, such as serum, leading to a false positive signal. Therefore, the possibility of improving the
stability and sensitivity of CrisprAIE in complex biological samples must be further explored. Integrating with nanoparticles is an important way to improve the stability and sensitivity of
linear nucleic acid probes34. Spherical nucleic acids (SNAs) combine the advantages of nucleic acid probes and the excellent properties of nanoparticles, including excellent nuclease
resistance, efficient fluorescence quench effect, adjustable detection sensitivity, and response range, and thus have been widely applied to enhance molecular diagnostics35,36,37,38.
Coupling Crispr/Cas and AIEgen-assisted SNA (SNA/AIEgen) reporters may provide a paradigm for the development of CrisprAIE with strong stability and high sensitivity. On this basis, we first
constructed an SNA/AIEgen reporter (Fig. 5a) consisting of a core-shell nanoquencher (Fe3O4@Au/PDA, Supplementary Fig. 30) with the surface modification of TPBT-loaded sulfhydryl dsDNA via
the Michael addition (Supplementary Fig. 31). Owing to the wide absorption of Fe3O4@Au/PDA in the 300–800 nm range that overlaps perfectly with the excitation and emission of TPBT molecules
(Supplementary Fig. 30d), the fluorescence of the obtained SNA/AIEgen probes can be effectively quenched by the fluorescence resonance energy transfer effect. To enable the SNA/AIEgens to be
degraded or cleaved by S1 nuclease and Cas nuclease, we introduced an ssDNA part (TTATTTTATT) as a linker to couple the dsDNA onto the surface of Fe3O4@Au/PDA. The successful synthesis and
characterization of the SNA/AIEgen probe were verified by zeta potential, dynamic light scattering measurement, and Fourier transform infrared spectroscopy (Supplementary Figs. 31, 32).
Prior to the development of CrisprAIE, we investigated the effect of the dsDNA length coated on the surface of Fe3O4@Au/PDA because of its close association with AIEgen loading and
fluorescence quenching efficiency. As shown in Supplementary Fig. 33, the labeling numbers of SH-dsDNA10, SH-dsDNA20, SH-dsDNA30, and SH-dsDNA40 on individual Fe3O4@Au/PDA were calculated as
733, 650, 629, and 607, respectively. The binding TPBT numbers on SH-dsDNA10, SH-dsDNA20, SH-dsDNA30, and SH-dsDNA40 were 8.3, 15.3, 23.7, and 32.7, respectively (Supplementary Fig. 34).
Thus, we successfully achieved the controlled regulation of the SNA/AIEgen probe by altering the dsDNA length. The binding TPBT numbers of the obtained four SNA/AIEgen probes were ~6084,
9945, 14907, and 19848, respectively, indicating that AIEgen loading gradually increased with the dsDNA length. Nonetheless, the SNA/AIEgen probes with the highest S/N ratio were obtained
with the desired combination of SH-dsDNA30 and 2.5 μM of TPBT (Supplementary Fig. 35). We then incorporated the constructed SNA/AIEgen reporter into a Crispr/Cas12a detection system for the
detection of synthetic DNA targets (Fig. 5b, c). The LOD of the improved CrisprAIE based on SNA/AIEgen reporters was as low as 55.7 fM, exhibiting about 3.3- and 270-fold improvement in
detection sensitivity compared with Crispr-Dx using Q-dsDNA/AIEgen-Q and Q-ssDNA-FAM reporters, respectively (Fig. 5c). Compared with the Q-dsDNA/AIEgen-Q and Q-ssDNA-FAM reporter probes,
the SNA/AIEgen reporter showed significantly enhanced stability and detection accuracy even in the presence of 5 U mL-1 DNase I and 100% fetal bovine serum (FBS), indicating the excellent
stability of SNA/AIEgens in complex biological systems (Fig. 5d–g). We further compared the amplification-free detection sensitivity of Crispr-Dx based on five different reporting probes.
Figure 5h demonstrates that the sensitivity of Crispr-Dx improved significantly with the evolution of signal reporters. To better evaluate the real-world application of the
SNA/AIEgen-enhanced CrisprAIE, we used it to detect a variety of clinically relevant nucleic acid targets, including norovirus, SARS-CoV-2, Ebola virus, HPV-16, HPV-18, and monkeypox virus
(MPXV). For comparison, Crispr/Cas12a detection using Q-dsDNA/AIEgens-Q and Q-ssDNA-FAM as signal reporters was simultaneously conducted. To simulate the direct detection of nucleic acid
targets from a clinically complex sample matrix, we added these synthetic DNA targets to the corresponding biological sample types comprising background nucleic acids. Prior to CrisprAIE
detection, the biological fluid samples were treated with the previously reported HUDSON method, which can simulate viral particles releasing DNA or RNA and eliminate nuclease activity from
the samples39. As revealed in Fig. 5i–n, the CrisprAIE assay based on Q-dsDNA/AIEgen-Q and SNA/AIEgen reporters exhibited significantly increased signal response in detecting all the gene
targets in the buffer alone and in the biological specimens, such as 8% vomitus, throat swab, 8% saliva, or 2.5% serum, compared with traditional Q-ssDNA-FAM. No significant difference in
signal response was observed in measuring the targets in buffer solutions and real samples when SNA/AIEgens were used as CrisprAIE reporters, indicating their negligible matrix interference.
These findings proved that our proposed CrisprAIE exhibited superior robustness in detecting nucleic acid targets in complex biological fluid samples, implying its broad application
prospects in field-deployable viral diagnostics by detecting various genetic targets. In addition to the above-mentioned amplification-free Crispr system, we explored the compatibility of
spherical SNA/AIEgen probes with amplification-based Crispr systems. As illustrated in Supplementary Fig. 36a, we integrated the SNA@AIEgen reporter into amplification-based Crispr systems
to detect SARS-CoV-2 pseudovirus with concentrations ranging from 0 to 1000 copies/µL. Supplementary Fig. 36b shows that the developed CrisprAIE, combined with RT-RPA and SNA@AIEgen
reporters, effectively detects virus concentrations as low as 0.1 copies/µL, representing a tenfold enhancement in sensitivity compared to the LOD of 1 copy/µL achieved by CrisprAIE combined
with RT-RPA and Q-dsDNA/AIEgen-Q reporters. Furthermore, to evaluate the repeatability and reproducibility of our spherical SNA@AIEgen probes, we conducted additional experiments.
Initially, we performed repeated detections of SARS-CoV-2 pseudovirus at a concentration of 1 copy/µL in quintuplicate on the same day. The results in Supplementary Fig. 36c showed
negligible changes in fluorescent signal response among the five replicates. Moreover, we synthesized three different batches of SNA@AIEgen probes (Supplementary Fig. 36d) and utilized them
to detect 1 copy/µL of SARS-CoV-2 pseudovirus. The results in Supplementary Fig. 36e indicated consistent fluorescence signal responses across the three different batches of probes. These
findings indicated the satisfactory reproducibility and repeatability of the SNA@AIEgen-combined CrisprAIE for quantifying SARS-CoV-2 pseudovirus. EXPANDING CRISPRAIE FOR THE DIRECT
DETECTION OF TARGET RNA The above results demonstrated that the coupling of DNA-enhanced AIEgen lighting-up readout with Crispr/Cas12a-assisted ssDNA cleavage reaction is promising for the
detection of genetic targets containing DNA and RNA targets when integrated with the isothermal amplification of RPA or RT-RPA. However, Cas12a can only specifically recognize DNA. Thus, the
direct detection of RNA virus using Cas12a usually requires a reverse transcription of RNA into DNA. By contrast, Cas13 can specifically recognize RNA and thus simplify its reverse
transcription. Compared with Cas12a enzymes involving the trans-cleavage of nontargeted ssDNA, the collateral activity of Cas13 enzymes involves the trans-cleavage of nontargeted
single-stranded RNA (ssRNA). Thus, to extend the DNA-enhanced AIEgen lighting-up readout into Crispr/Cas13-driven ssRNA cleavage reaction, we further developed a Cas13a-based CrisprAIE assay
using an ssRNA linker (T*A*rArUG*C*) as an alternative to an ssDNA linker (TTATT) in the Q-dsDNA/AIEgen-Q and SNA/AIEgen reporters for Cas13a trans-cleavage (Fig. 6a–c). For comparison,
conventional Q-ssRNA-FAM reporters were commercially synthesized. To further evaluate their detection performance, a series of RNA targets at different concentrations ranging from 0 to 10 nM
was prepared and analyzed using these three Crispr-Dx methods. Figure 6d shows that the Crispr-Dx based on Q-ssRNA-FAM reporters can only detect RNA targets at the 1 pM level without
amplification. Meanwhile, the CrisprAIE assay can sensitively respond to 100 and 10 fM RNA targets when using Q-dsDNA/AIEgens-Q and SNA/AIEgens as signal probes, respectively (Fig. 6e, f).
Our results indicated that the CrisprAIE based on Q-dsDNA/AIEgen-Q and SNA/AIEgen reporters provide 10- and 100-fold increased detection sensitivity relative to the conventional
Q-ssRNA-FAM-based Crispr-Dx. We further employed the developed CrisprAIE assay based on Cas13 nuclease for the direct detection of three synthetic RNA targets, namely, norovirus, SARS-CoV-2,
and Ebola virus, in their respective diagnostic samples. The synthesized RNA targets against norovirus, SARS-CoV-2, and Ebola virus were detected in target-activated collateral cleavage
reactions containing 8% vomitus, throat swad, 8% saliva, or 2.5% serum (Fig. 6g–i). The CrisprAIE using Q-dsDNA/AIEgen-Q and SNA/AIEgen reporters demonstrated significantly increased signals
relative to the control Q-ssRNA-FAM reporter even in complex biological fluid samples. We also found that the SNA/AIEgen reporter showed better resistance to sample matrix interference than
the Q-dsDNA/AIEgen-Q and Q-ssRNA-FAM reporters, indicating the excellent robustness and on-site diagnostic potential of the SNA/AIEgen reporter for detecting RNA targets in samples
containing biological fluids. ADAPTING CRISPRAIE FOR POC TESTING The above results demonstrated that the developed CrisprAIE technology had significantly improved detection sensitivity
compared with traditional Crispr-Dx. In addition, the CrisprAIE assay did not significantly increase the complexity of the testing procedure. These features make our single-tube isothermal
CrisprAIE assay suitable for POC testing applications. In this regard, we developed a portable cellphone-based CrisprAIE assay platform using a cellphone-assisted CrisprAIE fluorescence
readout device to output the detection signal. The prototype of this CrisprAIE device was assembled from 3D-printed main body parts (e.g., brackets and shells), an optical system, and an
electrical circuit system. Its core components and optical paths are shown in Fig. 7a. In the optical system, two 450 nm LED illuminators were used as the fluorescence excitation module, and
an imaging camera covered an optical filter (650 ± 20 nm) was fitted to the lateral position directly above the test tube to output visual photos. In the electrical circuit system, a 3.7 V
battery was used to power the LED light. The type-C port on the bottom of the device directly transmits the captured fluorescent images to the cellphone side for measuring the gray value and
report the test results. The CrisprAIE test can be run in about 30 min and displayed on the cellphone (Fig. 7b). This integrated CrisprAIE system was designed to use a straightforward
workflow, in which a sample was added in a tube prefilled with RPA reagents capped with a tube cap prefilled with CrisprAIE reagents and heated at 37 °C for 15 min. The tube was shaken to
mix the RPA and CrisprAIE components and then incubated at 37 °C for another 15 min. After the CrisprAIE test, the tube was placed in the sample tanks. The TEST button of the APP was
clicked, and the result was automatically reported on the display interface of cellphone. To investigate the feasibility and reliability of our CrisprAIE for POC testing, we assessed the POC
prototype for our isothermal CrisprAIE reaction and compared it with the commercial Multiskan GO multimode reader. In this case, the cellphone-based CrisprAIE device was applied to detect
three different synthetic RNA targets against norovirus, SARS-CoV-2, and Ebola virus with concentration gradients ranging from 10-15 to 10-10 M as shown in Fig. 7d–f. The findings suggested
that the cellphone-based CrisprAIE assay achieved comparable analytical performances in terms of detection sensitivity (LOD: 1 fM) and dynamic detection range (1–105 fM) for the three target
RNAs compared with the commercial multimode reader. A correlation analysis was conducted on the detected concentrations obtained by the cellphone-based CrisprAIE assay and the multimode
reader-based CrisprAIE assay. Figure 7g–j suggests highly linear correlations between the two assay modes, confirming the practicability and reliability of our cellphone-based CrisprAIE
assay for POC diagnostics. DISCUSSION The outbreak of emerging infectious diseases, such as COVID-19, urgently requires fast, sensitive, and reliable diagnostic technologies, especially
those that can be used in the field. In this regard, Crispr/Cas nucleases are attractive tools for field-deployable diagnostic applications due to their high programmability and ability to
directly detect viral nucleic acid sequences. However, the sensitivity and speed of Crispr/Cas-based diagnostic methods must be further improved to enable their extensive use as diagnostics.
In this study, we report the development of an improved Crispr/Cas-based diagnostic assay, named CrisprAIE, to sensitively detect nucleic acid targets by integrating DNA-enhanced AIEgen
lighting-up fluorescent signal readout. Compared with conventional Crispr-Dx assay using single-stranded nucleic acid-based fluorescence probes, the reported CrisprAIE assay possessed the
advantages of higher detection sensitivity and stronger stability in complex biofluid samples. Under optimized conditions, our CrisprAIE method can detect DNA and RNA targets at levels as
low as femtomolar concentrations without amplification and exhibits at least 80-fold enhancement in sensitivity than traditional Crispr-Dx assay. This LOD of our CrisprAIE was lower than
that of previously reported amplification-free Crispr-Dx detection techniques15,32,40,41 (Fig. 8a and Supplementary Table 5). The combination with isothermal amplification, such as RPA or
RT-RPA, can further increase the sensitivity to LOD of attomolar concentrations within 35 min from raw sample to result; this performance is comparable with or even better than that of
currently available amplification-enhanced Crispr-Dx methods10,11,12,13,14,42,43,44,45,46,47,48,49,50,51,52 (Fig. 8b and Supplementary Table 5). In summary, we believe that CrisprAIE
realized the amplification-free quantification of target sequences, and its combination with nucleic acid amplification techniques can accelerate the ultrasensitive quantitative assay of
trace targets in the attomolar range. To avoid the contamination risk of multiple specimen-transfer steps, we tried to introduce the single-tube Crispr diagnostic strategy for field
implementation through the simple spatial isolation of isothermal amplification reagents and Crispr/Cas reagents. This strategy can also effectively eliminate the uncapped operation and save
assay time. Despite the successful development, this approach was inconvenient and compromised the efficiency of Crispr/Cas. Fortunately, recent advances in the one-pot Crispr diagnostic
assay can further promote the adoption of CrisprAIE for POC applications53. For example, the use of a photocontrolled Crispr method and suboptimal protospacer adjacent motifs for Cas12a with
all components added to a closed system in one-pot assay improved the detection efficiency of Crispr diagnostic method with high sensitivity and fast speed48,52. We successfully
demonstrated the viability of CrisprAIE on Cas12a and Cas13. With the development of biotechnology, the number of Crispr/Cas systems are expanding rapidly. Various Crispr/Cas systems, such
as Cas9 and Cas14a, are being explored for the development of Crispr diagnostic methods. Further investigation on the compatibility of these Crispr/Cas systems with AIEgen lighting-up
fluorescent signal readout will help broaden the variety and application range of CrisprAIE. Given that CrisprAIE is independent of gRNA or crRNA design and specific Crispr/Cas enzyme, it
can be widely used to amplify all existing Crispr-Dx assays that rely on the collateral cleavage of single-stranded nucleic acid reporters, including the well-known SHERLOCK, DETECTOR, and
HOLMES. In addition, the possible application of CrisprAIE for the sensitive and accurate quantitative detection of nonnucleic acid targets is worthy of further exploration. The stability of
fluorescent reporting probes in complex biological systems is crucial for accurate Crispr/Cas detection. The widely used linear oligonucleotide reporters for Crispr/Cas usually run the risk
of being degraded by nucleases in biological samples, resulting in false-positive signals. Compared with such oligonucleotide reporters, our designed Q-dsDNA/AIEgens-Q reporter provided
better signal response with higher S/N ratio in the complex sample matrix. Nevertheless, the improvement in CrisprAIE’s sensitivity and stability by the Q-dsDNA/AIEgens-Q reporter remains
limited. Thus, to further enhance the biological stability of our CrisprAIE, we adopted the SNA strategy to improve the stability of AIEgen-modified nucleic acid reporters and generate a
sensitive and stable fluorescent signal output for CrisprAIE development. Figures 5–6 validate that the SNA/AIEgen signal reporter greatly improved the biostability and resistance to
background interference in complex systems. In addition, the CrisprAIE constructed using SNA/AIEgen reporters offered 3.3- and 270-fold increases in detection sensitivity relevant to
dsDNA/AIEgens-Q and conventional oligonucleotide reporters. With these characteristics, we can envision the SNA/AIEgens-based CrisprAIE enabling sensitive, accurate, and stable biodetection
in complex systems. The introduction of DNA-enhanced AIEgen lighting-up fluorescent signal readout is of great significance to improve the sensitivity of Crispr/Cas detection. The clinical
diagnostic efficiency and accuracy of our reported CrisprAIE system was well demonstrated. To extend its use in POC testing and commercial setting, we demonstrated the successful
construction of a portable cellphone-assisted CrisprAIE fluorescence readout device. The analytical performance of our CrisprAIE prototype cellphone device was comparable with that of the
commercial multimode fluorescence microplate reader in detecting different synthetic RNA targets on the Crispr/Cas13 platform, indicating the huge potential for the widespread use of our
cellphone-assisted CrisprAIE platform in screening and diagnostics in the field and even at home. While our proposed AIEgen probes offer numerous advantages, including “one to more” signal
output, label-free loading of AIEgens onto dsDNA, large Stokes shifts, excellent photostability, high signal-to-noise ratio, and minimal background interference, they also exhibit certain
limitations when compared to conventional FQ-based DNA probes. Unlike traditional FQ-based reporters, our AIEgen-loaded DNA reporters are prepared through the non-covalent binding of
cationic AIEgens and negatively charged DNA strands via electrostatic adsorption, hydrophobic action, and van der Waals forces. This feature renders our AIEgen probes more susceptible to
interference from complex sample matrices. Additionally, the proposed AIEgen probes cannot be directly applied to viral lysis amplification detection methods; instead, nucleic acid
extraction is required. Moreover, our CrisprAIE method is not yet field-deployable and still necessitates several sample-processing steps, along with temperature control for the storage of
reagents and the amplification process. Despite these limitations, ongoing research efforts continue to address these challenges and optimize AIEgen probes for various diagnostic
applications. By carefully considering the specific requirements of each assay and leveraging the unique properties of AIEgen probes, it is possible to overcome these limitations and harness
their full potential in advancing molecular diagnostics. In conclusion, this work is the first to report the AIEgen-enhanced Crispr-Dx assay for the sensitive and accurate detection of
nucleic acid targets and demonstrates its application potential in infectious disease diagnosis. Our CrisprAIE can be further improved by applying the recent achievements in
Crispr/Cas-related elements, such as Cas nuclease evolution, crRNA structure optimization, amplification primer screening, and reaction system optimization (e.g., ions, pH, and component
concentrations), and in signal reporter systems, such as AIEgens with high quantum yield, microfluidics, and digital reading device. METHODS MATERIALS Analytical-grade organic solvents, such
as methanol (cat. no. 10014118), ethanol (cat. no. 100091192), dimethyl sulfoxide (DMSO) (cat. no. 30072418), and acetonitrile (cat. no. 80000618), were purchased from Sinopharm Chemical
Reagent Co., Ltd. (Shanghai, China). The sequence information for all synthesized DNA or RNA, synthetic targets, RPA primers, Cas12a-crRNA, Cas13-crRNA, and fluorescent reporters used in
this work are shown in Supplementary Tables 1, 2. The SARS-CoV-2 N gene sequence was synthesized by Tsingke Biotechnology Co., Ltd. (Beijing, China). The DNA oligomers used for constructing
the reporter strand, norovirus ORF2 gene sequence, Ebola gene sequence, HPV-16 L1 gene sequence, HPV-18 L1 gene sequence, MPXV B6R gene sequence, and primers were synthesized by Sangon
Biotech (Shanghai) Co., Ltd. (Shanghai, China). S1 nuclease (cat. no. EN0321), gel loading buffer (cat. no. 10816015), and agarose (cat. no. R0492) were purchased from Thermo Fisher
Scientific Inc. (Waltham, MA). EnGen LbaCas12a (Cpf1, cat. no. M0653T) and NEBuffer 2.1 (cat. no. B6002S) were obtained from New England BioLabs (Ipswich, MA). The lyophilized TwistAmp®
Basic kit (product code: TABAS03KIT) comprising the Core Reaction Mix of recombinase, single-stranded binding protein, and strand-displacing polymerase, was acquired from TwistDx™ Limited
(Cambridge, UK, cat. no. TABAS03KIT). SARS-CoV-2 pseudovirus (cat. no. BDS-BW-118) was provided by Guangzhou BDS Biological Technology Co., Ltd. (Guangzhou, China). Express RNA rapid
extraction kit (cat. no. NR202) was purchased from GenDx Biotech Co., Ltd. (Suzhou, China). DNA gel extraction kit (cat. no. B110092-0050) was obtained from Sangon Biotech (Shanghai) Co.,
Ltd. (Shanghai, China). BUFFER SOLUTIONS Nuclease-free ultrapure distilled water (UPDW, Invitrogen) was used in all experiments. All buffers were prepared in RNase-free conditions. The
phosphate buffer saline (PBS) was 0.01 M phosphate buffer in a 0.8% w/v saline solution, pH 7.5. The 10X NEBuffer 2.1 contained 100 mM Tris-HCl, 500 mM NaCl, 100 mM MgCl2, pH 7.9. The
Tris-HCl buffer comprised 0.05 M Tris-HCl in RNase-free ultrapure distilled water, pH 8.5. The Tris-NaCl buffer was 50 mM Tris-HCl, 100 mM NaCl, pH 8.5. MOLECULAR DOCKING OF AIEGENS WITH
DSDNA USING DOCK 6.9 First, the 3D structures of dsDNA (receptor) were obtained from Xiao Lab (http://biophy.hust.edu.cn/new/), and the molecular structures of AIEgens (ligand) were obtained
with ChemDraw 20.0. The binding mode and interaction of nucleic acid (dsDNA) with small molecular AIEgens were performed using DOCK 6.9 program by YINFO TECHNOLOGY
(https://cloud.yinfotek.com/console/). The docking score of DOCK 6.9 is expressed as grid score. In general, the smaller the grid score, the stronger the binding force. Grid vdw and Grid es
represent the contribution of van der Waals force (nonpolar interaction) and electrostatic force (polar interaction), respectively. Internal energy refers to the repulsion between receptor
with ligand. SIMULATION SAMPLE PREPARATION Human saliva samples were supplemented with 1 M TCEP, 500 mM EDTA, and RNase inhibitor (40 U μL−1) at a volume ratio of 100:11.39:0.23:2.28 with
final concentrations of 87% biofluids, 100 mM TCEP, 1 mM EDTA, and 0.8 U μL−1 inhibitor. Human serum was diluted with PBS and supplemented with 1 M TCEP, 500 mM EDTA, and RNase inhibitor (40
U μL−1) at a volume ratio of 100:251.2:40:0.8:8 with final concentrations of 25% serum or plasma, 100 mM TCEP, 1 mM EDTA, and 0.8 U μL−1 inhibitor. Synthetic targets were spiked into the
biofluids and treated with the HUDSON method. These samples were incubated at 40 °C for 5 min and at 70 °C for another 5 min (or 5 min at 95 °C, if saliva) on a thermocycler. CLINICAL
SAMPLES Clinical samples were collected from Jiangxi Provincial Center for Disease Control and Prevention and Department of Laboratory Medicine, The First Affiliated Hospital of Nanchang
University. Among them, 15 clinical vomitus samples confirmed by RT-qPCR, including 9 vomitus samples from patients with Norovirus and 6 vomitus samples from patients without norovirus, were
provided by Jiangxi Provincial Center for Disease Control and Prevention. Meanwhile, 75 identified clinical nasopharyngeal swab samples with RT-qPCR-corroborated COVID-19, including 50
positive, 10 suspiciously positive, and 15 negative samples from patients in fever clinics, were provided by The First Affiliated Hospital of Nanchang University. ETHICAL STATEMENT This
study was approved by the Medical Ethics Committee of The First Affiliated Hospital of Nanchang University and the Jiangxi Provincial Center for Disease Control and Prevention Ethics
Committee (approval ID number 2020-Y114-01). Written informed consent was obtained from all participants before the study. PREPARATION AND CHARACTERIZATION OF AIEGEN REPORTERS Four types of
AIEgen reporters, namely, ssDNA/AIEgens, Q-dsDNA/AIEgens, Q-dsDNA/AIEgens, and Q-dsDNA/AIEgens-Q (collectively referred as linear AIEgen reporters), were prepared via the label-free coupling
of AIEgens and DNA. For ssDNA/AIEgen reporters, 10 µL of TPBT (250 µM) were mixed with 10 µL of ssDNA (20 µM) and supplemented with 180 µL of NaCl aqueous solution (200 mM). For
Q-dsDNA/AIEgen reporters, Q-dsDNA was obtained by mixed Q-ssDNAA15 (20 µM) with complementary ssDNAT15 (20 µM) at a volume ratio of 1:1 with final concentrations of 10 µM, followed by
annealing with 50 µL of TPBT aqueous solution (125 µM) and 50 µL of Q-dsDNA at 95 °C for 5 min. For Q-dsDNA/AIEgen-Q reporters, Q-dsDNA was obtained by mixed Q-ssDNAA30 (10 µM) with
complementary Q-ssDNAT30 (10 µM) at a volume ratio of 1:1 with final concentrations of 5 µM, followed by annealing with 50 µL of TPBT aqueous solution (125 µM) and 50 µL of Q-dsDNA-Q (5 µM)
at 95 °C for 5 min. The Q-dsDNA/AIEgen-Q reporter was separated and purified by agarose electrophoresis using a DNA gel extraction kit. Q-dsDNA/AIEgens-Q was separated from free AIEgens by
agarose electrophoresis, and the DNA fragment of interest (Q-dsDNA/AIEgens-Q) from agarose gel was excised with a clean, sharp scalpel. Afterward, the Q-dsDNA/AIEgen-Q reporter was recovered
in accordance with the operating procedure of the DNA gel extraction kit. The characterization and evaluation of AIEgen reporters were performed by adding 100 μL of 500 U mL−1 S1 nuclease
to 100 μL of prepared AIEgen reporters (200 nM). The fluorescent intensities were recorded every 5 min by Multiskan GO multimode reader. CRISPR/CAS12A FOR SYNTHETIC TARGET DNA DETECTION
Fluorescence assays were performed as described with modifications. The LbaCas12a protein stock (100 µM) was diluted to 1 µM using the storage buffer (50 mM Tris-HCl, 600 mM NaCl, 5%
glycerol, and 2 mM DTT, pH 7.5). The crRNA stock (100 µM) was diluted to 1 µM using nuclease-free water. The 20 μL of mixed solution per reaction contained 0.4 of LbaCas12a protein (1 μM),
0.4 μL crRNA (1 μM), 4 μL of linear AIEgen reporters (1 μM) or Q-ssDNA-FAM reporters (1 μM), 0.5 μL of RNase inhibitor (40 U μL−1), 2 μL of NEBuffer 2.1 (10×, 100 mM Tris-HCl, 500 mM NaCl,
100 mM MgCl2, pH 7.9), 2.7 μL of nuclease-free water, and 10 μL of synthetic DNA targets, unless otherwise indicated. All reactions were performed in a 384-well microplate (Corning) at 37
°C, with fluorescence monitored every 5 min over 0–60 min by a Multiskan GO multimode reader (excitation: 460 nm and emission: 650 nm for AIEgen reporters; or excitation: 490 nm, emission:
520 nm for Q-ssDNA-FAM reporters). RT-RPA Total virus RNA was extracted from the patient sample using the viral RNA extraction kit. The extracted and purified RNAs were aliquoted and stored
at −80 °C before use. cDNA was obtained by reverse transcription using the RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific, Vilnius, Lithuania). RPA pellet rehydration solution
was prepared by dissolving one RPA pellet into 29.5 µL of rehydration buffer. Afterward, 2.4 µL of 10 µM RPA forward/reverse primer, 1 µL of RNase inhibitor (40 U mL−1 stock), 1 µL of
reverse transcriptase, 1 µL of RNA extracts, and 10.2 µL of nuclease-free water were sequentially added into the prepared RPA pellet rehydration solution, followed by 2.5 µL of 280 mM
magnesium acetate to initiate the amplification. The amplification proceeded at 37 °C and was completed within 15 min. The amplification reaction tube was suspended in an ice-bath. The
amplification products were analyzed by gel electrophoresis. RNA QUANTIFICATION OF REAL SAMPLES USING CRISPRAIE COMBINED WITH RT-RPA The total virus RNA was extracted from the patient sample
using the viral RNA extraction kit, and cDNA was obtained by reverse transcription using the RevertAid First Strand cDNA Synthesis Kit. The above procedure was performed to add cDNA to the
RT-RPA system for isothermal amplification. The 20 μL of mixed solution per reaction contained 0.4 μL of LbaCas12a protein (1 μM), 0.4 μL of crRNA (1 μM), 4 μL AIEgen reporters (1 μM), 0.5
μL of RNase inhibitor (40 U μL−1), 2 μL of NEBuffer 2.1 (10X, 100 mM Tris-HCl, 500 mM NaCl, 100 mM MgCl2, pH 7.9), 2.7 μL of nuclease-free water, and 10 μL of RPA amplicons. All reactions
were performed in a 384-well microplate at 37 °C, with fluorescence monitored every 5 min over 0–60 min by a Multiskan GO multimode reader. RT-QPCR PrimeScript TM RT Reagent Kit (Takara,
Kusatsu, Japan) with g DNA Eraser was used for reverse transcription by the directions to create single-stranded cDNA. SYBR Premix Ex Taq II kit was used for quantitative real-time PCR
(qPCR). The sequences of primers used for real-time quantitative RT-qPCR are shown in Supplementary Table 2. The qPCR reactions were performed using a 7900HT fast real-time qPCR system
(Applied Biosystems, Foster City, CA) with the following cycling conditions: hold at 25 °C for 2 min, hold at 95 °C for 2 min, followed by 45 cycles with DNA denaturation at 95 °C for 3 s,
and annealing and elongation at 55 °C for 30 s. Data were analyzed by the 2−ΔΔCt method with CFX Maestro Software. SYNTHESIS AND CHARACTERIZATION OF SNA/AIEGEN REPORTERS Polydopamine and
AuNP-coated Fe3O4 nanocomplex (Fe3O4/Au/PDA) was selected as the nanoquencher and synthesized following a previously reported method with slight modifications. The 200 nm Fe3O4 core was
synthesized through the classical one-pot method. In brief, 667 mg of FeCl3•6H2O, 2.8 g of sodium acetate, and 40 mL of ethanediol were added to a three-neck flask. The mixture was then
stirred at room temperature for 30 min, transferred into a hydrothermal autoclave reactor, and reacted at 200 °C for 12 h. The obtained Fe3O4 NPs were washed with ethanol and water and
collected by vacuum drying. Meanwhile, 10 mg of prepared Fe3O4 NPs, 50 μL of 10% HAuCl4 aqueous solution, 10 mg of trisodium citrate, and 40 mL of ethanol were added into a round-bottom
flask, which was then placed at ultrasonic condition for 10 min. For polydopamine coating, a dopamine solution was first prepared by dissolving 50 mg of dopamine hydrochloride into 30 mL
0.05 M pH 8.5 Tris buffer. The freshly prepared dopamine solution was rapidly added into the round-bottom flask. The mixture was sonicated at room temperature for 3.5 h. The end-product was
washed with ultrapure water for five times, and the obtained 100 mg of Fe3O4/Au/PDA was resuspended in 10 mL of ultrapure water solution for further use. All batches of Fe3O4/Au/PDA were
diluted to 1 mg mL−1 (70 pM) in UPDW and characterized using Malvern Zetasizer Nano ZS ZEN3700 DLS system to measure the charge and particle size based on the zeta potential and DLS,
respectively. TEM imaging was performed using a high-resolution JEOL JEM 2100 microscope. Before the imaging and analyses, the samples were prepared by placing a 2 µL of droplet of the
nanoparticle dispersion on a 200-mesh carbon-coated copper grid (Electron Microscopy Science, USA). SH-dsDNA was obtained by mixing SH-ssDNATTATTTTATT-A30 with ssDNAT30 and anneal for 5 min
at 95 °C and then conjugated to the surface of Fe3O4/Au/PDA as described with modifications. In brief, 1.43 mL of Fe3O4/Au/PDA (70 pM) was moved from ultrapure water to 1 mL of 0.05 M pH 8.5
Tris buffer through magnetic separation (final concentration of 100 pM). Afterward, 10 μL of 10 μM HS-dsDNA was mixed with the suspended Fe3O4/Au/PDA solution. The mixture was shaken at
room temperature for 36 h. The excessive dsDNA was removed by using magnets to collect dsDNA-conjugated Fe3O4/Au/PDA (SNAs). The collected SNAs were washed with ultrapure water three times
and then suspended at 1 mL of ultrapure water. SNAs/AIEgens were prepared by directly mixing the SNASs with TPBT under optimized concentrations. The SNASs were resuspended in 900 μL of 0.05
M Tris-NaCl (50 mM Tris-HCl, 100 mM NaCl, pH 8.0), then added with 100 μL of TPBT (25 μM) mixed with SNAs. Free AIEgens were removed _via_ magnetic separation. The prepared SNA/AIEgen
reporters were dispersed in 0.05 M Tris-NaCl buffer for subsequent experiments. All batches of SNA/AIEgen reporters were characterized using Malvern Zetasizer Nano ZS ZEN3700 DLS system to
measure the charge based on the zeta potential. CRISPR/CAS13A FOR SYNTHETIC TARGET RNA DETECTION The LwaCas13a protein stock (100 µM) was diluted to 1 µM using the storage buffer (50 mM
Tris-HCl, 600 mM NaCl, 5% glycerol, and 2 mM DTT, pH 7.5). The crRNA stock (100 µM) was diluted to 1 µM using nuclease-free water. The 20 μL of mixed solution per reaction contained 0.4 μL
of LwaCas13a protein (1 μM), 0.4 μL of crRNA (1 μM), 4 μL of AIEgen reporter or Q-ssDNA-FAM reporters (1 μM), 0.5 μL of RNase inhibitor (40 U μL−1), 2 μL of NEBuffer 2.1 (10X, 100 mM
Tris-HCl, 500 mM NaCl, 100 mM MgCl2, pH 7.9), 2.7 μL of nuclease-free water, and 10 μL of synthetic RNA targets, unless otherwise indicated. All reactions were performed in a 384-well
microplate (Corning) at 37 °C, with fluorescence monitored every 5 min over 0–60 min by a Multiskan GO multimode reader. STATISTICAL ANALYSIS All data statistical analyses were conducted in
GraphPad Prism v.8.3.0. Data were expressed as means ± standard deviation. All sample sizes and statistical tests were specified in the figure legends. All experiments were independently
repeated at least thrice. Significant differences between groups were assessed by parametric and nonparametric comparisons, and multiple hypothesis tests were adjusted appropriately. For
statistical significance test, unpaired two-tailed t-test (parametric) was used for two-group comparisons, and ANOVA was used for multiple-group comparisons. REPORTING SUMMARY Further
information on research design is available in the Nature Portfolio Reporting Summary linked to this article. DATA AVAILABILITY The data generated in this study are provided in the
Supplementary Information/Source Data file. All relevant data are available from the corresponding author on request. The source data underlying Figs. 2a–d, f, h, j, l, 3b–e, 4a–c, 5b–n,
6d–i, and 7d–g are provided as a Source Data file. Source data are provided with this paper. REFERENCES * Wood, C. S. et al. Taking connected mobile-health diagnostics of infectious diseases
to the field. _Nature_ 566, 467–474 (2019). Article ADS CAS PubMed PubMed Central Google Scholar * Ravindran, S. Smartphone science: apps test and track infectious diseases. _Nature_
593, 302–304 (2021). * Wang, A. M., Doyle, M. V. & Mark, D. F. Quantitation of mRNA by the polymerase chain reaction. _Proc. Natl Acad. Sci. USA_ 86, 9717–9721 (1989). Article ADS CAS
PubMed PubMed Central Google Scholar * Milanez-Almeida, P., Martins, A. J., Germain, R. N. & Tsang, J. S. Cancer prognosis with shallow tumor RNA sequencing. _Nat. Med._ 26, 188–192
(2020). Article CAS PubMed Google Scholar * Notomi, T. et al. Loop-mediated isothermal amplification of DNA. _Nucleic Acids Res._ 28, e63–e63 (2000). Article CAS PubMed PubMed
Central Google Scholar * Tian, B., Minero, G. A. S., Fock, J., Dufva, M. & Hansen, M. F. CRISPR-Cas12a based internal negative control for nonspecific products of exponential rolling
circle amplification. _Nucleic Acids Res._ 48, e30–e30 (2020). Article CAS PubMed PubMed Central Google Scholar * Xing, S. et al. An ultrasensitive hybridization chain
reaction-amplified CRISPR-Cas12a aptasensor for extracellular vesicle surface protein quantification. _Theranostics_ 10, 10262 (2020). Article CAS PubMed PubMed Central Google Scholar *
Xu, Z. et al. Carrier strategies boost the application of CRISPR/Cas system in gene therapy. _Exploration_ 2, 20210081 (2022). * Marraffini, L. A. CRISPR-Cas immunity in prokaryotes.
_Nature_ 526, 55–61 (2015). Article ADS CAS PubMed Google Scholar * Gootenberg, J. S. et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. _Science_ 356, 438–442 (2017). Article ADS
CAS PubMed PubMed Central Google Scholar * Gootenberg, J. S. et al. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. _Science_ 360, 439–444
(2018). Article ADS CAS PubMed PubMed Central Google Scholar * Chen, J. S. et al. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. _Science_ 360,
436–439 (2018). Article ADS CAS PubMed PubMed Central Google Scholar * Broughton, J. P. et al. CRISPR–Cas12-based detection of SARS-CoV-2. _Nat. Biotechnol._ 38, 870–874 (2020).
Article CAS PubMed PubMed Central Google Scholar * Li, S. Y. et al. CRISPR-Cas12a-assisted nucleic acid detection. _Cell Discov._ 4, 20 (2018). Article PubMed PubMed Central Google
Scholar * Dai, Y. et al. Exploring the trans‐cleavage activity of CRISPR‐Cas12a (cpf1) for the development of a universal electrochemical biosensor. _Angew. Chem., Int. Ed._ 58, 17399–17405
(2019). Article CAS Google Scholar * Su, J. et al. CRISPR/Cas12a powered DNA framework‐supported electrochemical biosensing platform for ultrasensitive nucleic acid analysis. _Small
Methods_ 5, 2100935 (2021). Article CAS Google Scholar * Zhou, T. et al. CRISPR/Cas13a powered portable electrochemiluminescence chip for ultrasensitive and specific MiRNA detection.
_Adv. Sci._ 7, 1903661 (2020). Article CAS Google Scholar * Kim, H. et al. Clustered regularly interspaced short palindromic repeats-mediated surface-enhanced Raman scattering assay for
multidrug-resistant bacteria. _ACS Nano_ 14, 17241–17253 (2020). Article CAS PubMed Google Scholar * Yin, B. et al. A CRISPR-Cas12a integrated SERS nanoplatform with chimeric DNA/RNA
hairpin guide for ultrasensitive nucleic acid detection. _Theranostics_ 12, 5914 (2022). Article CAS PubMed PubMed Central Google Scholar * Yuan, C. et al. Universal and naked-eye gene
detection platform based on the clustered regularly interspaced short palindromic repeats/Cas12a/13a system. _Anal. Chem._ 92, 4029–4037 (2020). Article CAS PubMed Google Scholar *
Huang, D. et al. CRISPR-Cas12a-assisted multicolor biosensor for semiquantitative point-of-use testing of the nopaline synthase terminator in genetically modified crops by unaided eyes. _ACS
Synth. Biol._ 9, 3114–3123 (2020). Article CAS PubMed Google Scholar * Hajian, R. et al. Detection of unamplified target genes _via_ CRISPR–Cas9 immobilized on a graphene field-effect
transistor. _Nat. Biomed. Eng._ 3, 427–437 (2019). Article CAS PubMed PubMed Central Google Scholar * Li, H. et al. Amplification‐free detection of SARS‐CoV‐2 and respiratory syncytial
virus using CRISPR Cas13a and graphene field‐effect transistors. _Angew. Chem., Int. Ed._ 61, e202203826 (2022). Article ADS CAS Google Scholar * Chen, W. et al. A suite of PCR-LwCas13a
assays for detection and genotyping of Treponema pallidum in clinical samples. _Nat. Commun._ 13, 4671 (2022). Article ADS CAS PubMed PubMed Central Google Scholar * Naqvi, M. M., Lee,
L., Montaguth, O. E. T., Diffin, F. M. & Szczelkun, M. D. CRISPR–Cas12a-mediated DNA clamping triggers target-strand cleavage. _Nat. Chem. Biol._ 18, 1014–1022 (2022). Article CAS
PubMed PubMed Central Google Scholar * Yang, J. et al. Engineered LwaCas13a with enhanced collateral activity for nucleic acid detection. _Nat. Chem. Biol._ 19, 45–54 (2022). Article CAS
PubMed Google Scholar * Hu, R., Leung, N. L. & Tang, B. Z. AIE macromolecules: syntheses, structures and functionalities. _Chem. Soc. Rev._ 43, 4494–4562 (2014). Article CAS PubMed
Google Scholar * Tang, X., Zhu, Y., Guan, W. & Lu, C. Assembling aggregation‐induced emission with natural DNA to maximize donor/acceptor ratio for efficient light‐harvesting
antennae. _Aggregate_ 4, e348 (2023). * Li, H. et al. Activity‐based smart AIEgens for detection, bioimaging, and therapeutics: recent progress and outlook. _Aggregate_ 2, e51 (2021).
Article CAS Google Scholar * Gao, Y. et al. Dual-dolor emissive AIEgen for specific and label-free double-stranded DNA recognition and single-nucleotide polymorphisms detection. _J. Am.
Chem. Soc._ 141, 20097–20106 (2019). Article CAS PubMed Google Scholar * Wang, Y.-L. et al. Real-time fluorescence in situ visualization of latent fingerprints exceeding level 3 details
based on aggregation-induced emission. _J. Am. Chem. Soc._ 142, 7497–7505 (2020). Article CAS PubMed Google Scholar * Armbruster, D. A. & Pry, T. Limit of blank, limit of detection
and limit of quantitation. _Clin. Biochem. Rev._ 29, S49–S52 (2008). PubMed PubMed Central Google Scholar * Broto, M. et al. Nanozyme-catalysed CRISPR assay for preamplification-free
detection of non-coding RNAs. _Nat. Nanotechnol._ 17, 1120–1126 (2022). Article ADS CAS PubMed Google Scholar * Truong, P. L. et al. Advancement in COVID-19 detection using
nanomaterial-based biosensors. _Exploration_ 3, 20210232 (2023). Article CAS PubMed PubMed Central Google Scholar * Cutler, J. I., Auyeung, E. & Mirkin, C. A. Spherical nucleic
acids. _J. Am. Chem. Soc._ 134, 1376–1391 (2012). Article CAS PubMed Google Scholar * Huang, C., Han, Z., Evangelopoulos, M. & Mirkin, C. A. CRISPR spherical nucleic acids. _J. Am.
Chem. Soc._ 144, 18756–18760 (2022). Article CAS PubMed PubMed Central Google Scholar * Xiao, F. et al. Light‐harvesting fluorescent spherical nucleic acids self‐assembled from a
DNA‐grafted conjugated polymer for amplified detection of nucleic acids. _Angew. Chem., Int. Ed._ 61, e202115812 (2022). Article ADS CAS Google Scholar * Fu, X. et al. Exploring the
trans-cleavage activity of CRISPR/Cas12a on gold nanoparticles for stable and sensitive biosensing. _Anal. Chem._ 93, 4967–4974 (2021). Article CAS PubMed Google Scholar * Myhrvold, C.
et al. Field-deployable viral diagnostics using CRISPR-Cas13. _Science_ 360, 444–448 (2018). Article ADS CAS PubMed PubMed Central Google Scholar * Bruch, R. et al.
CRISPR/Cas13a‐powered electrochemical microfluidic biosensor for nucleic acid amplification‐free miRNA diagnostics. _Adv. Mater_. 31, 1905311 (2019). * East-Seletsky, A. et al. Two distinct
RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. _Nature_ 538, 270–273 (2016). Article ADS CAS PubMed PubMed Central Google Scholar * Ning, B. et al.
Rapid detection of multiple SARS-CoV-2 variants of concern by PAM-targeting mutations. _Cell Rep_. _Methods_ 2, 100173 (2022). * Park, J. S. et al. Digital CRISPR/Cas‐assisted assay for
rapid and sensitive detection of SARS‐CoV‐2. _Adv. Sci_. 8, 2003564 (2021). * Ackerman, C. M. et al. Massively multiplexed nucleic acid detection with Cas13. _Nature_ 582, 277–282 (2020).
Article ADS CAS PubMed PubMed Central Google Scholar * Guo, L. et al. SARS-CoV-2 detection with CRISPR diagnostics. _Cell Discov._ 6, 34 (2020). Article CAS PubMed PubMed Central
Google Scholar * Welch, N. L. et al. Multiplexed CRISPR-based microfluidic platform for clinical testing of respiratory viruses and identification of SARS-CoV-2 variants. _Nat. Med._ 28,
1083–1094 (2022). Article CAS PubMed PubMed Central Google Scholar * Mahas, A. et al. Characterization of a thermostable Cas13 enzyme for one-pot detection of SARS-CoV-2. _Proc. Natl
Acad. Sci. USA_ 119, e2118260119 (2022). Article CAS PubMed PubMed Central Google Scholar * Hu, M. et al. Photocontrolled crRNA activation enables robust CRISPR-Cas12a diagnostics.
_Proc. Natl Acad. Sci. USA_ 119, e2202034119 (2022). Article CAS PubMed PubMed Central Google Scholar * Chen, Y. et al. A Cas12a ortholog with distinct TTNA PAM enables sensitive
detection of HPV16/18. _Cell Rep. Methods_ 3, 100444 (2023). * Arizti-Sanz, J. et al. Streamlined inactivation, amplification, and Cas13-based detection of SARS-CoV-2. _Nat. Commun._ 11,
5921 (2020). Article ADS CAS PubMed PubMed Central Google Scholar * Ding, X. et al. Ultrasensitive and visual detection of SARS-CoV-2 using all-in-one dual CRISPR-Cas12a assay. _Nat.
Commun_. 11, 4711 (2020). * Lu, S. et al. Fast and sensitive detection of SARS-CoV-2 RNA using suboptimal protospacer adjacent motifs for Cas12a. _Nat. Biomed. Eng._ 6, 286–297 (2022).
Article CAS PubMed Google Scholar * Yan, H. et al. A one-pot isothermal Cas12-based assay for the sensitive detection of microRNAs. _Nat. Biomed. Eng_. 7, 1583–1601 (2023). Download
references ACKNOWLEDGEMENTS This work was supported by the National Natural Science Foundation of China (32172296 to X.L.H.), Key Research and Development Program of Jiangxi Province
(20232BBG70030 and 20232BCD44004 to Y.H.X.), Double-Thousand Plan of Jiangxi Province (jxsq2023201113 to Y.H.X.), Jiangxi Provincial Natural Science Foundation (20224ACB205012 to X.L.H.),
Jiangxi Medicine Academy of Nutrition and Health Management (2022-PYXM-03 to Y.H.X.), Shenzhen Key Laboratory of Functional Aggregate Materials (ZDSYS20211021111400001 to B.Z.T.), and the
Science Technology Innovation Commission of Shenzhen Municipality (KQTD20210811090142053 and JCYJ20220818103007014 to B.Z.T.). AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * State Key
Laboratory of Food Science and Resources, School of Food Science and Technology, Nanchang University, Nanchang, China Yuqian Guo, Yaofeng Zhou, Min Wei, Yuhao Wu, Xirui Chen, Xiaolin Huang
& Yonghua Xiong * Beijing Engineering and Technology Research Center of Food Additives, Beijing Technology & Business University, Beijing, China Hong Duan * The National Engineering
Research Center for Bioengineering Drugs and the Technologies, Institute of Translational Medicine, Nanchang University, Nanchang, China Derong Xu & Hongbo Xin * National Engineering
Research Center of Rice and Byproducts Deep Processing, College of Food Science and Engineering, Central South University of Forestry and Technology, Changsha, China Ying Xiong * Key
Laboratory of Agricultural Information Acquisition Technology, Ministry of Agriculture and Rural Affairs, China Agricultural University, Beijing, China Siyuan Wang * Jiangxi Provincial
Center for Disease Control and Prevention, Nanchang, China Daofeng Liu * School of Science and Engineering, Shenzhen Institute of Aggregate Science and Technology, The Chinese University of
Hong Kong, Shenzhen, Guangdong, China Ben Zhong Tang Authors * Yuqian Guo View author publications You can also search for this author inPubMed Google Scholar * Yaofeng Zhou View author
publications You can also search for this author inPubMed Google Scholar * Hong Duan View author publications You can also search for this author inPubMed Google Scholar * Derong Xu View
author publications You can also search for this author inPubMed Google Scholar * Min Wei View author publications You can also search for this author inPubMed Google Scholar * Yuhao Wu View
author publications You can also search for this author inPubMed Google Scholar * Ying Xiong View author publications You can also search for this author inPubMed Google Scholar * Xirui
Chen View author publications You can also search for this author inPubMed Google Scholar * Siyuan Wang View author publications You can also search for this author inPubMed Google Scholar *
Daofeng Liu View author publications You can also search for this author inPubMed Google Scholar * Xiaolin Huang View author publications You can also search for this author inPubMed Google
Scholar * Hongbo Xin View author publications You can also search for this author inPubMed Google Scholar * Yonghua Xiong View author publications You can also search for this author
inPubMed Google Scholar * Ben Zhong Tang View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS X.L.H., Y.H.X. and B.Z.T. conceived, designed, and
managed the project. Y.Q.G. performed most experiments and data analysis with the help of H.D., M.W., Y.H.W., Y.X., X.R.C., S.Y.W. D.R.X., D.F.L. and H.B.X. provided clinical real samples
and conducted relevant experiments. H.B.X., X.L.H., Y.H.X. and B.Z.T. provided critical insights on protocols, the experimental results, and the whole work. Y.Q.G. and Y.F.Z. completed the
first draft of the paper under the guidance from X.L.H. and Y.H.X. The manuscript was further revised by X.L.H., Y.H.X. and B.Z.T., who jointly supervised the project. All authors reviewed
the manuscript and approved it for publication. CORRESPONDING AUTHORS Correspondence to Xiaolin Huang, Yonghua Xiong or Ben Zhong Tang. ETHICS DECLARATIONS COMPETING INTERESTS Y.Q.G.,
X.L.H., and Y.H.X. are listed as inventors on a pending patent concerning the design and application of CrisprAIE. All other authors declared no conflict of interest. PEER REVIEW PEER REVIEW
INFORMATION _Nature Communications_ thanks Xiao-fan Jiang, Xiaoming Zhou and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is
available. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY
INFORMATION SUPPLEMENTARY INFORMATION REPORTING SUMMARY PEER REVIEW FILE SOURCE DATA SOURCE DATA RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons
Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give
appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission
under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons
licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit
http://creativecommons.org/licenses/by-nc-nd/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Guo, Y., Zhou, Y., Duan, H. _et al._ CRISPR/Cas-mediated “one to more”
lighting-up nucleic acid detection using aggregation-induced emission luminogens. _Nat Commun_ 15, 8560 (2024). https://doi.org/10.1038/s41467-024-52931-0 Download citation * Received: 07
December 2023 * Accepted: 24 September 2024 * Published: 03 October 2024 * DOI: https://doi.org/10.1038/s41467-024-52931-0 SHARE THIS ARTICLE Anyone you share the following link with will be
able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing
initiative