Play all audios:
ABSTRACT Excessive glucocorticoid (GC) action is linked to various metabolic disorders. Recent findings suggest that disrupting skeletal GC signaling prevents bone loss and alleviates
metabolic disorders in high-fat diet (HFD)-fed obese mice, underpinning the neglected contribution of skeletal GC action to obesity and related bone loss. Here, we show that the elevated
expression of 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1), the enzyme driving local GC activation, and GC signaling in osteoblasts, are associated with bone loss and obesity in
HFD-fed male mice. Osteoblast-specific 11β-HSD1 knockout male mice exhibit resistance to HFD-induced bone loss and metabolic disorders. Mechanistically, elevated 11β-HSD1 restrains glucose
uptake and osteogenic activity in osteoblast. Pharmacologically inhibiting osteoblastic 11β-HSD1 by using bone-targeted 11β-HSD1 inhibitor markedly promotes bone formation, ameliorates
glucose handling and mitigated obesity in HFD-fed male mice. Taken together, our study demonstrates that osteoblastic 11β-HSD1 directly contributes to HFD-induced bone loss, glucose handling
impairment and obesity. SIMILAR CONTENT BEING VIEWED BY OTHERS OSTEOBLASTIC GLUCOCORTICOID SIGNALING EXACERBATES HIGH-FAT-DIET- INDUCED BONE LOSS AND OBESITY Article Open access 01
September 2021 BONE CONTROLS BROWNING OF WHITE ADIPOSE TISSUE AND PROTECTS FROM DIET-INDUCED OBESITY THROUGH SCHNURRI-3-REGULATED SLIT2 SECRETION Article Open access 06 August 2024
D-GALACTOSE-INDUCED AGING AGGRAVATES OBESITY-INDUCED BONE DYSHOMEOSTASIS Article Open access 20 May 2022 INTRODUCTION High-fat diet (HFD) not only causes obesity1 and the related metabolic
complications, e.g., insulin resistance, glucose intolerance and dyslipidemia2, but also induces substantial bone loss that increases the fracture risk in obese individuals3. Therefore, it
is desirable to develop new therapeutic strategies for hitting two birds with one stone that combats HFD-induced obesity and bone loss simultaneously. Glucocorticoids (GCs) are steroid
hormones that regulate diverse physiological functions, including cardiovascular, metabolic, immune, and homeostatic activities4,5,6,7. However, exogenous (therapeutic) and endogenous GC
excess are both harmful to health by leading to maladaptive conditions such as Cushing’s syndrome8,9, hypertension10, central obesity11, insulin resistance12 and osteoporosis13 that
recapitulate the HFD-induced metabolic abnormalities. The local GC activity in cells and tissues is governed by two 11β-hydroxysteroid dehydrogenase isoforms (11β-HSDs), namely 11β-HSD1 and
11β-HSD214. 11β-HSD1 plays a pivotal role in converting intracellular inactive GC, i.e., cortisone in humans and 11-dehydrocorticosterone (11-DHC) in rodents, to their physiologically active
forms, i.e., cortisol in humans and corticosterone in rodents, while 11β-HSD2 catalyzes the reverse reaction for inactivating GC15,16. Noteworthily, 11β-HSD1 has been recognized as a
promising therapeutic target for obesity and diabetes in the past decade since the global 11β-HSD1 knockout mice were largely resistant to HFD-induced obesity and glucose handling
impairment. Intriguingly, a recent study found that disrupting skeletal GC signaling through genetic overexpression of the GC-inactivating enzyme 11β-HSD2 could not only attenuate obesity
and glucose handling impairment but also prevent bone loss in HFD-fed mice17. These findings underpin the potential contribution of skeletal GC action to the development of HFD-induced bone
loss and obesity, wherein the role of osteoblastic 11β-HSD1 still remains unexplored. In this study, we sought to explore the roles of skeletal 11β-HSD1 in HFD-induced bone loss and
metabolic disorders. Using genetic and pharmacological approaches, our findings illustrate that the elevated 11β-HSD1 in osteoblasts contributes to HFD-induced bone loss, glucose handling
impairment and obesity. This effect could be attributed to the GC-siganling-mediated suppression on Early Growth Response 2 (Egr2) in osteoblasts that restrains skeletal glucose uptake and
bone formation. Therefore, targeting osteoblastic 11β-HSD1 could be a potential therapeutic strategy for addressing bone loss and glucose handling impairment in obese individuals. RESULTS
INCREASED SKELETAL 11Β-HSD1 EXPRESSION AND GC SIGNALING ACTIVATION ARE ASSOCIATED WITH HIGH-FAT DIET-INDUCED SYSTEMIC METABOLIC DISORDERS AND TRABECULAR BONE LOSS We conducted a
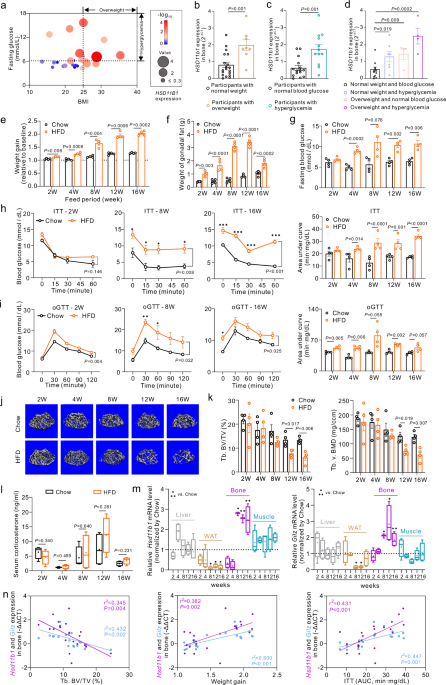
comprehensive analysis encompassing body mass index (BMI), blood glucose levels, and skeletal _HSD11B1_ (encoding 11β-HSD1) expression in 27 human participants (Fig. 1a). Interestingly, the
_HSD11B1_ expressions in bone specimens were significantly elevated in overweight participants (BMI > 25) than in the normal weight participants (BMI < 25, Fig. 1b), and in individuals
with hyperglycemia (fasting glucose >6.11 mmol/L) than those with normal blood glucose levels (fasting glucose<6.11 mmol/L, Fig. 1c). Moreover, either the normal weight participants
with hyperglycemia (BMI < 25 and fasting glucose > 6.11 mmol/L), or the overweight participants with normal blood glucose levels (BMI > 25 and fasting glucose <6.11 mmol/L), or
the overweight participants with hyperglycemia (BMI > 25 and fasting glucose > 6.11 mmol/L) exhibited significantly higher skeletal HSD11B1 expression compared to normal weight
participants without hyperglycemia (BMI < 25 and fasting glucose < 6.11 mmol/L, Fig. 1d). Notably, the overweight participants with hyperglycemia showed the highest levels of skeletal
HSD11B1 expression among these groups. These data hint at a negative association of the skeletal 11β-HSD1 with body weight and glycemia. To study the alteration of endogenous GC activation
and local tissue 11β-HSD1 expression in obesity, we established the diet-induced obesity model by feeding the C57BL/6 J mice with a high-fat diet (HFD, 60% kcal from fat, Figure S1a). As
expected, the mice became overweight with accumulated white adipose tissue (WAT) as early as 2 weeks of HFD induction (Fig. 1e, f, Figure S1c). Meanwhile, the fasting blood glucose of
HFD-fed mice was elevated compared to chow diet (Chow)-fed mice after 4-week HFD (Fig. 1g). These mice also developed insulin resistance and glucose intolerance after 4-week HFD as revealed
by the insulin tolerance test (ITT) and the oral glucose tolerance test (oGTT), respectively (Fig. 1h–i, Figure S1d). Intriguingly, these mice further developed significant trabecular bone
loss after 12 weeks of HFD feeding as revealed by the significant bone volume fraction (bone volume over total volume BV/TV), lower bone mineral density (v. BMD) and trabecular number (Tb.
N) of distal femoral metaphysis in HFD-fed mice compared to Chow-fed mice (Fig. 1j–k, Figure S1e). No significant difference was found in cortical bone parameters in mice of these two groups
(Figure S1f–g). We then assessed both the circulating GC levels, i.e., serum corticosterone concentration and the local GC activation, i.e., the mRNA expression of _Hsd11b1_, the gene
encoding 11β-HSD1, and _Tsc22d3_, the gene encoding glucocorticoid-induced leucine zipper (a GC target gene, hereafter _Gilz_), in the key metabolic tissues including liver, adipose tissue,
bone and skeletal muscle, respectively. The serum corticosterone concentration tended to be slightly higher in HFD-fed mice than Chow-fed mice at 8 and 12 weeks of HFD induction, but no
significant difference was observed at all the time points (Fig. 1l). Impressively, the expressions of _Hsd11b1_ and _Gilz_ mRNAs were coincidently and markedly upregulated only in bone of
mice after 8-week HFD compared to Chow-fed mice (Fig. 1m). Moreover, the protein expression levels of 11β-HSD1 and its colocalized enzyme H6PDH, which provides NADPH necessary for 11β-HSD1
to activate GC, in bone of HFD-fed mice also substantially increased from 8 to 16 weeks of feeding. But Chow-fed mice did not exhibit significant changes in these protein expression levels
during the feeding period (Figure S1h). Interestingly, the expressions of _Hsd11b1_ mRNA rather than _Gilz_ mRNA were slightly upregulated in skeletal muscle of HFD-fed mice at all the time
points, while both mRNAs were downregulated in gonadal WAT (gWAT) of HFD-fed mice at most of the time points (Fig. 1m). But the expressions of both mRNAs in liver HFD-fed mice were not
significantly altered at most of the time points except a slight upregulation of _Hsd11b1_ mRNA in mice with 4-week of HFD (Fig. 1m). All these findings suggest that the aberrantly increased
11β-HSD1 expression accompanied by the excessive activation of GC signaling dominantly occurs in the bone of HFD-fed mice, which follows the development of glucose handling impairment but
precedes the appearance of substantial bone loss. Importantly, linear regression analysis also revealed that the skeletal _Hsd11b1_ and _Gilz_ mRNA levels were both positively correlated
with the weight gain, area under curve (AUC) of ITT and trabecular BV/TV, respectively, in those mice after 8-week HFD (Fig. 1n, Figure S2). Together, it suggests that increased 11β-HSD1
expression and GC signaling activation in bone are associated with HFD-induced bone loss and systemic metabolic disorders. OSTEOBLAST-SPECIFIC _HSD11B1_ KNOCKOUT MICE ARE RESISTANT TO
HIGH-FAT DIET-INDUCED TRABECULAR BONE LOSS AND SYSTEMIC METABOLIC DISORDERS To explore which type of bone cells contribute to the elevated 11β-HSD1 expression and GC signaling activation in
bone, we analyzed 11β-HSD1 expression within osteoblast and osteoclast of either HFD-fed or Chow-fed mice. The immunofluorescence results showed a significantly higher incidence of 11β-HSD1
in osterix- / osteocalcin-positive (Osx+/Ocn+) osteoblast lineage cells and the comparable incidence of 11β-HSD1 in osteoclast-associated receptor- /cathepsin K- positive (Oscar+/Ctsk+)
osteoclast lineage cells at distal femoral metaphysis of HFD-fed mice compared to Chow-fed mice (Fig. 2a, Figure S3a). The osteoblast progenitors (Osx+Ocn-) and osteoblasts (Osx+Ocn+) were
further harvested from bone marrow for RT-qPCR analysis, which showed that the _Hsd11b1_ mRNA expression was upregulated dominantly in osteoblasts rather than osteoblast progenitors of
HFD-fed mice (Fig. 2b, Figure S3b). To determine the role of osteoblastic 11β-HSD1 in HFD-induced dysmetabolism, we established the osteoblast-specific _Hsd11b1_ conditional knockout mouse
line (Bglap-Cre; _Hsd11b1__fl/fl_, hereafter Ob-CKO mice) wherein the floxed allele of _Hsd11b1_ gene (_Hsd11b1__fl/fl_) was excised by the osteocalcin promoter-driven Cre recombination
(Bglap -Cre) (Fig. 2c, Figure S4a). The _Hsd11b1_ and _Gilz_ mRNA expressions were coincidently and drastically decreased in bone rather than the other non-bone tissues of Ob-CKO mice
compared to their wildtype littermates (_Hsd11b1__fl/fl_, hereafter WT mice) (Figure S4b). Moreover, the skeletal GC conversion ratio was significantly lower in Ob-CKO mice compared to WT
mice, but no significant differences in liver, fat and muscle were found between the two genotypes (Figure S4c). These data suggested that the skeletal 11β-HSD1 activity was specifically
inhibited in Ob-CKO mice. After HFD or Chow induction (Fig. 2d), there was no significant difference in the serum concentration of either adrenocorticotropic hormone (ATCH) or corticosterone
between Ob-CKO and WT mice, indicating no impact on the hypothalamic-pituitary-adrenal (HPA) axis after osteoblast-specific _Hsd11b1_ knockout (Fig. 2e). Subsequent results revealed that
the expressions of _Hsd11b1_ and _Gilz_ mRNA were coincidently upregulated in WT mice but unaltered in Ob-CKO mice when fed with HFD (Fig. 2f). Next, we examined the bone quality and bone
formation in these mice after diet induction. Interestingly, there was no significant difference in trabecular or cortical bone microarchitecture between Chow-fed Ob-CKO and WT mice (Fig.
2g, h, Figure S4d, f–g). Impressively, the trabecular microarchitecture as well as the levels of Tb. BV/TV and Tb. v. BMD in HFD-fed Ob-CKO mice resembled those in their Chow-fed controls,
whereas substantial trabecular bone deterioration was presented in HFD-fed WT mice (Fig. 2g–h, Figure S4d). In addition, there was no obvious difference in the mineral apposition rate (MAR),
bone formation rate per bone surface (BFR/BS), osteoblast number per bone surface (N.Ob/BS) and osteoclast number per bone surface (N.Oc/BS) at distal femoral metaphysis between the
Chow-fed Ob-CKO and WT mice, respectively (Fig. 2i–j, Figure S4e). However, the MAR, BFR/BS and N.Ob/BS at distal femoral metaphysis were significantly decreased in HFD-fed WT mice compared
to their Chow-fed controls, indicating reduced bone formation in WT mice after HFD feeding. Notably, the MAR, BFR/BS and N.Ob/BS all remained comparable between HFD-fed and Chow-fed Ob-CKO
mice, and the values in HFD-fed Ob-CKO mice were significantly higher than those observed in HFD-fed WT mice (Fig. 2i–j, Figure S4e). Interestingly, there was no obvious change of N.Oc/BS at
distal femoral metaphysis in either WT or Ob-CKO mice when fed with HFD (Figure S4e). Similarly, the mRNA expressions of bone formation-related marker genes, i.e., _Runx2_ (RUNX family
transcription factor 2) and _Bglap_ (bone gamma-carboxyglutamate protein, also known as _osteocalcin_), were comparable between HFD-fed and Chow-fed Ob-CKO mice, but significantly
downregulated in HFD-fed WT mice compared to their Chow-fed controls (Figure S4h). Interestingly, both Ob-CKO and WT mice had comparable energy intake and body weight gain when fed with
Chow, whereas Ob-CKO mice exhibited similar energy intake but a notable reduction of body weight gain compared to WT mice when fed with HFD (Fig. 2k, l, Figure S4i). The HFD-induced
increment of WAT weight was also attenuated in Ob-CKO mice (Fig. 2m, Figure S4j). Surprisingly, unlike WT mice, Ob-CKO mice didn’t develop obvious hyperglycemia, insulin resistance and
glucose intolerance after HFD feeding (Fig. 2n–p). Furthermore, HFD feeding promoted fat deposition in bone marrow of WT mice, which was markedly compromised in Ob-CKO mice (Figure S4k).
Meanwhile, as revealed by in vivo glucose uptake assay, the glucose uptake into WAT, skeletal muscle and bone were all markedly reduced in HFD-fed WT mice compared to their Chow-fed
controls, whereas the skeletal glucose uptake in HFD-fed Ob-CKO mice remained comparable to their Chow-fed controls (Figure S4l). In addition, as shown in the energy expenditure analysis
(Figure S4m), HFD-fed WT mice exhibited lower energy expenditure compared to Chow-fed WT mice. In contrast, Ob-CKO mice showed no significant difference in energy expenditure between those
fed HFD and those fed Chow. Notably, HFD-fed Ob-CKO mice showed significantly higher energy expenditure compared to HFD-fed WT mice (Figure S4m), this increased energy expenditure in HFD-fed
Ob-CKO mice likely contributes to their reduced weight gain. Furthermore, to validate if _Hsd11b1_ in osteoblast dominantly contributes to HFD-induced bone loss and systemic metabolic
disorders, we further compared the HFD-related phenotypes in either the osteoblast-specific (Ob-CKO), hepatocyte-specific (Alb creER; _Hsd11b1__fl/f_, hereafter Liver-CKO) or
adipocyte-specific (Adiponectin creER; _Hsd11b1__fl/f_, hereafter Adipo-CKO) _Hsd11b1_ knockout mice with WT _Hsd11b1__fl/f_ mice (Figure S5a, b). To ensure uniform experimental conditions
with simultaneous HFD induction, we only included male mice with the date of birth close to each other. Although the number of non-cre wild-type (WT) littermates (Hsd11b1flox/flox) obtained
from each CKO strain was limited (_n_ = 2 for Liver-CKO, _n_ = 3 for Ob-CKO, _n_ = 1 for Adipo-CKO), no heterogeneity of the _Hsd11b1_ and _Gilz_ mRNA expression in respective tissues was
found among the non-cre WT littermates of each CKO strain (Figure S5c). Therefore, we decided to pool all the WT littermates as the common WT control of all CKO strains. We further confirmed
the tissue-specific knockdown of _Hsd11b1_ mRNA expression accompanied by the downregulated _Gilz_ mRNA expression in each CKO strain relative to their corresponding non-cre WT littermates
(Figure S5d), and the pooled non-cre wild-type littermates (Figure S5e), respectively. In addition, the mRNA expressions of _Hsd11b1_ and _Gilz_ in bone were comparable between the Liver-CKO
mice and WT littermates, suggesting that the skeletal _Hsd11b1_ expression and GC signaling were not affected in Liver-CKO mice (Figure S5e). Interestingly, the skeletal mRNA expressions of
_Hsd11b1_ and _Gilz_ were higher in Adipo-CKO mice compared to WT mice (Figure S5e). After HFD feeding, the body weight gain and increment of visceral WATs were found to be significantly
lower, while the glucose intolerance reflected by OGTT and insulin resistance reflected by ITT were attenuated, in both Ob-CKO and Liver-CKO mice rather than Adipo-CKO mice compared to WT
mice when fed with HFD (Figure S5f–j). Nevertheless, the trabecular bone microarchitecture was largely preserved in Ob-CKO mice with the highest trabecular v. BMD and BV/TV levels compared
to the Liver-CKO, Adipo-CKO and WT mice, respectively, when fed with HFD (Figure S5k–m). Given the sensitivity of trabecular bone mass to slight changes in the background strains, we further
assessed the ratios of Tb. v. BMD and Tb. BV/TV in each type of CKO mice relative to their respective WT littermates and found that these ratios were markedly higher in Ob-CKO mice than
Liver-CKO mice, indicating that more bone mass was preserved in Ob-CKO mice as compared to Liver-CKO mice after HFD feeding. (Figure S5n). Taken together, it suggests that the elevated
11β-HSD1 was responsible for the excessive GC signaling activation in osteoblast and the impaired glucose uptake and osteoblastic bone formation in bone, thus, contributing to the
HFD-induced trabecular bone loss and systemic metabolic disorders. 11Β-HSD1-MEDIATED GC SIGNALING OVERACTIVATION RESTRAINS THE EGR2-GOVERNED OSTEOGENIC ACTIVITY AND GLUCOSE UPTAKE IN
OSTEOBLAST To explore the mechanism by which osteoblastic 11β-HSD1 regulates bone formation and glucose handling, we transfected the mouse MC3T3-E1 osteoblastic cells with a plasmid carrying
the exogenous _Hsd11b1_ or _EGFP_ gene to construct the 11β-HSD1-overexpressed and control osteoblastic cells (hereafter MC3T3-HSD1 and MC3T3-GFP cells), respectively (Figure S6a). RT-qPCR
and western blot analysis confirmed the substantial overexpression of _Hsd11b1_ but little upregulation of _Gilz_ mRNA expression in MC3T3-HSD1 cells compared to MC3T3-GFP cells without
feeding with 11-DHC (the inactivated form of mouse GC) (Figure S6b–d). When fed with 11-DHC, the _Gilz_ mRNA expression was drastically upregulated in MC3T3-HSD1 cells compared to 11-DHC-fed
MC3T3-GFP cells, indicating the overactivation of GC signaling in MC3T3-HSD1 cells (Figure S6e). During osteogenic differentiation, the MC3T3-HSD1 cells exhibited decreased alkaline
phosphatase (ALP) activity at 7 days after osteogenic induction and alizarin red-stained (ARS) calcium deposits at 14 and 21 days after osteogenic induction, respectively (Fig. 3a).
Consistently, the mRNA expressions of _Runx2_ and _Bglap_ were both notably downregulated in MC3T3-HSD1 cells during osteogenic induction (Fig. 3b). These data suggest that the elevated
11β-HSD1 over-activates GC signaling and suppresses osteogenic activity of osteoblasts. We then employed RNA sequencing (RNA-seq) and transposase-accessible chromatin with sequencing
(ATAC-seq, Figure S8) to examine the gene expression profile and chromatin accessibility between MC3T3-HSD1 and MC3T3-GFP cells. Combination analysis of the RNA-seq and ATAC-seq data
revealed 86 dysregulated genes with accessible chromatin in their genomes, wherein 10 genes were significantly downregulated while 76 genes were significantly upregulated in MC3T3-HSD1 cells
compared to MC3T3-GFP cells (Fig. 3c). Among the ten downregulated genes, we identified the _Egr2_, which encodes the transcriptional factor early growth factor 2, as the fourth most
significantly downregulated gene with accessible chromatin in its genome. Further bioinformatic analysis showed that Egr2 was enriched in a set of genes relevant to ‘response to insulin’,
‘regulation of osteoblast differentiation’ and ‘regulation of ossification’ (Fig. 3d, Figure S7). In line with the RNA-seq data, both mRNA and protein levels of Egr2 were markedly
downregulated in MC3T3-HSD1 cells compared to MC3T3-GFP cells either before or during osteogenic induction (Fig. 3e–f). A previous study documented that GC could inhibit osteocalcin
transcription in osteoblasts via suppressing Egr2/Krox20-binding enhancer18. Consistently, the _Egr2_ mRNA expressions were notably upregulated when we silenced _Gliz_ expression in
MC3T3-HSD1 cells by transfection of small interfering RNA (siRNA) targeting _Gilz_ (Figure S10a–c). To investigate if the GC-suppressed Egr2 expression contributes to the reduced osteogenic
activity in MC3T3-HSD1 cells, we transfected the MC3T3-E1 cells with another plasmid carrying both exogenous _Hsd11b1_ and _Egr2_ genes (MC3T3-HSD1+Egr2 cells) (Figure S6f, g). As compared
to the MC3T3-HSD1 cells, the MC3T3-HSD1+Egr2 cells exhibited increased calcium deposition as well as elevated _Bglap_, _Runx2_ and _Osterix_ mRNA expressions (Fig. 3g, h, Figure S6h, i).
Importantly, both the mRNA expressions of _Hsd11b1_ and _Gilz_ remained upregulated in MC3T3-HSD1+Egr2 cells that resembled MC3T3-HSD1 cells when fed with 11-DHC (Figure S6g). Collectively,
these data indicate that the elevated 11β-HSD1-mediated GC signaling overactivation suppresses Egr2 expression to impair osteogenic activity of osteoblasts. Thereafter, we examined the
effect of 11β-HSD1 overexpression on glucose uptake in 11-DHC-fed osteoblasts in vitro. As compared to MC3T3-GFP cells, the MC3T3-HSD1 cells showed markedly reduced glucose uptake capacity
before or during osteogenic induction (Fig. 3i). Consistently, the mRNA expressions of _Slc2a4_ (solute carrier family 2, facilitated glucose transporter member 4), i.e., the gene encoding
glucose transporter type 4 (_Glut4)_, but not _Slc2a1 and Slc2a3_, i.e., the gene encoding glucose transporter type 1 and 3 (_Glut1_ and _Glut3)_, were markedly downregulated in MC3T3-HSD1
cells (Fig. 3j). Then, we screened the mRNA expression of genes that encode the key pathway molecules involved in regulating the _Glut4_-mediated insulin-dependent glucose uptake, including
insulin receptor (_Ir_), insulin receptor substrate (_Irs_), phosphoinositide-3-kinase regulatory subunit 1 (_Pik3r1_), phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha
(_Pik3ca_) and phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit beta (_Pik3cb_). Interestingly, we only identified the mRNA expressions of _Pik3cb_ out of the other four
genes that were significantly downregulated in MC3T3-HSD1 cells before or during osteogenic induction (Fig. 3k). Importantly, the _Pik3cb_-encoded P110β, together with the _Pik3ca_-encoded
P110α, are the two critical subunits of phosphoinositide 3-kinase (PI3K) PI3Ks, the vital components of the PI3K/AKT signaling pathway that govern the insulin-dependent glucose uptake via
regulating Glut4. Western blot analysis further verified that the protein levels of P110β and Glut4, as well as the phosphorylated AKT (pAKT) rather than the total AKT, were coincidently
downregulated in MC3T3-HSD1 cells (Figure S6l). Moreover, the glucose uptake capacity was largely restored while the mRNA expressions of _Pik3cb and Glut4_ were markedly upregulated when we
silenced Gliz expression in MC3T3-HSD1 cells. Interestingly, the MC3T3-HSD1+Egr2 cells showed enhanced glucose uptake capacity and increased mRNA expressions of _Pik3cb_ and _Glut4_ either
before or during osteogenic induction compared to MC3T3-HSD1 cells (Fig. 3l, m, Figure S6j, k). The promoted PI3K/AKT signaling in MC3T3-HSD1+Egr2 cells was further confirmed by the
increased protein expressions of P110β, pAKT and Glut4 (Figure S6l). All these data suggest that the elevated 11β-HSD1-mediated GC signaling overactivation suppresses Egr2 expression to
impair glucose uptake capacity in osteoblasts. Next, we asked if the Egr2 directly regulates the expression of Pik3cb and Glut4. Analysis using the JASPAR online database of transcription
factor binding profiles revealed a high score for Egr2 binding to the promoter region of _Pik3cb_ and _Glut4_ genes (Table-S1). This observation suggests a substantial likelihood that Egr2
regulates Pik3cb and Glut4. Then, luciferase reporter assay revealed that Egr2 could directly regulate the promoter activity of both _Pik3cb_ and _Glut4_ genes (Fig. 3n). In addition,
chromatin immunoprecipitation (ChIP) in combination with RT-qPCR analysis confirmed that Egr2 could directly bind with the promoter of both _Pik3cb_ and _Glut4_ to promote their gene
expression (Fig. 3o). The binding sites at the promoter of each gene were further predicted by the open-access database of transcription factor binding profiles (JASPAR
https://jaspar.elixir.no/about/), among which two of the predicted sequences with the highest binding scores were selected for gene mutation and plasmid construction followed by luciferase
reporter assay (Table-S1). We found that the regulation of Egr2 on the promoter of _Pik3cb_ and _Glut4_ genes was only markedly interrupted when both two binding sites of each gene promoter
were simultaneously mutated (Fig. 3p), indicating that Egr2 alternatively binds with the two compensatory binding sites of each gene to regulate _Pik3cb_ and _Glut4_ gene expression,
respectively. Importantly, we further validated that the skeletal mRNA expressions of _Egr2_, _Pik3cb_ and _Glut4_ were all markedly downregulated in the HFD-fed WT mice, whereas their
expressions were all restored in the HFD-fed Ob-CKO mice, as compared to their Chow-fed controls (Figure S9). Collectively, all these data indicate that 11β-HSD1-mediated GC signaling
overactivation restrains the Egr2-governed glucose uptake and osteogenic activity in osteoblasts. INHIBITING 11Β-HSD1 ACTIVITY RESTORES IMPAIRED OSTEOGENIC ACTIVITY AND GLUCOSE UPTAKE IN
OSTEOBLASTS WITH GC SIGNALING OVERACTIVATION Next, we sought to investigate whether inhibiting 11β-HSD1 activity could rescue the impaired osteogenic activity and glucose uptake in
MC3T3-HSD1 cells by treating them with an 11β-HSD1 inhibitor (AZD8329). It was found that 11-DHC-fed MC3T3-HSD1 cells consumed a remarkably higher concentration of 11-DHC and produced a
notably higher concentration of corticosterone in their culture supernatant compared to MC3T3-GFP cells, which was substantially restricted when the 11β-HSD1 inhibitor was introduced in the
cell culture (Figure S10). The _Gilz_ mRNA expression was markedly downregulated in 11-DHC-fed MC3T3-HSD1 cells with 11β-HSD1 inhibitor treatment before or during osteogenic induction (Fig.
4a, Figure S10k). Interestingly, the _Hsd11b1_ mRNA expression was only slightly reduced by 11β-HSD1 inhibitor during osteogenic induction (Figure S10f, k). Together, it indicates that the
11β-HSD1-mediated GC signaling overactivation in osteoblast could be efficiently blocked by 11β-HSD1 inhibitor treatment in vitro. Moreover, the 11β-HSD1 inhibitor notably rescued the
downregulated expression of Egr2 in MC3T3-HSD1 cells before and during osteogenic induction (Fig. 4b–d). In addition, 11β-HSD1 inhibitor markedly promoted the calcium deposition as well as
upregulated the _Bglap_, _Runx2_ and _Osterix_ mRNA expressions in MC3T3-HSD1 cells during osteogenic induction (Fig. 4e–h). On the other hand, in line with the result of osteogenic activity
assay, 11β-HSD1 inhibitor drastically enhanced glucose uptake into MC3T3-HSD1 cells and upregulated _Glut4_ mRNA expressions in MC3T3-HSD1 cells before or during osteogenic induction (Fig.
4i, j, Figure S10h, i). Western blot analysis further verified that the downregulated protein levels of P110β and Glut4, as well as the phosphorylated AKT (pAKT) rather than the total AKT,
were coincidently upregulated in MC3T3-HSD1 cells with 11β-HSD1 inhibitor treatment (Figure S10j). Collectively, it suggests that inhibiting the 11β-HSD1 activity could restore osteogenic
activity and glucose uptake in osteoblasts with GC signaling overactivation. PHARMACOLOGICAL INHIBITION OF 11Β-HSD1 IN OSTEOBLASTS ATTENUATES HIGH-FAT DIET-INDUCED OBESITY, AND RELATED
GLUCOSE HANDLING IMPAIRMENT AND BONE LOSS As inspired by the protective effect of osteoblast-specific 11β-HSD1 knockout against HFD, we next sought to investigate whether pharmacological
inhibition of osteoblastic 11β-HSD1 activity could combat HFD-induced dysmetabolism. To facilitate the inhibition of 11β-HSD1 activity in osteoblasts, we developed a bone-targeted 11β-HSD1
inhibitor by conjugating the AZD8329 with our previously developed bone formation surface targeting moiety (DSS)6, i.e., (DSS)6-AZD8329 (Fig. 5a, Figure S11a, b). The control 11β-HSD1
inhibitor without bone targeting specificity was also generated by conjugating AZD8329 with the control moiety (RKK)6, i.e., (RKK)6-AZD8329 (Figure S11c, d). In vitro assay confirmed that
both inhibitors could significantly inhibit the conversion of 11-DHC to corticosterone in either MC3T3-HSD1 cells or MC3T3-GFP cells (Figure S12a). As revealed by IVIS imaging, there was
more fluorescent signal of Cy5-labelled AZD8329 distributed in bone, but less fluorescent signal distributed in liver and adipose tissues, from mice intravenously treated with one dose of
(DSS)6-AZD8329 compared to the mice with (RKK)6-AZD8329 treatment (Fig. 5b, Figure S12b). Consistently, confocal imaging showed more instances of colocalization of fluorescent signals of
Cy5-labelled AZD8329 with osteocalcin-expressing osteoblasts from mice intravenously treated with one dose of (DSS)6-AZD8329 compared to the mice with (RKK)6-AZD8329 treatment (Fig. 5c,
Figure S12c). Furthermore, 11β-HSD1 activity tests indicated that (DSS)6-AZD8329-treated mice showed significantly lower GC conversion ratio (Figure S12d) in bone, while
(RKK)6-AZD8329-treated mice only showed slightly lower GC conversion ratio in bone, as compared to vehicle-treated mice. However, the GC conversion ratio in liver, fat and muscle were all
significantly lower in (RKK)6-AZD8329-treated mice, while they were only slightly lower in (DSS)6-AZD8329-treated mice, as compared to vehicle-treated mice. These data suggest that the
(DSS)6-AZD8329 was superior to (RKK)6-AZD8329 in suppressing the skeletal 11β-HSD1 activity, whereas (RKK)6-AZD8329 showed a more profound effect on suppressing 11β-HSD1 activity in
non-skeletal tissues. Moreover, the data of _Gilz_ expression in the respective tissues paralleled the above 11β-HSD1 activity trends (Figure S12e), further advocating _Gilz_ as an indicator
of local GC signaling associated with 11β-HSD1 activity. In addition, the skeletal _Gilz_ mRNA expression was decreased by more than 50% from baseline in mice at three days after one dose
of intravenous administration of (DSS)6-AZD8329, while such inhibition on _Gilz_ mRNA expression was enhanced dose-dependently from 3 mg/kg to 10 mg/kg (Figure S12f). Therefore, we chose the
10 mg/kg dosage at a three-day interval as the regimen for the following in vivo experiments. We first tested the inhibitor in the mouse model induced by moderate HFD (43% kcal from fat,
hereafter mHFD), the same HFD formula used in the previous study for testing the role of osteoblastic GC action in HFD (Figure S13a)17. Interestingly, (DSS)6-AZD8329 rather than
(RKK)6-AZD8329 treatment even totally reversed the decrease of Tb. BV/TV and Tb. v. BMD, and microarchitecture deterioration (Figure S13b, c, e). In addition, (DSS)6-AZD8329 treatment
resulted in significantly lower percentage decreases in MAR and N. Ob/BS as compared to (RKK)6-AZD8329 treatment (Figure S13d, e). Neither of the inhibitor treatments affected osteoclast
formation (Figure S13d). Moreover, (DSS)6-AZD8329 treatment significantly alleviated body weight gain, reduced the WAT accumulation and improved glucose tolerance, while (RKK)6-AZD8329
treatment only modestly mitigated weight gain and reduced the weight of iWAT in mice with 24 weeks of mHFD (Figure S13f, h–j). Neither of the inhibitor treatments affected energy intake in
mice during mHFD feeding (Figure S13g). It was noted that (DSS)6 treatment alone did not affect weight gain, adipose weight, glucose tolerance, and bone mass and microarchitecture in
mHFD-fed mice, albeit they have similar mitigation on mHFD-induced bone formation reduction as (RKK)6-AZD8329 treatment. Collectively, these data suggest that pharmacological inhibition of
osteoblastic 11β-HSD1 could improve glucose tolerance and promote osteoblastic bone formation to attenuate mHFD-induced obesity. Given that the skeletal 11β-HSD1 expression became markedly
upregulated in mice after 8 weeks of induction by HFD containing 60% kcal from fat in our study, we next tested the inhibitor in the mice with established obesity after 8 weeks of HFD
induction (Fig. 5d). Impressively, we found that (DSS)6-AZD8329 treatment significantly mitigated the HFD-induced bone loss and microarchitecture deterioration while increasing bone
formation, which is reflected by significantly lower percentage decreases in Tb. BV/TV, Tb. v. BMD, MAR and N.Ob/BS in (DSS)6-AZD8329 treatment group compared to (RKK)6-AZD8329 treatment
group (Figure S14a–d, Fig. 5e–i). Neither of the inhibitor treatments affected the osteoclast formation (Figure S14c). Importantly, we found that the body weight gain was notably reduced in
the HFD-fed obese mice after treatment of (DSS)6-AZD8329 and (RKK)6-AZD8329, respectively (Fig. 5j, k). Either of the inhibitor treatments markedly reduced the WAT weight, attenuated
hyperglycemia and insulin resistance, and improved the glucose tolerance in HFD-fed obese mice after 8 weeks of treatment (Fig. 5l–o, Figure S14e). Impressively, as revealed in the glucose
uptake assay by IVIS imaging, only (DSS)6-AZD8329 treatment significantly enhanced the glucose uptake into bone, while neither of the inhibitor treatments affected the glucose uptake into
liver, WAT and skeletal muscle, respectively (Fig. 5p). Interestingly, (DSS)6-AZD8329 treatment not only significantly downregulated the skeletal mRNA expressions of _Hsd11b1_ and _Gilz_ but
also concurrently restored the mRNA expressions of _Egr2_, _Pik3cb_ and _Glut4_ that were suppressed in HFD-fed mice (Fig. 5q). In contrast, (RKK)6-AZD8329 treatment only marginally
inhibited the skeletal mRNA expressions of _Hsd11b1_ and _Gilz_, and slightly increased mRNA expressions of _Egr2_, _Pik3cb_, and _Glut4_ (Fig. 5q). Neither (DSS)6-AZD8329 nor (RKK)6-AZD8329
treatment altered the serum concentrations of ACTH and corticosterone (Figure S14f). On the other hand, we further tested the bone quality of the HFD-induced obese mice treated with
Semaglutide, a glucagon-like peptide 1 receptor agonist (GLP-1RA) approved for lowering blood sugar and reducing body weight. Surprisingly, we found that Semaglutide treatment even
exacerbated the HFD-induced trabecular bone loss, albeit it markedly diminished both body weight and adipose weight as well as improved glucose handling in HFD-fed mice (Figure S15). These
data reflect that weight loss and metabolic improvement may not necessarily protect bone, which in turn suggests that the bone benefits of 11β-HSD1 inhibition in osteoblast could be
independent of the protection from weight gain and associated metabolic benefits. Taken together, our data suggest that pharmacological inhibition of osteoblastic 11β-HSD1 could attenuate
the HFD-induced obesity, and related glucose handling impairment and bone loss. DISCUSSION The chronic consumption of HFD, particularly rich in saturated fat, has been associated not only
with the development of obesity and metabolic syndromes in both mice and humans19,20, but also with the induction of bone loss21,22. Previous studies suggest that diet-induced obesity is
accompanied by GC overproduction in key metabolic tissues17,23,24,25. Furthermore, elevated expression of 11β-HSD1, the principal regulator of local GC activation, has been observed in obese
individuals14,26,27,28,29. These observations hint at the contribution of 11β-HSD1 to the HFD-induced dysmetabolism. In our study on human specimens, we observed that the 11β-HSD1 mRNA
expression in bone was markedly elevated in overweight and hyperglycemic individuals. Consistently, we found that the expression of 11β-HSD1 and its colocalized enzyme H6PDH in bone were
both significantly upregulated, accompanied by the elevated skeletal GC signaling in mice after 8 weeks of HFD feeding, which were closely associated with the development of HFD-induced bone
loss, obesity and glucose handling impairment. Our findings, which coincide with the previous report showing the elevated 11β-HSD1 expressions and GC signaling activation in bones of mice
on CD1 background fed with 18-week HFD17, challenge the preconceived notions considering liver and adipose tissues as the primary organs with elevated 11β-HSD1 and excessive GC action that
majorly contribute to the adverse metabolic consequences of HFD16,30,31,32. Surprisingly, we found that the 11β-HSD1 expressions and GC signaling activation didn’t significantly elevate in
the liver, WATs, and skeletal muscles of HFD-fed mice compared to Chow-fed mice. This finding aligns with a previous study reporting an early-stage reduction of 11β-HSD1 expression in the
liver and WAT of HFD-fed rats, which normalized after prolonged diet induction33. Similarly, Morton et al. found that both adipose and hepatic 11β-HSD1 were downregulated after 18 weeks of
HFD feeding in A/J mice and C57BL/6 J mice34. This downregulation may represent an adaptive mechanism in response to the chronic stress caused by HFD. In our study, skeletal 11β-HSD1
expression in HFD-fed mice initially decreased at the early stages of obesity but subsequently increased after 8 weeks. This biphasic response suggests that skeletal 11β-HSD1 may fail to
adapt to the prolonged HFD, thereby contributing to exacerbating metabolic abnormalities, although further investigation on the dysregulation of skeletal 11β-HSD1 is needed. We identified
that the 11β-HSD1 expressions were dominantly upregulated in osteoblasts of HFD-fed mice. Previous studies have shown that global deletion of 11β-HSD1 can significantly prevent exogenous GC
excess-induced and trabecular bone loss35, whereas globally transgenic overexpression of 11β-HSD1 in mice resulted in osteoporosis compared to wildtype mice36, highlighting its involvement
in regulating bone metabolism. Our in vivo data of Ob-CKO mice demonstrate that conditional deletion of osteoblastic 11β-HSD1 could enhance osteoblastic bone formation and prevent
HFD-induced bone loss. These findings align with the previously reported bone phenotype of mice with global35 or osteoblastic/osteocytic GC signaling blockage17,37. On the other hand,
previous studies also allude to the involvement of osteoblastic GC action in regulating systemic metabolism across diverse conditions, including aging38, long-term exogenous GC
administration39 and HFD17. Noteworthily, conditional deletion of osteoblastic 11β-HSD1 also protected against the HFD-induced obesity and glucose handling impairment, which recapitulates
the protective phenotype of 11β-HSD1 global knockout mice with chronic high-fat feeding40,41. The Ob-CKO mice exhibited higher energy expenditure, but similar energy intake compared to WT
mice after HFD feeding, which could be explained by the increased energy consumption in bone as the Ob-CKO mice exhibited improved glucose uptake dominantly in bone and enhanced osteogenic
activity compared to WT mice after HFD feeding. Glucose uptake in osteoblasts is essential for osteogenesis42, while this activity reversely affects whole-body glucose metabolism because
skeletal glucose uptake accounting for approximately 15% of total systemic glucose uptake43,44. Therefore, the protection from weight gain and metabolic benefits observed in our Ob-CKO mice
following HFD exposure could be driven by the increased consumption of energy, e.g., glucose, in bone42. Our study substantiates that the 11β-HSD1-mediated GC signaling overactivation in
osteoblasts could not only inhibit bone formation to contribute to HFD-induced bone loss, but also dominantly participate in the HFD-related adverse metabolic consequence. We also
acknowledge that the skeletal 11β-HSD1 has been implicated in mediating the systemic anti-inflammatory action of GC45,46,47. In murine models of polyarthritis treated with GC, global
deletion of 11β-HSD1 led to resistance against the anti-inflammatory effects of GC. This resistance was partially replicated in myeloid (including osteoclasts) but not mesenchymal (including
osteoblasts) 11β-HSD1 deletion45,46. It will be worth determining the specific contribution of osteoblastic 11β-HSD1 to the systemic anti-inflammatory action of GC using our Ob-CKO model in
our future study. Noteworthily, our Liver-CKO mice rather than the Adipo-CKO mice also protected against HFD-induced obesity and glucose handling impairment. Interestingly, a previous study
showed that liver-specific 11β-HSD1 knockout only mildly improved glucose tolerance without affecting fat and weight accumulation after HFD48. This discrepancy may be attributed to the
different knockout strategies used in their study, i.e., Alb-Cre-mediated 11β-HSD1 knockout since embryos compared to our study, i.e., Alb-Cre ERT-mediated knockout induced before HFD
feeding. Regarding the bone benefit observed in Liver-CKO mice, it could be partially explained by the metabolic improvement instead of the protection of weight gain in these mice after
HFD-feeding, as the improved systemic glucose handling has been found to positively influence bone homeostasis49,50 whereas the body weight gain is primarily beneficial to bone by increasing
mechanical stimulation51. It should be noted that the bone benefits in Liver-CKO mice were inferior to those observed in Ob-CKO mice, hinting the distinct underlying mechanisms that drive
the bone benefits after osteoblastic and hepatic 11β-HSD1 knockout, respectively. Nevertheless, future studies in our Liver-CKO mice with enlarged sample size are still required for
validation on the metabolic and bone benefits of liver-specific 11β-HSD1 knockout or inhibition. Our mechanistic study further identified _Egr2_ as one of the most downregulated genes in
MC3T3-HSD1 cells compared to control cells after induction with inactivated GC 11-DHC. Existing evidence indicates that Egr2 is a transcription factor directly repressed by the GC-targeted
transcription factor Gliz52,53. Egr2 has been previously identified as a component of strain-sensitive pathways in osteoblasts that is responsive to anabolic signaling and mechanical
loading54. It was also reported that GC could inhibit the Egr2-binding enhancer to downregulate the transcription of _Bglap_53. Consistently, our study demonstrated that 11β-HSD1-mediated GC
signaling overactivation directly repressed Egr2 to restrain glucose uptake and osteogenic activities in osteoblasts. Further, we showed that Egr2 could directly govern the gene expression
of the crucial mediators, i.e., _Pik3cb_ and _Glut4_, involved in insulin-dependent glucose uptake. The _Pik3cb_-encoded p110β is the key component of insulin-driven PI3K-AKT pathways55,56,
while Glut4 is the key glucose transporter for insulin-dependent glucose uptake57,58. Noteworthily, mice with conditional deletion of _Pik3cb_ in osteoblast had osteopenia with reduced bone
mass and impaired osteoblastic function59, while mice with conditional deletion of _Glut4_ in osteoblast developed peripheral insulin resistance with impaired osteoblast maturation60.
Consistently, the skeletal expressions of _Egr2_, _Pik3cb_ and _Glut4_ were also drastically downregulated in WT mice with chronic high-fat feeding, while their expression were maintained in
HFD-fed Ob-CKO mice. Such evidence endorses our mechanistic findings showing that 11β-HSD1-mediated GC signaling overactivation impairs glucose uptake via repressing the Egr2-promoted
_Pik3cb_ and _Glut4_ expression. Although 11β-HSD1 inhibition has long been recognized as a promising therapeutic strategy for obesity and metabolic syndrome, the clinical development of
11β-HSD1 inhibitor remains challenging and unsatisfactory61,62,63,64,65,66. Identifying the precise targeting tissues or cells might be a new direction for 11β-HSD1 inhibitor development.
For instance, Liu et al. have developed an adipose tissue-targeted 11β-HSD1 inhibitor67, which exhibited improved glucose tolerance but no impact on insulin resistance. However, this
inhibitor hindered glucose uptake in skeletal muscle, potentially leading to adverse effects when targeting 11β-HSD1 in adipose tissues to counteract diet-induced obesity. Our animal study
suggested that targeted inhibition of osteoblastic 11β-HSD1 could be an alternative and desirable strategy for clinical translation. The peptide (DSS)6 preferentially binds to the lowly
crystallized hydroxyapatite and amorphous calcium phosphonate of bone formation surface68, thus, can serve as the targeting moiety for facilitating 11β-HSD1 inhibitor approaching the
osteoblasts residing at the bone formation surface in vivo68. Accordingly, we developed the bone-targeted 11β-HSD1 inhibitor (DSS)6-AZD8329 that exhibited superior osteoblast-targeting
specificity than the nonspecific control inhibitor (RKK)6-AZD8329 in vivo. When evaluated in HFD-fed mice with two regimens, i.e., full course treatment during moderate HFD (43% kcal from
fat) and treatment initiated when obesity was established and skeletal 11β-HSD1 was upregulated after 8 weeks of HFD (60% kcal from fat), we are encouraging to find that, in no matter which
regiments, (DSS)6-AZD8329 treatment drastically improved bone formation and preserved bone quality in HFD-fed mice that was superior to (RKK)6-AZD8329 treatment. Previous clinical studies
have investigated the effects of 11β-HSD1 inhibitors on bone remodeling. In clinical observation, the 11β-HSD1 inhibitor AZD4017 showed no effect on bone turnover markers in postmenopausal
osteopenic women69. However, in another clinical study, AZD4017 prevented prednisolone-induced decreased bone turnover in men, suggesting that the effects of 11β-HSD1 inhibition on bone
turnover may vary across different disease conditions70. (DSS)6-AZD8329 treatment could also effectively restore the skeletal glucose uptake as well as ameliorate the development of obesity
and glucose handling impairment in HFD-fed mice that was comparable to or even better than (RKK)6-AZD8329 treatment. Importantly, the GLP-1RA experiment reflects that weight loss and
metabolic improvement may not necessarily protect bone, which in turn suggests that the bone benefits of 11β-HSD1 inhibition in osteoblast could drive the protection from weight gain and
associated metabolic benefits in Ob-CKO mice and (DSS)6-AZD8329-treated mice. It should be noted that we did not observe significant changes in HPA axis in either (DSS)6-AZD8329 or
(RKK)6-AZD8329 treatment, although previous studies indicated that long-term use of 11β-HSD1 inhibitors might activate the HPA axis because of negative feedback71,72,73,74,75,76.
Nonetheless, the potential side effects of long-term inhibition of osteoblastic 11β-HSD1 need to be further investigated before clinical translation. In summary, we found that elevated
11β-HSD1 in osteoblast could mediate the GC overactivation in osteoblast to restrain skeletal glucose uptake and bone formation, thus contributing to HFD-induced bone loss, obesity, and
glucose handling impairment (Fig. 6). Our study also suggests that targeting osteoblastic 11β-HSD1 could be a promising strategy for reducing body weight, improving glucose metabolism and
strengthening bone in obese individuals. METHODS All animal experiments were conducted in compliance with approved protocols by the Hong Kong Baptist University animal care committees. All
research complies with relevant ethical regulations. IN VIVO STUDY MOUSE HOUSING AND HFD FEEDING This study was approved by the Committees of Animal Ethics and Experimental Safety of Hong
Kong Baptist University (Ethical Permission NO: REC/22-23/0593 and REC/21-22/0404), demonstrating compliance with ethical standards. Bglap-Cre mice (Strain NO. 019509) were purchased from
Jackson Lab. The Hsd11b1fl/fl mice (Strain NO. T008729), Alb creER mice (Strain NO. T017784) and Adiponectin creER mice (Strain NO. T052679) were purchased from GemPharmatech Co. Ltd
(China). To generate the Ob-CKO mice (Bglap-Cre; _Hsd11b1__fl/fl_ mice) required for the experiment, _Hsd11b1__fl/+_ mice were firstly crossed with Bglap-Cre mice to produce Bglap-Cre;
_Hsd11b1__fl/+_ mice. Then, Bglap-Cre; _Hsd11b1__fl/+_ mice were crossed with _Hsd11b1__fl/fl_ mice to generate Bglap-Cre; _Hsd11b1__fl/fl_ mice (Ob-CKO mice). These Bglap-Cre;
_Hsd11b1__fl/fl_ mice were crossed with _Hsd11b1__fl/fl_ mice to generate CKO mice (Bglap-Cre; _Hsd11b1__fl/fl_ mice) and littermates (_Hsd11b1__fl/fl_ mice) for conducting experiments.
Liver-CKO mice (Alb-CreER; _Hsd11b1__fl/fl_ mice) and Adipose-CKO mice (Adiponectin-CreER; _Hsd11b1__fl/fl_ mice) were also generated by crossing Hsd11b1flox/flox mice with each type of cre
mice separately. The corresponding non-cre littermates (_Hsd11b1__fl/fl_ mice) generated during the breeding of each strain served as controls in the respective animal experiments. The male
mice were housed in the animal house with a 12 h light and 12 h dark cycle and were given libitum access to food and water. At 2-month old, male mice were started diet induction. The HFD
(60% kcal from fat, Research Diet #D12492, 5.24 kcal/g) or Chow diet (10% kcal from fat, Research Diet #D12450J, 3.76 kcal/g) were purchased from Research Diet (NY, USA). The moderate HFD
(43 % energy from fat, #SF14-144, 16.3 MJ/kg) and relative Chow diet (14% energy from fat, #SF14-008, 13.8 MJ/kg) was purchased from Specialty Feeds (WA, Australia). The weight and food
intake were examined every week. Energy intake was calculated based on the caloric values obtained from Research Diets, which were 5.24 kcal/g for the HFD and 3.76 kcal/g for Chow. At
designated time points, the mice were euthanized humanely for sample collection and testing. OGTT AND ITT OGTT and ITT were performed on mice after a period of fasting. Prior to OGTT, the
mice underwent a 12-hour fast and a baseline blood glucose reading was taken. Subsequently, the mice were administered orally administered with 20% glucose at 2 g/kg, and blood glucose
levels were measured at intervals of 15, 30, 60, 90, and 120 min after the glucose administration. For ITT, the mice were subjected to a 4 h fast. Insulin (00169-1834-11, Novo Nordisk,
Denmark) was injected with 0.75 U/kg body weight in mice, and blood glucose levels were measured at 0, 15, 30 and 60 min after the injection. Blood samples were collected from a tail prick
and blood glucose levels were determined using glucose strips and an Accu-check glucometer (Roche)39. The total glucose areas under the curve (AUC) were calculated to assess the extent of
glucose response. Three technical replicates were performed for each test. MICRO-COMPUTED TOMOGRAPHY (MICRO-CT) Micro-CT (VivaCT 40, SCANCO Medical AG, Switzerland) was utilized to evaluate
trabecular and cortical bone of distal and midshaft femur on the left side according to the manufacturer’s instructions60,77. The femur and tibia images were reconstructed with voxel sizes
of 12.5 μm, utilizing an integration time of 200 ms, 70 kVp, 114 μA, and 260 thresholds. For trabecular bone, the central region of 70% vertebral height was selected, extending from the
proximal growth plate towards the vertebral body. Regions of interest (ROI) of trabecular bone from 100 consecutive layers were selected. The trabecular bone volume/total volume (Tb. BV/TV),
trabecular bone density (Tb. v. BMD), thickness (Tb. Th) and number (Tb. N) were measured. The automatic threshold algorithm was employed to analyze the cortical bone of 100 slices in the
distal 50% of the femur, and cortical bone parameters, such as cortical bone volume/total volume (Ct. BV/TV), bone density (Ct. BMD), porosity (Ct. porosity) and thickness (Ct. Th) were
analyzed. SERUM CORTICOSTERONE AND ACTH MEASUREMENT Serum concentrations of mouse corticosterone (ab108821, Abcam, USA) and ACTH (ab263880, Abcam, USA) were measured by enzyme-linked
immunosorbent assay (ELISA) kits in accordance with the manufacturer’s guidelines. Three technical replicates were performed for each test. PARAFFIN SECTION AND H&E STAINING Following
sacrifice, the tibia was fixed in 4% paraformaldehyde for 48 h. Subsequently, the samples underwent complete decalcification in 10% EDTA solution. After decalcification, tissues were
embedded in paraffin and sectioned into 5 μm thick slices. These sections were then stained using H&E staining working solution according to standard procedures. FROZEN SECTION AND
IMMUNOHISTOCHEMISTRY Following sacrifice, the distal end of the right femur was fixed in 4% paraformaldehyde for 48 h. The specimen was then dehydrated in sucrose solutions, with increasing
concentrations of 10%, 20%, and 30% sucrose in phosphate-buffered saline (PBS) for 24 h and embedded in a compound (Sakura Finetek, Japan) at the optimal cutting temperature without
decalcification. Using a CryoStar NX50 (Thermo Fisher Scientific, USA), longitudinal sections of proximal region of the specimen were obtained with a thickness of 5 μm. Fluorescence
immunostaining was performed on the obtained sections. After treating the blocking buffer for one hour, samples kept at a temperature of 4 °C overnight with primary antibodies specifically
targeting osteocalcin (1:100; bs-4917R, Bioss, USA), osterix (1:200; ab22552, Abcam, UK), oscar (1:500; Cell Signaling; MAB1633, R&D system), CTSK (1:100; ab19027, Abcam, UK), and
11β-HSD1 (1:100; 52041, BiCell Scientific, USA). The sections were then washed and incubated with fluorescent secondary antibodies (1:1000; A78946; A-31573; A32849TR; Thermo Fisher
Scientific, USA) for 45 min, then washed three times and mounted on coverslips using a Fluoroshield mounting medium containing DAPI (ab104139, Abcam, UK)78. The mouse IgG (ab37355, Abcam,
UK) was used as isotype control. The dyed sections were captured utilizing a confocal laser scanning microscope (Leica, Germany). The fluorescence images were processed by Las X software
(Leica). FLUORESCENCE ACTIVE CELL SORTING (FACS) Cells in femurs and tibias were first blown off with a syringe. Then, cells were washed with PBS/1% BSA and directly incubated with the first
antibody for osteocalcin (1:50; bs-4917R, Bioss, USA) and stained with donkey anti-rabbit IgG-FITC (1:100; ab6717, Abcam, UK). Subsequently, cells were exposed to the primary antibody
against osterix (1:50, sc-393325, Santa Cruz, USA) and then subjected to staining using donkey anti-mice IgG-APC (1:100; ab130782, Abcam, UK). FACS (BD FACSVia™ flow cytometry system, Becton
Dickinson, USA) was then performed on the stained cells population60,79. The sorted cell population was used for qRT-PCR analysis. Three technical replicates were performed for each of the
four biological replicates. BONE HISTOMORPHOMETRY Prior to the administration of euthanasia, all mice were injected intraperitoneally with calcein at doses of 10 mg/kg on two separate
occasions, 10 and 2 days before the Micro-CT scanning procedure. After Micro-CT scanning, femurs were extracted and histologically sectioned. Additionally, modified Tartrate-resistant acid
phosphatase (TRAP; 387A-1KT, Sigma, USA) staining was conducted to examine the activity of osteoclasts. Bone static histomorphometric analyses were performed to determine the osteoblast
surface (Ob.N/BS) and osteoblast surface (Oc.N/BS), while bone dynamic histomorphometric analyses were conducted to determine the mineral apposition rate (MAR) and bone formation rate per
bone surface (BFR/BS). These analyses were carried out using professional image analysis software (Image J, USA) following the standardized nomenclature for bone histomorphometry80. Three
technical replicates were performed for each biological sample. AGAROSE GEL ELECTROPHORESIS OF GENOTYPES The small piece of tail of newborn mice was collected at about 3 weeks of age. The
genotypes of mice were determined by PCR analyses of genomic DNA isolated from mouse tails. The floxed Hsd11b1 allele was identified with primers: upper primers,
5’-GGAGTGACACAACAGGCAACTTC-3’ and P2 (5’-AGAGACCAGACATTAGGACACCAG-3’) and lower primers, 5’-TGGTGACTGGATAAAGGGACAG-3’ and 5’-TTTCAGCTCAGCAGGTCTGTG-3’. The genotyping for Bglap Cre transgene
was performed by PCR with the primers Cre F (5’-CAAATAGCCCTGGCA GATTC-3’) and Cre R (5’-TGATACAAGGGACATCTT CC-3’). IN VIVO GLUCOSE UPTAKE The glucose uptake of mice was assessed using the
XenoLight RediJect 2-DeoxyGlucosone (2-DG)-750 kit (760567, PerkinElmer, USA), in accordance with established protocol81. Briefly, the mice were fasted for 6 h, following which DG-750 was
administered via the tail vein. After a period of 12 h, the ex vivo glucose uptake of liver, fat, muscle, and bone was assessed post-dissection, utilizing an In Vivo Imaging System (IVIS,
PerkinElmer, USA) with excitation and emission wavelengths of Ex745 nm/Em820 nm. The obtained data were analyzed by using the Living Image software. Three technical replicates were performed
for each biological sample. ENERGY EXPENDITURE Ob-CKO mice were acclimated in the Comprehensive Lab Animal Monitoring System (Columbus Instruments) for 48 h. Then, mice were monitored
continuously for 24 h to measure heat generation, oxygen consumption, and carbon dioxide production82. 11Β-HSD1 ACTIVITY TESTS AND LC/MS/MS ANALYSIS For tissue sample preparation, tissues
were promptly snap-frozen in liquid nitrogen and subsequently homogenized in 0.5 mL KHBR buffer for 25 min. To this homogenate, 1 mM cofactor NADPH, 10 μM 11-DHC, and 1.5 mL KHBR buffer were
added. The tissue samples without adding NADPH and 11-DHC were also extracted to measure the basal level of 11-DHC and corticosterone. The mixture was then incubated in a 37 °C water bath
for 2 h for liver and fat tissues, and 4 h for muscle and bone tissues. Following this incubation period, 1 mL chloroform was added to the tissue mixture, and the resulting suspension was
sonicated for 15 min and centrifuged at 4000 rpm for 5 min to collect the organic layer. This extraction process was repeated twice. The resulting liquid was evaporated under a nitrogen
stream, redissolved in 1 mL methanol, and prepared for LC/MS/MS analysis. LC/MS/MS analysis was employed to quantify the concentrations of 11-DHC and corticosterone in both tissue
homogenates and cell culture media from in vitro studies. Isocratic conditions were used for the separation of 11-DHC and corticosterone83. LC/MS/MS with a TurboIonSpray ionization source
operating in positive ion mode at 500 °C was utilized to simultaneously monitor 11-DHC and corticosterone. Multiple reaction monitoring (MRM) techniques were applied for specific
transitions: m/z 363.2-121.2 for 11-DHC and m/z 361.2-163.2 for corticosterone. Conversion ratio was normalized to tissue weight. Each biological replicate was analyzed with three technical
replicates to ensure robustness and reliability of the data. HUMAN BONE SPECIMEN COLLECTION The cancellous bone of human femoral head was procured during orthopaedic surgery from the
patients who had experienced femoral neck fractures and received artificial hip replacement at the First Affiliated Hospital of Shantou University (Ethical Permission NO: B-2024-121) and the
Third Affiliated Hospital of Southern Medical University (Ethical Permission NO: 2024-ER-010). The collection of samples was conducted ethically, with clinical approval, and explicit
informed consent was obtained from all participating patients. IN VITRO STUDY CELL CULTURE AND OSTEOGENIC DIFFERENTIATION The mouse pre-osteoblastic MC3T3-E1 cells were cultured in complete
α-MEM (32571-036, Gibco, USA) that contained 10% fetal bovine serum (10270-106, Gibco, USA) and 1% streptomycin/penicillin (15140-122, Gibco, USA) and incubated at 37 °C with 5% CO2. The
osteogenic medium used in this study was composed of completed α-MEM supplemented with 50 µg/ml L-ascorbic acid (A4544, Sigma-Aldrich, USA) and 10 mM β-glycerophosphate (ST637, Beyotime,
China). Additionally, 1 μM of 11-DHC (32907, Cayman Chemical, USA) was included in the culture medium, as specified in experimental designs. PLASMID AND SIRNA TRANSFECTION Plasmids
expressing GFP, _Hsd11b1_ and _Egr2_ were provided by BGscience Co., LTD, and transfected into MC3T3-E cells via Lipofectamine™ 3000 Transfection Reagent (L3000015, Thermo Fisher Scientific,
USA). The commercially validated siRNAs (50 nM; siNT, siGilz) were purchased from RiboBio (China) by using Lipofectamine™ 2000 Transfection Reagent (11668027, Thermo Fisher Scientific,
USA). Initially, MC3T3-E1 osteoblastic cells were seeded in a culture plate and cultured with a complete medium for 24 h until they reached 80% confluency. Subsequently, the culture medium
was replaced with a fresh complete medium, and the prepared complexes were combined and incubated for 15 min at ambient temperature. Then, the complex was added to the wells and culture for
48 h. RT-QPCR The Trizol reagent (Invitrogen, USA) was utilized to extract Total RNA from the tissues. Next, purified RNA was subjected to cDNA synthesis through a HiScript III RT SuperMix
kit (R323, Vazyme, China). Real-time PCR reactions were conducted on the 7900 HT Sequence Detection System (Applied Biosystems), employing the ChamQ Universal SYBR gPCR Master Mix (Q711,
Vazyme, China). The primers were synthesized by the Sangon Biotech Co., Ltd (China). The gene expression levels were measured and normalized against the endogenous control Actin. The 2−ΔΔCT
method was employed to calculate the relative fold changes. The primer sequences are listed in Table S1. WESTERN BLOT Cellular protein extraction was performed using a Tissue Protein
Extraction Reagent (78510, Thermo Scientific, USA) and the protein concentration was determined using a BCA Protein Detection Kit (23225, Thermo Scientific, USA). Equal amounts of protein
(30 µg) from HSD1, GFP, and MC3T3-E1 osteoblastic cells were resolved by SDS-_P_AGE using a 12% polyacrylamide gel and transferred onto a PVDF membrane (Millipore, USA). Following blocking
with 5% nonfat milk in TBST, the membrane was incubated overnight at 4 °C with primary antibodies (1:500) against Actin (4967, Cell Signaling, USA), p110β (ab151549, abcam, UK), AKT (4691 T,
Cell Signaling, USA), phosphorylated AKT (4060 T, pAKT, Cell Signaling, USA), Glut4 (21619, SAB, USA), 11β-HSD1 (AF3397, R&D system, USA), Egr2 (EPR4004, Abcam, UK), H6PDH (EPR12338,
Abcam, UK). After incubation with appropriate secondary antibodies (Goat Anti-Rabbit IgG, L3012; Rabbit Anti-Goat IgG, L3042; 1:1000, SAB, USA) conjugated with horseradish peroxidase for 1
hour, the blots were developed using an ECL kit (Thermo Scientific, USA) and exposed to film. Three technical replicates were performed for each of the three biological replicates. WB
results were collected using BioRad ChemiDoc XRS chemiluminescence imaging system. The protein levels were quantified by ImageJ v2.0.0 software. ALP STAINING After treatment, the cells in
each experimental group were gently washed with PBS and then fixed using 4% paraformaldehyde for 30 min. Following fixation, the cells were incubated with a working solution of BCIP/NBT ALP
staining kit (C3206, Beyotime, China) for an additional hour. The resultant images were captured using a camera for further analysis. The OD value was measured at a wavelength of 500 nm for
quantitative analysis. Three technical replicates were performed for each of the three biological replicates. ALIZARIN RED STAINING After fixation, the cells were treated with Alizarin red S
staining solution (C0148S, Beyotime, China) and incubated for 30 min. The resulting images were acquired using a camera. The OD value was measured at a wavelength of 500 nm for quantitative
analysis. Three technical replicates were performed for each of the three biological replicates. IN VITRO GLUCOSE UPTAKE Glucose uptake analysis was carried out utilizing a colorimetric
glucose uptake kit (36503, ATT Bioquest, USA) according to manufacturer’s instructions84. The experimental protocol involved washing the cells twice with Krebs-Ringer-Phosphate-HEPES buffer
and incubating them in Glucose Uptake Buffer for one hour. Subsequently, the cells were subjected to insulin stimulation for 20 min. The cells were then exposed to 2-Deoxyglucose (2-DG) for
40 min. After the treatment, Acidic Lysis Buffer was utilized to lyse the cells. The resulting lysate was subjected to the addition of a 2-DG Uptake Assay working solution, followed by
incubation for 2 h. Finally, the absorbance ratio at 570/610 nm was recorded using an absorbance plate reader. Three technical replicates were performed for each of the three biological
replicates. RNA-SEQ The RNA-Seq and ATAC-Seq were performed by Guangzhou Epibiotek Co., Ltd. For RNA-Seq analysis, a total of 3 µg RNA was extracted from HSD1 and Con cells treated with
11-DHC for 24 h. The VAHTS Stranded mRNA-seq Library Prep Kit for Illumina V2 (Vazyme Biotech, China) was used to prepare the libraries in accordance with the manufacturer’s instructions.
The resulting sequencing reads were aligned to the mouse Ensemble genome GRCm38 with the Hisat2 aligner (version 2.1.0) and the “rna-strandness RF” parameter. The quantification of
genome-mapped reads was performed using FeatureCounts (version 1.6.3). The DESeq2 R-package was used to conduct, with a significance threshold of _p_ < 0.05 for differential expression
analysis. Three technical replicates were performed for each of the three biological replicates. ATAC-SEQ To perform ATAC-Seq analysis, Cutadapt (v2.5) was utilized for adapter trimming and
sequence filtering. The resulting reads were then aligned to the mouse Ensemble genome GRCm38 using bowtie2 (v2.11.1) with default parameters. Only the paired reads that mapped uniquely were
sorted using SAMtools and retained for further analysis. To generate viewable bigwig files, deepTools (v3.2.0) was employed. Genomic regions of the identified peaks were annotated using the
ChIPseeker R package. Differential sites were detected from ATAC-Seq experiments using the DiffBind R package. For motif analysis, homer (v4.10.4) was used to select the ATAC peaks that
were identified, then employing a significance threshold of _p_ < 0.05 to select open chromatin regions and target genes. The Integrative Genomics Viewer (IGV) screenshots of 10 GC
responded genes are shown in Figure S8. Three technical replicates were performed for each of the three biological replicates. COMBINATION ANALYSIS OF RNA-SEQ AND ATAC-SEQ The results from
both sequencing techniques were integrated to identify overlapping differentially expressed genes. Following the identification of these overlapping differentially expressed genes, pathway
enrichment analysis was conducted using the KEGG pathway and GO enrichment analysis to uncover the pathways associated with these differentially expressed genes. Biological processes were
annotated for the top 10 differentially expressed genes. To provide a more intuitive representation, processes related to osteogenesis and glucose metabolism are highlighted, and the
complete version can be found in Figure S7. LUCIFERASE REPORTER ASSAY The pGL3-basic plasmids were used to construct the pGL3-_Pik3cb_ and pGL3-_Glut4_ vectors by cloning DNA fragments from
mouse _Pik3cb_ and _Glut4_ promotors, respectively. Subsequently, Lipofectamine 3000 reagent was used to transfect MC3T3-E1 osteoblastic cells with 0.5 μg of reporter luciferase vectors, 0.5
μg of Egr-2 plasmids, and 0.1 μg of pRL-TK. After a 48 h incubation period, the cells were detected luciferase activity through Dual-Luciferase Reporter Assay System (E1910, Promega, USA).
Three technical replicates were performed for each of the three biological replicates. CHROMATIN IMMUNOPRECIPITATION-QUANTITATIVE PCR (CHIP-QPCR) 293 T cells were transfected with
pCDNA3.1-3*flag-tagged plasmids encoding _Egr2_ and control proteins. ChIP assays (Millipore, USA) were carried out following established protocols. Cells were stimulated in 10 cm dishes,
fixed with 1% formaldehyde, and sonicated in PBS supplemented with a protease inhibitor. Immunoprecipitation was performed with anti-FLAG antibody-conjugated protein A agarose beads. DNA
purification was carried out using the phenol/chloroform/isoamyl alcohol (Sigma, USA) method, and precipitation was done using 3 M sodium acetate, followed by elution in DEPC water. Finally,
the eluted DNA (5–10 ng) was subjected to qPCR to evaluate the _Pik3cb_ promoter and _Glut4_ promoter enrichment. STATISTICAL ANALYSIS The study variables were presented as mean ± standard
error of the mean. Unpaired tests, One-way ANOVA with Tukey’s post-hoc tests, or Two-way ANOVA with Sidak’s multiple comparisons tests were conducted to evaluate intergroup variations.
Statistical analysis and linear regression analysis were performed using GraphPad Prism 8.3.0 (GraphPad Software, USA) and _P_ < 0.05 was regarded as statistically significant68.
REPORTING SUMMARY Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article. DATA AVAILABILITY Source data are provided with this
paper. Raw data of RNA-seq and ATAC-seq in this study can be accessed through following link in NCBI websites: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE274790(RNA-seq) and
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE274974(ATAC-Seq). Source data are provided with this paper. REFERENCES * Li, J. et al. An obligatory role for neurotensin in
high-fat-diet-induced obesity. _Nature_ 533, 411–415 (2016). Article ADS CAS PubMed PubMed Central Google Scholar * Hancock, C. R. et al. High-fat diets cause insulin resistance
despite an increase in muscle mitochondria. _Proc. Natl. Acad. Sci._ 102, 7815–7820 (2008). Article ADS Google Scholar * Patsch, J. M. et al. Increased bone resorption and impaired bone
microarchitecture in short-term and extended high-fat diet–induced obesity. _Metabolism_ 60, 243–249 (2011). Article CAS PubMed Google Scholar * Cain, D. W. & Cidlowski, J. A. Immune
regulation by glucocorticoids. _Nat. Rev. Immunol._ 17, 233–247 (2017). Article CAS PubMed PubMed Central Google Scholar * Quax, R. A. et al. Glucocorticoid sensitivity in health and
disease. _Nat. Rev. Endocrinol._ 9, 670–686 (2013). Article CAS PubMed Google Scholar * Pianca, N. et al. Glucocorticoid receptor antagonization propels endogenous cardiomyocyte
proliferation and cardiac regeneration. _Nat. Cardiovasc. Res._ 1, 617–633 (2022). Article PubMed Google Scholar * Jeanneteau, F. D. et al. BDNF and glucocorticoids regulate
corticotrophin-releasing hormone (CRH) homeostasis in the hypothalamus. _Proc. Natl Acad. Sci._ 109, 1305–1310 (2012). Article ADS CAS PubMed PubMed Central Google Scholar * Mitchell,
F. Glucocorticoid-induced Cushing syndrome increases the risk of cardiovascular events. _Nat. Rev. Endocrinol._ 8, 563–563 (2012). Article PubMed Google Scholar * Lodish, M. &
Stratakis, C. A. A genetic and molecular update on adrenocortical causes of Cushing syndrome. _Nat. Rev. Endocrinol._ 12, 255–262 (2016). Article CAS PubMed Google Scholar * Melander, O.
et al. Association between a variant in the 11 beta-hydroxysteroid dehydrogenase type 2 gene and primary hypertension. _J. Hum. Hypertens._ 14, 819–823 (2000). Article CAS PubMed Google
Scholar * Peckett, A. J., Wright, D. C. & Riddell, M. C. The effects of glucocorticoids on adipose tissue lipid metabolism. _Metabolism_ 60, 1500–1510 (2011). Article CAS PubMed
Google Scholar * Caratti, G. et al. Glucocorticoid activation of anti-inflammatory macrophages protects against insulin resistance. _Nat. Commun._ 14, 2271 (2023). Article ADS CAS PubMed
PubMed Central Google Scholar * Rizzoli, R. & Biver, E. Glucocorticoid-induced osteoporosis: who to treat with what agent? _Nat. Rev. Rheumatol._ 11, 98–109 (2014). Article PubMed
Google Scholar * Morgan, S. A. et al. 11beta-HSD1 is the major regulator of the tissue-specific effects of circulating glucocorticoid excess. _Proc. Natl Acad. Sci._ 111, 2482–2491 (2014).
Article Google Scholar * Lindroos, J. et al. Human but Not Mouse Adipogenesis Is Critically Dependent on LMO3. _Cell Metab._ 18, 62–74 (2013). Article CAS PubMed PubMed Central Google
Scholar * Masuzaki, H. et al. A transgenic model of visceral obesity and the metabolic syndrome. _Science_ 294, 2166–2170 (2001). Article ADS CAS PubMed Google Scholar * Kim, S. et al.
Osteoblastic glucocorticoid signaling exacerbates high-fat-diet- induced bone loss and obesity. _Bone Res_ 9, 40 (2021). Article CAS PubMed PubMed Central Google Scholar * Gabet, Y. et
al. Krox20/EGR2 deficiency accelerates cell growth and differentiation in the monocytic lineage and decreases bone mass. _Blood_ 116, 3964–3971 (2010). Article CAS PubMed PubMed Central
Google Scholar * Teijeiro, A., Garrido, A., Ferre, A., Perna, C. & Djouder, N. Inhibition of the IL-17A axis in adipocytes suppresses diet-induced obesity and metabolic disorders in
mice. _Nat. Metab._ 3, 496–512 (2021). Article CAS PubMed Google Scholar * Ng, S. F. et al. Chronic high-fat diet in fathers programs beta-cell dysfunction in female rat offspring.
_Nature_ 467, 963–966 (2010). Article ADS CAS PubMed Google Scholar * Chen, F. et al. Flaxseed oil ameliorated high-fat-diet-induced bone loss in rats by promoting osteoblastic function
in rat primary osteoblasts. _Nutr. Metab._ 16, 71 (2019). Article ADS Google Scholar * Urs, S., Henderson, T., Le, P., Rosen, C. J. & Liaw, L. Tissue-specific expression of Sprouty1
in mice protects against high-fat diet-induced fat accumulation, bone loss and metabolic dysfunction. _Br. J. Nutr._ 108, 1025–1033 (2012). Article CAS PubMed Google Scholar * Geer, E.
B., Islam, J. & Buettner, C. Mechanisms of glucocorticoid-induced insulin resistance: focus on adipose tissue function and lipid metabolism. _Endocrinol. Metab. Clin. North Am._ 43,
75–102 (2014). Article CAS PubMed PubMed Central Google Scholar * Tsai, S. F. et al. High-fat diet-induced increases in glucocorticoids contribute to the development of non-alcoholic
fatty liver disease in mice. _FASEB J._ 36, e22130 (2022). Article CAS PubMed Google Scholar * Dunford, E. C. & Riddell, M. C. The Metabolic Implications of Glucocorticoids in a
High-Fat Diet Setting and the Counter-Effects of Exercise. _Metabolites_ 6, 44 (2016). Article PubMed PubMed Central Google Scholar * Tomlinson, N. D. et al. Absence of Cushingoid
Phenotype in a Patient with Cushing’s Disease due to Defective Cortisone to Cortisol Conversion. _J. Clin. Endocrinol. Metab._ 87, 57–62 (2002). CAS PubMed Google Scholar *
Espíndola-Antunes, Daniela CEK. Adipose Tissue Expression of 11β-Hydroxysteroid Dehydrogenase Type 1 in Cushing’s Syndrome and in Obesity. _Arq. Bras. Endocrinol. Metab._ 51, 1397–1403
(2007). Article Google Scholar * Desbriere, R. et al. 11β-Hydroxysteroid Dehydrogenase Type 1 mRNA is Increased in Both Visceral and Subcutaneous Adipose Tissue of Obese Patients.
_Obesity_ 14, 794–798 (2012). Article Google Scholar * Baudrand, R. et al. Overexpression of 11beta-hydroxysteroid dehydrogenase type 1 in hepatic and visceral adipose tissue is associated
with metabolic disorders in morbidly obese patients. _Obes. Surg._ 20, 77–83 (2010). Article PubMed Google Scholar * Frayn, K. N., Arner, P. & Yki-Jarvinen, H. Fatty acid metabolism
in adipose tissue, muscle and liver in health and disease. _Essays Biochem_ 42, 89–103 (2006). Article CAS PubMed Google Scholar * Lundbom, J. Adipose tissue and liver. _J. Appl
Physiol._ 124, 162–167 (2018). Article CAS PubMed Google Scholar * Paterson, J. M. et al. Metabolic syndrome without obesity: Hepatic overexpression of 11β-hydroxysteroid dehydrogenase
type 1 in transgenic mice. _Proc. Natl Acad. Sci._ 101, 7088–7093 (2004). Article ADS CAS PubMed PubMed Central Google Scholar * Drake, A. J. et al. Reduced adipose glucocorticoid
reactivation and increased hepatic glucocorticoid clearance as an early adaptation to high-fat feeding in Wistar rats. _Endocrinology_ 146, 913–919 (2005). Article CAS PubMed Google
Scholar * Morton, N. M., Ramage, L. & Seckl, J. R. Down-regulation of adipose 11beta-hydroxysteroid dehydrogenase type 1 by high-fat feeding in mice: a potential adaptive mechanism
counteracting metabolic disease. _Endocrinology_ 145, 2707–2712 (2004). Article CAS PubMed Google Scholar * Fenton, C. G. et al. 11beta-HSD1 plays a critical role in trabecular bone loss
associated with systemic glucocorticoid therapy. _Arthritis Res Ther._ 21, 188 (2019). Article CAS PubMed PubMed Central Google Scholar * Li, H. et al. 11beta-Hydroxysteroid
Dehydrogenase Type 1 Facilitates Osteoporosis by Turning on Osteoclastogenesis through Hippo Signaling. _Int J. Biol. Sci._ 19, 3628–3639 (2023). Article ADS CAS PubMed PubMed Central
Google Scholar * Yang, J. et al. Blocking glucocorticoid signaling in osteoblasts and osteocytes prevents mechanical unloading-induced cortical bone loss. _Bone_ 130, 115108 (2020). Article
CAS PubMed Google Scholar * Henneicke, H. et al. Skeletal glucocorticoid signalling determines leptin resistance and obesity in aging mice. _Mol. Metab._ 42, 101098 (2020). Article CAS
PubMed PubMed Central Google Scholar * Brennan-Speranza, T. C. et al. Osteoblasts mediate the adverse effects of glucocorticoids on fuel metabolism. _J. Clin. Invest_ 122, 4172–4189
(2012). Article CAS PubMed PubMed Central Google Scholar * Morton, N. M. et al. Improved lipid and lipoprotein profile, hepatic insulin sensitivity, and glucose tolerance in
11beta-hydroxysteroid dehydrogenase type 1 null mice. _J. Biol. Chem._ 276, 41293–41300 (2001). Article CAS PubMed Google Scholar * Morton, N. M. et al. Novel Adipose Tissue–Mediated
Resistance to Diet-Induced Visceral Obesity in 11beta-Hydroxysteroid Dehydrogenase Type 1–Deficient Mice. _Diabetes_ 53, 931–938 (2004). Article CAS PubMed Google Scholar * Wei, J. et
al. Glucose Uptake and Runx2 Synergize to Orchestrate Osteoblast Differentiation and Bone Formation. _Cell_ 161, 1576–1591 (2015). Article CAS PubMed PubMed Central Google Scholar *
Dirckx, N. et al. Vhl deletion in osteoblasts boosts cellular glycolysis and improves global glucose metabolism. _J. Clin. Invest_ 128, 1087–1105 (2018). Article PubMed PubMed Central
Google Scholar * Fulzele, K. et al. Insulin receptor signaling in osteoblasts regulates postnatal bone acquisition and body composition. _Cell_ 142, 309–319 (2010). Article CAS PubMed
PubMed Central Google Scholar * Fenton, C. et al. Local steroid activation is a critical mediator of the anti-inflammatory actions of therapeutic glucocorticoids. _Ann. Rheum. Dis._ 80,
250–260 (2021). Article CAS PubMed Google Scholar * Fenton, C. G. et al. 11beta-Hydroxysteroid Dehydrogenase Type 1 within Osteoclasts Mediates the Bone Protective Properties of
Therapeutic Corticosteroids in Chronic Inflammation. _Int. J. Mol. Sci._ 23, 7334 (2022). * Hardy, R. S. et al. 11 Beta-hydroxysteroid dehydrogenase type 1 regulates synovitis, joint
destruction, and systemic bone loss in chronic polyarthritis. _J. Autoimmun._ 92, 104–113 (2018). Article CAS PubMed PubMed Central Google Scholar * Lavery, G. G. et al. Lack of
significant metabolic abnormalities in mice with liver-specific disruption of 11beta-hydroxysteroid dehydrogenase type 1. _Endocrinology_ 153, 3236–3248 (2012). Article CAS PubMed PubMed
Central Google Scholar * Ducy, P. et al. Leptin Inhibits Bone Formation through a Hypothalamic Relay: A Central Control of Bone Mass. _Cell_ 100, 197–207 (2000). Article CAS PubMed
Google Scholar * Yang, J., Ueharu, H. & Mishina, Y. Energy metabolism: A newly emerging target of BMP signaling in bone homeostasis. _Bone_ 138, 115467 (2020). Article CAS PubMed
PubMed Central Google Scholar * Gao, H. et al. Bone marrow adipoq(+) cell population controls bone mass via sclerostin in mice. _Signal Transduct. Target Ther._ 8, 265 (2023). Article CAS
PubMed PubMed Central Google Scholar * Mittelstadt, P. R. & Ashwell, J. D. Inhibition of AP-1 by the glucocorticoid-inducible protein GILZ. _J. Biol. Chem._ 276, 29603–29610 (2001).
Article CAS PubMed Google Scholar * Leclerc, N., Noh, T., Khokhar, A., Smith, E. & Frenkel, B. Glucocorticoids inhibit osteocalcin transcription in osteoblasts by suppressing
Egr2/Krox20-binding enhancer. _Arthritis Rheum._ 52, 929–939 (2005). Article CAS PubMed Google Scholar * Zaman, G. et al. Loading-related Regulation of Transcription Factor EGR2/Krox-20
in Bone Cells Is ERK1/2 Protein-mediated and Prostaglandin, Wnt Signaling Pathway-, and Insulin-like Growth Factor-I Axis-dependent. _J. Biol. Chem._ 287, 3946–3962 (2012). Article CAS
PubMed Google Scholar * Molinaro, A. et al. Insulin-Driven PI3K-AKT Signaling in the Hepatocyte Is Mediated by Redundant PI3Kα and PI3Kβ Activities and Is Promoted by RAS. _Cell Metab._
29, 1400–1409.e1405 (2019). Article CAS PubMed Google Scholar * He, Y. et al. Targeting PI3K/Akt signal transduction for cancer therapy. _Signal Transduct. Target Ther._ 6, 425 (2021).
Article CAS PubMed PubMed Central Google Scholar * Leto, D. & Saltiel, A. R. Regulation of glucose transport by insulin: traffic control of GLUT4. _Nat. Rev. Mol. Cell Biol._ 13,
383–396 (2012). Article CAS PubMed Google Scholar * Zisman, A. et al. Targeted disruption of the glucose transporter 4 selectively in muscle causes insulin resistance and glucose
intolerance. _Nat. Med._ 6, 924–928 (2000). Article CAS PubMed Google Scholar * Gámez, B., Rodríguez‐Carballo, E., Graupera, M., Rosa, J. L. & Ventura, F. Class I PI‐3‐Kinase
Signaling Is Critical for Bone Formation Through Regulation of SMAD1 Activity in Osteoblasts. _J. Bone Min. Res_ 31, 1617–1630 (2016). Article Google Scholar * Li, D. et al.
Osteoclast-derived exosomal miR-214-3p inhibits osteoblastic bone formation. _Nat. Commun._ 7, 10872 (2016). Article ADS CAS PubMed PubMed Central Google Scholar * Gregory, S. et al.
11beta-hydroxysteroid dehydrogenase type 1 inhibitor use in human disease-a systematic review and narrative synthesis. _Metabolism_ 108, 154246 (2020). Article CAS PubMed Google Scholar
* Gathercole, L. L. et al. 11beta-Hydroxysteroid dehydrogenase 1: translational and therapeutic aspects. _Endocr. Rev._ 34, 525–555 (2013). Article CAS PubMed Google Scholar * Masuzaki,
H. F. & Jeffrey, S. Tissue-Specific Glucocorticoid Reactivating Enzyme, 11β-Hydroxysteoid Dehydrogenase Type 1 (11β-HSD1) - A Promising Drug Target for the Treatment of Metabolic
Syndrome. _Curr. Drug Targets: Immune, Endocr. Metab. Disord._ 3, 255–262 (2003). CAS Google Scholar * Tomlinson, J. W. & Stewart, P. M. Mechanisms of disease: Selective inhibition of
11beta-hydroxysteroid dehydrogenase type 1 as a novel treatment for the metabolic syndrome. _Nat. Clin. Pr. Endocrinol._ 1, 92–99 (2005). Article CAS Google Scholar * Scott, J. S.,
Goldberg, F. W. & Turnbull, A. V. Medicinal chemistry of inhibitors of 11beta-hydroxysteroid dehydrogenase type 1 (11beta-HSD1). _J. Med Chem._ 57, 4466–4486 (2014). Article CAS PubMed
Google Scholar * Chuanxin, Z. et al. Progress in 11beta-HSD1 inhibitors for the treatment of metabolic diseases: A comprehensive guide to their chemical structure diversity in drug
development. _Eur. J. Med Chem._ 191, 112134 (2020). Article PubMed Google Scholar * Liu, J. et al. Adipose tissue-targeted 11β-hydroxysteroid dehydrogenase type 1 inhibitor protects
against diet-induced obesity. _Endocr. J._ 58, 199–209 (2011). Article CAS PubMed Google Scholar * Zhang, G. et al. A delivery system targeting bone formation surfaces to facilitate
RNAi-based anabolic therapy. _Nat. Med._ 18, 307–314 (2012). Article PubMed Google Scholar * Abbas, A. et al. Effect of AZD4017, a Selective 11beta-HSD1 Inhibitor, on Bone Turnover
Markers in Postmenopausal Osteopenia. _J. Clin. Endocrinol. Metab._ 107, 2026–2035 (2022). Article PubMed PubMed Central Google Scholar * Othonos, N. et al. 11β-HSD1 inhibition in men
mitigates prednisolone-induced adverse effects in a proof-of-concept randomised double-blind placebo-controlled trial. _Nat. Commun._ 14, 1025 (2023). * Oda, S. et al. An Open-label Phase
I/IIa Clinical Trial of 11beta-HSD1 Inhibitor for Cushing’s Syndrome and Autonomous Cortisol Secretion. _J. Clin. Endocrinol. Metab._ 106, e3865–e3880 (2021). Article PubMed Google Scholar
* Yadav, Y. et al. Inhibition of 11beta-Hydroxysteroid dehydrogenase-1 with AZD4017 in patients with nonalcoholic steatohepatitis or nonalcoholic fatty liver disease: A randomized,
double-blind, placebo-controlled, phase II study. _Diabetes Obes. Metab._ 24, 881–890 (2022). Article CAS PubMed PubMed Central Google Scholar * Markey, K. et al. 11beta-Hydroxysteroid
dehydrogenase type 1 inhibition in idiopathic intracranial hypertension: a double-blind randomized controlled trial. _Brain Commun._ 2, fcz050 (2020). Article PubMed PubMed Central Google
Scholar * Rosenstock, J. et al. The 11-beta-hydroxysteroid dehydrogenase type 1 inhibitor INCB13739 improves hyperglycemia in patients with type 2 diabetes inadequately controlled by
metformin monotherapy. _Diabetes Care_ 33, 1516–1522 (2010). Article CAS PubMed PubMed Central Google Scholar * Feig, P. U. et al. Effects of an 11beta-hydroxysteroid dehydrogenase type
1 inhibitor, MK-0916, in patients with type 2 diabetes mellitus and metabolic syndrome. _Diabetes Obes. Metab._ 13, 498–504 (2011). Article CAS PubMed Google Scholar * Shah, S. et al.
Efficacy and safety of the selective 11beta-HSD-1 inhibitors MK-0736 and MK-0916 in overweight and obese patients with hypertension. _J. Am. Soc. Hypertens._ 5, 166–176 (2011). Article CAS
PubMed Google Scholar * Liang, C. et al. Aptamer-functionalized lipid nanoparticles targeting osteoblasts as a novel RNA interference-based bone anabolic strategy. _Nat. Med_ 21, 288–294
(2015). Article PubMed PubMed Central Google Scholar * Liu, J. et al. Exosomal transfer of osteoclast-derived miRNAs to chondrocytes contributes to osteoarthritis progression. _Nat.
Aging_ 1, 368–384 (2021). Article PubMed Google Scholar * Zhao, S. et al. Wound dressings composed of copper-doped borate bioactive glass microfibers stimulate angiogenesis and heal
full-thickness skin defects in a rodent model. _Biomaterials_ 53, 379–391 (2015). Article CAS PubMed Google Scholar * Dempster, D. W. et al. Standardized nomenclature, symbols, and units
for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. _J. Bone Min. Res_ 28, 2–17 (2013). Article Google Scholar * Lim, S. et al.
Immunoregulatory Protein B7-H3 Reprograms Glucose Metabolism in Cancer Cells by ROS-Mediated Stabilization of HIF1alpha. _Cancer Res_ 76, 2231–2242 (2016). Article CAS PubMed PubMed
Central Google Scholar * Song, R. et al. Central role of E3 ubiquitin ligase MG53 in insulin resistance and metabolic disorders. _Nature_ 494, 375–379 (2013). Article ADS CAS PubMed
Google Scholar * Xu, R., Sang, B. C., Navre, M. & Kassel, D. B. Cell-based assay for screening 11beta-hydroxysteroid dehydrogenase inhibitors using liquid chromatography/tandem mass
spectrometry detection. _Rapid Commun. Mass Spectrom._ 20, 1643–1647 (2006). Article ADS CAS PubMed Google Scholar * Rao, X. S. et al. AMPK-mediated phosphorylation enhances the
auto-inhibition of TBC1D17 to promote Rab5-dependent glucose uptake. _Cell Death Differ._ 28, 3214–3234 (2021). Article CAS PubMed PubMed Central Google Scholar Download references
ACKNOWLEDGEMENTS This work was supported by Theme-based Research Scheme (T12-201/20-R, A.P.L.) and General Research Fund (12106924 J.L., 12136616 J.L., 12103519 J.L., 12102120 Y.Y.Y.,
12102322 Y.Y.Y., 12100921 G.Z., and 12102223 G.Z.) and Early Career Scheme (22102823 J.L.) of Research Grants Council of Hong Kong SAR. This work is also supported by 2020 Guangdong
Provincial Science and Technology Innovation Strategy Special Fund (Guangdong-Hong Kong-Macau Joint Lab, No: 2020B1212030006 A.P.L.), Interdisciplinary Research Clusters Matching Scheme of
Hong Kong Baptist University (RC-IRCs/17-18/02 G.Z.), Research Matching Grant Scheme of Hong Kong Baptist University (RMGS2023 _15_03 G.Z.), Inter-institutional Collaborative Research Scheme
from Hong Kong Baptist University (Project No. RC-ICRS/19-20/01 G.Z.), International Science and Technology Corporation Key Program of Jiangxi Province (20232BBH80012 A.P.L.& J.L.), The
National Key R&D Program of China (2018YFA0800804 G.Z.), Youth’s Project of Guangdong Basic and Applied Basic Research Fund (2022A1515110044 D.J.L.). This work is also supported by The
Young Scientists Fund of the National Natural Science Foundation of China (82304378 S.F.Y.), Guangdong-Hong Kong Technology Cooperation Funding Scheme (GHP/149/21GD, 2023A0505010015 G.Z.),
The National Natural Science Foundation of China (52371251 F.Z.R.), The Fundamental and Applied Fundamental Research Fund of Guangdong Province (2022B1515120082 F.Z.R.), and The National
Natural Science Foundation of China (82373733 S.Z.W.). AUTHOR INFORMATION Author notes * These authors contributed equally: Chuanxin Zhong, Nanxi Li, Shengzheng Wang. AUTHORS AND
AFFILIATIONS * Law Sau Fai Institute for Advancing Translational Medicine in Bone and Joint Diseases, School of Chinese Medicine, Hong Kong Baptist University, Hong Kong SAR, China Chuanxin
Zhong, Nanxi Li, Dijie Li, Haitian Li, Wing Sze Yeung, Shuting Ma, Zhuqian Wang, Zhanghao Li, Xiaoxin Wen, Xiaohui Tao, Duoli Xie, Yihao Zhang, Zefeng Chen, Zhengming Liang, Luyao Wang, Yuan
Ma, Yuanyuan Yu, Fangfei Li, Aiping Lyu, Jin Liu & Ge Zhang * Department of Materials Science and Engineering, Southern University of Science and Technology, Shenzhen, Guangdong, China
Chuanxin Zhong, Shan He, Junqin Wang, Jianfeng Yan & Fuzeng Ren * Guangdong-Hong Kong-Macao Greater Bay Area International Research Platform for Aptamer-based Translational Medicine and
Drug Discovery, Hong Kong SAR, China Chuanxin Zhong, Luyao Wang, Sifan Yu, Yuan Ma, Yuanyuan Yu, Fangfei Li, Baoting Zhang, Aiping Lyu, Jin Liu & Ge Zhang * Institute of Systems Medicine
and Health Sciences, School of Chinese Medicine, Hong Kong Baptist University, Hong Kong SAR, China Chuanxin Zhong, Nanxi Li, Luyao Wang, Yuanyuan Yu, Fangfei Li, Aiping Lyu, Jin Liu &
Ge Zhang * Department of Medicinal Chemistry, School of Pharmacy, Fourth Military Medical University, Xi’an, Shaanxi, China Shengzheng Wang * Guangxi Universities Key Laboratory of Stem cell
and Biopharmaceutical Technology, College of Life Sciences, Guangxi Normal University, Gui Lin, China Dijie Li * The Second Affiliated Hospital of Guangzhou University of Chinese Medicine,
Guangzhou, China Zhihua Yang * Sports Medicine Center, The First Affiliated Hospital of Shantou University Medical College, Shantou, China Lin Du, Haorui Li & Zhigang Zhong * Department
of Joint Surgery, The Third Affiliated Hospital of Southern Medical University, The Third School of Clinical Medicine, Southern Medical University, Guangzhou, China Guangxin Huang * School
of Chinese Medicine, Faculty of Medicine, The Chinese University of Hong Kong, Hong Kong SAR, China Hewen Jiang, Huarui Zhang, Zongkang Zhang, Sifan Yu & Baoting Zhang * Department of
Sports Medicine and Rehabilitation, Peking University Shenzhen Hospital, Shenzhen, China Song Xue * International Medical Service Center, The First Affiliated Hospital of Shantou University
Medical College, Shantou, China Zeting Wu * Key Laboratory of Regenerative Medicine, Ministry of Education, School of Biomedical Sciences, Faculty of Medicine, The Chinese University of Hong
Kong, Hong Kong, China Chao Wan * Department of Biology, Southern University of Science and Technology, Shenzhen, China Chao Liang * Key Laboratory of Phytochemistry and Natural Medicines,
Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, China Yang Chen & Jin Liu * Guangdong-Hong Kong-Macau Joint Lab on Chinese Medicine and Immune Disease
Research, Hong Kong, China Aiping Lyu * Bone Research Program, ANZAC Research Institute, The University of Sydney, Sydney, Australia Hong Zhou Authors * Chuanxin Zhong View author
publications You can also search for this author inPubMed Google Scholar * Nanxi Li View author publications You can also search for this author inPubMed Google Scholar * Shengzheng Wang
View author publications You can also search for this author inPubMed Google Scholar * Dijie Li View author publications You can also search for this author inPubMed Google Scholar * Zhihua
Yang View author publications You can also search for this author inPubMed Google Scholar * Lin Du View author publications You can also search for this author inPubMed Google Scholar *
Guangxin Huang View author publications You can also search for this author inPubMed Google Scholar * Haitian Li View author publications You can also search for this author inPubMed Google
Scholar * Wing Sze Yeung View author publications You can also search for this author inPubMed Google Scholar * Shan He View author publications You can also search for this author inPubMed
Google Scholar * Shuting Ma View author publications You can also search for this author inPubMed Google Scholar * Zhuqian Wang View author publications You can also search for this author
inPubMed Google Scholar * Hewen Jiang View author publications You can also search for this author inPubMed Google Scholar * Huarui Zhang View author publications You can also search for
this author inPubMed Google Scholar * Zhanghao Li View author publications You can also search for this author inPubMed Google Scholar * Xiaoxin Wen View author publications You can also
search for this author inPubMed Google Scholar * Song Xue View author publications You can also search for this author inPubMed Google Scholar * Xiaohui Tao View author publications You can
also search for this author inPubMed Google Scholar * Haorui Li View author publications You can also search for this author inPubMed Google Scholar * Duoli Xie View author publications You
can also search for this author inPubMed Google Scholar * Yihao Zhang View author publications You can also search for this author inPubMed Google Scholar * Zefeng Chen View author
publications You can also search for this author inPubMed Google Scholar * Junqin Wang View author publications You can also search for this author inPubMed Google Scholar * Jianfeng Yan
View author publications You can also search for this author inPubMed Google Scholar * Zhengming Liang View author publications You can also search for this author inPubMed Google Scholar *
Zongkang Zhang View author publications You can also search for this author inPubMed Google Scholar * Zhigang Zhong View author publications You can also search for this author inPubMed
Google Scholar * Zeting Wu View author publications You can also search for this author inPubMed Google Scholar * Chao Wan View author publications You can also search for this author
inPubMed Google Scholar * Chao Liang View author publications You can also search for this author inPubMed Google Scholar * Luyao Wang View author publications You can also search for this
author inPubMed Google Scholar * Sifan Yu View author publications You can also search for this author inPubMed Google Scholar * Yuan Ma View author publications You can also search for this
author inPubMed Google Scholar * Yuanyuan Yu View author publications You can also search for this author inPubMed Google Scholar * Fangfei Li View author publications You can also search
for this author inPubMed Google Scholar * Yang Chen View author publications You can also search for this author inPubMed Google Scholar * Baoting Zhang View author publications You can also
search for this author inPubMed Google Scholar * Aiping Lyu View author publications You can also search for this author inPubMed Google Scholar * Fuzeng Ren View author publications You
can also search for this author inPubMed Google Scholar * Hong Zhou View author publications You can also search for this author inPubMed Google Scholar * Jin Liu View author publications
You can also search for this author inPubMed Google Scholar * Ge Zhang View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS Conceptualization:
G.Z., H.Z, and J.L., In vitro Investigation: C.X.Z., S.H., W.S.Y., Y.H.Z., Z.F.C., J.Q.W., J.F.Y., D.L.X., Z.M.L. In vivo Investigation: C.X.Z., N.X.L., D.J.L., S.T.M., H.W.J., H.R.Z.,
Z.H.L., X.X.W., H.T.L., S.X., Z.Q.W., X.H.T., Chemical synthesis and modification: S.Z.W., Clinical sample collection: L.D., G.X.H., H.R.L., Z.G.Z., Z.T.W., Maintenance and breeding of
genetically engineered mouse model: Z.Z.K., S.F.Y., C.W., Data analysis: C.X.Z., N.X.L., Z.H.Y., C.L., Y.C., L.Y.W., Y.M., Y.Y.Y., and F.F.L., Funding acquisition: A.P.L., S.Z.W., F.Z.R.,
J.L., and G.Z., Project administration: G.Z., J.L., B.T.Z. and A.P.L., Supervision: G.Z., J.L., H.Z. and F.Z.R., Manuscript draft: C.X.Z., and J.L. Manuscript review & editing: J.L.,
H.Z., and G.Z. CORRESPONDING AUTHORS Correspondence to Aiping Lyu, Fuzeng Ren, Hong Zhou, Jin Liu or Ge Zhang. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing
interests. PEER REVIEW PEER REVIEW INFORMATION _Nature Communications_ thanks Jan Tuckermann and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A
peer review file is available. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION REPORTING SUMMARY SOURCE DATA SOURCE DATA PEER REVIEW FILE RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative
Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as
you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have
permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s
Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not
permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit
http://creativecommons.org/licenses/by-nc-nd/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Zhong, C., Li, N., Wang, S. _et al._ Targeting osteoblastic 11β-HSD1 to
combat high-fat diet-induced bone loss and obesity. _Nat Commun_ 15, 8588 (2024). https://doi.org/10.1038/s41467-024-52965-4 Download citation * Received: 16 March 2024 * Accepted: 27
September 2024 * Published: 04 October 2024 * DOI: https://doi.org/10.1038/s41467-024-52965-4 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content:
Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative