Play all audios:
ABSTRACT Enhancing the efficacy of immunotherapy in brain metastases (BrM) requires an improved understanding of the immune composition of BrM and how this is affected by radiation and
dexamethasone. Our two-arm pilot study (NCT04895592) allocated 26 patients with BrM to either low (Arm A) or high (Arm B) dose peri-operative dexamethasone followed by pre-operative
stereotactic radiosurgery (pSRS) and resection (_n_= 13 per arm). The primary endpoint, a safety analysis at 4 months, was met. The secondary clinical endpoints of overall survival, distant
brain failure, leptomeningeal disease and local recurrence at 12-months were 66%, 37.3%, 6%, and 0% respectively and were not significantly different between arms (_p_= 0.7739, _p_= 0.3884,
_p_= 0.3469). Immunological data from two large retrospective BrM datasets and confirmed by correlates from both arms of this pSRS prospective trial revealed that BrM CD8 T cells were
composed of predominantly PD1+ TCF1+ stem-like and PD1+ TCF1-TIM3+ effector-like cells. Clustering of TCF1+ CD8 T cells with antigen presenting cells in immune niches was prognostic for
local control, even without pSRS. Following pSRS, CD8 T cell and immune niche density were transiently reduced compared to untreated BrM, followed by a rebound 6+ days post pSRS with an
increased frequency of TCF1- effector-like cells. In sum, pSRS is safe and therapeutically beneficial, and these data provide a framework for how pSRS may be leveraged to maximize
intracranial CD8 T cell responses. SIMILAR CONTENT BEING VIEWED BY OTHERS EVALUATION OF THE IMPACT OF PRE-OPERATIVE STEREOTACTIC RADIOTHERAPY ON THE ACUTE CHANGES IN HISTOPATHOLOGIC AND
IMMUNE MARKER PROFILES OF BRAIN METASTASES Article Open access 16 March 2022 A RETROSPECTIVE STUDY OF RADIOTHERAPY COMBINED WITH IMMUNOTHERAPY FOR PATIENTS WITH BASELINE BRAIN METASTASES
FROM NON-SMALL CELL LUNG CANCER Article Open access 27 February 2025 STEREOTACTIC RADIOSURGERY FOR PATIENTS WITH BRAIN METASTASES: CURRENT PRINCIPLES, EXPANDING INDICATIONS AND OPPORTUNITIES
FOR MULTIDISCIPLINARY CARE Article 19 March 2025 INTRODUCTION CD8 T cell infiltration in primary and metastatic malignancies is associated with improved outcomes and superior responses to
immunotherapy1,2,3,4. Studies of the CD8 T cell response to tumors have highlighted the importance of a TCF1+ stem-like subset. This cell self-renews and gives rise to cytotoxic effector
daughter cells in the setting of chronic antigen exposure5 and across several human tumor types6,7,8,9,10. Functional experiments and TCR sequencing analysis indicate stem-like CD8 T cells
retain proliferative capacity and are likely the source of anti-tumor effector CD8 T cells8,11,12. Our prior work showed these cells reside in close proximity to densely clustered MHC-II+
antigen-presenting cells (APCs) in the tumor, which we termed antigen-presenting niches, or immune niches13. We also recently reported that these stem-like TCF1+ CD8 T cells in head and neck
tumors are specific for HPV antigens, and upon restimulation with their cognate antigen, proliferate and differentiate to the effector state8. Furthermore, numerous studies correlate the
presence of this CD8 T cell population in the tumor with response to PD-1 blockade, highlighting their importance in the immune response to cancer10,14,15. Brain metastases (BrM), despite
residing in an immune-privileged organ, have notable immunologic similarities to other metastatic sites, including the presence of antigen-experienced TCF1+ CD8 T cells16,17,18,19. A recent
study also suggested that these cells may have overlapping TCR clonotypes with a more exhausted pool of TOX+ CD8 T cells present in BrM19. Furthermore, the specific spatial organization of
BrM immune infiltrates appears to influence prognosis20. However, whether the TCF1+ CD8 T cell population is organized into antigen-presenting niches and whether this also has prognostic
value is unknown. Importantly, for patients with a limited number of large or symptomatic BrM, surgery followed by stereotactic radiosurgery (SRS) (post-operative) is frequently
recommended21,22. Our group has been a pioneer in the pre-operative delivery of SRS23. Given the known immunostimulatory activity of radiation, pre-operative SRS (pSRS) may potentiate the
intracranial activity of immunotherapy24,25. In this work, we performed both a retrospective analysis and completed a prospective clinical trial evaluating the safety, efficacy, and
immunomodulatory activity of pSRS and peri-operative steroid dosing. We found that pSRS can be delivered safely, and that dexamethasone dose did not have a significant impact on clinical
outcomes. For the immunological correlates, we evaluated the organization of TCF1+ PD1+ T cells in BrM under baseline conditions and the association of this biology with patient outcomes
finding that increased BrM niche density was associated with longer local BrM control. We also assessed the impact of pSRS on the intracranial CD8 T cell immune response, finding that pSRS
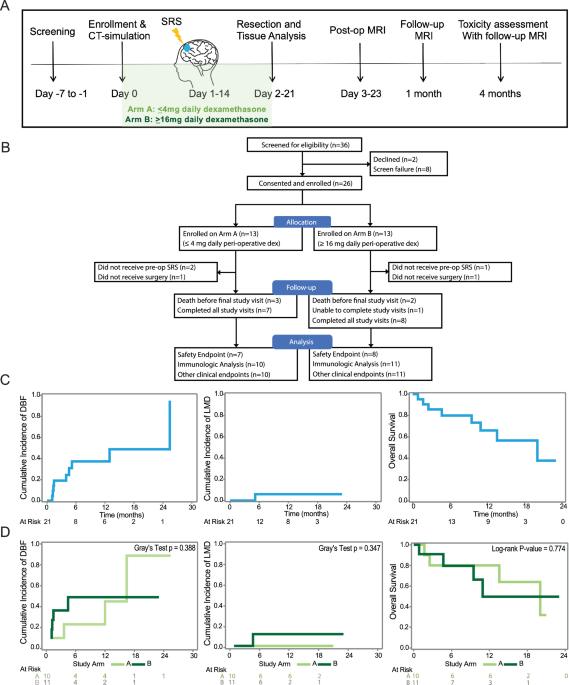
is immunomodulatory, altering both the T cell and APC compartments, while the overall immune organization is maintained. RESULTS STUDY DESIGN AND PATIENT DISPOSITION To prospectively
investigate the impact of both pre-operative SRS (pSRS) and dexamethasone on brain metastases (BrM) clinical outcomes and CD8 T cell/APC subsets, a total of 36 patients with BrM were
screened and 26 were enrolled on a pSRS clinical trial between August 2021 and April 2023 (Fig. 1A, B). Due to common dexamethasone administration for patients with BrM and unknown effects
on clinical outcomes and the tumor immune microenvironment, enrollees were allocated to Arm A ( ≤ 4 mg of peri-operative dexamethasone daily) or Arm B ( ≥ 16 mg daily) in an alternating
fashion, followed by pSRS and surgical resection for both arms. The characteristics for the 26 patients enrolled are shown by Arm in Supplementary Table 1. A total of 21 patients completed
all therapy and were included in our analysis (Table 1). PRIMARY AND SECONDARY OUTCOMES For these 21 patients, there were 3 deaths in Arm A prior to the final study visit, 2 in Arm B, and 1
patient in Arm B was unable to complete the final study visit leading to a final total of 15 patients reaching the pre-specified safety analysis endpoint of 4 months (Fig. 1B). The median
follow-up time was 10.32 months (range: 0.84-23.04). Toxicities for the entire treated cohort are summarized in Table 2, with only two grade 3 or greater toxicities observed and attributable
to therapy (altered mental status and cerebral edema, both in the same patient on Arm B) during the trial. Clinical endpoints including the cumulative incidence of local recurrence, distant
brain failure (DBF), leptomeningeal disease (LMD), and overall survival (OS) were also assessed. The 12-month local recurrence was 0%. The 12-month cumulative incidence of DBF and LMD
competing with death were 37.3% and 6.0%, respectively (Fig. 1C). The 12-month cumulative incidence of DBF was 23.1% for Arm A and 49.1% for Arm B (Gray test _p_= 0.3884; 95% CI:
0.033-0.533) (Fig. 1D). The 12-month cumulative incidence of LMD was 0.0% for Arm A and 11.4% for Arm B (Gray test _p_= 0.3469; 95% CI: N/A-0.394) (Fig. 1D). The 12-month overall survival
was 66.0% (Fig. 1C). The 12-month overall survival was 80.5% for Arm A and 49.7% for Arm B (_p_= 0.7739; 95% CI: 0.20-3.33) (Fig. 1D). This trial met its primary safety endpoint.
Additionally, the secondary clinical outcomes, similar to results seen on other retrospective cohort studies for pre-operative SRS26, compare favorably in both safety and outcomes to
historic controls of upfront resection23. EXPLORATORY IMMUNOLOGIC OUTCOMES WITH COMPARISON DATASETS TCF1+ PD1+ STEM-LIKE T CELLS ARE FOUND IN BRAIN METASTASES AND RESIDE IN IMMUNOLOGICAL
NICHES To characterize the CD8 T cell response in untreated BrM, we first performed flow cytometry analysis on freshly resected tissue from six patients in an independent cohort not treated
on our prospective trial (Fig. 2A, Supplementary Fig. 1A). The total CD8 T cell infiltration was variable across patients (Supplementary Fig. 1B), with most tumor infiltrating CD8 T cells
expressing PD1 and negative for CD45RA, indicative of antigen reactivity (Fig. 2B). Among PD1 + CD8 T cells, we identified an effector-like population expressing inhibitory molecules (TIM3
and CD39), as well as the residency marker CD69 (Fig. 2B, C). In contrast, a second CD8 T cell population, which made up around 30% of the PD1+ response, was negative for effector and
inhibitory molecules but retained high expression of the transcription factor TCF1 in BrM across patients (Fig. 2B, C). Furthermore, these cells also expressed high levels of the
co-stimulatory molecule CD28, and the IL-7 receptor compared to TCF1- effector-like CD8 T cells (Fig. 2B, C). As the phenotype of this population is consistent with previously defined
stem-like CD8 T cells in other tumor types outside of the CNS and chronic infections5,7,12, we also refer to these cells as “stem-like”. To further characterize and acquire spatial
information of the BrM immune landscape, we evaluated 67 up-front resected samples at Winship Cancer Institute of Emory University and performed quantitative multiparametric
immunofluorescence (Fig. 2D, E, Supplementary Tables 2-3). This retrospective cohort of BrM, which were not treated on our prospective study, underwent upfront resection (Res) and included
non-small cell lung cancer (NSCLC), melanoma, or breast cancer; none of which had previously received immunotherapy (Supplementary Table 2). We found a high degree of variation between
patients in the infiltration of CD8 and CD4 T cells, as well as of MHC-II+ antigen presenting cells (APCs) (Fig. 2E). Consistent with the flow cytometry results, the majority of CD8 T cells
expressed PD1 (95%) and were found throughout the tumor tissue (Supplementary Fig. 1C–F). Additionally, approximately 40% of the total CD8 T cell population were TCF1+ (stem-like) and the
remainder TCF1- cells (effector-like) (Supplementary Fig. 1F, G, Fig. 2E). It is worth noting that the frequency of TCF1+ CD8 T cells (of DAPI+ cells) correlated with the frequency of total
CD8 T cells (R2 = 0.6233, p < 0.001) suggesting that this population could sustain the overall CD8 T cell response in BrM (Supplementary Fig. 1H). In our prior work, we found that
stem-like TCF1+ CD8 T cells were not randomly distributed in kidney tumors, but instead resided in densely clustered APC immune niches7. Here, we found a correlation between the density of
TCF1+ CD8 T cells and MHC-II+ APCs (Supplementary Fig. 1H) in BrM, suggesting a similar relationship. To quantitively assess the proximity of CD8 T cell populations with APCs, we generated
contour maps of the density of MHC-II+ cells and overlayed the x, y location of TCF1+ CD8 T cells (green) and of TCF1- CD8 T cells (red) (Fig. 2F). We found that TCF1+ CD8 T cells resided in
areas of higher MHC-II+ cell density (Fig. 2F), and on average were significantly closer to their nearest MHC-II+ cell neighbor than TCF1- CD8 T cells (Fig. 2G). In contrast, TCF1- CD8 T
cells were distributed throughout the tumor without any preference for APC zones, consistent with the biology of PD1+ TCF1+ CD8 T cells specifically requiring these organized immune niches
within the tumor. Prior data suggested that immune niches were important to sustain the CD8 T cell response within non-CNS tumors13. Notably, our BrM cohort showed a correlation between
immune niche density and the frequency of total CD8 T cell infiltration (Fig. 2H), suggesting a similar supportive role for immune niches in BrM (Supplementary Fig. 2A–F). Although our
patient cohort was comprised of a diverse set of primary tumor types, we did not find significant differences in number of total CD8 T cells, TCF1+ , TCF1- CD8 T cell subsets/mm2 or niche
density by tumor histology (Supplementary Table 3, Supplementary Fig. 2G, K). Moreover, we did not find any significant association between dexamethasone dose and CD8 cells per mm2, TCF1+
frequency or immune niche proportion, despite the known effects of glucocorticoids on CD8 T cells27,28 (Supplementary Table 3). Overall, these data indicate that CD8 T cell responses are
successfully mounted in BrM, despite being present in an immune privileged site. Moreover, these PD1+ TCF1+ CD8 T cells reside within APC-dense immune niches, which are maintained in the
presence of dexamethasone. HIGH IMMUNE NICHE DENSITY IS ASSOCIATED WITH LONGER BRM LOCAL CONTROL CD8 T cell infiltration and a high density of local immune niches have previously been
associated with improved clinical outcomes in solid tumors1,2,3,4. Thus, we next examined whether high CD8 T cell density or immune niches are associated with a longer time to BrM local
recurrence in our retrospective dataset. We performed a competing risk analysis in a patient cohort that did not receive pSRS, allowing us to account for the competing events of death and
local recurrence. Although total CD8 T cell density was not associated with disease progression as previously seen for other solid malignancies (Supplementary Fig. 2L), BrM tumors with a
greater frequency of PD1+ TCF1+ CD8 T cells or a higher density of immune niches were significantly associated with longer time to local recurrence (Fig. 2I). A representative patient with a
high density of TCF1+ CD8 T cells and immune niches distributed throughout the BrM is highlighted, where the resection cavity remained free of local recurrence at ten years (Fig. 2J). In
contrast, a representative patient with low TCF1+ CD8 T cell infiltration and scant immune niche formation developed local recurrence within six months after treatment (Fig. 2K). Together,
these data suggest that the formation of immune niches appears to capture a link between the immune microenvironment and patient outcomes. These favorable responses are not evident with bulk
CD8 T cell analysis alone, highlighting the importance of stem-like CD8 T cells and immune niche organization in restraining tumor growth and local recurrence. THE IMPACT OF PRE-OPERATIVE
SRS ON T CELL SUBSETS AND THE IMMUNE NICHE In preclinical studies, focal radiation stimulates innate immune responses via the c-GAS-STING pathway and increases type I interferon
signaling29,30. Additionally, we previously found focal radiation increased stem-like CD8 T cell infiltration into the tumor in murine cancer models25. However, whether pSRS has similar
immunostimulatory activity in the brain and an analogous impact on the BrM immune response in patients is not known. To examine the impact of pSRS on BrM, we performed quantitative IF on 76
patients who were treated with pSRS 0-11 days prior to surgery from our retrospective dataset. Relevant patient, BrM, and pSRS characteristics are detailed in Supplementary Table 4. As a
comparison, we included the retrospective upfront resection cohort (Res) for this analysis. Importantly, the pSRS and Res cohort characteristics were overall very similar, including in BrM
volume (Fig. 3A, Supplementary Table 5). The pSRS BrM had a wide range of T infiltration, with CD4 and CD8 T cells ranging from 8 to over 100 cells per mm2 of tissue, which was significantly
lower than Res BrM tumors (Fig. 3B and Supplementary Fig. 3A–D). Although total CD8 T cell numbers were reduced, the majority of infiltrating CD8 T cells in pSRS tumors exhibited a PD1+
phenotype similar to that of the Res cohort, with comparable proportions of stem-like (TCF1+) and effector-like (TCF1-) populations (Supplementary Fig. 3E, F). However, although similar in
frequency, the numbers of both TCF1+ and TCF1- CD8 T cells were reduced in pSRS tumors (Fig. 3B). We validated these results by flow cytometry, with a total reduction in CD8 T cells across
pSRS BrM tumors (Supplementary Fig. 4A-B), while exhibiting similar PD1+ TCF1+ and TCF1- CD8 T cell phenotypes compared to Res BrM counterparts (Supplementary Fig. 4C-F). Interestingly, the
number of MHC-II+ APCs were not significantly different between the two cohorts (Fig. 3B), indicating that pSRS may differentially impact immune populations in the brain. Given the
maintenance of MHCII+ cells, we next assessed whether immune niche organization was impacted by pSRS. We found immune niche density in pSRS tumors was significantly reduced compared to Res
tumors (Fig. 3C), while the positive correlation between CD8 T cell density and immune niches was maintained (Fig. 3D). Importantly, this relationship was not impacted by dexamethasone dose
(_p_= 0.990) or BrM histologies (Supplementary Fig. 3G, K). In correlates from our prospective trial (Supplementary Fig. 5A–N) for the high dose (Arm B) and low dose (Arm A) of
dexamethasone, we observed no significant differences in the density of total CD8 T cells or PD1+ CD8 T cell subsets per mm2 between the study arms (Supplementary Fig. 6A–D). Nor was there a
significant difference between MHC-II+ APC infiltration or immune niche density (Supplementary Fig. 6E–I). Overall, these data indicate that pSRS reduces local CD8 T cell numbers in BrM
independent of dexamethasone dose, with limited effect on MHCII+ APCs. Importantly, PD1+ TCF1+ stem-like CD8 T cells and intratumoral immune niche organization were maintained. We then
assessed the temporal effects of pSRS on the immune infiltrates in our retrospective cohort. In patients who had BrM resected on the same day as pSRS (Day 0), there was no significant
difference in the number of total CD8, TCF1+ , or TCF1- cells in the BrM compared to Res patients (Supplementary Fig. 7A–C). In contrast, tissue resected between 1-5 days post pSRS had
significantly lower numbers per mm2 of total CD8 T cells, as well as reduced TCF1+ , and a trend for lower TCF1- effector-like CD8 T cells (Fig. 3E–J, Supplementary Fig. 7D). Interestingly,
this difference was not seen in BrM which were resected 6 or more days after pSRS. These patients exhibited a rebound of TCF1- effector-like CD8 T cells to baseline numbers, while PD1+ TCF1+
CD8 T cells remained depressed (Fig. 3I, J). Notably, these changes were also reflected in the frequency distribution of PD1+ CD8 T cell populations, with an increase in TCF1- CD8 T cell
frequency and a concomitant reduction in the TCF1+ CD8 T cells (Fig. 3K, L). Although MHCII+ APC numbers did not significantly change over the treatment time course (Supplementary Fig. 7E,
F), there was a trend for a reduced immune niche density that persisted through the 6+ timepoint (Fig. 3M, Supplementary Fig. 7G). This effect is consistent with the reduction in the TCF1+
CD8 T cell population,which primarily resides within these organized clusters. Overall, these data show a broad and rapid decrease in CD8 T cell numbers in BrM following pSRS. Although CD8 T
cell numbers were reduced, BrM retained immune niche composition with MHC-II+ APCs following pSRS therapy. Additionally, our data suggest that TCF1- CD8 T cells rapidly rebound. Given the
extensive data in pre-clinical models of TCF1+ CD8 T cells being the precursor to the TCF1- populations, we hypothesize that the relative abundance of TCF1- CD8 T cells at day 6+ following
pSRS may be driven by differentiation of the TCF1+ population within the BrM. PSRS SHIFTS THE DENDRITIC CELL SIGNATURE TOWARDS A PRO-INFLAMMATORY PHENOTYPE AND PROMOTES THE ACCUMULATION OF
THE TD1 GZMB HIGH EFFECTOR SUBSET To further examine the impact of pSRS on immune cells in BrMs, we performed single cell RNAseq on 13 pSRS BrM from the prospective trial and 18 Res BrM
collected prospectively off trial. For this analysis, we separately sorted CD8 T cells and the remaining CD45+ cells from patients’ tumors and mixed them 1:1 to enrich for infiltrating CD8 T
cells (Supplementary Fig. 8A). Flow cytometry quantification showed a wide range of CD45+ cell infiltration, with pSRS patients exhibiting a lower frequency of total CD45+ cells in their
tumors, which was consistent across pSRS dosage and fractionation (Supplementary Fig. 8B, C, Table 1). Transcriptional analysis identified six clusters of CD45+ immune populations within our
BrM cohort of multiple histologies, including CD8 T cells _(CD3, CD8B)_, CD4 T cells _(CD3, CD4, FOXP3)_, activated B cells _(CD79A, MS4A1, IGHM)_, antibody secreting cells (ASCs; _MZB11,
IGKC)_, plasma cells _(MZB1, PRDM1, IGKC)_, and a grouped myeloid antigen presenting cell population (Macs/DCs; _CD68, ITGAX, ITGAM)_ (Supplementary Fig. 8D–F). We found that pSRS BrM were
enriched for activated B cells and consistently had a reduced frequency of CD4 T cells, indicating that pSRS may have differential effects on various immune populations (Supplementary Fig.
8G). Given that immune niches are primarily composed of myeloid APCs, we next examined whether pSRS impacts their transcriptional program. After sub-clustering the myeloid APCs, we found
four distinct populations, including macrophages (_ITGAM, CD68, FCGR3A, LYZ)_, activated dendritic cells (_ITGAX, LYZ, IL1B, HSP1AB_), classical dendritic cells type 2 (cDC2s) (_ITGAX,
SIRPA, CD109, CD300E)_, and monocytes (_ITGAM, CD14, C1QA)_ (Supplementary Fig. 8H). We focused on activated DCs, given their consistent presence across patient BrMs, whereas the other APC
population frequencies were highly variable and driven by single patient samples. Activated DCs were characterized by high expression of antigen processing machinery, co-stimulation markers,
as well as cytokines and chemokines related to immune cell recruitment _(IL1B, CXCL8, CXCL16)_ across both groups (Supplementary Fig. 8I). Interestingly, activated DCs from pSRS BrM had
significantly higher expression of chemokines _CXCL3_ and _CXCL2_, which are associated with inflammatory recruitment of innate immune cells, and higher expression of complement associated
genes (Supplementary Fig. 8J). In agreement, activated DCs from pSRS enriched for pathways related to inflammatory immune response, recruitment of innate immune populations, and complement
cascades (Supplementary Fig. 8K), suggesting pSRS may shift the myeloid populations towards a more inflammatory state. Given that pSRS reduces local BrM CD8 T cell numbers (Fig. 4A), we next
examined whether the transcriptional program of CD8 T cell populations was altered by pSRS. Single cell RNAseq analysis identified four populations of activated CD8 T cells, including
stem-like, transitory, GZMB high effector (TD1), and GZMB low effector (TD2) cells, as well as a population of naive/memory CD8 T cells across both groups (Fig. 4B, C, Supplementary Fig. 9A,
B). Stem-like CD8 T cells (cluster 1) were characterized by high expression of the transcription factor _TCF7_, encoding TCF1, and had low levels of effector and cytotoxic molecules (Fig.
4C and Supplementary Fig. 9B). These _TCF7_+ CD8 T cells transcriptionally differed from naive/memory CD8 T cell subsets, with low expression of _SELL_ and increased expression of activation
associated genes _CXCR3, GPR183_ and LGALS3 (Fig. 4C). Cluster 1, _TCF7_+ CD8 T cells enriched for the human stem-like signature, previously defined in antigen specific CD8 T cells in HPV+
head and neck tumors8 (Supplementary Fig. 9C), further supporting a precursor-like phenotype. Cluster 2 was defined as a transitory population, characterized by high expression of _CD69_,
the transcription factors _JUNB_ and _FOS_, TCR signaling dependent gene _NR4A2_, and effector cytokine _IFNG_ (Fig. 4C and Supplementary Fig. 9B). The remaining activated CD8 T cells had an
effector transcriptional profile with low expression of _TCF7_, high levels of _PRDM1_, along with various cytotoxic molecules and inhibitory receptors (Supplementary Fig. 9B). While both
cluster 3 and cluster 4 populations enriched for an effector signature (Supplementary Fig. 9D), we detected a distinct pattern of expression across cytotoxic molecules, transcription
factors, and inhibitory receptors. A subset of effector CD8 T cells (TD1) expressed high levels of _GZMB, PRF1, CX3CR1_, and _GNLY_ while a second population (TD2) expressed _GZMK, EOMES,
TIGIT_, and _LAG3_ (Fig. 4C and Supplementary Fig. 9B), suggesting distinct differentiation states. Notably, although both Res and pSRS CD8 T cells clustered together (Fig. 4D, E and
Supplementary Fig. 9A), pSRS BrM exhibited a significant enrichment of CD8 T cells in cluster 3 (TD1 GZMB high) and a trend towards a lower proportion of cells within clusters 2 (Transitory)
and 4 (TD2 GZMB low) (Fig. 4D). Evaluating these populations as a function of time from pSRS to surgery, we found a trend towards an increase in TD1 _GZMB_+ CD8 T cells with a concomitant
reduction in _TCF7+_ stem-like CD8 T cells frequency at 6+ days after pSRS (Supplementary Fig. 9E). In the overall pSRS group, these CD8 T cell populations were not significantly altered by
high relative to low dose dexamethasone (Supplementary Fig. 9F). Furthermore, these differences in frequency within cluster were no longer significant when comparing CD8 T cell distribution
as a proportion of total CD45+ cells, due to the reduced overall CD8 T cell infiltration in pSRS BrM (Fig. 4E). To gain deeper insight into the transcriptional changes induced by pSRS on CD8
T cell subsets, we performed differential gene expression (DEG) analysis across treatment groups. Stem-like, _GZMB_ high TD1, _GZMB_ low TD2, and transitory CD8 T cells exhibited 74, 135,
194, and 118 upregulated genes in pSRS BrMs, respectively (Supplementary Fig. 9G). The stem-like, TD2 and transitory clusters had enrichment in pathways related to apoptosis, DNA damage and
cellular response to stress (Fig. 4F, Supplementary Fig. 9G-I). In contrast, _GZMB_ high TD1 effector cells showed enrichment for pathways related to cytotoxicity and effector
differentiation (Fig. 4G, Supplementary Fig. 9G). Among the top upregulated genes in this population were _KLF2_ and natural killer (NK) receptors _KLRG1, KIR3DL1_, which have previously
been associated with exhausted CD8 T cells and enhanced effector functions (Fig. 4H, Supplementary Fig. 9G). In contrast, CD8 T cells from Res BrMs showed enhanced expression of chemokine
receptors _(CXCR3, CXCR6)_, cytokine signaling pathways _(STAT1)_, interferon induced genes (_IFITM1, OASL)_, and inhibitory receptors (_TIGIT)_ (Fig. 4H, Supplementary Fig. 9G). Overall,
these data indicate that pSRS induces a reduction in total CD8 T cell infiltration and promotes transcriptional changes associated with cellular stress, DNA damage repair, and cytotoxicity
among PD1+ CD8 T cell subsets. Given the co-localization of CD8 T cells near APC-rich immune niches, we next explored the possible interactions between these cells and how they might be
modulated by pSRS. We performed Niche-Net sender-receiver analysis between the top differentially expressed ligands on activated DCs and differentially expressed receptors on CD8 T cell
subsets from Res and pSRS groups. Res BrM DCs expressed high levels of lymphotoxin alpha (_LTA)_ and other TNF family members, which were predicted to interact with TNF receptors across CD8
T cell populations (Fig. 4I). Moreover, DC _CXCL9_ and _CCL2_ were predicted as top chemokine receptor pairs to recruit _CXCR3_ and _CCR5_ expressing CD8 T cell subsets, which might
facilitate co-localization of these populations in immune niches in the BrM (Fig. 4I). Notably, pSRS altered the array of ligand-receptor pairs expressed among these populations. Chemokine
receptor CXCR3 was no longer among the top predicted interactions, partially due to reduced expression across CD8 T cell populations in pSRS BrM (Fig. 4J). Instead, TNF family members
related to apoptotic pathways and intrinsic inhibitory signals, including _FASLG, TNFSF10_ (TRAIL), and _TNFSF14_ were among the top predicted interactions (Fig. 4J). Moreover, this
increased expression of ligands related to TNF family members in pSRS DCs was accompanied by an increased expression of lipid transporter apolipoprotein E (_APOE)_ (Fig. 4J)_. APOE4_ is
associated with neuro-degenerative tauopathy, and a recent study demonstrated that overexpressing human _APOE4_ intracranially in mice lead to significant increases in neuroinflammation and
cytotoxic T cell31. Together, these data indicate that pSRS shifts the transcriptional signatures of dendritic cells towards both a pro-inflammatory and an apoptosis-inducing phenotype.
These activated DCs may deliver these signals to PD1+ CD8 T cells that promote both the accumulation of the TD1 cytotoxic subset and cell death, contributing to the overall reduction in the
CD8 T cell population across BrM tumors. IMMUNODOMINANT CLONOTYPIC OVERLAP IS OBSERVED BETWEEN THE CD8 T CELL SUBSETS IN RES AND PSRS BRM Our immunofluorescence, flow cytometry, and
transcriptional data suggest that PD1+ TCF1+ CD8 T cells can act as tumor-reactive stem-like precursors to the TD1 and TD2 effector populations in BrM. To gain further insight into the
relationship between these CD8 T cell subsets, we performed matched VDJ sequencing analysis from the single cell RNAseq above on pSRS samples from our prospective trial or Res samples
collected prospectively off trial. We first assessed the clonal expansion of each CD8 T cell subset across groups. Consistent with their respective phenotypes, PD1+ TCF1+ stem-like,
transitory, and TD effector populations shared immunodominant and subdominant expanded clones in both Res and pSRS BrM (Fig. 5A, B and Supplementary Fig. 10A, B), indicative of lineage
relationship and tumor reactivity. Additionally, all the activated stem-like, transitory, TD1, and TD2 CD8 T cell populations had a lower diversity TCR score when compared to the
naive/memory cluster (Supplementary Fig. 10C). Furthermore, there were fewer unique clones along the effector differentiation pathway (stem-like to effector) with a high degree of clonal
overlap between stem-like and TD1/TD2 in both Res and pSRS (Supplementary Fig. 10D, Fig. 5C). When accounting for the number of unique clones and clone size in the stem-like T cell
repertoire, there was around 40% TCR clonal overlap with the TD1 population and 50% overlap with the TD2 population in both groups (Fig. 5C). In agreement, stem-like and TD populations
shared a high degree of TCR similarity as measured by the Morisita-Horn index (Supplementary Fig. 10E), further indicating the _TCF7_ expressing stem-like CD8 T cell population acts as a
precursor to TD1 and TD2 CD8 T cells within BrM. TD1 and TD2 populations also had a very high degree of TCR similarity (Supplementary Fig. 10E) suggesting that TD2 may reflect a more
exhausted differentiation state of TD1. Phenotypically this was also apparent, given the higher expression of exhaustion markers (_TOX, PDCD1, LAG3_) and reduced expression of _GZMB_ (Fig.
4C, Supplementary Fig. 9B). Interestingly, there was a trend for a greater proportion of dominant TCR clones in the TD1 population in the pSRS group when compared to Res (Fig. 5D). This may
indicate that pSRS boosts differentiation to the TD1 _GZMB_ high effector population or preferentially affects subdominant clones. Together, these data support that _TCF7_+ stem-like CD8 T
cells are the precursors to effector CD8 T cells within BrMs. Importantly, the lineage of differentiation progressing from the TCF1+ stem-like to effector clusters remained intact following
pSRS treatment. DISCUSSION In this study, we sought to understand the clinical and immunologic impact of pSRS for BrM. We found that pSRS could be delivered safely with clinical outcomes
similar to BrM receiving post-operative SRS irrespective of dexamethasone dose (Fig. 1). Using multiple data sets including samples from our prospective trial, we found that immune niches
comprised of TCF1 + CD8 T cells and antigen presenting cells were present at varying densities in Res and pSRS BrM. Importantly, these immune niches were associated with longer local control
of disease (Fig. 2)7, which were not affected by peri-operative dexamethasone dosage (Fig. 1, Supplementary Fig. 6). In the pSRS cohort, while there was a transient reduction in BrM total
and CD8 T cell subsets relative to Res, CD8 T cell populations appeared to rebound by day 6 post pSRS, driven by an increase in the frequency of effector-like TCF1- CD8 T cells (Fig. 3).
Single cell RNAseq and VDJseq analysis found robust TCR clonal overlap between TCF1+ and effector CD8 T cell subsets across both Res and pSRS patients, indicating these cells have stem-like
properties and may serve as the precursors to effector CD8 T cell populations within BrM. Finally, transcriptional analysis showed increased expression of inflammatory and apoptotic genes in
pSRS dendritic cells and CD8 T cell subsets that could lead to an accumulation of GZMB high effector CD8 T cells within BrM (Figs. 4, 5). The clinical trial had a few limitations. First,
long term safety evaluation are needed to draw more extensive clinical conclusions. Second, it was not powered for secondary outcomes including comparing the low and high dose steroid Arms.
Therefore, although no significant differences in outcomes were observed, validation on a larger trial will be required. Taken together, these immunological data fit into and build upon an
expanding body of work evaluating the tumor-microenvironment in BrM19,26,32,33,34. Prior investigation of the spatial immune landscape of BrM has demonstrated a wide variety of immune
clusters, some of which appear to confer prognostic significance20. One particularly elegant study compared the microenvironment of melanoma BrM and extracranial metastases and found that
the BrM had higher frequencies of monocyte-derived macrophages and of exhausted TOX + CD8 T cells19. With our study, we did not seek to verify these comparisons, but rather to deeply
interrogate the BrM CD8 T cell populations under both baseline (Res) and pSRS conditions. Our study is hypothesis-generating but does have a few limitations. Due to the sample size, a
multivariate analysis for immune niche density and local control was not possible. Additionally, we did not find that measuring bulk CD8 T cells had prognostic value, which may be due to a
less robust association of CD8 T cells alone with outcomes, leading to underpowering. Importantly, we selected local control as an end point as it has fewer confounders than distant brain
failure (which is impacted by systemic disease burden) or overall survival (which has innumerable confounders). Validation of the immune niche in an independent BrM cohort, as well as
further expanding the number and diversity of histologies of the BrM evaluated is the focus of our ongoing studies and will help determine whether it is a clinically useful biomarker. Future
investigation of other endpoints beyond local control will also be important, as BrM CD8 T cell infiltration has been shown to be associated with improved survival35. Given the focus of
this study was the CD8 T cell response, a deeper analysis of APC subtypes in the immune niche also requires further investigation. Here, by scRNA seq, we confirmed there are macrophages,
dendritic cells, and B cells in the BrM, as also shown by other investigators19,32 and the transcriptional profile of dendritic cells is impacted by pSRS towards an inflammatory state. The
pSRS findings were unexpected. We had hypothesized, based on our preclinical studies using non-CNS tumors, that pSRS would increase the density of stem-like CD8 T cells and the presence of
immune niches around day 7 following radiation25. However, in our time from pSRS to surgery analysis in Fig. 3, we did not find an increase above baseline by day 6 + , but we did find a
rebound in total CD8 T cell numbers that appeared to be driven by an increase in the frequency of the TCF1- subsets, particularly the TD1 GZMB + CD8 T cells. These results suggest that SRS,
like anti-PD1 therapy, may drive both the proliferation and differentiation of stem-like CD8 T cells into more terminally differentiated, effector-like cells5,36. This mechanism would
account for the rebound in total CD8 T cells and the relative changes in the frequency of these populations. The in-situ expansion and differentiation model is supported by the sustained
clonotypic overlap between stem and terminal effector cells observed in the pSRS group. Alternatively, although less likely given these clonotypic data, SRS may promote the infiltration of
terminal effector cells which differentiate from stem-like T cells at sites outside the CNS, including draining lymph nodes12,25,37. The differences between these human results and our prior
pre-clinical work suggest that pre-clinical models may not fully recapitulate the human CNS immune response following SRS25. Further studies of in vivo and in vitro systems are necessary to
interrogate these different possibilities and develop a more complete understanding of the similarities and differences between model systems and human patients. Radiation has been shown to
also induce dendritic cell maturation via the release of endogenous adjuvants (i.e., damage associated molecular patterns, or DAMPs)38,39. This stimulatory effect may explain the enhanced
inflammatory response in DCs as well an immune effector process in the TD1 cells by single cell RNA seq in the pSRS group. The immunogenicity of radiation is well known to depend on both the
total dose and number of fractions40. In the 76 patients receiving pSRS analyzed by immunofluorescence, 97% received a single fraction with a median and mode of 15 Gy. This relatively
homogeneous data set allows for a more uniform analysis of the impact of SRS on T cells subsets, but it also makes it more difficult to draw conclusions regarding the immunogenicity of other
dose/fraction regimens in the CNS. This was investigated in our smaller prospective clinical trial dataset without any statistically significant results. This is a critically important
question and will be the focus of future studies. Additionally, glucocorticoids, including dexamethasone, are known to impact T cell survival and CD8 T cell differentiation27,28. Here, we
did not find a significant effect of dexamethasone dose on BrM T cell numbers in either the 67-patient retrospective Res cohort or in the clinical trial, which is consistent with other
studies41. Importantly, despite this high dexamethasone dose, there was still significant BrM infiltration detected, demonstrating that dexamethasone does not eliminate nor appear to
substantially impair the intracranial T cell response at the time points evaluated. Broadly, these findings confirm that, despite the brain having several barriers to T cell infiltration, an
immune response is capable of being mounted that resembles that of many other tumor types/locations and benefits patient outcomes. Clinically, these data suggest that the immune niche may
not only be an important prognostic factor for outcomes, but also be predictive of an intracranial response to immunotherapy given the known importance of both stem-like CD8 T cells and
co-stimulation for a robust response to anti-PD1 therapy5,36,42. These results also provide some insight into the optimal timing of surgical resection or immunotherapy strategies following
SRS, while complementing the existing compelling data that combination SRS+immunotherapy may improve clinical outcomes43,44. Our data suggest that resection of BrM <6 days following
pre-operative SRS limits any immunostimulatory benefit of SRS, due to the acute decline in CD8 T cells. Furthermore, anti-PD1 administration at a time point where CD8 T cell numbers are
limited likely also blunts potential synergy of combinatory SRS and anti-PD1 therapy. These data can, therefore, inform investigation into optimal sequencing and combination of multiple
therapeutic modalities. Future clinical studies are needed to further reveal the immunostimulatory potential of SRS, validate the prognostic value of immune niches, and determine whether an
intra-cranial abscopal response can be generated. METHODS The research complies with all relevant ethical regulations. Both the prospective trial and the retrospective tissue analysis were
approved by the Emory University Institutional Review Board. Informed consent was obtained for all patients enrolled on the prospective trial and prior to banking of tissue for those in the
retrospective cohort. SOURCES OF SAMPLES The source of all samples are detailed in the respective figures/figure legends as well as Supplementary Table 6. Broadly, there are two
retrospective cohorts: upfront resection (Res) and pre-operative SRS (pSRS) used for IF, and two prospective cohorts (also Res and pSRS) used for the remaining analyzes. The pSRS prospective
cohort samples were all collected on the Emory University Winship Cancer Institute institutional pre-operative SRS clinical trial (NCT04895592). The tissue originated from two institutions
(Emory University and the Levine Cancer Institute (LCI)) and are as follows: (1) Upfront resection (Res) immunofluorescence samples in Figs. 2, 3 are from the Emory University brain tumor
bank (Supplementary Table 2); (2) pre-operative SRS (pSRS) immunofluorescence samples used in Fig. 3 are from the LCI brain tumor bank (Supplementary Table 4); (3) Res flow cytometry and
scRNA seq samples in Figs. 2, 4, 5 were collected at Emory University Winship Cancer Institute utilizing our tissue collection protocol IRB #00045732; (4) pSRS immunofluorescence, flow and
scRNA seq samples in Figs. 4, 5 were collected on pre-operative SRS clinical trial (NCT04895592) (Supplementary Table 1). EMORY UNIVERSITY PRE-OPERATIVE SRS CLINICAL TRIAL NCT04895592 is a
two-arm non-randomized pilot study allocating patients with BrM to low dose Arm A (≤ 4 mg dexamethasone) or high dose Arm B (≥ 16 mg dexamethasone) in an alternating fashion. Both arms
receive pSRS followed by resection. This trial accrued patient exclusively at Emory University Winship Cancer Institute. Eligible patients were ≥ 18 years of age, had a prior pathologically
confirmed or suspected extracranial diagnosis of malignancy, brain metastases were visible on MRI, ECOG ≤ 2, a life expectancy >12 weeks and willingness and ability to tolerate both
pre-operative SRS and surgical resection. Primary safety endpoint: <33% of patients develop grade ≥ 3 adverse event at 4 months. The sample size was determined based off a true toxicity
event rate of 10%. This study design and conduct complied with all relevant regulations regarding the use of human study participants and was conducted in accordance with the criteria set by
the Declaration of Helsinki. Additional details are available on clinicaltrials.gov. Time zero for all clinical outcomes was the date of pre-operative SRS delivery. First patient was
enrolled on 8/11/2021, last treated patient enrolled on 4/21/2023. The protocol was amended on 4/11/23 to allow for a minimum of 10 evaluable patients per arm. PATIENT OUTCOMES Records of
patients treated at two institutions (Emory University and the LCI) between 2007-2021 were evaluated and reviewed. Data were de-identified according to the Health Insurance Portability and
Accountability Act, and all investigations were performed in accordance with the relevant guidelines and regulations. Patient characteristics of each cohort are shown in Supplementary Tables
2 and 4. Emory University comprised the Res cohort, and LCI comprised the pSRS cohort. All clinical outcomes data are derived from the immunofluorescence samples from Emory University brain
tumor bank as these patients had not received pSRS and had long-term follow-up available. Patient samples were selected which had a pathologic diagnosis of metastatic cancer to the brain
and no prior immunotherapy. Immune biomarkers evaluated by patient characteristics are shown in Supplementary Table 3. DEFINING LOCAL RECURRENCE Local recurrence was defined based on the
development of a contrast-enhancing mass within or adjacent to the prior resection cavity on MRI. If a re-resection was performed, pathologic confirmation was also utilized, although not
required. If there was a question of the enhancement representing local recurrence vs. radiation necrosis, additional advanced imaging (e.g., MR perfusion, MR spectroscopy, or brain positron
emission tomography) was obtained, and consensus was reached in a multidisciplinary neuro-oncology tumor board. Additionally, the lesion was followed over time, and persistence or
resolution with observation or glucocorticoids further assisted with the differentiation of local recurrence vs. radiation necrosis. FFPE SAMPLES Formalin fixed paraffin embedded tissue
samples from these patients were stained and analyzed. 67 standard of care samples were obtained from the Emory Brain Tumor Bank, and 76 pre-operative SRS samples were acquired from LCI.
FFPE SAMPLE PREPARATION Sections were deparaffinized in successive incubations with xylene and decreasing concentrations (100, 95, 75, 50, 0%) of ethanol. Antigen retrieval was achieved
using Abcam 100x TrisEDTA Antigen Retrieval Buffer (pH = 9) heated under high pressure. Sections were then washed in PBS + 0.1% Tween20 before antibody staining. IMMUNOFLUORESCENCE STAINING
Immunofluorescence antibody staining was done using two different techniques: (1) Sections were blocked for 30 min with 10% goat serum in 1x PBS + 0.1% Tween20. Sections were then stained
with appropriate primary and secondary antibodies. Primary antibodies were incubated for 1 h at room temperature. Secondary antibodies were incubated for 30 min at room temperature. Detailed
information about antibodies used are listed in Supplementary Table 7. Sections were counterstained with DAPI according to manufacturer instructions (Thermo-Fisher). (2) Immunofluorescence
antibody staining was performed using the Opal 7-color immunofluorescence kit (Akoya Biosciences) endogenous peroxidase activity was quenched by microwave treatment of the slides with AR
buffer. Non-specific binding was blocked with blocking/antibody diluent. After incubation with the primary antibody, the slides were incubated with HRP Mouse+Rabbit secondary antibody and
then incubated in the appropriate opal fluorophore for 10 min. The slides were counterstained with DAPI. IMAGE CAPTURE AND ANALYSIS The selected fluorophore panel (1) allowed for
simultaneous visualization of three targets and a nuclear stain (DAPI) using a Zeiss Axio Scan.Z1 Slide Scanner equipped with a Colibri 7 Flexible Light Source. Zeiss ZenBlue software was
used for post-acquisition image processing. Slides stained with the Opal IHC Kit (2) were scanned using a Perkin Elmer Vectra Polaris and allowed for simultaneous visualization of six
targets and a nuclear stain. For brightfield imaging, slides were scanned using a Hamamatsu’s Nanozoomer slide scanner. Images were analysed using CellProfiler, QuPath, and custom R and
python scripts, as previously described7. This analysis pipeline allowed for determination of the x, y location of each cell in each image, as well as the quantitation of the distance
between cells and the density of each cell type. Immune niches were defined as 100um x 100um cellular neighborhoods where both TCF1+ CD8 T cells and MHC-II+ antigen presenting cells
co-localized. Proportion of tumor with immune niches was defined as the percentage of tumor tissue (percentage of 10,000um2 neighborhoods) occupied by immune niches (where TCF1+ CD8 T cells
and MHC-II+ cells co-localize in a local 10,000um2 cellular neighborhood). FRESH HUMAN SAMPLE COLLECTION, PROCESSING AND FLOW STAINING BrM samples were collected after patients underwent
craniotomy and surgical BrM resection. For the pSRS group, SRS was administered prior to craniotomy on our institutional clinical trial, NCT04895592. All fresh samples (Res and pSRS) for
flow analysis were treated and acquired at Winship Cancer Institute. Samples were collected directly after resection into Phosphate Buffered Saline. The samples were cut into small pieces,
digested with a MACS enzyme cocktail, and homogenized using a MACS Dissociator. The digested tumor was washed then through a 70um filter to obtain a single cell suspension. Samples were then
preserved in freezing media (FBS + 10% DMSO) at −80°C. Single cell suspensions from processed human tumor samples were stained with antibodies listed Supplementary Table 7. Live/dead
staining was done using fixable near-IR or aqua dead cell staining kit (Invitrogen). Cells were permeabilized using the FOXP3 Fixation/Permeabilization kit (eBioscience) for 45 min at 4°C
and stained with intracellular antibodies in permeabilization buffer for 30 mins at 4°C. Samples were acquired on a BD FACSymphony instrument and analyzed using FlowJo (v10). SCRNA/VDJ SEQ
Single cell suspensions were stained and sorted on the Beckton Dickinson FACS Aria II Cell Sorter to acquire CD45+ CD8- and CD45 + CD8+ populations. These two sorted populations were then
mixed 1:1 with the goal of enriching for the infiltrating CD8 T cell population. Single cell RNAseq libraries were made using the Chromium single cell 5’ Library and Gel Bead Kit (10x
Genomics) and captured into the Gel Beads-in-emulsion (GEMs). _N_ = 34 BrM were submitted for sorting, and sufficient cells were captured for _n_= 31 (_n_= 18 Res, _n_= 13 pSRS). After the
reverse transcription GEMs were disrupted and cDNA was isolated and pooled. The barcoded cDNA was fragmented, and end repair and A-tailing was done, followed by sample index PCR. The
purified libraries were sequenced to 50,000 reads/cell on a HisSeq300 (Illumina) with 26 cycles for read 1, 8 cycles for index (i7) and 91 cycles for read 2. Paired VDJ sequencing was also
performed. Filtered contig annotation files were compiled for each sample, and raw consensus ids were used to delineate clonotype. 72 cells with multiple clonotypes were removed. Samples
with >50% of cells with missing TCF1 information were removed (Res1, pSRS4, pSRS13). Samples with dominant clonotype present in less than 20 cells were removed (Res1, pSRS13, Res5, pSRS7)
leaving _n_= 15 for Res and _n_= 11 for pSRS. Cellranger v3.1 was used to align, filter, and count the barcodes and unique molecular identifiers (UMI). Data was then analyzed using Seurat
v3.0. Briefly, cells with less than 7% mitochondrial genes were used. Cells that expressed less than 500 genes or more than 6000 were excluded from analysis. Samples with at least 3 cells
and 100 features were analyzed. Raw counts were then normalized for each UMI based on total expression, scaled by multiplying by 10,000, and then log transformed. Variable genes were
determined based on average expression and dispersion, then used to perform principal component (PCA) analysis. Selected PCAs were used to generate clusters and UMAP plots. Heatmaps were
generated using scaled expression data of marker genes, obtained using the FindAllMarkers function in Seurat. Normalized gene expression data was also shown as feature plots. Gene set
scoring was performed using VISION R package V2.1. For clonotype analysis, MiXCR was run on raw fasq for Res and pSRS. The clonotypes identified were overlaid on the UMAP of the T cell
subsets. The proportion of cells with top 3 dominant clonotypes were calculated for each CD8 T cell subset. The number of unique clonotypes were calculated for each CD8 T cell subset.
STATISTICAL ANALYSIS The optimal cutoff values for stem-like T cells and immune niche proportion was determined by bias-adjusted log-rank test45. Local recurrence and death were regarded as
two competing events. Cumulative incidence plots were created based on the proportional sub-distribution hazards model, and Gray’s test was performed to analyze the differences between high
and low groups. SAS software version 9.4 (SAS Institute, Inc. Cary, NC) was utilized for the data analyzes. REPORTING SUMMARY Further information on research design is available in the
Nature Portfolio Reporting Summary linked to this article. DATA AVAILABILITY Study protocol (for NCT04895592) is available in the Supplementary Information file. Additional de-identified
participant data including dosimetric parameters will be made available upon request. Appropriate inquiries should be directed to the corresponding authors. scRNA-seq data are available in
the NCBI Gene Expression Omnibus (GEO) database under the accession number GSE275929. The remaining data are available within the Article, Supplementary Information or Source Data file.
Source data are provided in this paper. Source data are provided with this paper. REFERENCES * Naito, Y. et al. CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in
human colorectal cancer. _Cancer Res_. 58, 3491–3494 (1998). PubMed CAS Google Scholar * Sharma, P. et al. CD8 tumor-infiltrating lymphocytes are predictive of survival in muscle-invasive
urothelial carcinoma. _Proc. Natl Acad. Sci. USA_ 104, 3967–3972 (2007). Article ADS PubMed PubMed Central CAS Google Scholar * Hiraoka, K. et al. Concurrent infiltration by CD8+ T
cells and CD4+ T cells is a favourable prognostic factor in non-small-cell lung carcinoma. _Br. J. Cancer_ 94, 275–280 (2006). Article PubMed PubMed Central CAS Google Scholar * Dieci,
M. V. et al. Immune characterization of breast cancer metastases: prognostic implications. _Breast Cancer Res_. 20, 62 (2018). Article ADS PubMed PubMed Central Google Scholar * Im, S.
J. et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. _Nature_ 537, 417–421 (2016). Article ADS PubMed PubMed Central CAS Google Scholar * Luoma, A.
M. et al. Tissue-resident memory and circulating T cells are early responders to pre-surgical cancer immunotherapy. _Cell_ 185, 2918–2935.e2929 (2022). Article PubMed PubMed Central CAS
Google Scholar * Jansen, C. S. et al. An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. _Nature_ 576, 465–470 (2019). Article ADS PubMed PubMed Central CAS
Google Scholar * Eberhardt, C. S. et al. Functional HPV-specific PD-1(+) stem-like CD8 T cells in head and neck cancer. _Nature_ 597, 279–284 (2021). Article ADS PubMed PubMed Central
CAS Google Scholar * Krishna, S. et al. Stem-like CD8 T cells mediate response of adoptive cell immunotherapy against human cancer. _Science_ 370, 1328–1334 (2020). Article ADS PubMed
PubMed Central CAS Google Scholar * Sade-Feldman, M. et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. _Cell_ 175, 998–1013.e1020 (2018).
Article PubMed PubMed Central CAS Google Scholar * Brummelman, J. et al. High-dimensional single cell analysis identifies stem-like cytotoxic CD8(+) T cells infiltrating human tumors.
_J. Exp. Med._ 215, 2520–2535 (2018). * Prokhnevska, N. et al. CD8(+) T cell activation in cancer comprises an initial activation phase in lymph nodes followed by effector differentiation
within the tumor. _Immunity_ 56, 107–124.e105 (2023). Article PubMed CAS Google Scholar * Jansen, C. S. et al. An intra-tumoral niche maintains and differentiates stem-like CD8 T cells.
_Nature_ 576, 465–470 (2019). * Miller, B. C. et al. Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. _Nat. Immunol._ 20, 326–336
(2019). Article PubMed PubMed Central CAS Google Scholar * Tabanelli, V. et al. The identification of TCF1+ progenitor exhausted T cells in THRLBCL may predict a better response to
PD-1/PD-L1 blockade. _Blood Adv._ 6, 4634–4644 (2022). Article PubMed PubMed Central CAS Google Scholar * Alvarez, J. I. et al. The Hedgehog pathway promotes blood-brain barrier
integrity and CNS immune quiescence. _Science_ 334, 1727–1731 (2011). Article ADS PubMed CAS Google Scholar * Engelhardt, B. & Ransohoff, R. M. Capture, crawl, cross: the T cell
code to breach the blood-brain barriers. _Trends Immunol._ 33, 579–589 (2012). Article PubMed CAS Google Scholar * Shechter, R., London, A. & Schwartz, M. Orchestrated leukocyte
recruitment to immune-privileged sites: absolute barriers versus educational gates. _Nat. Rev. Immunol._ 13, 206–218 (2013). Article PubMed CAS Google Scholar * Biermann, J. et al.
Dissecting the treatment-naive ecosystem of human melanoma brain metastasis. _Cell_ 185, 2591–2608.e2530 (2022). Article PubMed PubMed Central CAS Google Scholar * Karimi, E. et al.
Single-cell spatial immune landscapes of primary and metastatic brain tumours. _Nature_ 614, 555–563 (2023). Article ADS PubMed PubMed Central CAS Google Scholar * Brown, P. D. et al.
Postoperative stereotactic radiosurgery compared with whole brain radiotherapy for resected metastatic brain disease (NCCTG N107C/CEC.3): a multicentre, randomised, controlled, phase 3
trial. _Lancet Oncol._ 18, 1049–1060 (2017). Article PubMed PubMed Central Google Scholar * Mahajan, A. et al. Post-operative stereotactic radiosurgery versus observation for completely
resected brain metastases: a single-centre, randomised, controlled, phase 3 trial. _Lancet Oncol._ 18, 1040–1048 (2017). Article MathSciNet PubMed PubMed Central Google Scholar *
Prabhu, R. S. et al. Single-Fraction Versus Fractionated Preoperative Radiosurgery for Resected Brain Metastases: A PROPS-BM International Multicenter Cohort Study. _Int. J. Radiat. Oncol.
Biol. Phys_. https://doi.org/10.1016/j.ijrobp.2023.09.012 (2023). * Tawbi, H. A. et al. Combined Nivolumab and Ipilimumab in Melanoma Metastatic to the Brain. _N. Engl. J. Med_. 379, 722–730
(2018). Article PubMed PubMed Central CAS Google Scholar * Buchwald, Z. S. et al. Tumor-draining lymph node is important for a robust abscopal effect stimulated by radiotherapy. _J
Immunother Cancer_ 8. https://doi.org/10.1136/jitc-2020-000867 (2020). * Sudmeier, L. J. et al. Distinct phenotypic states and spatial distribution of CD8(+) T cell clonotypes in human brain
metastases. _Cell Rep. Med_. 3, 100620 (2022). Article PubMed PubMed Central CAS Google Scholar * Acharya, N. et al. Endogenous glucocorticoid signaling regulates cd8(+) t cell
differentiation and development of dysfunction in the tumor microenvironment. _Immunity_ 53, 658–671.e656 (2020). Article PubMed PubMed Central CAS Google Scholar * Giles, A. J. et al.
Dexamethasone-induced immunosuppression: mechanisms and implications for immunotherapy. _J. Immunother. Cancer_ 6, 51 (2018). Article PubMed PubMed Central Google Scholar * Deng, L. et
al. STING-dependent cytosolic DNA sensing promotes radiation-induced type i interferon-dependent antitumor immunity in immunogenic tumors. _Immunity_ 41, 843–852 (2014). Article PubMed
PubMed Central CAS Google Scholar * Twyman-Saint Victor, C. et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. _Nature_ 520, 373–377 (2015).
Article ADS PubMed CAS Google Scholar * Chen, X. et al. Microglia-mediated T cell infiltration drives neurodegeneration in tauopathy. _Nature_ 615, 668–677 (2023). Article ADS PubMed
PubMed Central CAS Google Scholar * Gonzalez, H. et al. Cellular architecture of human brain metastases. _Cell_ 185, 729–745.e720 (2022). Article PubMed PubMed Central CAS Google
Scholar * Klemm, F. et al. Interrogation of the microenvironmental landscape in brain tumors reveals disease-specific alterations of immune cells. _Cell_ 181, 1643–1660.e1617 (2020).
Article PubMed PubMed Central CAS Google Scholar * Friebel, E. et al. Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. _Cell_
181, 1626–1642.e1620 (2020). Article PubMed CAS Google Scholar * Berghoff, A. S. et al. Density of tumor-infiltrating lymphocytes correlates with extent of brain edema and overall
survival time in patients with brain metastases. _Oncoimmunology_ 5, e1057388 (2016). Article PubMed Google Scholar * Siddiqui, I. et al. Intratumoral Tcf1(+)PD-1(+)CD8(+) T Cells with
Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. _Immunity_ 50, 195–211.e110 (2019). Article PubMed CAS Google Scholar *
Connolly, K. A. et al. A reservoir of stem-like CD8(+) T cells in the tumor-draining lymph node preserves the ongoing antitumor immune response. _Sci. Immunol._ 6, eabg7836 (2021). Article
PubMed PubMed Central CAS Google Scholar * Formenti, S. C. & Demaria, S. Combining radiotherapy and cancer immunotherapy: a paradigm shift. _J. Natl Cancer Inst._ 105, 256–265
(2013). Article PubMed PubMed Central CAS Google Scholar * Golden, E. B., Demaria, S., Schiff, P. B., Chachoua, A. & Formenti, S. C. An abscopal response to radiation and ipilimumab
in a patient with metastatic non-small cell lung cancer. _Cancer Immunol. Res_. 1, 365–372 (2013). Article PubMed PubMed Central Google Scholar * Demaria, S. et al. Radiation dose and
fraction in immunotherapy: one-size regimen does not fit all settings, so how does one choose? _J Immunother Cancer_ 9. https://doi.org/10.1136/jitc-2020-002038 (2021). * Berghoff, A. S. et
al. Tumour-infiltrating lymphocytes and expression of programmed death ligand 1 (PD-L1) in melanoma brain metastases. _Histopathology_ 66, 289–299 (2015). Article PubMed Google Scholar *
Kamphorst, A. O. et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. _Science_ 355, 1423–1427 (2017). Article ADS PubMed PubMed Central CAS Google
Scholar * Minniti, G. et al. Stereotactic radiosurgery combined with nivolumab or Ipilimumab for patients with melanoma brain metastases: evaluation of brain control and toxicity. _J.
Immunother. Cancer_ 7, 102 (2019). Article PubMed PubMed Central Google Scholar * Lussier, D. M. et al. Radiation-induced neoantigens broaden the immunotherapeutic window of cancers with
low mutational loads. _Proc Natl Acad Sci USA_ 118. https://doi.org/10.1073/pnas.2102611118 (2021). * Contal, C. & O’Quigley, J. An application of changepoint methods in studying the
effect of age on survival in breast cancer. _Comput. Stat. Data Anal._ 30, 253–270 (1999). Article Google Scholar Download references ACKNOWLEDGEMENTS This work was primarily supported by
the American Cancer Society grant CSDG-22-049-01-IBCD to ZSB. Additional support came from National Institute of Health grants 1-K12-CA-237806-01 and 1-K08-CA-270401-01A1 to ZSB as well as
National Cancer Institute grant 1-F30-CA-243250 for CSJ, National Cancer Institute grant 1-R01-CA280069 to HK, ASTRO-MRA #816282 for ZSB, DOD grant W81XWH-20-1-0525 to HK and VM, and Cancer
Research Institute funding to HK. We would like to acknowledge the Yerkes NHP Genomics Core (P51 OD011132, NIH S10 OD026799) and the Emory Flow Cytometry Core (UL1TR002378). AUTHOR
INFORMATION Author notes * These authors contributed equally: Caroline S. Jansen, Meghana S. Pagadala, Maria A. Cardenas. * These authors jointly supervised this work: Haydn Kissick and
Zachary S. Buchwald. AUTHORS AND AFFILIATIONS * Department of Urology, Emory University, Atlanta, USA Caroline S. Jansen, Maria A. Cardenas, BaoHan T. Vo, Chengyu Ye, Nataliya Prokhnevska,
Luke del Balzo & Haydn Kissick * Department of Medicine, Memorial Sloan Kettering Cancer Center, New York, USA Meghana S. Pagadala * Southeast Radiation Oncology Group, Levine Cancer
Institute, Atrium Health, Charlotte, USA Roshan S. Prabhu & Stuart H. Burri * Department of Biostatistics and Bioinformatics, Rollins School of Public Health, Emory University, Atlanta,
USA Subir Goyal & Jeffrey M. Switchenko * Department of Radiation Oncology, Emory University, Atlanta, USA Chengjing Zhou, Prasanthi Chappa, Benjamin Hopkins, Jim Zhong, Taylor Daniels,
Maedot Admassu, India Green, Mylin A. Torres, Aparna H. Kesarwala, Kristin A. Higgins, Pretesh Patel, Vishal Dhere, Mohammad K. Khan & Zachary S. Buchwald * Winship Cancer Institute,
Emory University, Atlanta, USA Chengjing Zhou, Prasanthi Chappa, Benjamin Hopkins, Jim Zhong, Taylor Daniels, Maedot Admassu, India Green, Kimberly B. Hoang, Mylin A. Torres, Jeffrey J.
Olson, Edjah K. Nduom, Aparna H. Kesarwala, Kristin A. Higgins, Pretesh Patel, Vishal Dhere, Mohammad K. Khan & Zachary S. Buchwald * Biomedical Sciences Program, University of
California San Diego, La Jolla, USA Adam Klie * Department of Radiation Oncology, University of Alabama Birmingham, Birmingham, AL, USA Neil T. Pfister * Department of Pathology, Emory
University, Atlanta, USA Stewart G. Neill * Department of Neurosurgery, Emory University, Atlanta, USA Kimberly B. Hoang, Jeffrey J. Olson & Edjah K. Nduom * Department of Pathology,
Nationwide Children’s Hospital, Columbus, USA Suzanna Logan * Kaiser Permanente, Atlanta, USA Kirtesh Patel * Neuroscience Institute, Atrium Health, Charlotte, USA Anthony L. Asher *
Genitourinary Malignancies Branch, National Cancer Institute, Bethesda, USA Scott Wilkinson & Adam G. Sowalsky * Laboratory of Cancer Biology and Genetics, National Cancer Institute,
Bethesda, USA Ross Lake * Department of Medicine, Division of Medical Genetics, University of California San Diego, La Jolla, USA Hannah Carter * Department of Microbiology and Immunology,
Emory University, Atlanta, USA Haydn Kissick Authors * Caroline S. Jansen View author publications You can also search for this author inPubMed Google Scholar * Meghana S. Pagadala View
author publications You can also search for this author inPubMed Google Scholar * Maria A. Cardenas View author publications You can also search for this author inPubMed Google Scholar *
Roshan S. Prabhu View author publications You can also search for this author inPubMed Google Scholar * Subir Goyal View author publications You can also search for this author inPubMed
Google Scholar * Chengjing Zhou View author publications You can also search for this author inPubMed Google Scholar * Prasanthi Chappa View author publications You can also search for this
author inPubMed Google Scholar * BaoHan T. Vo View author publications You can also search for this author inPubMed Google Scholar * Chengyu Ye View author publications You can also search
for this author inPubMed Google Scholar * Benjamin Hopkins View author publications You can also search for this author inPubMed Google Scholar * Jim Zhong View author publications You can
also search for this author inPubMed Google Scholar * Adam Klie View author publications You can also search for this author inPubMed Google Scholar * Taylor Daniels View author publications
You can also search for this author inPubMed Google Scholar * Maedot Admassu View author publications You can also search for this author inPubMed Google Scholar * India Green View author
publications You can also search for this author inPubMed Google Scholar * Neil T. Pfister View author publications You can also search for this author inPubMed Google Scholar * Stewart G.
Neill View author publications You can also search for this author inPubMed Google Scholar * Jeffrey M. Switchenko View author publications You can also search for this author inPubMed
Google Scholar * Nataliya Prokhnevska View author publications You can also search for this author inPubMed Google Scholar * Kimberly B. Hoang View author publications You can also search
for this author inPubMed Google Scholar * Mylin A. Torres View author publications You can also search for this author inPubMed Google Scholar * Suzanna Logan View author publications You
can also search for this author inPubMed Google Scholar * Jeffrey J. Olson View author publications You can also search for this author inPubMed Google Scholar * Edjah K. Nduom View author
publications You can also search for this author inPubMed Google Scholar * Luke del Balzo View author publications You can also search for this author inPubMed Google Scholar * Kirtesh Patel
View author publications You can also search for this author inPubMed Google Scholar * Stuart H. Burri View author publications You can also search for this author inPubMed Google Scholar *
Anthony L. Asher View author publications You can also search for this author inPubMed Google Scholar * Scott Wilkinson View author publications You can also search for this author inPubMed
Google Scholar * Ross Lake View author publications You can also search for this author inPubMed Google Scholar * Aparna H. Kesarwala View author publications You can also search for this
author inPubMed Google Scholar * Kristin A. Higgins View author publications You can also search for this author inPubMed Google Scholar * Pretesh Patel View author publications You can also
search for this author inPubMed Google Scholar * Vishal Dhere View author publications You can also search for this author inPubMed Google Scholar * Adam G. Sowalsky View author
publications You can also search for this author inPubMed Google Scholar * Hannah Carter View author publications You can also search for this author inPubMed Google Scholar * Mohammad K.
Khan View author publications You can also search for this author inPubMed Google Scholar * Haydn Kissick View author publications You can also search for this author inPubMed Google Scholar
* Zachary S. Buchwald View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS C.J., H.K., R.S.P., Z.S.B., and M.K.K. conceived of the study. P.C.,
B.T.V., C.Z., S.W., and R.L. acquired the immunofluorescence, single cell sequencing and flow cytometry data. P.C., C.S.J., and L.D.B. analyzed the immunofluorescence. B.H., T.D., M.A.,
I.G., and V.D. acquired the patient data. S.G.N., S.L. provided annotation of the pathology slides. K.B.H., J.Z., J.J.O., E.K.N., K.A.H., and P.P. enrolled patients on study and assisted
with tissue acquisition. R.S.P., K.P., S.H.B., and A.L.A. provided tissue and assisted with pSRS retrospective dataset. S.G. and J.M.S. performed all clinical statistical analyzes. M.S.P.,
N.T.P., C.Y., M.A.C., H.C., N.P., and A.K. analyzed and interpreted the scRNA seq data. All authors contributed to writing and editing the manuscript including M.A.T., A.G.S., and A.H.K.
CORRESPONDING AUTHORS Correspondence to Haydn Kissick or Zachary S. Buchwald. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. PEER REVIEW PEER REVIEW
INFORMATION _Nature Communications_ thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available. ADDITIONAL INFORMATION
PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION
PEER REVIEW FILE REPORTING SUMMARY SOURCE DATA SOURCE DATA RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which
permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to
the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless
indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or
exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints
and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Jansen, C.S., Pagadala, M.S., Cardenas, M.A. _et al._ Pre-operative stereotactic radiosurgery and peri-operative dexamethasone for
resectable brain metastases: a two-arm pilot study evaluating clinical outcomes and immunological correlates. _Nat Commun_ 15, 8854 (2024). https://doi.org/10.1038/s41467-024-53034-6
Download citation * Received: 01 July 2024 * Accepted: 29 September 2024 * Published: 14 October 2024 * DOI: https://doi.org/10.1038/s41467-024-53034-6 SHARE THIS ARTICLE Anyone you share
the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer
Nature SharedIt content-sharing initiative