Play all audios:
ABSTRACT Arterial thrombosis is a leading cause of death and disability worldwide with no effective bioassay for clinical prediction. As a symbolic feature of arterial thrombosis, severe
stenosis in the blood vessel creates a high-shear, high-gradient flow environment that facilitates platelet aggregation towards vessel occlusion. Here, we present a thrombus profiling assay
that monitors the multi-dimensional attributes of thrombi forming in such biomechanical conditions. Using this assay, we demonstrate that different receptor–ligand interactions contribute
distinctively to the composition and activation status of the thrombus. Our investigation into hypertensive and older individuals reveals intensified biomechanical thrombogenesis and
multi-dimensional thrombus profile abnormalities, endorsing the diagnostic potential of the assay. Furthermore, we identify the hyperactivity of GPIbα-integrin αIIbβ3 mechanosensing axis as
a molecular mechanism that contributes to hypertension-associated arterial thrombosis. By studying drug-disease interactions and inter-individual variability, our work reveals a need for
personalized anti-thrombotic drug selection that accommodates each patient’s pathological profile. SIMILAR CONTENT BEING VIEWED BY OTHERS BIORHEOLOGY OF OCCLUSIVE THROMBI FORMATION UNDER
HIGH SHEAR: _IN VITRO_ GROWTH AND SHRINKAGE Article Open access 29 October 2020 PLATELET-PRIMED INTERACTIONS OF COAGULATION AND ANTICOAGULATION PATHWAYS IN FLOW-DEPENDENT THROMBUS FORMATION
Article Open access 17 July 2020 AUTOANTIBODIES IMMUNO-MECHANICALLY MODULATE PLATELET CONTRACTILE FORCE AND BLEEDING RISK Article Open access 25 November 2024 INTRODUCTION Arterial
thrombosis, which describes the formation of pathological blood clots in the artery, is one of the leading causes of mortality and morbidity worldwide1,2. Pathological conditions such as
hypertension, diabetes, metabolic syndrome, and aging not only increase thrombotic risks but also foster resistance to conventional antiplatelets that target soluble agonists (e,g., ADP,
thrombin)-induced platelet activation and aggregation3,4,5,6, contributing to high incidence and recurrence rates of cardiovascular diseases (CVD)7. However, the associated mechanisms are
not fully elucidated. The current clinical paradigm is further challenged by the lack of a standard bioassay for evaluating thrombotic risks: while conventional coagulation assays and
aggregometry assays were indicated to be unreliable in predicting thrombosis or major adverse cardiovascular events, the new generation of hematological function assays (e.g., global
coagulation assays and seer sonorheometry) also have limited evidence supporting their performance, and contain major drawbacks such as high cost, low sensitivity, and lack of
standardization8,9. As an understudied but symbolic mechanism of arterial thrombosis, discoid platelets can be mechanically driven by the elevated shear stress and shear gradient caused by
vessel stenosis to form large aggregates5,10—a phenomenon we termed “biomechanical platelet aggregation”11,12. The biomechanical platelet aggregation process is composed of two steps, mainly
involving three molecular interactions. Firstly, shear-induced von Willebrand factor (VWF) activation13 and glycoprotein (GP) Ibα (GPIbα)–VWF catch bond14 together facilitate GPIbα–VWF
binding under force, which initiates the aggregation of platelets in an activation-independent manner. Then, the GPIbα–VWF binding under force triggers GPIbα mechanosignaling that activates
integrin αIIbβ3 to reach an intermediate affinity and an extended-close (E+Act.-) conformation12, which subsequentially binds to fibrinogen (Fg) and VWF to allow more stable thrombus
development. Notably, E+Act.- integrin αIIbβ3 is only achievable via GPIbα mechanosignaling but not soluble agonist-induced platelet activation12. Biomechanical platelet aggregation cannot
be effectively inhibited by conventional antiplatelets or amplification loop blockers (ALBs), but is strongly impeded by shear rate decrease and reversible upon the release of vessel
stenosis10,12. Unfortunately, platelet mechanobiology was barely investigated in pathological contexts15,16. It remains unclear whether biomechanical platelet aggregation is intensified by
any thrombotic risk factor and contributes to a higher incidence of CVD in certain human populations. Also, existing methods for observing biomechanical platelet aggregation10,12,17 cannot
provide all-around information and quantitative analysis regarding the composition of the thrombus and the activation status of platelets within, which hinders our understanding of arterial
thrombosis and improvement of anti-thrombotic treatment. For example, it remains elusive how the three molecular interactions mediating platelet crosslinking in biomechanical platelet
aggregation, i.e., GPIbα–VWF, integrin αIIbβ3–VWF and integrin αIIbβ3–Fg10,12,18, respectively mediate the VWF and Fg levels and platelet activation in the biomechanical thrombus, and
whether they are dysregulated by thrombotic risk factors to cause abnormal biomechanical thrombogenesis. To address these outstanding clinical and scientific needs, we develop a thrombus
profiling assay that combines a standard stenosis microfluidics setup with multi-color thrombus staining. It enables quick and all-around thrombus characterization under conditions that
mimic the biorheological settings of arterial thrombosis. Using this assay, we delineated the differential roles of complex platelet crosslinking mechanisms in biomechanical thrombogenesis.
We identified exacerbated biomechanical thrombogenesis and multi-dimensional thrombus abnormality associated with hypertension and aging, unraveling a clinical linkage between mechanobiology
and arterial thrombosis. With complementary data from other experimental approaches, we further demonstrated that GPIbα and integrin αIIbβ3 receptors on hypertension patients’ platelets
have endogenous hyperactivity. By using the thrombus profiling assay to study drug–disease interactions and acquire personal thrombus profiles, we identified a gap in standard approaches of
anti-thrombotics evaluation, which urges a re-evaluation of the efficacy and safety of anti-thrombotics using the “thrombus profile” and in the context of different pathology models. All the
above results also showcase the potential of our thrombus profiling assay for anti-thrombotic drug screening, diagnosis of thrombotic risks, and personalized anti-thrombotic regimen
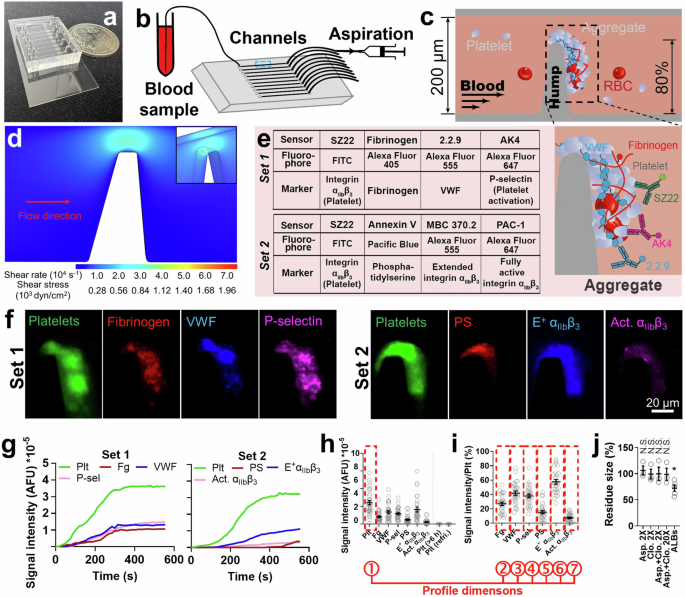
selection. RESULTS THROMBUS PROFILING ASSAY: DEVELOPMENT AND VALIDATION Our microfluidic chip is composed of ten rectangular (width × height: 200 μm × 50 μm) channels with respective inlets
and outlets for tubing connection (Fig. 1a, b). A pump drives a syringe to perfuse heparinized blood (0.5 mL) through the channel pre-coated with VWF. An 80% stenosis site stimulates
biomechanical thrombogenesis (Fig. 1c). A perfusion rate of 18 µL/min was selected, which creates a wall shear stress (WSS) of 857 dyn/cm² at the stenosis site according to fluid dynamics
simulation (Fig. 1d and Supp. Fig. 1a, f). The same stenosis site WSS is achieved by an inlet wall shear rate of 1485 s−1 in a circular vessel with the same cross-sectional area (Supp. Fig.
1b, d, f), mimicking human arterioles19, human arteries during systole, and mouse arteries20. The calculated Reynolds numbers in channels of different shapes are within the same scale (Supp.
Fig. 1f). With the above settings, platelet thrombi can be consistently observed within the channel, which is primarily driven by shear force because no external agonist is added to the
blood and the high-speed perfusion prevents the localized accumulation of agonists released from attached platelets and red blood cells. Due to the high-shear force, most thrombi have a
tendency to grow toward the downstream side of the stenosis. Nonetheless, most thrombi (>85%) cover the whole stenosis apex, and most (>85%) thrombi have the point in their contour
most close to the opposing channel wall positioned above the stenosis apex (Supp. Fig. 2), making the stenosis apex still the most likely position for occlusion. Replacing VWF with collagen
for channel coating did not significantly affect thrombus formation. However, with collagen coating, thrombus formation was basically eliminated by antibody RU5 which blocks plasma VWF
binding to collagen, reflecting an indispensable role of VWF on the hump for platelet attachment (Supp. Fig. 3a). Replacing heparin with citrate or ethylenediaminetetraacetic acid (EDTA) for
anticoagulation attenuated thrombus formation (Supp. Fig. 3a), because the latter two chelate calcium from the blood and inhibit platelet activation, while EDTA also eliminates integrin
αIIbβ3 activity. These results validate the use of VWF and heparin for channel coating and blood anticoagulation, respectively. To comprehensively characterize the thrombus, 7 biomarkers
with their respective molecular sensors were selected (Fig. 1e). Platelets are reported by SZ22, a monoclonal antibody (mAb) against integrin αIIbβ312. Fg is reported by purified Fg that
spikes the blood at 2% of plasma concentration. VWF is reported by non-inhibitory mAb 2.2.921. P-selectin is reported by AK4 to indicate platelet α-granule release22. Phosphatidylserine (PS)
exposure in the membrane is reported by Annexin V to signify platelet procoagulant function23. Conformationally extended (E+) and fully activated (Act.) integrin αIIbβ3 are detected by mAbs
MBC 370.2 and PAC-1, respectively, which together report integrin αIIbβ3 activation status12. The above sensors were grouped into two sets for fluorophore conjugation (Fig. 1e), where SZ22
appears in both sets for reference. All sensors have negligible influence on thrombogenesis (Supp. Fig. 3b–d). Fluorescent signals were observed from all seven biomarkers (Fig. 1f). Agreeing
with previous observations, real-time tracking showed rapid thrombogenesis in the first 300–400 s followed by a quasi-steady phase, in which the thrombus reaches a relative equilibrium
between platelet aggregation and disaggregation10,24 (Fig. 1g). Thus, we selected 450 s after the onset as the time point for quantitating fluorescent signals so as to assess the thrombus in
the fully developed status while avoiding unnecessary waiting (Fig. 1h). Signal intensities of Fg, VWF, P-selectin, PS, and E+ and Act. αIIbβ3 were normalized by platelet signal to assess
their enrichment (Fig. 1i), where the high E+ αIIbβ3 signal and low Act. αIIbβ3 signal agrees with our previous discovery that biomechanical platelet aggregation is mainly mediated by an
intermediate activation state of αIIbβ3 integrins12. P-selectin expression and low-level PS exposure observed here (Fig. 1f, i; further confirmed using different microscope setup, staining
agents, and microfluidic channel design (Supp. Fig. 4)) should be induced by GPIbα and/or integrin αIIbβ3 mechanosignaling25,26,27,28,29. The total signal intensity of platelets (first
dimension, indicating thrombus size) and the normalized signal intensities of Fg, VWF, P-selectin, PS, E+, and Act. αIIbβ3 (2nd–7th dimensions) are summarized into a seven-dimension thrombus
profile (Fig. 1f, i). Blood stored for >6 h or refrigerated overnight failed to generate visible thrombi (Fig. 1h), likely due to a loss of platelet activity during room temperature
storage and GPIbα shedding during cold storage30, respectively. Activated platelets can release and/or help produce soluble agonists such as thromboxane A2 and ADP to further activate
themselves and recruit surrounding platelets to the growing thrombus, wherein the activation signaling processes are called amplification loops. However, conventional antiplatelet aspirin
(targeting thromboxane A2 (TXA2)) and clopidogrel (targeting P2Y12–ADP interaction) rendered negligible inhibition to the biomechanical thrombogenesis both separately and combined at twice
or 20 times of human plasmatic concentrations31 (Fig. 1j). In contrast, both drugs can significantly inhibit ADP or collagen-induced platelet aggregation at much lower concentrations31.
Also, a platelet ALB cocktail (including apyrase, MRS2179 and 2-MeSAMP to block ADP, indomethacin to block TXA2, and hirudin to block thrombin, all at saturating concentrations) only reduced
the thrombus size by ~20% (Fig. 1j). These results corroborate the previous observations that inhibiting platelet amplification loops is ineffective in suppressing biomechanical platelet
aggregation10,12,32, endorsing a secondary role of soluble agonists in biomechanical thrombogenesis. DELINEATING THE CONTRIBUTION OF DIFFERENT RECEPTOR–LIGAND INTERACTIONS Biomechanical
platelet aggregation is mainly mediated by a mechanosensing axis on the platelet surface composed of two mechanoreceptors: GPIbα and integrin αIIbβ3. GPIbα first binds to VWF to initiate
platelet crosslinking, during which GPIbα mechanosignaling induces integrin αIIbβ3 intermediate activation (E+Act.-). The activated integrin αIIbβ3 binds to its ligands VWF and Fg to
reinforce the platelet crosslinking process and also trigger its further activation towards the fully activated state (E+Act.+)12. To investigate how the above platelet-crosslinking
mechanisms, namely, GPIbα–VWF, integrin αIIbβ3–VWF, and integrin αIIbβ3–Fg interactions, respectively mediate the growth, composition, and activation status of biomechanical thrombi, blood
was treated with a panel of highly specific inhibitory mAbs to inhibit the interactions one at a time (Fig. 2a). AK2 and NMC4 both inhibit GPIbα–VWF interaction, with AK2 targeting GPIbα,
and NMC4, previously shown to have anti-thrombotic effects33, targeting VWFA1 domain (VWFA1) which binds to GPIbα33,34. LJ-P5 and 152B6 both inhibit integrin αIIbβ3–VWF interaction, with
LJ-P5 blocking integrin αIIbβ3 binding to VWF but not Fg35, and 152B6 blocking VWF binding to integrin αIIbβ3 but not GPIbα36. 7E9, LJ-155B39, and LJ-134B29 inhibit integrin αIIbβ3–Fg
interaction by respectively blocking one of the three integrin-binding sites in Fg: γ408-411 (AGDV), Aα95-98 (RGDF), and Aα572-575 (RGDS)37,38. Single fluorescence imaging was first used to
measure the dose-dependency of the above mAbs in inhibiting thrombogenesis (Fig. 2b–e). Only AK2 and NMC4, but not the other mAbs, eliminated thrombogenesis (Fig. 2c–e), which agrees with
previous findings that GPIbα–VWF interaction serves as the initiator of biomechanical platelet aggregation10,12. At high concentrations, both LJ-P5 and 152B6 reduced the thrombus size to
<20% (Fig. 2d), and the cocktail of 7E9, LJ-155B39, and LJ-134B29 also reduced the thrombus size to ~5% (Fig. 2e), indicating comparable importance of integrin αIIbβ3–VWF and αIIbβ3–Fg
interactions. 7E9 alone achieved a strong inhibitory effect comparable to the cocktail, which corroborates the primary role of AGDV in Fg for integrin αIIbβ3 binding39,40. However, LJ-155B39
and LJ-134B29 also manifested considerable inhibition (Fig. 2e). Half-maximal inhibitory concentrations (IC50) were acquired for these mAbs via model fitting (Supp. Table 1), which were
then used in thrombus profiling. Both AK2 and NMC4 significantly decreased VWF, P-selectin, and E+ and Act. αIIbβ3 levels in the thrombus (Fig. 2f, g). In comparison, LJ-P5 and 152B6 only
reduced VWF enrichment, while 7E9, LJ-155B39, and LJ-134B29 only reduced Fg enrichment; neither set of mAbs inhibited PS exposure, P-selectin expression, or integrin αIIbβ3 activation (Fig.
2h–l). None of the above mAbs affected the average signal intensity of SZ22-FITC, ruling out the possibility that the reduced VWF and Fg signals were due to increased platelet density (Supp.
Fig. 5a). Altogether, our results indicate that different platelet-crosslinking mechanisms cooperatively mediate biomechanical thrombogenesis, with each having a distinct focus in their
contribution to the thrombus composition and activation status. To succinctly express the effects of different factors on biomechanical platelet aggregation, we created an “effect barcode”
system with seven columns, each corresponding to one dimension of the thrombus profile. A positive, neutral, or negative effect of a factor on a dimension is respectively represented by a
bar at the top, middle, or bottom of the column, also numerically expressed as “+”, “0”, or “−”. Using this system, the effects of AK2 and NMC4 on the thrombus profile are both summarized as
[- 0 - - 0 - -], those of LJ-P5 and 152B6 as [- 0 - 0 0 0 0], and those of 7E9, LJ-155B39, and LJ-134B29 as [- - 0 0 0 0 0] (Fig. 2m). IDENTIFYING AN “ADDITION RULE” IN THE EFFECT BARCODE
SYSTEM Intrigued by how different receptor–ligand interactions synergize in mediating biomechanical thrombogenesis, we tested inhibitors with combinational effects. 7E3 (prototype of the
antiplatelet abciximab) and 10E5 are mAbs that block integrin αIIbβ3 binding to both Fg and VWF12,41,42. Unlike specific inhibitors of integrin αIIbβ3–VWF or αIIbβ3–Fg, both 7E3 and 10E5
eliminated thrombogenesis at high concentrations (Fig. 3a). At IC50, both mAbs reduced Fg and VWF levels in the thrombus without affecting platelet activation markers, rendering an effect
barcode of [- - - 0 0 0 0] (Fig. 3c, d). Interestingly, this barcode equals the add-up of those of integrin αIIbβ3–VWF ([- 0 - 0 0 0 0]) and αIIbβ3–Fg ([- - 0 0 0 0 0]) inhibitors (Fig. 3f).
Negatively charged nanoparticles inhibit platelet aggregation at high-shear rates due to their inhibition of VWF extension and, therefore, VWF–platelet interactions43. We tested two sizes
of polystyrene negatively charged nanoparticles (PS-CNP) (50 and 510 nm), both showing biphasic dose-dependency in thrombus inhibition (Fig. 3b), consistent with the original report43. A
concentration that decreases the thrombus size by ~50% was estimated for the 510-nm PS-CNP to perform thrombus profiling (Fig. 3e), which derived an effect barcode of [- 0 - - 0 - -]. Again,
this barcode equals the add-up of those of GPIbα–VWF ([- 0 - - 0 - -]) and integrin αIIbβ3–VWF ([- 0 - 0 0 0 0]) inhibitors (Fig. 3f). The above results demonstrate that the mathematical
addition rule applies to the effect barcode system. This addition rule will be further validated below in drug–disease interactions. MULTI-DIMENSIONAL THROMBUS PROFILE ABNORMALITY IN
HYPERTENSION AND AGING Aging and hypertension are strong risk factors for thrombosis44,45. To test the performance of our assay in identifying risks of arterial thrombosis, we first compared
the thrombus size of healthy adults at different ages and identified that older ages (≥50) significantly increase the thrombus size (Fig. 4a). Furthermore, we tested blood samples from
primary hypertension patients, which formed much larger biomechanical thrombi than healthy young subjects (Fig. 4b). By fitting the “thrombus size _versus_ time” curves with the sigmoidal
model, it was observed that unlike the growth of healthy young subjects’ thrombi which approached a plateau at ~400 s, hypertension patients’ thrombi remained in the rapid development phase
until ~500 s, again indicating a prothrombotic tendency (Fig. 4b). Characterizing the thrombus profile revealed that aging and hypertension, either alone or together, significantly increased
the thrombus size, Fg level as well as integrin αIIbβ3 activation in the thrombi, rendering the same effect barcode of [+ + 0 0 0 + +] (Fig. 4c). Two-way ANOVA with variance heterogeneity
identified a bi-directional cooperation between hypertension and aging in increasing the thrombus size and E+ αIIbβ3 level, indicating synergy between these two risk factors (Fig. 4d). None
of the above abnormalities was contributed by platelet density changes in the thrombus or hematocrit changes or platelet count increase in the blood (Supp. Fig. 5b–e). We then evaluated the
inter-correlation of the different biomarkers and their performance in distinguishing different cohorts. To address the scattering patterns of the signal intensities (Fig. 4c), which is
likely due to inter-individual variability, multiple statistical analyses were performed for cross-checking. Firstly, by using the linear regression model, Spearman rank correlation
coefficient46 and Kendall’s tau correlation coefficient47, a positive correlation was consistently identified between thrombus size and Fg, E+ αIIbβ3, and Act. αIIbβ3 levels but not the
other factors (Fig. 4e and Supp. Fig. 6), with E+ αIIbβ3 being the strongest correlating factor (Supp. Table 2). Secondly, among all markers, E+ αIIbβ3 has the best performance in separating
healthy young from hypertensive and/or older age groups (Fig. 4e and Supp. Fig. 6a–e), with specificity and sensitivity respectively reaching 86 and 85%, comparable to the performance of
thrombus size (Supp. Table 2). The consistency of E+ αIIbβ3 level with thrombus size in group separation also reached 81%. Altogether, these results unraveled intensified biomechanical
thrombogenesis and multi-dimensional thrombus profile abnormality associated with hypertension and aging, and suggest E+ αIIbβ3 as a potential biomarker for intensified biomechanical
thrombogenesis. Most hypertension patients enrolled in this study had their blood pressure well controlled by medication (systolic/diastolic <140/90 mmHg, respectively) and had hemoglobin
A1C (HbA1C), body mass index (BMI) and cholesterol levels within the healthy range (Fig. 4f and Supp. Table 3). Furthermore, neither the size nor the E+ αIIbβ3 level of these patients’
thrombi has a significant correlation with the disease duration, systolic or diastolic blood pressure, or the sum of the two, or the patients’ HbA1C level, BMI, total cholesterol,
low-density lipoprotein cholesterol (LDL-C), high-density lipoprotein cholesterol (HDL-C), or triglyceride levels (Fig. 4f and Supp. Fig. 7a, b). Also, the thrombi of hypertensive subjects
who have systolic and diastolic blood pressures and HbA1C, BMI, and cholesterol levels all in the normal ranges still have larger sizes and higher E+ αIIbβ3 levels than healthy young
subjects, regardless of aging (Supp. Fig. 7c). These results indicate that hypertension can independently cause intensified biomechanical thrombogenesis and thrombus profile abnormality even
with relatively short disease duration and effective antihypertensive medication. Nonetheless, we cannot exclude the likelihood that poorly controlled blood pressure, diabetes (high HbA1C
level), obesity (high BMI) or dyslipidemia (abnormal cholesterol levels) can have extra contributions to the thrombus profile abnormality, especially considering that the latter three
diseases are known risk factors of CVD. Next, we inspected whether demographics other than age affect the thrombus profile. Within healthy young as well as hypertensive and/or older
subjects, no significant difference in the thrombus size or E+ αIIbβ3 level was found between males and females or among different races/ethnicities (Fig. 4g–l). Seemingly in discrepancy
with previous reports of a higher prevalence of CVD in males than in females and slight prevalence differences in different ancestries, these results corroborate more careful cohort studies
demonstrating that the correlation of gender and ancestry with thrombotic risks is mainly due to the differential prevalence of social determinants of health and cardiovascular risk
factors45,48,49. Due to size variations, different human arteries and arterioles have distinct Reynolds numbers (affecting flow patterns such as laminar versus turbulent) and shear rates in
the blood flow50,51, together resulting in a certain extent of diversification in the shear stress. However, changing the perfusion rate in our assay from 18 to 13.5, 27, and 36 μl/min
(respectively changing the shear stresses to 0.75, 1.5, and 2 times of the original) did not significantly affect the thrombus profiling outcome (Supp. Fig. 8), wherein significantly larger
thrombus size (Supp. Fig. 8a) and higher E+ and Act. αIIbβ3 levels (Supp. Fig. 8f, g), marginally higher Fg level (Supp. Fig. 8b) but comparable VWF, P-selectin, and PS levels (Supp. Fig.
8c–e) were consistently observed in the thrombi of hypertensive young subjects than healthy young subjects. These results validated that our assay could assess the general shear-driven
platelet “aggregatability” of blood samples. HYPERTENSION CAUSES HYPERACTIVITY OF THE GPIBΑ-INTEGRIN ΑIIBΒ3 MECHANOSENSING AXIS We previously identified that the intermediate activation
state of αIIbβ3 integrin with an extended-close conformation (E+Act.−) plays a crucial role in biomechanical platelet aggregation12. Thus, the over-expressed E+ αIIbβ3, predominantly E+Act.−
αIIbβ3 in the thrombi of hypertensive patients (Fig. 4c and Supp. Fig. 6f) should directly contribute to their intensified biomechanical thrombogenesis. We hypothesize that the E+ αIIbβ3
over-expression is possibly due to (1) hyperactivity in GPIbα, with triggers stronger mechanosignaling for integrin activation12 and/or (2) integrin αIIbβ3 pre-activation in the patients. To
test these two hypotheses, we used four complementary approaches to investigate the activities of GPIbα and integrin αIIbβ3 in hypertension patients. Firstly, a conventional laminar flow
chamber assay was used to assess the overall ligand binding activity of the two receptors. Unlike the stenosis assay, here, the channels adopt a plain surface pre-coated with VWFA1 or Fg to
engage GPIbα and integrin αIIbβ3, respectively. Plasma in the blood was depleted and replaced with buffer to remove endogenous VWF and Fg and prevent platelet aggregation. By perfusing blood
through the channels under varied shear rates, it was found that platelets from hypertensive young, hypertensive older, and healthy older groups all achieved much higher surface coverage
and slower rolling on VWFA1 than the healthy young group (Fig. 5a–c and Supp. Fig. 9a, b). On the other hand, only hypertensive young and hypertensive older groups achieved high surface
coverage on Fg (Fig. 5a, d). These results indicate that both hypertension and aging cause GPIbα hyperactivity, but only hypertension induces hyperactivity in integrin αIIbβ3 at the same
time. Considering that the activities of GPIbα and integrin αIIbβ3 in hypertensive young and hypertensive older subjects were comparable (Fig. 5b–d), mechanistic studies below combined young
and older hypertensive subjects into a single cohort to compare with healthy young subjects. However, this does not exclude the possibility that aging can influence hypertensive patients’
GPIbα and integrin αIIbβ3 as a secondary factor, which shall be inspected in future studies. Secondly, a single-molecule force spectroscopy technique, biomembrane force probe (BFP)52, was
used to measure the ligand binding of single platelets. A micropipette-aspirated biotinylated human red blood cell (RBC) was used as an ultrasensitive force transducer, and a probe bead
co-functionalized with streptavidin and VWFA1 or Fg was glued to the RBC apex. A platelet was aspirated by an opposing micropipette and driven to repeatedly contact the bead, which induced
adhesion events to measure the receptor–ligand binding kinetics (Fig. 5e). Adhesion frequency assay was first deployed to enumerate the absence or presence of adhesion events after long
contacts to calculate the steady-state adhesion frequency, _P_a53. The _P_a of hypertensive subjects’ platelets adhering to the same batch of VWFA1 and Fg beads were significantly higher
than healthy young (Fig. 5f, i), reflecting a significantly higher effective avidity (ligand-binding capability of each unit of platelet surface area) of both GPIbα and integrin αIIbβ3 (Fig.
5g, j, left), consistent with the platelets’ enhanced capability of engaging VWFA1 and Fg in the flow chamber (Fig. 5a–d). Dividing effective avidities by the receptors’ surface densities
showed that the average effective affinities of GPIbα and integrin αIIbβ3 on the hypertensive subjects’ platelets were also significantly enhanced (Fig. 5g, j, right). Then, the BFP
force-clamp assay was used to measure the stability of single GPIbα–VWFA1 and single integrin αIIbβ3–Fg bonds under force. This was achieved by adjusting the contact time between the bead
and the platelet to achieve _P_a ≈ 20%, thereby realizing a ~90% probability of single bonds53. The GPIbα–VWFA1 bond lifetime of hypertensive subjects’ platelets manifested a ‘slip bond’
instead of a triphasic ‘slip-catch-slip’ trend seen on healthy young subjects’ platelets54, resulting in a substantial prolongation of bond lifetimes under forces <20 pN (Fig. 5h). On the
other hand, hypertension caused a substantial rightward and upward shift of the integrin αIIbβ3–Fg catch bond12, so that the peak force increased from ~15 to ~35 pN, the peak lifetime
increased from ~5 to ~10 s, and the force range where lifetime events were observable was widened from 0-40 to 0-65 pN (Fig. 5k). Notably, this lifetime curve from hypertensive subjects’
platelets also resembles healthy young subjects’ E+Act.- integrin αIIbβ3–Fg lifetime curve characterized before12. Altogether, our BFP results indicate that hypertension increases not only
the avidity, but also the affinity and force-regulated ligand binding strength of platelet GPIbα and integrin αIIbβ3. Thirdly, we combined fluorescence imaging with BFP (fBFP) to study
whether the increased affinity and ligand binding strength of GPIbα in hypertension patients can result in stronger mechanosignaling to better induce integrin αIIbβ3 activation. Platelets
pre-loaded with a Ca2+ dye (Fura-2) were repeatedly stimulated by a VWFA1-coated bead in force-clamp cycles at a fixed 2-s contact time for 5 min (Fig. 5l), while the normalized
intraplatelet Ca2+ level was monitored (Fig. 5m). Agreeing with our hypothesis, hypertension patients’ platelets fluxed stronger Ca2+ signals—reflected by higher Ca2+ peak increase—than
healthy young subjects’ platelets under a wide force range (Fig. 5n). Unlike healthy young subjects’ platelets where the Ca2+ signal intensity first increases and then decreases as clamping
force increases, mirroring their lifetime’s ‘catch-slip’ trend, the Ca2+ signal intensity of hypertension patients’ platelets manifested a gradual decline, also consistent with the shape of
their GPIbα–VWFA1 lifetime slip bond (Fig. 5n). This corroborates our previous finding that the GPIbα mechanosignaling intensity, manifested by both Ca2+ flux and integrin αIIbβ3 activation,
heavily relies on the duration of force pulling on GPIbα55. Fourthly, flow cytometry was used to investigate whether the αIIbβ3 integrins on hypertension patients’ platelets are
pre-activated. While similar high expression of integrin αIIbβ3 and baseline expression of Act. αIIbβ3 and P-selectin were detected on the platelets of healthy young and hypertensive
subjects, the expression of E+ αIIbβ3 in the hypertensive group was much higher than in the healthy young group (Fig. 5o–s). Although hypertension patients’ platelets are slightly larger
than healthy young subjects’, a positive correlation between E+ αIIbβ3 signal and platelet volume was found only in the hypertensive group but not the healthy young group (Supp. Fig. 9c, d).
These results indicate that hypertension patients’ platelets are pre-activated, with integrin αIIbβ3 up-regulated to the intermediate activation state (E+Act.-) and minimal P-selectin
expression. Altogether, our results indicate that two mechanisms work in parallel to induce E+ αIIbβ3 over-expression in the biomechanical thrombi of hypertensive patients (Fig. 5t): (1)
some αIIbβ3 integrins already adopt a native E+ status rather than remaining inactive as on healthy platelets; and (2) hyperactive GPIbα triggers stronger mechanosignaling upon VWF binding,
inducing more αIIbβ3 integrins to undergo E+ activation than on healthy platelets. EXPANDING THE ADDITION RULE TO DRUG–DISEASE INTERACTIONS Using the thrombus profiling assay, we tested how
anti-thrombotic inhibitors affect the thrombus profile of hypertension patients. Consistent with our results on healthy subjects, the combination of aspirin and clopidogrel at twice their
human plasmatic concentrations in clinical practice31 showed no effect on hypertension patients’ thrombi (Fig. 6a). In contrast, at IC50, NMC4 reduced the thrombus size and E+ αIIbβ3 and
Act. αIIbβ3 expressions to healthy levels, but also lowered VWF and P-selectin levels that were unaffected by hypertension (Fig. 6a). The resulting effect barcode, [0 + - - 0 0 0], equals
the add-up of those of NMC4 and hypertension (Fig. 6b). Similarly, adding 7E3 to hypertension patients’ blood resulted in an effect barcode of [0 0 - 0 0 + +], equaling the add-up of the
effect barcodes of 7E3 and hypertension (Fig. 6a, b). These results indicate that the addition rule of the barcode system can also be applied to predict drug–disease interactions. Neither
NMC4 nor 7E3 completely corrected the effect barcode of hypertension, with 7E3 even incapable of suppressing the integrin αIIbβ3 over-activation, implying a treatment mismatch between the
inhibitors and the patients. INTER-INDIVIDUAL VARIABILITY IN PERSONAL THROMBUS BARCODES Lastly, to evaluate the normality and abnormalities of individuals’ thrombus profiles, we created the
concept of “personal thrombus barcodes”. From the thrombus profiles of healthy young subjects, values of each dimension were fitted to a Gaussian distribution, of which the mean ± 2 s.d.
(~95% confidence interval) was defined as the reference range (“0”) (Supp. Fig. 10), and values lower or higher were defined as abnormally low (“−”) and high (“+”), respectively (Fig. 6c).
Applying this system to healthy young subjects rendered all dimensions of thrombus profiles being dominated by normal values, with only very small fractions being abnormally low or high,
which is consistent with the definition of the reference ranges (Fig. 6d). In contrast, much larger fractions of healthy older, hypertensive young, and hypertensive older subjects had
abnormally large thrombi and high E+ and Act. αIIbβ3 levels, with moderately higher fractions also having abnormally high Fg levels (Fig. 6d). Most of these subjects (26/36) have abnormally
high values in thrombus size and E+ αIIbβ3 level, yet 3 subjects with abnormally large thrombi have a normal E+ αIIbβ3 level. An abnormally high Act. αIIbβ3 level was observed in half of the
subjects with large thrombi (14/29), but also in two subjects with normal-sized thrombi (Supp. Fig. 11). Most subjects in these three groups (23/36) have abnormal VWF, P-selectin, and PS
levels, which may or may not co-exist with abnormally high values of thrombus size and E+ αIIbβ3 level. Of all the 69 subjects, a total of 30 different personal thrombus barcodes were
identified (Supp. Fig. 11). Overall, the above results indicate strong inter-individual variability in the personal thrombus barcode that cannot be ascribed to only disease and aging, and
demonstrate obvious decoupling of the different dimensions in the thrombus profile. Notably, we repeated our test on 14 randomly picked subjects after different time intervals (from 2 weeks
to 9 months). Among a total of 21 re-tests, only two showed changes in the personal thrombus barcodes, which were associated with the longest time intervals (7 and 9 months, respectively)
(Supp. Fig. 11). This reflects the high reliability of our assay and indicates that the personal thrombus profiles of individuals are relatively stable but can still vary over time.
Inter-individual variability was also observed in the subjects’ responses to anti-thrombotic inhibitors. While NMC4 effectively corrected the size and E+ αIIbβ3 level of most hypertension
patients’ thrombi (Fig. 6d), it did not uniformly modify their personal thrombus barcodes, but instead produced three different barcodes in five patients’ blood samples (Fig. 6e). Similar
diversification was found in 7E3, despite its consistent negative effect on the thrombus size and neutral effect on E+ αIIbβ3 level (Fig. 6d, e). These diversifications cannot be completely
ascribed to differences in the patients’ original personal thrombus barcodes (Fig. 6e). DISCUSSION The methodology framework developed in this study includes not only the experimental setup
itself, but also the ‘thrombus profile’, the barcode systems, and the “addition rule” as conceptual elements. Unlike conventional laboratory and point-of-care assays, our thrombus profiling
assay mainly assesses biomechanical platelet aggregation. Because the high-shear, high-gradient blood flow associated with arterial thrombosis reinforces biomechanical platelet
aggregation10,32 and, at the same time, impedes soluble agonist-induced platelet aggregation and coagulation by limiting the local accumulation of soluble substances56, biomechanical
platelet aggregation should be one, and possibly the most, essential mechanism of arterial thrombosis. This rationalizes the outstanding performance of our thrombus profiling assay in
testing clinical subjects associated with higher risks of arterial thrombosis, demonstrating its potential to clinically assess thrombotic risks in general populations. In this context, the
detection of integrin αIIbβ3 over-activation and the identification of “treatment mismatch” further showcase the assay’s ability in identifying the mechanisms of prothrombotic tendency and
in evaluating prevention/treatment strategies. Meanwhile, cost-effectiveness and low sample volume represent additional advantages. Hardware upgrades, e.g., using a multi-channel syringe
pump and a motorized stage or a multi-camera array to reach relatively high throughput, and/or system automation, will enable the current setup to become more suitable for clinical practice.
To provide more accurate diagnosis and treatment suggestions, the assay can benefit from more detailed segmentation (e.g., borderline, stage-I, and stage-II abnormal) in judging normal
_versus_ abnormal thrombus barcodes, and can be combined with other existing diagnostic approaches, e.g., risk score assessment57,58. Notably, the assay also has the potential of evaluating
bleeding tendency in humans and the bleeding side effect of anti-thrombotic agents24, which warrants future investigation. As a limitation, our assay cannot recapitulate the biomechanical
scenarios of thrombosis in all different arteries and arterioles, especially in large stenotic arteries where the Reynolds number can reach sufficiently high to trigger turbulence50.
Nonetheless, by replicating critical aspects of thrombosis, the assay allows the evaluation of the general prothrombotic tendency of blood samples. Using a panel of mAbs with highly specific
targets, we showed that GPIbα–VWF, integrin αIIbβ3–VWF, and integrin αIIbβ3–Fg interactions all contribute to the size growth of biomechanical thrombi. However, suggesting a central role of
GPIbα–VWF interaction in biomechanical thrombogenesis, only its blockage but not the blockage of integrin αIIbβ3–VWF or integrin αIIbβ3–Fg interaction can eliminate thrombus formation.
Also, only blocking GPIbα–VWF interaction inhibits integrin αIIbβ3 activation while blocking integrin αIIbβ3–ligand interactions failed so, reflecting a primary role of GPIbα for integrin
αIIbβ3 activation in the GPIbα-integrin αIIbβ3 mechanosensing axis12. We showed that GPIbα–VWF and integrin αIIbβ3–VWF interactions both modulate the deposition of VWF into the thrombus,
while integrin αIIbβ3–Fg only modulates that of Fg, which seems intuitive because both VWF and Fg need to be bound to their respective platelet receptors to maintain their presence in the
thrombus. However, the fact that inhibiting either VWF or Fg binding to integrin αIIbβ3 does not enrich the other ligand, but both reduce the thrombus size, suggests that VWF and Fg
cooperate, rather than mutually compensate, in integrin αIIbβ3 crosslinking for biomechanical platelet aggregation. Our observation that inhibiting the two RGD sequences in Fg effectively
reduces the thrombus size contrasts with the previous report that mutating either of these two sequences did not impair Fg function in mediating ADP-induced platelet aggregation59. This is
likely because ADP activates integrin αIIbβ3 to the fully active state, while biomechanical platelet aggregation is mainly driven by intermediate state integrin αIIbβ312, so that the RGD
sequences in Fg are redundant in the former scenario for platelet crosslinking but become a useful supplement to the Fg AGDV sequences in the latter. This suggests mechanistic distinctions
when Fg mediates biomechanical _versus_ biochemical platelet aggregation and unravels an underestimated contribution of the Fg RGD sequences to arterial thrombosis. On the other hand,
previous works showed that when the shear rate increases, the dependency of shear-induced platelet aggregation on GPIbα and integrin αIIbβ3 becomes progressively stronger and weaker,
respectively60. It will be interesting to test whether changing the shear rate in our assay affects how the three receptor–ligand interactions contribute to the thrombus profile. Lastly, our
results appear to indicate that the effect barcode of each anti-thrombotic agent is dictated by its target rather than its pharmacological design. Moreover, the observed “addition rule”
suggests a lack of synergy or discord when multiple targets are concurrently inhibited, indicating that different molecular interactions and signaling pathways function in relatively
independent and parallel ways. These principles are potentially useful for drug screening, enabling us to quickly narrow down the possible target(s) of uncharacterized anti-thrombotic agents
using their effect barcode. To serve the above purpose, inhibitors of all other contributing factors of biomechanical platelet aggregation, e.g., mechanosignaling of GPIbα, integrin αIIbβ3,
and Piezo116,61,62, need to be tested to acquire their effect barcodes. Hypertension is the leading cause of CVD and is also closely associated with antiplatelet (e.g., aspirin and
clopidogrel) resistance3,4. Among multiple postulated mechanisms, abnormal platelet activation has been identified as a central contributor to the prothrombotic status of hypertension
patients, where changes in platelet morphology and biochemical activities (e.g., elevated sensitivity to soluble agonists, reduced sensitivity to exogenous nitric oxide) were reported63. In
comparison, we discover that GPIbα in hypertension patients are hyperactive and can induce stronger mechanosignaling, while a substantial amount of integrin αIIbβ3 molecules are already in
the E+ status, which together results in an over-expression of E+ integrin αIIbβ3 in the patients’ biomechanical thrombi. Considering the central roles of GPIbα and E+ integrin αIIbβ3 in
biomechanical platelet aggregation12, these results explain the intensified biomechanical thrombogenesis observed in hypertension patients’ blood, and suggest that GPIbα-integrin αIIbβ3
mechanosensing axis hyperactivity directly contributes to the high incidence rate of CVD in hypertension patients. On the other hand, antiplatelet resistance is conventionally believed to be
due to patients’ lack of sensitivity to antiplatelets in inhibiting platelet amplification loops64. However, we found that biomechanical thrombogenesis is essentially “immune” to aspirin
and clopidogrel in both healthy young subjects and hypertension patients (Supp. Fig. 12). These, together with similar observations by other works10,12,32, indicate a new mechanism of
antiplatelet resistance: biomechanical platelet aggregation can mediate arterial thrombosis independent of platelet amplification mechanisms, and therefore the sole inhibition of platelet
amplification loops allows thrombotic risks to persist by leaving biomechanical platelet aggregation active. Altogether, our results strongly advocate the development of GPIbα and/or
integrin αIIbβ3 targeting anti-thrombotic “mechanomedicines” that can work complementarily with conventional antiplatelets for enhanced treatment efficacy. The results also underscore the
pathophysiological relevance of E+-closed integrin αIIbβ3, which should inspire future investigations on the importance of the E+-closed conformation in other integrins and in the context of
other diseases. The causes of GPIbα and integrin αIIbβ3 hyperactivity in hypertension patients as well as the similar trend of platelet hyperreactivity in older people warrant further
investigation, which are possibly relevant to hypertension/aging-associated oxidative stress and inflammation that cause platelet pre-activation65,66,67, dysregulated glycosylation of GPIbα
and integrin αIIbβ3 by metabolic disorders68,69, and/or the activation of mechanosensitive ion channel Piezo1 that causes platelet hyper-sensitivity to shear force16. On the other hand, the
slightly higher Fg level in hypertensive and older subjects’ thrombi is likely due to the elevated Fg plasma concentration in these populations70,71 as well as integrin αIIbβ3 hyperactivity
that more efficiently recruits Fg. P-selectin and Act. αIIbβ3 are widely used markers of platelet activation, but their performance in diagnosing thrombosis is unsatisfactory due to low
sensitivity72. We show that E+ αIIbβ3 has a much better performance than P-selectin and Act. αIIbβ3 in correlating with the biomechanical thrombus size and in separating healthy young
subjects and subjects carrying thrombotic risk factors. Furthermore, only E+ αIIbβ3, but not P-selectin or Act. αIIbβ3, was detected on platelets freshly isolated from hypertension patients.
These results underscore the accuracy and sensitivity of E+ αIIbβ3 in detecting platelet hyperreactivity, suggesting its use as an independent biomarker for predicting arterial thrombosis
in certain populations. To validate this application requires an investigation on the correlation between native E+ αIIbβ3 expression (assessed by flow cytometry) and thrombus size (assessed
by thrombus profiling assay) in different patient cohorts. Over the past decades, a routine has formed to evaluate the efficacy of new anti-thrombotic strategies solely based on thrombus
size reduction and without considering inter-individual variability11,73. Our work demonstrates that thrombi possess multi-dimensional characteristics that can be orthogonal, which should be
summarized as a “profile” or a “barcode”. Because different individuals have differential personal thrombus barcodes, and different anti-thrombotics have differential effect barcodes, a
treatment mismatch can easily occur. Conceptually distinguished from antiplatelet resistance, a drug with treatment mismatch is still effective in reducing the thrombus size, but has limited
or undesired effects on changing the thrombus composition and/or activation status (Supp. Fig. 12). The life-threatening danger of treatment mismatch has been documented in multiple phase
III trials where conventional integrin αIIbβ3 antagonists (e.g., orbofiban), despite high potency in inhibiting soluble agonist-induced platelet aggregation (and also biomechanical platelet
aggregation as demonstrated in this work), paradoxically increased patient mortality by enhancing the risk of myocardial infarction74,75. It was later realized that the failure of these
drugs was associated with their effect of stimulating integrin αIIbβ3 activation76. Addressing this issue, a chemical principle was recently discovered to develop anti-thrombotic candidates
that lock integrin αIIbβ3 in the inactive state77. Developing diversified anti-thrombotics and determining their effect barcodes and their interactions with thrombosis-exacerbating factors
can help avoid treatment mismatch, in which the “addition rule” could be helpful for prediction. The inter-individual variability in drug efficacy further urges the personalized selection of
anti-thrombotics for treatment optimization. METHODS REAGENTS SZ22-FITC and P2-Alexa Fluor 488 (Beckman Coulter), Type I collagen, AK4-Alexa Fluor 647 and PAC-1-Alexa Fluor 647 (BioLegend),
AK2, HIP-8-Alexa Fluor 488, Annexin V-Pacific Blue, Annexin V-Alexa Fluor 488, heparin, DiOC6(3), and Alexa Fluor 405, 555, and 647 conjugation kits (Thermo Fisher Scientific), MBC 370.2
(Kerafast), fibrinogen (Innovative Research), NMC4, 2.2.9, LJ-P5, 152B6, LJ-155B39, LJ-134B29 and VWFA178 (MERU VasImmune), VWF monomer (Sino Biological), RU5 (Creative Biolabs), and PS-CNP
beads (Bangs Laboratories) were purchased. 7E9, 7E3, and 10E5 were gifts from Barry S. Coller (Rockefeller University). HUMAN SUBJECTS All procedures involving human subjects were approved
by the Institutional Review Board of the University of Texas Medical Branch (protocol number: 22-0015) and the University of Sydney (ethics reference number: 2023/582). Informed consent was
obtained from all subjects to allow the publishing of data acquired from their blood samples and their demographic information that is relevant to research while protecting their privacy.
All subjects were compensated for their participation. Number (_n_) and age (mean ± s.d.) of subjects who participated in the thrombus profiling assay: healthy young: _n_ = 33, age = 34.0 ±
6.3; healthy older: _n_ = 14, age = 62.1 ± 8.9; hypertensive young: _n_ = 9, age = 36.1 ± 8.7; hypertensive older: _n_ = 13, age = 60.2 ± 9.4. All groups contained both male and female
subjects with multiple races and both non-Hispanic/Latino and Hispanic/Latino ethnicities. All hypertension patients were taking prescribed hypertension medications (e.g., prazosin,
amlodipine, and enalapril). Patients taking other medications or under treatment for other diseases within 2 weeks before the blood draw were excluded from this study. BLOOD COLLECTION,
RECONSTITUTION, AND PLATELET ISOLATION For whole blood stenosis assay, blood was slowly drawn from the vein of a volunteer into a syringe pre-loaded with heparin (20 U/mL). In some control
experiments, sodium citrate (4%) or EDTA (1.5 mg/ml) was used as the anticoagulant instead. For laminar flow chamber assay, BFP assays, and flow cytometry, blood was drawn into a syringe
pre-loaded with ACD buffer. Then blood reconstitution78 was performed for laminar flow chamber assay to deplete plasma and reach a hematocrit of 45% and platelet count of 20,000 µL−1. Or,
platelet isolation was performed for BFP assays and flow cytometry12, with platelets finally resuspended in modified Tyrode’s buffer (135 mM NaCl, 11.9 mM NaHCO3, 2.9 mM KCl, 0.42 mM
NaH2PO4, 10 mM Hepes, 5.5 mM dextrose, and pH 7.4). MICROFLUIDIC DEVICE PREPARATION Polydimethylsiloxane (PDMS) was applied on a silicon mold (1-μm resolution), which was heated at 75 °C for
1 h for curing, peeled off, and cut into single pieces. Holes were drilled to create outlets and inlets. The devices then underwent plasma treatment and were bonded to glass coverslips.
MICROFLUIDIC STENOSIS ASSAY Microfluidic channels were coated with VWF monomer (2 μg/mL) for 1 h. In some control experiments, the coating was done with 100 µg/ml collagen instead. Blood was
incubated with DiOC6(3) (5 μM) for 1 min, or with Sensor Set 1 (SZ22-FITC (0.5 μg/mL), Fg-Alexa Fluor 405 (60 μg/mL), 2.2.9-Alexa Fluor 555 (1 μg/mL) and AK4-Alexa Fluor 647 (1 μg/mL)), or
Set 2 (SZ22-FITC (0.5 μg/mL), Annexin V-Pacific Blue (1 μg/mL), MBC 370.2-Alexa Fluor 555 (1 μg/mL), and PAC-1-Alexa Fluor 647 (1 μg/mL)) for 10 min, and perfused through the channel.
Thrombus formation was observed using a Leica DM IL LED microscope (camera: Leica DFC360 FX; objective lens: air, 20×; acquisition software: LAS X). No bleed-through between fluorescence
channels was observed. Platelet autofluorescence was detected in 391-nm channel79, which was subtracted when calculating signals. Data analysis was performed using ImageJ 1.53 (Fiji,
National Institutes of Health). In some experiments, different concentrations of aspirin (with 15 µg/mL defined as 2×) and/or clopidogrel (with 6 µg/mL defined as 2×) or ALB cocktail (1 U/mL
apyrase, 100 mM MRS2179, 10 mM 2-MeSAMP, 10 μM indomethacin, 800 U mL−1 hirudin) were added into blood to inhibit platelet amplification loops. Hill equation was used to derive IC50 of
inhibitors: $${{{\rm{Residue}}}}\; {{{\rm{size}}}}=R+\left(100-R\right)/ \left(1+\left({{{\rm{IC}}}}50/C\right)^{{{\rm{HillSlope}}}}\right)$$ (1) wherein _C_ is the inhibitor concentration,
_R_ is the residue size when the effect of the inhibitor saturates, and HillSlope is a constant. For subjects who were tested multiple times, average values of these test results were used
for data presentation and statistical analyses. MICROFLUIDIC LAMINAR FLOW CHAMBER ASSAY Reconstituted blood added with DiOC6(3) (10 μM) was perfused at different shear rates over straight
channels pre-coated with VWFA1 or fibrinogen. After 5 min, fluorescent signals from platelets were recorded at 40 frame s−1. Data analysis was performed using ImageJ 1.53 (Fiji, National
Institutes of Health). BIOMEMBRANE FORCE PROBE (BFP) AND FLUORESCENCE BFP (FBFP) In a chamber filled with modified Tyrode’s buffer + 0.5% BSA (plus 1 mM Ca2+/Mg2+ when interrogating
platelets with Fg beads), a streptavidin-coated glass probe bead was glued to the apex of a biotinylated RBC, which is aspirated by a micropipette to form an ultrasensitive force probe52.
The probe bead was also coated with VWFA1 or Fg. On the opposing target side, a freshly isolated platelet was aspirated by a second micropipette, which was driven by a piezoelectric
translator (Physical Instrument) to repeatedly bring the platelet in and out of contact with the bead to form adhesion events. The bead was monitored under an inverted microscope (IX83,
Olympus) by a high-speed camera. A custom image analysis LabView (National Instrument) program tracks the bead position with 3 nm precision in real time. The BFP spring constant _k_ was
determined by the suction pressure inside the probe pipette and the geometric parameters of the force transducer assembly80. For adhesion frequency assay, the platelet was repeatedly brought
into contact with the probe bead for 2 seconds and retracted. Adhesion events were signified by the elongation of the RBC upon platelet retraction, which yielded a tensile force signal on
the bead. Adhesion and non-adhesion events in 30 cycles were enumerated to calculate adhesion frequency, _P_a. The effective avidity (_A_c_K_a_m_r) and affinity (_A_c_K_a) were derived by
the following equation53, $${P}_{a}=1-\exp \left\{{-{m}_{r}{m}_{l}A}_{c}{K}_{a}\right\}$$ (2) where _m_r and _m_l are the receptor and ligand surface densities derived from flow cytometry.
For the force-clamp assay, contact time was shortened until achieving infrequent (~20%) adhesion, which ensures that most (~90%) of the adhesion events are mediated by single receptor–ligand
bonds. Once an adhesion event was observed, the platelet would be held at a desired clamping force to wait for the bond to dissociate52. Lifetime was determined as the time from the instant
when the force reached the desired level to the instant of bond dissociation. The collected lifetimes were categorized into bins that cover successive force ranges. The average lifetime in
each force bin was calculated to plot the “lifetime vs. clamping force” curve. For fBFP, platelets were pre-loaded with Fura-2-AM and interrogated by VWFA1 beads with the force-clamp assay
mode, but the contact time was kept at 2 s. Ratiometric imaging with a light source that alternates between 340 nm (to excite Ca2+-engaged Fura-2) and 380 nm (to excite Ca2+-free Fura-2) was
used to measure the Ca2+ level in the aspirated platelet55. Matlab R2020b was used to analyze fluorescence images from fBFP experiments. Signal intensity from the 340-nm channel was divided
by that from the 380-nm channel and then normalized by the average value of the first 10 frames to derive the normalized Ca2+ level. FLOW CYTOMETRY ASSAY Platelet suspension was incubated
with 2 μg/mL of HIP-8-Alexa Fluor 488, MBC 370.2-Alexa Fluor 555, PAC-1-Alexa Fluor 647, or AK4-Alexa Fluor 647 for 10 min, diluted with Hepes-Tyrode buffer by ten times, and immediately
analyzed by flow cytometry. STATISTICAL ANALYSIS GraphPad Prism 10 was used for data plotting and statistical analysis. The statistical significance of the differences between the two groups
was determined by a two-sided Student's _t_-test or multiple t-test. For the test of drug effects, multiple _t_-test assuming paired experimental design was used. For multi-group
analysis, one-way or two-way ANOVA was used. When significant differences were shown, data was subjected to the Tukey test for multiple comparisons. A regression slope test was used to
assess whether the slope of a linear fitting is significantly non-zero. Spearman rank correlation coefficient46 and Kendall’s tau correlation coefficient47 were also used to test whether a
positive correlation exists between different readouts of the thrombus profile. _P_ values <0.05 were considered significant. REPORTING SUMMARY Further information on research design is
available in the Nature Portfolio Reporting Summary linked to this article. DATA AVAILABILITY All data supporting the findings of this study are available within the article and its
supplementary files. Source data of fluid dynamics simulation generated in this study have been deposited in the Harvard Dataverse repository under accession code
(https://doi.org/10.7910/DVN/D4SJIP). Source data of human blood sample experiments are protected and unavailable for public deposition or upon request in accordance with the signed consent
of study subjects. Any additional requests for information can be directed to, and will be fulfilled by, the corresponding authors. Source data are provided with this paper. REFERENCES *
Benjamin, E. J. et al. Heart disease and stroke statistics-2019 update: a report from the American Heart Association. _Circulation_ 139, e56–e528 (2019). Article PubMed Google Scholar *
Springer, T. A. von Willebrand factor, Jedi knight of the bloodstream. _Blood_ 124, 1412–1425 (2014). Article PubMed PubMed Central CAS Google Scholar * Akturk, I. F. et al.
Hypertension as a risk factor for aspirin and clopidogrel resistance in patients with stable coronary artery disease. _Clin. Appl. Thromb. Hemost._ 20, 749–754 (2014). Article PubMed CAS
Google Scholar * Liu, X. F. et al. Prevalence of and risk factors for aspirin resistance in elderly patients with coronary artery disease. _J. Geriatr. Cardiol._ 10, 21–27, (2013). PubMed
PubMed Central Google Scholar * Jackson, S. P. Arterial thrombosis–insidious, unpredictable and deadly. _Nat. Med._ 17, 1423–1436 (2011). Article PubMed CAS Google Scholar * Previtali,
E., Bucciarelli, P., Passamonti, S. M. & Martinelli, I. Risk factors for venous and arterial thrombosis. _Blood Transfus._ 9, 120–138, (2011). PubMed PubMed Central Google Scholar *
Govender, R. D., Al-Shamsi, S., Soteriades, E. S. & Regmi, D. Incidence and risk factors for recurrent cardiovascular disease in middle-eastern adults: a retrospective study. _BMC
Cardiovasc. Disord._ 19, 253 (2019). Article PubMed PubMed Central Google Scholar * Zhang, Y. et al. Emerging microfluidic approaches for platelet mechanobiology and interplay with
circulatory systems. _Front. Cardiovasc. Med._ 8, 766513 (2021). Article PubMed PubMed Central CAS Google Scholar * Zhang, Y., Jiang, F., Chen, Y. & Ju, L. A. Platelet
mechanobiology inspired microdevices: from hematological function tests to disease and drug screening. _Front. Pharmacol._ 12, 779753 (2021). Article PubMed CAS Google Scholar * Nesbitt,
W. S. et al. A shear gradient-dependent platelet aggregation mechanism drives thrombus formation. _Nat. Med._ 15, 665–673 (2009). Article PubMed CAS Google Scholar * Chen, Y. & Ju,
L. A. Biomechanical thrombosis: the dark side of force and dawn of mechano-medicine. _Stroke Vasc. Neurol._ 5, 185–197 (2020). Article PubMed Google Scholar * Chen, Y. et al. An integrin
alphaIIbbeta3 intermediate affinity state mediates biomechanical platelet aggregation. _Nat. Mater._ 18, 760–769 (2019). Article ADS PubMed PubMed Central CAS Google Scholar * Fu, H.
et al. Flow-induced elongation of von Willebrand factor precedes tension-dependent activation. _Nat. Commun._ 8, 324 (2017). Article ADS PubMed PubMed Central Google Scholar * Yago, T.
et al. Platelet glycoprotein Ibalpha forms catch bonds with human WT vWF but not with type 2B von Willebrand disease vWF. _J. Clin. Investig._ 118, 3195–3207 (2008). PubMed PubMed Central
CAS Google Scholar * Ju, L. et al. Compression force sensing regulates integrin alphaIIbbeta3 adhesive function on diabetic platelets. _Nat. Commun._ 9, 1087 (2018). Article ADS PubMed
PubMed Central Google Scholar * Zhao, W. et al. Piezo1 initiates platelet hyperreactivity and accelerates thrombosis in hypertension. _J. Thromb. Haemost._
https://doi.org/10.1111/jth.15504 (2021). * Ju, L. A. et al. Microfluidic post method for 3-dimensional modeling of platelet-leukocyte interactions. _Analyst_ 147, 1222–1235 (2022). Article
ADS PubMed CAS Google Scholar * Zhang, X. F. & Cheng, X. Platelet mechanosensing axis revealed. _Nat. Mater._ 18, 661–662 (2019). Article ADS PubMed CAS Google Scholar *
Yakusheva, A. A. et al. Traumatic vessel injuries initiating hemostasis generate high shear conditions. _Blood Adv._ 6, 4834–4846 (2022). Article PubMed PubMed Central Google Scholar *
Panteleev, M. A. et al. Wall shear rates in human and mouse arteries: standardization of hemodynamics for in vitro blood flow assays: communication from the ISTH SSC subcommittee on
biorheology. _J. Thromb. Haemost._ 19, 588–595 (2021). Article PubMed Google Scholar * Dent, J. A., Galbusera, M. & Ruggeri, Z. M. Heterogeneity of plasma von Willebrand factor
multimers resulting from proteolysis of the constituent subunit. _J. Clin. Investig._ 88, 774–782 (1991). Article PubMed PubMed Central CAS Google Scholar * Kamath, S., Blann, A. D.
& Lip, G. Y. Platelet activation: assessment and quantification. _Eur. Heart J._ 22, 1561–1571 (2001). Article PubMed CAS Google Scholar * Schoenwaelder, S. M. et al. Two distinct
pathways regulate platelet phosphatidylserine exposure and procoagulant function. _Blood_ 114, 663–666 (2009). Article PubMed CAS Google Scholar * Brazilek, R. J. et al. Application of a
strain rate gradient microfluidic device to von Willebrand’s disease screening. _Lab Chip_ 17, 2595–2608 (2017). Article PubMed CAS Google Scholar * Merten, M., Chow, T., Hellums, J. D.
& Thiagarajan, P. A new role for P-selectin in shear-induced platelet aggregation. _Circulation_ 102, 2045–2050 (2000). Article PubMed CAS Google Scholar * Deng, W. et al. Platelet
clearance via shear-induced unfolding of a membrane mechanoreceptor. _Nat. Commun._ 7, 12863 (2016). Article ADS PubMed PubMed Central CAS Google Scholar * Hu, H. et al.
Platelet-leukocyte aggregation under shear stress: differential involvement of selectins and integrins. _Thromb. Haemost._ 90, 679–687 (2003). Article PubMed CAS Google Scholar *
Roka-Moiia, Y. et al. Platelet activation via shear stress exposure induces a differing pattern of biomarkers of activation versus biochemical agonists. _Thromb. Haemost._ 120, 776–792
(2020). Article PubMed PubMed Central Google Scholar * Pang, A. et al. Shear-induced integrin signaling in platelet phosphatidylserine exposure, microvesicle release, and coagulation.
_Blood_ 132, 533–543 (2018). Article PubMed PubMed Central CAS Google Scholar * Chen, W. et al. Inhibiting GPIbalpha shedding preserves post-transfusion recovery and hemostatic function
of platelets after prolonged storage. _Arterioscler. Thromb. Vasc. Biol._ 36, 1821–1828 (2016). Article PubMed PubMed Central Google Scholar * Arrebola, M. M. et al. In vitro effects of
clopidogrel on the platelet-subendothelium interaction, platelet thromboxane and endothelial prostacyclin production, and nitric oxide synthesis. _J. Cardiovasc. Pharm._ 43, 74–82 (2004).
Article Google Scholar * Li, M., Hotaling, N. A., Ku, D. N. & Forest, C. R. Microfluidic thrombosis under multiple shear rates and antiplatelet therapy doses. _PLoS ONE_ 9, e82493
(2014). Article ADS PubMed PubMed Central Google Scholar * Kanaji, S. et al. Humanized GPIbalpha-von Willebrand factor interaction in the mouse. _Blood Adv._ 2, 2522–2532 (2018).
Article PubMed PubMed Central CAS Google Scholar * Yuan, Y. et al. The von Willebrand factor-glycoprotein Ib/V/IX interaction induces actin polymerization and cytoskeletal
reorganization in rolling platelets and glycoprotein Ib/V/IX-transfected cells. _J. Biol. Chem._ 274, 36241–36251 (1999). Article PubMed CAS Google Scholar * De Marco, L., Girolami, A.,
Zimmerman, T. S. & Ruggeri, Z. M. von Willebrand factor interaction with the glycoprotein IIb/IIa complex. Its role in platelet function as demonstrated in patients with congenital
afibrinogenemia. _J. Clin. Investig._ 77, 1272–1277 (1986). Article PubMed PubMed Central Google Scholar * Berliner, S., Niiya, K., Roberts, J. R., Houghten, R. A. & Ruggeri, Z. M.
Generation and characterization of peptide-specific antibodies that inhibit von Willebrand factor binding to glycoprotein IIb-IIIa without interacting with other adhesive molecules.
Selectivity is conferred by Pro1743 and other amino acid residues adjacent to the sequence Arg1744-Gly1745-Asp1746. _J. Biol. Chem._ 263, 7500–7505 (1988). Article PubMed CAS Google
Scholar * Lengweiler, S. et al. Preparation of monoclonal antibodies to murine platelet glycoprotein IIb/IIIa (alphaIIbbeta3) and other proteins from hamster-mouse interspecies hybridomas.
_Biochem. Biophys. Res. Commun._ 262, 167–173 (1999). Article PubMed CAS Google Scholar * Felding-Habermann, B., Ruggeri, Z. M. & Cheresh, D. A. Distinct biological consequences of
integrin alpha v beta 3-mediated melanoma cell adhesion to fibrinogen and its plasmic fragments. _J. Biol. Chem._ 267, 5070–5077 (1992). Article PubMed CAS Google Scholar * Rooney, M.
M., Farrell, D. H., van Hemel, B. M., de Groot, P. G. & Lord, S. T. The contribution of the three hypothesized integrin-binding sites in fibrinogen to platelet-mediated clot retraction.
_Blood_ 92, 2374–2381 (1998). Article PubMed CAS Google Scholar * Liu, Q., Matsueda, G., Brown, E. & Frojmovic, M. The AGDV residues on the gamma chain carboxyl terminus of
platelet-bound fibrinogen are needed for platelet aggregation. _Biochim. Biophys. Acta_ 1343, 316–326 (1997). Article PubMed CAS Google Scholar * Coller, B. S. A new murine monoclonal
antibody reports an activation-dependent change in the conformation and/or microenvironment of the platelet glycoprotein IIb/IIIa complex. _J. Clin. Investig._ 76, 101–108 (1985). Article
PubMed PubMed Central CAS Google Scholar * Kaul, D. K. et al. Monoclonal antibodies to alphaVbeta3 (7E3 and LM609) inhibit sickle red blood cell-endothelium interactions induced by
platelet-activating factor. _Blood_ 95, 368–374 (2000). Article PubMed CAS Google Scholar * Griffin, M. T., Zhu, Y., Liu, Z., Aidun, C. K. & Ku, D. N. Inhibition of high shear
arterial thrombosis by charged nanoparticles. _Biomicrofluidics_ 12, 042210 (2018). Article PubMed PubMed Central Google Scholar * Wilkerson, W. R. & Sane, D. C. Aging and
thrombosis. _Semin. Thromb. Hemost._ 28, 555–568 (2002). Article PubMed CAS Google Scholar * Tsao, C. W. et al. Heart disease and stroke statistics-2022 update: a report from the
American Heart Association. _Circulation_ 145, e153–e639 (2022). Article PubMed Google Scholar * Charles, S. The proof and measurement of association between two things. _Am. J. Psychol._
15, 72–101 (1904). Article Google Scholar * Kendall, M. G. A new measure of rank correlation. _Biometrika_ 30, 81–93 (1938). Article Google Scholar * Safford, M. M. et al. Association
of race and sex with risk of incident acute coronary heart disease events. _JAMA_ 308, 1768–1774, (2012). Article PubMed PubMed Central CAS Google Scholar * Colantonio, L. D. et al.
Black-White differences in incident fatal, nonfatal, and total coronary heart disease. _Circulation_ 136, 152–166 (2017). Article PubMed PubMed Central CAS Google Scholar * Mahalingam,
A. et al. Numerical analysis of the effect of turbulence transition on the hemodynamic parameters in human coronary arteries. _Cardiovasc. Diagn. Ther._ 6, 208–220 (2016). Article PubMed
PubMed Central Google Scholar * Ku, D. N. Blood flow in arteries. _Annu. Rev. Fluid Mech._ 29, 399–434 (1997). Article ADS MathSciNet Google Scholar * Chen, Y. et al. Fluorescence
biomembrane force probe: concurrent quantitation of receptor-ligand kinetics and binding-induced intracellular signaling on a single cell. _J. Vis. Exp._ https://doi.org/10.3791/52975
(2015). * Chesla, S. E., Selvaraj, P. & Zhu, C. Measuring two-dimensional receptor-ligand binding kinetics by micropipette. _Biophys. J._ 75, 1553–1572 (1998). Article PubMed PubMed
Central CAS Google Scholar * Ju, L. et al. Von Willebrand factor-A1 domain binds platelet glycoprotein Ibalpha in multiple states with distinctive force-dependent dissociation kinetics.
_Thromb. Res._ 136, 606–612 (2015). Article PubMed PubMed Central CAS Google Scholar * Ju, L., Chen, Y., Xue, L., Du, X. & Zhu, C. Cooperative unfolding of distinctive
mechanoreceptor domains transduces force into signals. _eLife_ 5, e15447 (2016). Article PubMed PubMed Central Google Scholar * Du, J., Kim, D., Alhawael, G., Ku, D. N. & Fogelson,
A. L. Clot permeability, agonist transport, and platelet binding kinetics in arterial thrombosis. _Biophys. J._ 119, 2102–2115 (2020). Article ADS PubMed PubMed Central CAS Google
Scholar * Wong, N. D. et al. Atherosclerotic cardiovascular disease risk assessment: an American Society for Preventive Cardiology clinical practice statement. _Am. J. Prev. Cardiol._ 10,
100335 (2022). Article ADS PubMed PubMed Central Google Scholar * Anjum, M. et al. Stroke and bleeding risk in atrial fibrillation with CHA2DS2-VASC risk score of one: the Norwegian
AFNOR study. _Eur. Heart J._ 45, 57–66 (2024). Article PubMed Google Scholar * Farrell, D. H., Thiagarajan, P., Chung, D. W. & Davie, E. W. Role of fibrinogen alpha and gamma chain
sites in platelet aggregation. _Proc. Natl Acad. Sci. USA_ 89, 10729–10732 (1992). Article ADS PubMed PubMed Central CAS Google Scholar * Jackson, S. P. The growing complexity of
platelet aggregation. _Blood_ 109, 5087–5095 (2007). Article PubMed CAS Google Scholar * Dai, K., Bodnar, R., Berndt, M. C. & Du, X. A critical role for 14-3-3zeta protein in
regulating the VWF binding function of platelet glycoprotein Ib-IX and its therapeutic implications. _Blood_ 106, 1975–1981 (2005). Article PubMed PubMed Central CAS Google Scholar *
Shen, B. et al. A directional switch of integrin signalling and a new anti-thrombotic strategy. _Nature_ 503, 131–135 (2013). Article ADS PubMed PubMed Central CAS Google Scholar *
Gkaliagkousi, E., Passacquale, G., Douma, S., Zamboulis, C. & Ferro, A. Platelet activation in essential hypertension: implications for antiplatelet treatment. _Am. J. Hypertens._ 23,
229–236 (2010). Article PubMed CAS Google Scholar * Cattaneo, M. Resistance to anti-platelet agents. _Thromb. Res._ 127, S61–S63 (2011). Article PubMed CAS Google Scholar *
Griendling, K. K. et al. Oxidative stress and hypertension. _Circ. Res._ 128, 993–1020 (2021). Article PubMed PubMed Central CAS Google Scholar * Scherlinger, M., Richez, C., Tsokos, G.
C., Boilard, E. & Blanco, P. The role of platelets in immune-mediated inflammatory diseases. _Nat. Rev. Immunol._ 23, 495–510 (2023). Article PubMed CAS Google Scholar * Li, X. et
al. Inflammation and aging: signaling pathways and intervention therapies. _Signal Transduct. Target. Ther._ 8, 239 (2023). Article PubMed PubMed Central Google Scholar * Schjoldager, K.
T., Narimatsu, Y., Joshi, H. J. & Clausen, H. Global view of human protein glycosylation pathways and functions. _Nat. Rev. Mol. Cell Biol._ 21, 729–749 (2020). Article PubMed CAS
Google Scholar * Palmer, A. K. & Jensen, M. D. Metabolic changes in aging humans: current evidence and therapeutic strategies. _J. Clin. Invest._ https://doi.org/10.1172/JCI158451
(2022). * Shankar, A., Wang, J. J., Rochtchina, E. & Mitchell, P. Positive association between plasma fibrinogen level and incident hypertension among men: population-based cohort study.
_Hypertension_ 48, 1043–1049 (2006). Article PubMed CAS Google Scholar * Hager, K., Felicetti, M., Seefried, G. & Platt, D. Fibrinogen and aging. _Aging_ 6, 133–138 (1994). * Chung,
T. et al. Platelet activation in acute pulmonary embolism. _J. Thromb. Haemost._ 5, 918–924 (2007). Article PubMed CAS Google Scholar * Gawaz, M., Geisler, T. & Borst, O. Current
concepts and novel targets for antiplatelet therapy. _Nat. Rev. Cardiol._ 20, 583–599 (2023). Article PubMed Google Scholar * Topol, E. J. et al. Randomized, double-blind,
placebo-controlled, international trial of the oral IIb/IIIa antagonist lotrafiban in coronary and cerebrovascular disease. _Circulation_ 108, 399–406 (2003). Article PubMed CAS Google
Scholar * Chew, D. P., Bhatt, D. L., Sapp, S. & Topol, E. J. Increased mortality with oral platelet glycoprotein IIb/IIIa antagonists: a meta-analysis of phase III multicenter
randomized trials. _Circulation_ 103, 201–206 (2001). Article PubMed CAS Google Scholar * Cox, D. et al. Evidence of platelet activation during treatment with a GPIIb/IIIa antagonist in
patients presenting with acute coronary syndromes. _J. Am. Coll. Cardiol._ 36, 1514–1519 (2000). Article PubMed CAS Google Scholar * Lin, F. Y. et al. A general chemical principle for
creating closure-stabilizing integrin inhibitors. _Cell_ 185, 3533–3550.e3527 (2022). Article PubMed PubMed Central CAS Google Scholar * Reininger, A. J. et al. Mechanism of platelet
adhesion to von Willebrand factor and microparticle formation under high shear stress. _Blood_ 107, 3537–3545 (2006). Article PubMed PubMed Central CAS Google Scholar * Lohmann, W.
& Lohmann, C. Native fluorescence of platelets from patients with occlusive arterial disease. _Biochem. Biophys. Res. Commun._ 152, 1410–1415 (1988). Article PubMed CAS Google Scholar
* Evans, E., Ritchie, K. & Merkel, R. Sensitive force technique to probe molecular adhesion and structural linkages at biological interfaces. _Biophys. J._ 68, 2580–2587 (1995).
Article ADS PubMed PubMed Central CAS Google Scholar Download references ACKNOWLEDGEMENTS We thank B. S. Coller (Rockefeller University) for sharing precious reagents, F. Ola-Daniel,
Y. Wang (The University of Texas Medical Branch), and A. Dupuy and Y. C. Zhao (The University of Sydney) for technical support, and Z. M. Ruggeri (The Scripps Research Institute) for
providing valuable suggestions. This work was supported by the following funding sources: National Heart, Lung, and Blood Institute grant R00HL153678 (Y.C.). National Institute on Aging the
Claude D. Pepper Older Americans Independence Center Award #P30-AG024832 (Y.C.). UT System Rising STARs award (Y.C.). American Heart Association Postdoctoral Fellowship 20POST35080023
(Y.C.). National Institute of General Medical Sciences grant 1R01GM152812 (L.X.). National Science Foundation grants DMS-1953189, CCF-2007823, and DMS-2210775 (L.X.). MRFF Cardiovascular
Health Mission Grants APP2016165 and APP2023977 (L.A.J.). National Heart Foundation Future Leader Fellowship Level 2 (105863) (L.A.J.). Snow Medical Research Foundation Fellowship 2022SF176
(L.A.J.). AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Biochemistry and Molecular Biology, The University of Texas Medical Branch, Galveston, TX, 77555, USA Misbahud Din,
Souvik Paul, Sana Ullah, Rong-Guang Xu, Bari Chowdhury, Stephenie Rogers, Mariel Miller, Atreyee Biswas & Yunfeng Chen * Department of Pathology, The University of Texas Medical Branch,
Galveston, TX, 77555, USA Misbahud Din, Souvik Paul, Sana Ullah, Rong-Guang Xu, Bari Chowdhury, Mariel Miller, Atreyee Biswas, Christopher Zahner & Yunfeng Chen * Department of
Statistics, The Pennsylvania State University, University Park, Pennsylvania, PA, 16802, USA Haoyi Yang & Lingzhou Xue * Division of Thoracic Surgery, Brigham and Women’s Hospital,
Harvard Medical School, Boston, MA, 02115, USA Rong-Guang Xu & Zi Chen * School of Biomedical Engineering, The University of Sydney, Darlington, NSW, 2008, Australia Nurul Aisha Zainal
Abidin, Allan Sun, Yiyao Catherine Chen, Rui Gao & Lining Arnold Ju * Charles Perkins Centre, The University of Sydney, Camperdown, NSW, 2006, Australia Allan Sun & Lining Arnold Ju
* Heart Research Institute, Newtown, NSW, 2042, Australia Allan Sun & Lining Arnold Ju * The University of Sydney Nano Institute (Sydney Nano), The University of Sydney, Camperdown, NSW,
2006, Australia Allan Sun, Rui Gao & Lining Arnold Ju * Coulter Department of Biomedical Engineering, Georgia Institute of Technology, Atlanta, GA, 30332, USA Fangyuan Zhou * School of
Integrative Medicine, Shanghai University of Traditional Chinese Medicine, Shanghai, 201203, China Liang Hu * Department of Immunology, School of Medicine, UConn Health, Farmington, CT,
06030, USA Zhichao Fan * Department of Mechanical Engineering, The City University of New York - City College, New York, NY, 10031, USA Jing Fan * Department of Internal Medicine, The
University of Texas Medical Branch, Galveston, TX, 77555, USA Megan Berman Authors * Misbahud Din View author publications You can also search for this author inPubMed Google Scholar *
Souvik Paul View author publications You can also search for this author inPubMed Google Scholar * Sana Ullah View author publications You can also search for this author inPubMed Google
Scholar * Haoyi Yang View author publications You can also search for this author inPubMed Google Scholar * Rong-Guang Xu View author publications You can also search for this author
inPubMed Google Scholar * Nurul Aisha Zainal Abidin View author publications You can also search for this author inPubMed Google Scholar * Allan Sun View author publications You can also
search for this author inPubMed Google Scholar * Yiyao Catherine Chen View author publications You can also search for this author inPubMed Google Scholar * Rui Gao View author publications
You can also search for this author inPubMed Google Scholar * Bari Chowdhury View author publications You can also search for this author inPubMed Google Scholar * Fangyuan Zhou View author
publications You can also search for this author inPubMed Google Scholar * Stephenie Rogers View author publications You can also search for this author inPubMed Google Scholar * Mariel
Miller View author publications You can also search for this author inPubMed Google Scholar * Atreyee Biswas View author publications You can also search for this author inPubMed Google
Scholar * Liang Hu View author publications You can also search for this author inPubMed Google Scholar * Zhichao Fan View author publications You can also search for this author inPubMed
Google Scholar * Christopher Zahner View author publications You can also search for this author inPubMed Google Scholar * Jing Fan View author publications You can also search for this
author inPubMed Google Scholar * Zi Chen View author publications You can also search for this author inPubMed Google Scholar * Megan Berman View author publications You can also search for
this author inPubMed Google Scholar * Lingzhou Xue View author publications You can also search for this author inPubMed Google Scholar * Lining Arnold Ju View author publications You can
also search for this author inPubMed Google Scholar * Yunfeng Chen View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS Methodology: M.D., S.P.,
R.-G.X., F.Z., Z.C., and Y.C. Experiments: M.D., S.P., S.U., B.C., N.A.Z.A., A.S., Y.C.C., R.G., S.R., A.B., and Y.C. Data analysis: M.D., S.P., S.U., B.C., N.A.Z.A., A.S., Y.C.C., S.R.,
A.B., H.Y., L.X., R.-G.X., F.Z., J.F., and Y.C. Donor recruitment and blood collection: M.D., S.P., S.U., A.B., and M.B. Funding acquisition: L.X., L.A.J., and Y.C. Supervision: L.A.J. and
Y.C. Writing: M.D., H.Y., R.-G.X., F.Z., N.A.Z.A., M.M., L.H., Z.F., C.Z., M.B., L.X., L.A.J., and Y.C. CORRESPONDING AUTHOR Correspondence to Yunfeng Chen. ETHICS DECLARATIONS COMPETING
INTERESTS The authors declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Nature Communications_ thanks Tzung Hsiai, and the other, anonymous, reviewer(s) for their
contribution to the peer review of this work. A peer review file is available. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in
published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION PEER REVIEW FILE REPORTING SUMMARY SOURCE DATA SOURCE DATA RIGHTS AND PERMISSIONS OPEN
ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format,
as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third
party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the
article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the
copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Din, M., Paul, S., Ullah,
S. _et al._ Multi-parametric thrombus profiling microfluidics detects intensified biomechanical thrombogenesis associated with hypertension and aging. _Nat Commun_ 15, 9067 (2024).
https://doi.org/10.1038/s41467-024-53069-9 Download citation * Received: 03 January 2024 * Accepted: 30 September 2024 * Published: 21 October 2024 * DOI:
https://doi.org/10.1038/s41467-024-53069-9 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative