Play all audios:
ABSTRACT The response of childhood acute lymphoblastic leukemia (ALL) to dexamethasone predicts the long-term remission outcome. To explore the mechanisms of dexamethasone resistance in B
cell ALL (B-ALL), we generated dexamethasone-resistant clones by prolonged treatment with dexamethasone. Using RNA-sequencing and high-throughput screening, we found that
dexamethasone-resistant cells are dependent on receptor tyrosine kinases. Further analysis with phosphokinase arrays showed that the type III receptor tyrosine kinase FLT3 is constitutively
active in resistant cells. Targeted next-generation and Sanger sequencing identified an internal tandem duplication mutation and a point mutation (R845G) in FLT3 in dexamethasone-resistant
cells, which were not present in the corresponding sensitive clones. Finally, we showed that resistant cells displayed sensitivity to second-generation FLT3 inhibitors both in vitro and in
vivo. Collectively, our data suggest that long-term dexamethasone treatment selects cells with a distinct genetic background, in this case oncogenic FLT3, and therefore therapies targeting
FLT3 might be useful for the treatment of relapsed B-ALL patients. SIMILAR CONTENT BEING VIEWED BY OTHERS GLUCOCORTICOIDS PARADOXICALLY PROMOTE STEROID RESISTANCE IN B CELL ACUTE
LYMPHOBLASTIC LEUKEMIA THROUGH CXCR4/PLC SIGNALING Article Open access 29 May 2024 DASATINIB OVERCOMES GLUCOCORTICOID RESISTANCE IN B-CELL ACUTE LYMPHOBLASTIC LEUKEMIA Article Open access 22
May 2023 MOLECULAR AND PHARMACOLOGICAL HETEROGENEITY OF _ETV6_::_RUNX1_ ACUTE LYMPHOBLASTIC LEUKEMIA Article Open access 29 January 2025 INTRODUCTION Acute lymphoblastic leukemia (ALL) is
one of the most common childhood cancers and can originate both from the B-lineage (B-ALL) and the T-lineage (T-ALL). Glucocorticoids, such as dexamethasone and prednisolone, are important
drugs for the treatment of ALL.1 In combination with chemotherapeutic agents, glucocorticoids help to achieve clinical remission, and sensitivity to glucocorticoids is considered as a
positive prognostic indicator. Patients unresponsive to glucocorticoids often relapse and display poor prognosis. Therefore, understanding the mechanisms behind glucocorticoid insensitivity
is important and will help us to develop novel therapeutic modalities. In ALL glucocorticoids induce apoptosis, which is mediated through binding to the glucocorticoid receptor (GR). GR is a
nuclear receptor that also acts as a transcription factor. Upon glucocorticoid binding, GR inhibits activator protein-1 (AP-1)- and nuclear factor-κB (NF-κB)-regulated gene transcription,
and at the same time promotes glucocorticoid-responsive element-driven gene transcription.2 Besides, inhibition of AP-1- and NF-κB-regulated gene transcription, cooperation between AP-1 and
GR in transcription,3 and crosstalk between NF-κB and GR4,5 have been reported, which suggests a context-dependent regulation of AP-1 and NF-κB rather than only inhibitory effects.
Glucocorticoids are useful drugs to induce apoptosis in ALL and have also been widely used to treat inflammatory disorders. However, prolonged use leads to the emergence of glucocorticoid
resistance.6 The mechanisms of glucocorticoid resistance in leukemia have been studied extensively. Both regulation of expression and function of GR can contribute to glucocorticoid
resistance. For instance, activation of NOTCH1 signaling inhibits auto-upregulation of GR expression. Therefore, pharmacological inhibition of NOTCH1 restores glucocorticoid sensitivity.7
The relapse-associated mutation in _NR3C1_ results in the expression of a non-functional receptor and thereby impairs glucocorticoid sensitivity.8 Furthermore, aberrant activation of the
PI3K/mTOR pathway has been linked to glucocorticoid resistance in T-ALL.9 This is partially mediated by AKT, which phosphorylates GR on S134 and thereby blocks nuclear localization of GR.10
Mutations in the transcriptional coactivator CREBBP transcriptionally regulates glucocorticoid-responsive genes, suggesting that functional CREBBP is required for glucocorticoid
sensitivity.11 Inhibition of glutathione synthesis restored dexamethasone sensitivity in the dexamethasone-resistant B-ALL cell line 697,12 suggesting the existence of additional mechanisms
of dexamethasone resistance. In this report, we show that cells resistant to dexamethasone harbor activating mutations in the receptor tyrosine kinase FLT3. RESULTS PROLONGED DEXAMETHASONE
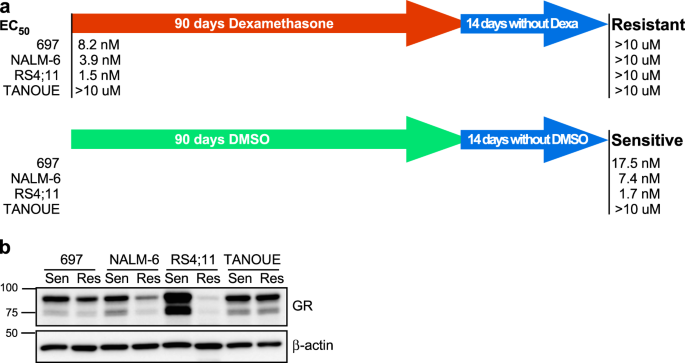
TREATMENT INDUCES DEXAMETHASONE RESISTANCE IN B-ALL CELLS In order to understand how long-term dexamethasone treatment affects B-ALL cells, we used three dexamethasone-sensitive cell lines:
697 (half-maximal effective concentration (EC50) = 8.2 nM), NALM-6 (EC50 = 3.9 nM), and RS4;11 (EC50 = 1.5 nM), and the dexamethasone-insensitive cell line TANOUE (EC50 >10 µM). These
cell lines were cultured with an increasing concentration of dexamethasone for 90 days. In parallel, another set of cell lines was cultured with an equivalent amount of dimethyl sulfoxide
(DMSO) (which was used to dilute dexamethasone). After 90 days, cells were cultured in normal growth medium for 2 weeks and EC50 was measured. We observed that all three
dexamethasone-sensitive cell lines cultured in the presence of dexamethasone became highly resistant to dexamethasone, while DMSO-treated cells were still sensitive (Fig. 1a). The relation
between dexamethasone sensitivity and GR expression does not always correlate.13,14 Therefore, we first checked the GR expression in both dexamethasone-sensitive and -resistant cell lines.
The expression of GR remained unchanged in TANOUE cells, while it was reduced in 697 and NALM-6 cells (Fig. 1b). However, while the most sensitive cell line, RS4;11, showed strong GR
expression, its expression was completely lost in the corresponding resistant cells (Fig. 1b). These data are in line with previous reports that GR expression is one of the factors relating
to dexamethasone sensitivity but not the only factor.13,14 DEXAMETHASONE-RESISTANT RS4;11 CELLS ARE SENSITIVE TO RTK INHIBITORS To understand the molecular differences between
dexamethasone-sensitive and -resistant cell lines, we used RNA-sequencing (RNAseq). We observed that the gene expression patterns were mostly identical between sensitive and resistant lines
of 697, NALM-6, and TANOUE cells, whereas RS4;11 cells showed a more scattered expression pattern indicative of differences in gene expression between the two types of cells (Fig. 2a). These
data suggest that both dexamethasone-sensitive and -resistant lines of 697, NALM-6, and TANOUE cells keep similar gene expression pattern, while RS4;11-resistant cells show a major
difference compared to its parental cell line. As we observed a major variation in gene expression of RS4;11, we checked the pathway enrichment in resistant cells using RNAseq data. We
observed enrichment of several kinase and cytokine signaling pathways in resistant RS4;11 cell line (Fig. 2b). Since we observed enrichment of kinase and cytokine signaling pathways in the
dexamethasone-resistant RS4;11 cell line, we hypothesized that there is a switch in the dependency of RS4;11 cells from dexamethasone to kinase-related signaling. To identify the possible
kinase dependency of RS4;11 cells, we used a panel of 378 inhibitors against different kinases. Both sensitive and resistant lines of NALM-6, 697, and TANOUE cells displayed similar response
to the inhibitors, but the resistant RS;411 cell line displayed increased sensitivity to several receptor tyrosine kinase (RTK) inhibitors compared to the corresponding sensitive cell line
(Fig. 2c). Taken together, these data suggest that the mechanism behind the resistant phenotype of RS4;11 is different from that of NALM-6, 697, and TANOUE cell lines.
DEXAMETHASONE-RESISTANT RS4;11 CELLS DISPLAY TYROSINE PHOSPHORYLATION OF FLT3 Since we did not observe any major differences between the gene expression and kinase inhibitor response, we
suggest that the resistance of 697 and NALM-6 is probably mediated by reduced expression of GR or due to a loss-of-function mutation in GR. Several other mechanisms have also been described
and discussed in the Introduction section.7,8,9,10,11,12 However, the difference in gene expression in the two RS4;11 cell lines and their differential response to kinase inhibitors evoked
our interest. Coinciding with the development of a resistant phenotype, the RS4;11 cells completely lost GR expression. Most likely this is due to the fact that a small fraction of cells
that initially were lacking GR expression were selected for during the long-term exposure to dexamethasone, and that selected for cells that carry different genetic mutations. Since we
observed that dexamethasone-resistant RS4;11 cells are sensitive to several RTK inhibitors, we checked for activation of RTKs in this cell line using a human proteome phospho-RTK array.
Surprisingly, we observed strong tyrosine phosphorylation of FLT3 and weak tyrosine phosphorylation of AXL in resistant cells, which could not be seen in sensitive cells (Fig. 3a).
Furthermore, using a phosphokinase array we observed that phosphorylation of ERK1/2 and of CREB at S133 was enhanced in resistant cells (Fig. 3b). Collectively, our data suggest that
dexamethasone-resistant RS4;11 cells display dependency of constitutively active RTK signaling. DEXAMETHASONE-RESISTANT RS4;11 CELLS CARRY ONCOGENIC MUTANTS OF FLT3 AND RESPOND TO FLT3
INHIBITION We then checked the expression of FLT3 and AXL in RS4;11 cell lines. We observed strong expression of FLT3 in dexamethasone-sensitive RS4;11, where the fully glycosylated, mature
FLT3 band was stronger than the partially glycosylated, immature band (Fig. 4a), which is a characteristic of cells expressing wild-type FLT3. The observation that the partially
glycosylated, immature FLT3 band was stronger in dexamethasone-resistant RS4;11 cells (Fig. 4a) raised the possibility that the resistant cells carry an oncogenic internal tandem duplication
(ITD) mutation in FLT3, which typically gives this pattern of expression.15,16 This is also supported by the fact that resistant RS4;11 cells showed constitutive tyrosine phosphorylation of
FLT3 as well as constitutive STAT5 phosphorylation (Fig. 4b) and that the second-generation FLT3 inhibitor AC220 could block tyrosine phosphorylation of both FLT3 and STAT5 (Fig. 4c).
DEXAMETHASONE-RESISTANT RS4;11 CELLS CARRY FLT3-ITD AND FLT3-R845G MUTATIONS To verify the presence of FLT3 mutations and also in order to see whether any other oncogenic mutations exist in
RS4;11 cells, we used targeted sequencing of 600 cancer-related genes. We identified a FLT3 point mutation (c.2533A>G, R845G, ratio 65%, coverage 1504×) and an FLT3-ITD mutation
(p.E598_Y599insFDFREYE 22%, coverage 487×) (Fig. 5a). The point mutation was further confirmed by Sanger sequencing (Fig. 5b). FLT3-ITD is a well-studied oncogenic mutation and R845G has
also been shown to be a constitutively activating mutation.17 DEXAMETHASONE-RESISTANT RS4;11 CELLS ARE SENSITIVE TO THE SECOND-GENERATION FLT3 INHIBITORS AC220 AND CRENOLANIB Since
dexamethasone-resistant RS4;11 cells harbor oncogenic mutations in FLT3, we have tested the possibility of using the second-generation FLT3 inhibitors AC220 and crenolanib. Both the
inhibitors significantly reduced the growth of dexamethasone-resistant RS4;11 cells, while the growth of dexamethasone-sensitive RS4;11 and 697 cells or dexamethasone-resistant 697 cells
remained unchanged (Fig. 6a, b). Furthermore, in a mouse xenograft model, crenolanib delayed tumor formation of dexamethasone-resistant RS4;11 cells (Fig. 6c, d). Taken together, data
suggest that dexamethasone-resistant RS4;11 cells are dependent on the activity of oncogenic FLT3 signaling. DISCUSSION In this study, we used three dexamethasone-sensitive B-ALL cell lines
from three different genetic backgrounds to generate dexamethasone-resistant cell lines. Although the 697 cell line carries an _E2A-PBX1_ (_TCF3-PBX1_) fusion, RS4;11 carry an _MLL-AF4_
fusion and NALM-6 carry an _NRAS_ mutation, all three cell lines displayed similar sensitivity to dexamethasone (EC50 <10 nM). E2A-PBX1 fusion acts as a constitutively active
transcription factor that downregulates the expression of _CDKN2A_.18 _CDKN2A_ encodes two distinct proteins (p16INK4A and p14ARF), which are well-known regulators of the cell cycle.
E2A-PBX1 does not act as a transcriptional repressor, but this fusion protein enhances expression of _BMI-1_,18 which is known to be a lymphoid oncogene and functions as a transcriptional
repressor. On the other hand, MLL-AF4 suppresses the expression of another cell cycle regulatory protein, p27KIP1 through direct transcriptional repression of _CDKN1B_.19 Furthermore, ALL
patients with _MLL-AF4_ rearrangements overexpress _HOXA9_,20 which is a transcription factor that has been shown to be important for the proliferation and survival of _MLL_-rearranged
leukemias.21 HOXA9 mediates upregulation _BCL2_ expression, which in turn provides survival signals to the leukemic cells.22 Therefore, combinatorial use of dexamethasone and BCL2 inhibitor
displayed a synergistic effect in inhibition of leukemia.23 Although patients with _MLL-AF4_ fusion respond to glucocorticoid-based chemotherapy, this group of patients are considered to
have a poor prognosis and have about 60% disease-free survival.24 Current studies suggest that this group of patients shows resistance to glucocorticoids, which has been shown to be at least
partially mediated by constitutive activation of mitogen-activated protein kinase signaling.25,26,27 Here we provide evidence that cells that are resistant to dexamethasone display
constitutive activation of RTK signaling. All three dexamethasone-sensitive B-ALL cell lines became resistant during 90 days treatment of dexamethasone, suggesting that prolonged
treatment-induced resistance to dexamethasone in vitro. This was independent of the GR expression levels as dexamethasone-resistant 697 cells also express a similar level of GR as sensitive
cells. However, while both 697 and NALM-6 cells kept a certain level of GR expression after 90 days of dexamethasone treatment, expression was almost lost in RS4;11 cells. Besides that,
RS4;11 cells displayed a major deviation with respect to gene expression and kinase inhibitor sensitivity when comparing the dexamethasone-sensitive and -resistant cells, suggesting that
this cell line harbors a different mechanism of dexamethasone resistance than the other two cell lines. Furthermore, RS4;11 cells resistant to dexamethasone showed constitutive activation of
FLT3. A relationship between _MLL_ rearrangement and FLT3 expression has been established in several studies. For example, FLT3 expression was found to be consistently higher in ALL
patients positive for _MLL_ rearrangement,28 and mutations in the activation loop of FLT3 that confer constitutive activation of FLT3 was identified in 17% of _MLL_-rearranged ALL
patients.29 However, higher FLT3 expression and oncogenic mutations are not exclusive to _MLL_-rearranged ALL, while it occurs frequently in hyperdiploid ALL and less frequently in
_TEL-AML1_ fusion ALL.30,31 We observed that the 697 and RS4;11 cell lines express higher levels of FLT3, while its expression was undetectable in TANOUE cells and low in NALM-6 cells. Since
dexamethasone-sensitive RS4;11 cells do not have any FLT3 activation, and since it is unlikely that dexamethasone will induce mutation in _FLT3_, it seems that a small fraction of RS4;11
cells carry FLT3 mutations from the beginning. Dexamethasone selection probably selects for cells that are dependent on FLT3 signaling. Collectively, our data suggest that
dexamethasone-resistant RS4;11 cells are a subpopulation of B-ALL cells that carry FLT3-ITD or R845G mutations, and therefore it could prove useful to screen B-ALL patients who are resistant
to dexamethasone for mutations in FLT3, which then could be targeted with FLT3 inhibitors that are already available on the market. METHODS ANTIBODIES AND CHEMICALS Anti-GR (sc-8992; 1:1000
dilution), anti-β-actin (sc-47778; 1:1000 dilution), anti-AXL (sc-1096; 1:1000 dilution), anti-FLT3 (sc-479; 1:1000 dilution), and anti-STAT5 (sc-835; 1:1000 dilution) were from Santa Cruz
Biotechnology (Dallas, TX, USA). Anti-phospho-tyrosine antibody 4G10 (05-321; 1:1000 dilution) was from Millipore. Dexamethasone (D4902, Sigma-Aldrich, St. Louis, MI, USA) was dissolved in
DMSO. All uncropped blots are available in supplementary figure. CELL CULTURE AND GENERATION OF DEXAMETHASONE-RESISTANT B-ALL CELL LINES The B-ALL cell lines RS4;11, 697, NALM-6, and TANOUE
were purchased from the DSMZ (Braunschweig, Germany). B-ALL cell lines were cultured in RPMI-1640 medium supplemented with 10% heat-inactivated fetal bovine serum (FBS), 100U/ml penicillin
and 100 µg/ml streptomycin. All B-ALL cell lines were treated with dexamethasone and doses were doubled when the treated cells started to proliferate at an equal rate to the untreated
parental cells. The doses were increased at regular intervals until 5 µM concentration was reached. The resistant cells were further grown for 2 weeks in the absence of inhibitors. DRUG
SENSITIVITY ASSAY Sensitive and resistant B-ALL cells were grown in RPMI-1640 medium supplemented with 10% FBS, 100U/ml penicillin, and 100 μg/ml streptomycin. Cells were then seeded in
96-well plates (10,000 cells per well) in the presence of different concentrations of dexamethasone. After 48 h, 10 μl of PrestoBlue was added to each well, followed by 2 h of incubation.
Cell viability was calculated according to the manufacturer’s protocol. A kinase inhibitor library including 378 kinase inhibitors was obtained from Selleck Chemicals (Houston, TX, USA).
Stock solutions of 10 mM inhibitor were diluted to 100 nM using the cell culture medium. Cell viability assays using PrestoBlue were used to examine the effect of inhibitors. PHOSPHOKINASE
ARRAYS Proteome Profiler Human Phospho-RTK Array Kit (ARY001B) and Proteome Profiler Human Phospho-Kinase Array Kit (ARY003B) were obtained from R&D System (Minneapolis, MN, USA).
Dexamethasone-sensitive and -resistant cells were lysed and the lysates were processed according to the manufacturer’s protocols. TARGETED SEQUENCING OF CANCER PANEL INVIEW Oncopanel
All-in-one service from Eurofins Genomics provided analysis of 597 key cancer-specific genes. Total genomic DNA from dexamethasone-sensitive and -resistant cells was purified using Qiagen
DNeasy Blood and Tissue Kit (69504), and then sent to Eurofins Genomics for processing. MOUSE XENOGRAFT STUDIES Ten female non-obese diabetic/severe combined immunodeficiency gamma (NSG)
mice (each weighing ~20 g and housed by the Laboratory Animal Facilities at Medicon Village, Lund University) were injected with 2,000,000 cells with 1:1 Matrigel subcutaneously. Mice were
then divided into two groups and treated either with crenolanib or with the vehicle. One week after injection of cells, mice were treated alternative days by intravenous injection of 12
mg/kg crenolanib or vehicle for additional days until the tumor reached a size of 1 cm3 (8th to 16th day). Drug efficacy was checked by monitoring the tumor growth in both groups and by
regularly measuring the body weight and tumor volume of the mice. Mice were sacrificed after the size of the tumors had reached about 1 cm3. ETHICAL CONSIDERATION Mice were maintained by
following regulation approved by the Lund University. All animal experiments were performed under an ethical permit from the Swedish Animal Welfare Authority. RNASEQ ANALYSIS The RNA quality
was analyzed using a Bioanalyzer (Agilent) and samples with an RNA integrity greater than seven were further analyzed. Subsequently, RNAseq was performed using TruSeq Stranded mRNA Kit for
NeoPrep from Illumina and the sequencing was performed using Illumina NextSeq 500 instrument. RNAseq data analysis was performed using a pipeline where Demultiplexing step involves in
reorganizing the FASTQ files based on the sample index information, and generating the statistics and reporting files, which was performed using the bcl2fastq2 software (Illumina), Masking
step involves filtering ribosomal RNA (GenBank loci NR_023363.1, NR_003285.2, NR_003286.2, NR_003287.2, X12811.1, U13369.1), PhiX (phiX174), and Illumina control (NC_001422.1), and repeat
sequence analysis was performed using the bowtie2 program. Mapping steps, after filtration of remaining reads, were aligned to the human genome reference (UCSC hg38 build), performed using
TopHat2 program. Expression count step, expression levels, and fragments per kilobase of exon per million mapped reads were calculated using Cufflinks 2.2.1 program. STATISTICAL ANALYSIS
Statistical analysis was performed using GraphPad Prism 5.0. One-way analysis of variance or Student’s _t_ test was used where needed. REPORTING SUMMARY Further information on research
design is available in the Nature Research Reporting Summary. DATA AVAILABILITY All raw data are available upon request. RNAseq data are available at the ArrayExpress (E-MTAB-7781). Raw data
supporting FLT3 mutations in RS4;11 cells have been deposited to figshare.com https://doi.org/10.6084/m9.figshare.7828172. REFERENCES * Inaba, H., Greaves, M. & Mullighan, C. G. Acute
lymphoblastic leukaemia. _Lancet_ 381, 1943–1955 (2013). Article Google Scholar * Ratman, D. et al. How glucocorticoid receptors modulate the activity of other transcription factors: a
scope beyond tethering. _Mol. Cell Endocrinol._ 380, 41–54 (2013). Article CAS Google Scholar * Biddie, S. C. et al. Transcription factor AP1 potentiates chromatin accessibility and
glucocorticoid receptor binding. _Mol. Cell_ 43, 145–155 (2011). Article CAS Google Scholar * Altonsy, M. O., Sasse, S. K., Phang, T. L. & Gerber, A. N. Context-dependent cooperation
between nuclear factor kappaB (NF-kappaB) and the glucocorticoid receptor at a TNFAIP3 intronic enhancer: a mechanism to maintain negative feedback control of inflammation. _J. Biol. Chem._
289, 8231–8239 (2014). Article CAS Google Scholar * Rao, N. A. et al. Coactivation of GR and NFKB alters the repertoire of their binding sites and target genes. _Genome Res_ 21, 1404–1416
(2011). Article CAS Google Scholar * Norman, M. & Hearing, S. D. Glucocorticoid resistance—what is known? _Curr. Opin. Pharmacol._ 2, 723–729 (2002). Article CAS Google Scholar *
Real, P. J. et al. Gamma-secretase inhibitors reverse glucocorticoid resistance in T cell acute lymphoblastic leukemia. _Nat. Med._ 15, 50–58 (2009). Article CAS Google Scholar * Oshima,
K. et al. Mutational landscape, clonal evolution patterns, and role of RAS mutations in relapsed acute lymphoblastic leukemia. _Proc. Natl Acad. Sci. USA_ 113, 11306–11311 (2016). Article
CAS Google Scholar * Hall, C. P., Reynolds, C. P. & Kang, M. H. Modulation of glucocorticoid resistance in pediatric T-cell acute lymphoblastic leukemia by increasing BIM expression
with the PI3K/mTOR inhibitor BEZ235. _Clin. Cancer Res._ 22, 621–632 (2016). Article CAS Google Scholar * Piovan, E. et al. Direct reversal of glucocorticoid resistance by AKT inhibition
in acute lymphoblastic leukemia. _Cancer Cell_ 24, 766–776 (2013). Article CAS Google Scholar * Mullighan, C. G. et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia.
_Nature_ 471, 235–239 (2011). Article CAS Google Scholar * Inoue, H. et al. Dexamethasone-resistant human Pre-B leukemia 697 cell line evolving elevation of intracellular glutathione
level: an additional resistance mechanism. _Jpn J. Cancer Res._ 93, 582–590 (2002). Article CAS Google Scholar * Tissing, W. J., Meijerink, J. P., den Boer, M. L. & Pieters, R.
Molecular determinants of glucocorticoid sensitivity and resistance in acute lymphoblastic leukemia. _Leukemia_ 17, 17–25 (2003). Article CAS Google Scholar * Bachmann, P. S., Gorman, R.,
Mackenzie, K. L., Lutze-Mann, L. & Lock, R. B. Dexamethasone resistance in B-cell precursor childhood acute lymphoblastic leukemia occurs downstream of ligand-induced nuclear
translocation of the glucocorticoid receptor. _Blood_ 105, 2519–2526 (2005). Article CAS Google Scholar * Moharram, S. A., Shah, K., Khanum, F., Ronnstrand, L. & Kazi, J. U. The ALK
inhibitor AZD3463 effectively inhibits growth of sorafenib-resistant acute myeloid leukemia. _Blood Cancer J._ 9, 5 (2019). Article Google Scholar * Chougule, R. A. et al. Expression of
GADS enhances FLT3-induced mitogenic signaling. _Oncotarget_ 7, 14112–14124 (2016). PubMed PubMed Central Google Scholar * Smith, C. C. et al. Characterizing and overriding the structural
mechanism of the quizartinib-resistant FLT3 “Gatekeeper” F691L mutation with PLX3397. _Cancer Discov._ 5, 668–679 (2015). Article CAS Google Scholar * Smith, K. S. et al. Bmi-1
regulation of INK4A-ARF is a downstream requirement for transformation of hematopoietic progenitors by E2a-Pbx1. _Mol. Cell_ 12, 393–400 (2003). Article CAS Google Scholar * Xia, Z. B. et
al. The MLL fusion gene, MLL-AF4, regulates cyclin-dependent kinase inhibitor CDKN1B (p27kip1) expression. _Proc. Natl Acad. Sci. USA_ 102, 14028–14033 (2005). Article CAS Google Scholar
* Rozovskaia, T. et al. Upregulation of Meis1 and HoxA9 in acute lymphocytic leukemias with the t(4: 11) abnormality. _Oncogene_ 20, 874–878 (2001). Article CAS Google Scholar * Faber,
J. et al. HOXA9 is required for survival in human MLL-rearranged acute leukemias. _Blood_ 113, 2375–2385 (2009). Article CAS Google Scholar * Brumatti, G. et al. HoxA9 regulated Bcl-2
expression mediates survival of myeloid progenitors and the severity of HoxA9-dependent leukemia. _Oncotarget_ 4, 1933–1947 (2013). Article Google Scholar * Benito, J. M. et al.
MLL-rearranged acute lymphoblastic leukemias activate BCL-2 through H3K79 methylation and are sensitive to the BCL-2-specific antagonist ABT-199. _Cell Rep._ 13, 2715–2727 (2015). Article
CAS Google Scholar * Winters, A. C. & Bernt, K. M. MLL-rearranged leukemias—an update on science and clinical approaches. _Front. Pediatr._ 5, 4 (2017). Article Google Scholar *
Driessen, E. M. et al. Frequencies and prognostic impact of RAS mutations in MLL-rearranged acute lymphoblastic leukemia in infants. _Haematologica_ 98, 937–944 (2013). Article CAS Google
Scholar * Jerchel, I. S. et al. RAS pathway mutations as a predictive biomarker for treatment adaptation in pediatric B-cell precursor acute lymphoblastic leukemia. _Leukemia_ 32, 931–940
(2018). Article CAS Google Scholar * Kerstjens, M. et al. MEK inhibition is a promising therapeutic strategy for MLL-rearranged infant acute lymphoblastic leukemia patients carrying RAS
mutations. _Oncotarget_ 8, 14835–14846 (2017). Article Google Scholar * Armstrong, S. A. et al. MLL translocations specify a distinct gene expression profile that distinguishes a unique
leukemia. _Nat. Genet._ 30, 41–47 (2002). Article CAS Google Scholar * Armstrong, S. A. et al. Inhibition of FLT3 in MLL. Validation of a therapeutic target identified by gene expression
based classification. _Cancer Cell_ 3, 173–183 (2003). Article CAS Google Scholar * Yeoh, E. J. et al. Classification, subtype discovery, and prediction of outcome in pediatric acute
lymphoblastic leukemia by gene expression profiling. _Cancer Cell_ 1, 133–143 (2002). Article CAS Google Scholar * Armstrong, S. A. et al. FLT3 mutations in childhood acute lymphoblastic
leukemia. _Blood_ 103, 3544–3546 (2004). Article CAS Google Scholar Download references ACKNOWLEDGEMENTS We would like to thank Professor Lars Rönnstrand for comments on the manuscript.
This research was the Kungliga Fysiografiska Sällskapet i Lund (S.A.M. and K.S.), Ollie and Elof Ericsson’s Stiftelse (J.U.K.), the Crafoord Foundation (J.U.K.), Magnus Bergvalls Stiftelse
(J.U.K.) and the Swedish Childhood Cancer Foundation (J.U.K.). J.U.K. is a recipient of an Assistant Professorship (forskarassistenttjänst) grant from the Swedish Childhood Cancer
Foundation. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Division of Translational Cancer Research, Department of Laboratory Medicine, Lund University, Lund, Sweden Rohit A. Chougule,
Kinjal Shah, Sausan A. Moharram & Julhash U. Kazi * Division of Oncology and Pathology, Department of Clinical Sciences Lund, Lund University, Lund, Sweden Johan Vallon-Christersson
Authors * Rohit A. Chougule View author publications You can also search for this author inPubMed Google Scholar * Kinjal Shah View author publications You can also search for this author
inPubMed Google Scholar * Sausan A. Moharram View author publications You can also search for this author inPubMed Google Scholar * Johan Vallon-Christersson View author publications You can
also search for this author inPubMed Google Scholar * Julhash U. Kazi View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS R.A.C., K.S.,
S.A.M., and J.V.-C. performed the experiments. J.U.K. supervised the research. R.A.C., K.S., S.A.M., J.V.-C., and J.U.K. analyzed the data. J.U.K., R.A.C., S.A.M., J.V.-C., and K.S. wrote
the manuscript. All authors read and approved the final manuscript. The authors declare that they have no conflict of interests. CORRESPONDING AUTHOR Correspondence to Julhash U. Kazi.
ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION PUBLISHER’S NOTE: Springer Nature remains neutral with regard to jurisdictional
claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION UNCROPPED IMAGES OF ALL BLOTS REPORTING SUMMARY RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed
under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give
appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in
this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative
Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a
copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Chougule, R.A., Shah, K., Moharram, S.A. _et al._
Glucocorticoid-resistant B cell acute lymphoblastic leukemia displays receptor tyrosine kinase activation. _npj Genom. Med._ 4, 7 (2019). https://doi.org/10.1038/s41525-019-0082-y Download
citation * Received: 31 December 2018 * Accepted: 13 March 2019 * Published: 04 April 2019 * DOI: https://doi.org/10.1038/s41525-019-0082-y SHARE THIS ARTICLE Anyone you share the following
link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature
SharedIt content-sharing initiative