Play all audios:
ABSTRACT The κ-opioid receptor (KOR) represents a highly desirable therapeutic target for treating not only pain but also addiction and affective disorders1. However, the development of KOR
analgesics has been hindered by the associated hallucinogenic side effects2. The initiation of KOR signalling requires the Gi/o-family proteins including the conventional (Gi1, Gi2, Gi3, GoA
and GoB) and nonconventional (Gz and Gg) subtypes. How hallucinogens exert their actions through KOR and how KOR determines G-protein subtype selectivity are not well understood. Here we
determined the active-state structures of KOR in a complex with multiple G-protein heterotrimers—Gi1, GoA, Gz and Gg—using cryo-electron microscopy. The KOR–G-protein complexes are bound to
hallucinogenic salvinorins or highly selective KOR agonists. Comparisons of these structures reveal molecular determinants critical for KOR–G-protein interactions as well as key elements
governing Gi/o-family subtype selectivity and KOR ligand selectivity. Furthermore, the four G-protein subtypes display an intrinsically different binding affinity and allosteric activity on
agonist binding at KOR. These results provide insights into the actions of opioids and G-protein-coupling specificity at KOR and establish a foundation to examine the therapeutic potential
of pathway-selective agonists of KOR. SIMILAR CONTENT BEING VIEWED BY OTHERS MOLECULAR MECHANISMS OF INVERSE AGONISM VIA Κ-OPIOID RECEPTOR–G PROTEIN COMPLEXES Article Open access 07 January
2025 MOLECULAR MECHANISM OF BIASED SIGNALING AT THE KAPPA OPIOID RECEPTOR Article Open access 11 March 2023 LIGAND RECOGNITION AND ALLOSTERIC MODULATION OF THE HUMAN MRGPRX1 RECEPTOR Article
27 October 2022 MAIN Opioid receptors are G-protein-coupled receptors (GPCRs) that have important roles in pain sensation. Almost all clinically used opioids act through the μ-opioid
receptor (MOR). However, their use is associated with severe side effects, including a high potential for abuse, addiction and death due to respiratory depression in overdose3. The magnitude
of these problems has led to a search for opioid alternatives for the treatment of pain and related conditions4. The activation of opioid receptors recruits downstream effectors, including
heterotrimeric G proteins (including Gα, Gβ and Gγ subunits) and β-arrestins. Specifically, opioid receptors primarily couple to the Gαi/o family (Gi1, Gi2, Gi3, GoA, GoB, Gz and gustducin
(Gg)) (Extended Data Fig. 1a). Some of these subtypes can mediate non-overlapping signalling pathways depending on the GPCR involved5,6,7,8. Whether signalling through individual pathways
has redundant roles or separately drives the therapeutic efficacy and side effects of opioids remains mostly unclear. KOR is a highly desirable therapeutic target for treating not only pain
but also addiction and affective disorders. KORs have gained increasing attention owing to their unique analgesic activity—they are predominantly expressed in pain-related neurons, and drugs
that target KOR do not lead to addiction or cause death due to overdose as observed for MOR agonists1. The lack of rewarding/euphorigenic effects initially encouraged the development of
KOR-agonist drugs as non-addictive analgesics9. Potent and selective KOR agonists have been developed, and these agonists produce effective peripheral and central analgesia. However, mood
disorders such as dysphoria and psychotomimesis have been frequently observed as side effects of KOR agonists, which has limited their therapeutic application2. Here we determined the atomic
structures of KOR in complex with different G-protein transducers and hallucinogenic ligands to help to elucidate the actions of opioids and the molecular basis for Gαi/o subtype
selectivity. OVERALL STRUCTURES OF KOR–G-PROTEIN COMPLEXES Although many efforts have been dedicated to the structural and molecular basis underlying the differences between G-protein and
arrestin signalling, the roles of individual G-protein subtypes and the molecular determinants of subtype selectivity remain largely unclear. Sequence alignment of the seven Gi/o subtypes
suggests that they could be further grouped into four subclasses on the basis of sequence identity (Gi1, Gi2 and Gi3; GoA and GoB; and Gz and Gg) (Extended Data Fig. 1b). To further
understand the role of KOR–G-protein coupling and signalling, we determined the structures of KOR in complexes with four representative Gi/o subtypes (Gi1, GoA, Gz and Gg) at nominal
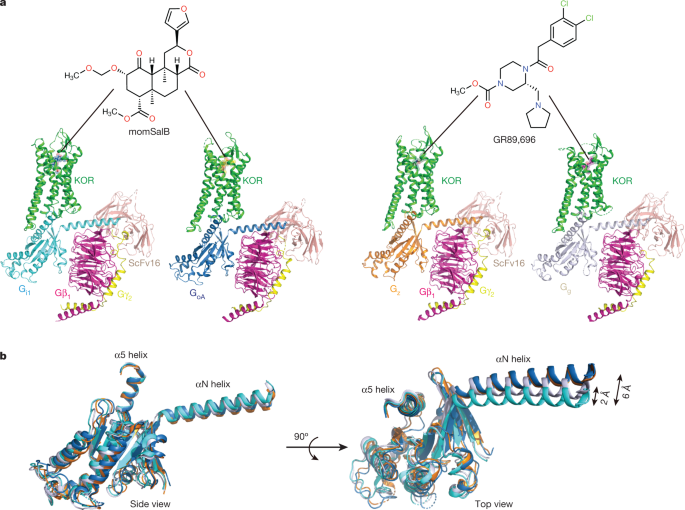
resolutions of 2.71 Å, 2.82 Å, 2.65 Å, and 2.61 Å, respectively, using single-particle cryo-electron microscopy (cryo-EM; Fig. 1a, Supplementary Fig. 1 and Extended Data Table 1). In
particular, KOR–Gi1 and KOR–GoA are bound to a psychotropic salvinorin analogue, methoxymethyl-salvinorin B (momSalB)10. However, cryo-EM experiments of KOR–Gz or KOR–Gg bound to momSalB
yielded only low-resolution reconstructions (resolution of around 4.5–5 Å) that prevented the delineation of detailed molecular interactions. Thus, we leveraged another highly potent KOR
agonist, GR89,696 (ref. 11), to obtain high-resolution structures of KOR–Gz and KOR–Gg. The high-resolution maps of the four structures enabled unambiguous modelling of the agonist-bound
heterotrimeric complexes (Supplementary Fig. 2). The overall differences between the four structures are subtle (root mean square deviations (r.m.s.d.) of 0.5 Å), with the exception of the
Gα subunit in each complex (Fig. 1b). G-protein interactions with the receptor are canonically driven by the α5 and the N-terminal (αN) helices of the Gα subunit. The overlay of the four
different G-protein subtypes showed that they adopt similar conformations in the α5 helix but differ in the extent of movement in the αN helix (Fig. 1b). In particular, relative to Gi1, both
GoA and Gz exhibit a 6 Å displacement in the αN helix, whereas Gg has a smaller 2 Å displacement. Notably, alignments of the MOR–Gi1 structure12 with KOR–Gi1 indicate that the αN helix of
Gi1 in MOR displays a position that is distinct from that of KOR–Gi1, whereas the α5 helix shows an orientation and interaction pattern similar to those in KOR (Extended Data Fig. 1c). The
overall structures of KOR in the Gi1/oA/z/g-bound states are similar to the previously reported nanobody-stabilized active conformation (KOR–Nb39)13 (r.m.s.d., 0.8 Å) (Extended Data Fig.
1d). Notably, a comparison of these two structures reveals that the intracellular end of transmembrane helix 6 (TM6) in the KOR–Gi1 protein complex moves 2 Å closer to TM7. Nb39 stabilizing
a different receptor conformation is further supported by its positive allosteric ability to enhance agonist binding affinity (Extended Data Fig. 1e). Another feature unique to
G-protein-bound KOR is the presence of a well-defined intracellular loop 3 (ICL3) conformation that is absent in the Nb39-stabilized KOR, presumably due to its inherent flexibility (Extended
Data Fig. 1d). Similar differences have also been captured between MOR–Gi112 or β2AR–Gs14 and their corresponding nanobody-stabilized active states15,16, which further corroborate that a
nanobody can stabilize a conformational state that mimics but is not identical to the G-protein-coupled state. INTERACTIONS OF KOR WITH HALLUCINOGENIC SALVINORINS KORs have a prominent role
in the modulation of human perception. Salvinorins, such as salvinorin A (SalA)17,18, are a group of naturally occurring hallucinogens with dissociative effects elicited by activating the
central KORs. momSalB is a semi-synthetic analogue of SalA and displays similar in vivo pharmacology compared to SalA19,20. GR89,696 is a potent and long-lasting KOR agonist that produces
antinociception and dysphoria but with unknown hallucinogenic properties21. Different binding poses of momSalB and GR89,696 were observed in the KOR orthosteric pocket. This is consistent
with their divergent chemical structures—GR89,696 is an alkaloid (containing basic nitrogen atoms) and momSalB is a terpenoid (lacking basic nitrogen atoms) (Fig. 1a). The pyrrolidine
nitrogen atom in GR89,696, as well as many other ligands including KOR’s endogenous dynorphin ligands22, is essential for the binding to KOR and enables the ligand to act as a hydrogen-bond
(H-bond) donor and forms a salt bridge with the carboxylate side chain of Asp1383.32 in the binding pocket (where the superscript values indicate Ballesteros–Weinstein numbering for GPCRs23)
(Fig. 2a). As salvinorin ligands (such as momSalB) lack the basic nitrogen atom, there are no attractive electrostatic interactions observed between the salvinorins and Asp1383.32. Indeed,
neither D1383.32A nor D1383.32N (the mutation in KOR DREADD24) showed detrimental effects in the binding affinity or agonistic potency of SalA, whereas both mutants abolished the interaction
with endogenous dynorphin ligands24,25,26. The mutation D1383.32N resulted in a significant loss of potency in U50,488 and GR89,696, but had minimal effects on momSalB (Fig. 2b). The side
chain of Asp1383.32 pointing to the methoxymethyl group of momSalB also explains an interesting observation that D1383.32N could further enhance the binding affinity and potency of SalA and
salvinorin B (SalB)24, probably due to the switch from the unfavourable acceptor–acceptor interaction to attraction resulting from the new H-bond interactions between the side chain of
mutated asparagine and methoxy oxygen of the ligand. Both momSalB and GR89,696 are highly selective and potent agonists at KOR (Fig. 2b and Extended Data Fig. 2a), making them ideal
templates to investigate the molecular determinants for ligand selectivity and efficacy. Although the two agonists overlap in the orthosteric binding pocket of KOR, the core rings occupy
different planes that are perpendicular to each other (Extended Data Fig. 2b). As a result, the subgroups of the two ligands form different interactions with residues in the corresponding
subpockets (Fig. 2c). Mutations of the majority of residues in these subpockets reduced the agonist activity of momSalB or GR89,696, but with different amplitudes (for example, Val1082.53,
Gln1152.60, Met1423.36, Val2305.42 or His2916.52) (Fig. 2d and Extended Data Fig. 2c–e). The observation that the binding-pocket mutations have greater effects on momSalB-mediated cAMP
inhibition than GR89,696 (for example, for H2916.52A, Δlog[median effective concentration (EC50)mutant−WT] = 2.23 ± 0.25 (momSalB) and 0.78 ± 0.27 (GR89,696)) is probably due to the lack of
the anchoring interactions with Asp1383.32, which makes salvinorins more sensitive to other residue contacts. The double mutation in KOR (for example, D1383.32N and H2916.52A, pEC50 = 9.95 ±
0.06) displays a less deleterious effect on the potency of momSalB than H2916.52A does (pEC50 = 9.08 ± 0.06) alone (Extended Data Fig. 3a). A 2-fold to 2.5-fold improvement in potency was
also observed from other mutations (Q1152.60N or V2305.42A) in combination with D1383.32N when compared with the single mutation without D1383.32N (Extended Data Fig. 3a). This effect might
be specific to momSalB or salvinorin ligands as the double mutations (V2305.42A/D1383.32N or H2916.52A/D1383.32N) led to an inactive U50,488 or a further loss of potency for
GR89,696-mediated cAMP inhibition in V2305.42A/D1383.32N (9,120-fold) or H2916.52A/D1383.32N (1,288-fold) compared with the respective single mutation (Extended Data Fig. 3a). Another major
difference between momSalB and GR89,696 is that momSalB mainly forms hydrophobic interactions with residues that specifically contribute to the high potency of momSalB, such as Val1082.53,
Val1343.28, Val2305.42 and Ile3167.39. In particular, Val1082.53 has also been indicated as a determinant of ligand selectivity between KOR and MOR or DOR, as the latter two opioid receptors
have an Ala2.53 at the corresponding position27. Another hydrophobic pocket formed by the side chains of Val1082.53 and Tyr3207.43 and the backbone of Gly3197.42 appears to be a key
determinant for agonist activity and receptor activation, as mutations of these residues significantly decreased or eliminated signal transduction of momSalB with a threefold reduction in
its ligand-binding affinity (Fig. 2d and Extended Data Fig. 3b). Notably, the amplitude of interactions with residues in this hydrophobic pocket positively correlates with agonist potency
because ligands with more extended interacting groups—such as SalB (-O-H), momSalB (-O-CH2-O-CH3) and ethoxymethyl SalB (-O-CH2-O-CH2-CH3) (Extended Data Fig. 3c,d)—have displayed increased
potency in activating KOR19. This subpocket at the bottom of the ligand-binding pocket acts as a potential allosteric connector to initiate the conformational changes of other microswitch
motifs, including the sodium site, CW6.48xxP and Pro5.50-Ile3.40-Phe6.44 motifs28,29. The overall binding pose of GR89,696 in KOR–Gz is similar to that in KOR–Gg. One notable difference is
that GR89,696 forms stronger salt-bridge interactions with Asp1383.32 (2.9 and 3.4 Å) in KOR–Gz than those in KOR–Gg (3.5 and 3.9 Å) (Fig. 2a and Supplementary Fig. 3), which probably
contributes to the higher potency in activating Gz compared with Gg (ref. 30). Mapping the atomic distances between the ligand and receptor showed that GR89,696 makes closer contact with
residues in the KOR–Gz structure than in the KOR–Gg structure (in terms of distance), whereas momSalB in KOR–Gi1 and KOR–GoA largely overlaps and displays similar strength (Supplementary
Fig. 3). For example, GR89,696 in KOR–Gz also forms H-bond interactions with Gln1152.60 (2.8 Å) and His2916.52 (3.3 Å), and, in KOR–Gg, Gln1152.60 (3.9 Å) and His2916.52 (4.0 Å). This
suggests that GR89,696 leads to more contractions of the ligand-binding pocket in the presence of Gz compared with Gg. STRUCTURAL BASIS OF G-PROTEIN SUBTYPE SELECTIVITY Similar to other
opioid receptors, KOR exclusively couples to the Gi/o family30, including the canonical Gi/o subtypes (Gi1, Gi2, Gi3, GoA and GoB) and the noncanonical Gz and Gg. Whereas Gz is predominantly
expressed in the central nervous system, Gg is the endogenous transducer of taste receptors, such as the bitter taste receptor 2 (TAS2R). Mice expressing engineered KORs in bitter-receptor
cells show a strong aversion to a designed KOR agonist (inert to endogenous wild-type KOR but active in engineered KOR)31, suggesting that the KOR–Gg interaction and signalling may also
occur in vivo. Using bioluminescence resonance energy transfer (BRET)-based transducerome profiling (Fig. 3a), we confirmed that both momSalB and GR89,696 could activate all four G-protein
subtypes, although with different potencies (Fig. 3b). The primary interaction sites in KOR bound to different Gi/o subtypes involve nearly the entire intracellular regions of the receptor
(ICL2, ICL3, TM3, TM5, TM6, TM7 and helix 8) and the αN and α5 helices of the Gα subunits (Extended Data Fig. 4). The key residues involved in KOR–G-protein interactions were mapped
(Extended Data Fig. 4) and screened by alanine substitutions. In this section, we first report the effects of interface residues from the KOR side and then the residues from the Gα protein
side. Although the KOR conformations in each G-protein-bound structure are similar to each other, notable differences were observed for the KOR residues involved in receptor–G-protein
interactions. Mutagenesis screening using G-protein-mediated cAMP inhibition assays suggested that almost all of the residues on the KOR side contribute to KOR–G-protein signalling (Extended
Data Fig. 5a,b). That was further confirmed by the BRET-based transducer profiling, which showed that mutations of these residues on the intracellular KOR side decreased agonist-mediated
Gi1, GoA, Gz and Gg coupling (Extended Data Fig. 5c and Supplementary Fig. 4). Although most of the residues in the KOR interface affect the four G-protein couplings in a similar manner
(Supplementary Fig. 4), some display subtype selectivity. Arg1563.50 is a highly conserved residue in the classic Asp3.49-Arg3.50-Tyr3.51 motif that has been implicated in having an
important role in receptor activation and signal transduction (Fig. 3c). An ‘ionic lock’ has been frequently observed between Arg3.50 and Glu6.30 in class A GPCRs, keeping the receptor in an
inactive state with TM3 and TM6 in close proximity. Thus, the breaking of this ionic lock is an important step towards the coupling of G proteins, as the TM6 movement away from TM3 is
critical for penetration of the G-protein α5 helix into the cytoplasmic pocket12. The R1563.50A mutation significantly reduced the potency of agonist-mediated activation by momSalB or
GR89,696 (Fig. 3d). Furthermore, R1563.50A specifically reduced the efficacy of agonist-mediated Gg activation, momSalB (WT (106 ± 3%) versus R1563.50A (38 ± 5%)) or GR89,696 (WT (103 ± 3%)
versus R1563.50A (46 ± 7%)) (Fig. 3d and Extended Data Fig. 5d). In the inactive-state KOR32, the partially formed ionic lock is between Arg1563.50 and Thr2736.34; in the fully active
KOR–agonist–G-protein states, this interaction is broken due to the insertion of the α5 helix of Gα protein, leading to the release of the side chain of Arg1563.50 to extend towards TM7 and
form hydrophobic interactions with the second-to-last leucine (Leu353H5.25 in Gαi1, GαoA or Gαg; Leu354H5.25 in Gαz) of the Gα subunits (superscript notes for G proteins represent the CGN
numbering system33) (Fig. 3c). This is further supported by the molecular dynamics simulations showing that the KOR-Arg1563.50 can form hydrophobic interactions with Gα-L353H5.25 or
Leu354H5.25 maintaining <4 Å distances with these side chains (Extended Data Fig. 6). In this extended conformation, the KOR-Arg1563.50 guanidine group also forms a persistent H bond with
Tyr2465.58 observed in all four complexes (Fig. 3c and Extended Data Fig. 6). These data suggest that the KOR-Arg1563.50 has an important role in KOR activation by directly interacting with
the G proteins. A recent study also suggested that G proteins might need to precouple to the receptor and break the Arg1563.50-mediated ionic interaction before agonist binding and
signalling34. Other important interactions are formed by residue Asn3368.49 in helix 8 of KOR, which engages different H-bond interactions with the backbone of the α5 helix in each Gα
protein, such as Lys/ArgH5.21 and Gly/Asp/TyrH5.22 (Fig. 3e and Extended Data Fig. 4). The molecular dynamics simulations also provide support for these interactions, suggesting dynamic
patterns of switches between specific interaction pairs (Extended Data Fig. 7). The mutation N3368.49A completely abolished KOR–Gg coupling (for example, momSalB), and a 2-fold loss in
potency in Gi1, 14-fold in GoA or 9-fold in Gz coupling (Fig. 3f). Together with the effects observed from Arg1563.50 and Asn3368.49, these data indicate that these residues have
differential roles in G-protein association, probably by engaging at different intermediate stages. The observation that several mutations have the largest effect on Gg compared with the
other Gi/o subtypes suggests a non-canonical role of Gg in KOR-mediated signalling. Next, we examined the Gα subunit by mutating the non-conserved residues in the αN or α5 helix to alanine
(Extended Data Fig. 8a). However, we did not observe significant changes in the potency of agonist-mediated G-protein activation in BRET2 assays (Extended Data Fig. 8b–f and Supplementary
Fig. 5). One exception is that C351H5.23A in Gαi1/oA/g or I352H5.23A in Gαz led to a significant decrease in potency for momSalB or GR89,696-mediated G-protein activation. This Ile352H5.23
in Gαz, compared with the corresponding Cys351H5.23 in other Gi/o subtypes, is known as the site that makes Gαz insensitive to pertussis toxin. The relative conformation of the α5 helix has
been implicated as a key determinant between Gs and Gi specificity, in which the α5 helix adopts distinct positions and results in a larger outward movement of TM6 (13 Å in β2AR–Gs versus 9
Å in MOR–Gi1)12,35,36. The subtle differences in the α5 helix conformation of Gαi1/oA/z/g and the mutational evidence suggest that the G-protein-coupling specificity in the Gi/o family is
probably determined by a more complex and/or dynamic three-dimensional network interaction37,38. The overall interfaces of Gαi1, GαoA, Gαz and Gαg with KOR are highly conserved (Extended
Data Fig. 4), but there are critical differences in the α5 and αN helices. The major contacts made by GαoA with KOR are through residues in the α5 helix (Extended Data Fig. 4b), whereas
contacts made by Gαi1, Gαz and Gαg involve regions in both the α5 and αN helices (Extended Data Fig. 4a,c,d). Similarly, the 5HT1BR–Go39 interaction is also mediated solely by the α5 helix,
but a structural comparison between KOR–GαoA and 5HT1BR–Go shows that the α5 helix in 5HT1BR–Go tilts an additional 9°, leading to a larger 3 Å outward movement of TM6 (Extended Data Fig.
9a). Alignment of the cytoplasmic regions of KOR–Gαi1, β2AR–Gαs and 5HT2AR–Gαq shows that the α5 helices are positioned differently. There are 6° and 12° tilts of the C-terminal end away
from the plane of the membrane compared with Gαq and Gαs, respectively, leading to different magnitudes of outward movement of TM6 (Extended Data Fig. 9b). As a result of intracellular
conformational differences, the KOR–Gαi1 forms an interface area of 1,219 Å2 (Extended Data Fig. 4a), compared with a slightly larger area of β2AR–Gαs (1,260 Å2) and a much smaller area of
5HT2AR–Gαq (1,077 Å2) (Extended Data Fig. 9c). Whereas Gαi1, Gαz and Gαg have similar interface areas (1,219, 1,262 and 1,221 Å2, respectively) (Extended Data Fig. 4a,c,d), GαoA has a much
smaller area (1,096 Å2) (Extended Data Fig. 4b). Notably, the 822 Å2 surface area of Go in contact with 5HT1BR39 is closer to that of KOR and GαoA compared with other G-protein subtypes,
suggesting a shared mechanism between different GPCRs and the same G protein. INTRINSIC DIFFERENCES IN G-PROTEIN SUBTYPES GPCR signalling is transduced through the allosteric changes between
the extracellular ligand pocket and the intracellular G-protein-binding pocket. Conformational changes induced by agonist binding can enhance the binding affinity of G-protein
heterotrimers. Conversely, G protein acts as a positive allosteric modulator and further enhances agonist-binding affinity by stabilizing the ternary complex40 formed by the receptor, ligand
and G proteins (Fig. 4a). We next sought pharmacological evidence to test whether G-protein subtypes have intrinsic differences, including binding affinity and allosteric activity at KOR in
the presence of agonists. On the basis of the ternary complex model41, the high-affinity agonist-binding states should increase in the presence of G-protein heterotrimers, as the latter can
stabilize the active-state receptor favouring agonist binding. We performed saturation binding assays to test the binding of agonist radioligand 3H-U69,593 to KOR in the presence of Gi1,
GoA, Gz and Gg. Notably, the four G proteins display substantial differences in the allosteric enhancement of agonist binding (Fig. 4b,c). Compared with the wild type alone (_B_max = 1,350 ±
116), the high-affinity binding sites for 3H-U69,593 were increased 62-, 38-, 14- and 7-fold in the presence of Gi1 (_B_max = 84,324 ± 4,214), GoA (_B_max = 52,086 ± 2,465), Gz (_B_max =
18,623 ± 1,468) and Gg (_B_max = 9,866 ± 3,493), respectively. These data are consistent with the ternary model that at least two binding states predominate in the unliganded receptor42—a
high-affinity (G-protein-coupled) and a low-affinity (G-protein-uncoupled) binding state. We also compared the wild-type G proteins with the engineered G proteins used in our structural
determination and observed similar patterns of _B_max increases (Extended Data Fig. 10b). The different magnitudes of _B_max increases among G-protein subtypes suggest that individual G
proteins have different allosteric effects on ligand binding. To test this hypothesis, we next compared the cooperativity of the four G-protein subtypes by radioactive competition binding
assays designed to quantify the G-protein binding affinity (_K_B) and cooperativity (_α_, G-protein cooperativity value, _α_ > 1 indicates positive effects in increasing agonist
affinity)43. The G-protein-insensitive antagonist radioligand 3H-JDTic was used for the following series of experiments. The inhibition of 3H-JDTic binding at KOR by GR89,696 progressively
improved as the concentration of G protein increased, indicating positive cooperativity between G proteins and agonist binding (Fig. 4d). The calculated _K_B and _α_-cooperativity displayed
a pattern similar to that observed in the saturation binding, in which Gi1 has the highest binding affinity and allosteric effects at KOR in the presence of agonists, and Gg has the least
(Fig. 4d). The different binding affinities could have a role in G-protein-subtype selectivity, as G-protein subtypes with higher affinity can outcompete other G-protein subtypes, depending
on subtype abundance, especially in cells expressing several or all Gi/o family subtypes. Guanosine diphosphates (GDPs) or guanosine triphosphates (GTPs) are important regulators of
GPCR–G-protein assembly and signalling44. We therefore examined whether the presence of GDP or GTP can affect the allosteric activity of G-protein subtypes. The specific binding of
3H-U69,593 was significantly reduced in the presence of GDP or GTP compared with the nucleotide-free state (Fig. 4e,f). The four G proteins exhibited a uniform pattern, showing similar
responses to GDP or GTP (Extended Data Fig. 10c). These nucleotide-specific effects are consistent with the results of single-molecule studies of the β2AR–Gs complex showing that the
presence of GDP or GTP accelerates the dissociation of β2AR and the Gs heterotrimer45, which is achieved through a sequential conformational change in the Gα subunit after the binding of GDP
or GTP44,46. Note that the dominant negative Gi2 has been reported to abolish GTP binding and GTPase activity47; however, the engineered G proteins in this study appear to maintain the GTP
binding affinity and GTP turnover activity, although at weaker levels compared with the wild type (Extended Data Fig. 10a). DISCUSSION After activation, KOR can interact with up to seven G
proteins, the coupling of which determines the direction of ligand-induced signalling. The seven G-protein subtypes are highly homologous but not structurally or functionally identical. The
binding of signal transducers is coupled with specific receptor conformational changes, such as the magnitude of TM6 displacement. Such conformational differences have been observed in GPCRs
bound to the Gs, Gi and Gq families. However, analysis of the four structures of KOR in complex with different Gi/o subtypes shows that the receptor adopts a similar conformation. In
particular, the receptor conformations in KOR–Gz and KOR–Gg are nearly identical, although the bound agonist GR89,696 activates the two G proteins with a 100-fold difference in potency. This
structural observation agrees with an original postulation that the cross-reactivity between receptors and G proteins speaks to the conservation of structure among the receptor-binding
domains of the G proteins and the G-protein-binding domains of the receptors48. This conformational similarity, irrespective of transducer subtypes, has also been observed in other GPCRs
engaging G proteins versus arrestins49. These subtle differences could be due to the limitation of the structures that reveal a well-resolved population of the nucleotide-free
G-protein-bound conformational state of the receptor. These nucleotide-free states of Gα subunits (Gαi1, GαoA, Gαz and Gαg) tend to stabilize a specific conformational state of KOR. In the
absence of G proteins or in the presence of nucleotide-bound G proteins, the receptor can adopt dynamic conformations that are different from that captured by nucleotide-free Gα45. Other
approaches, including nuclear magnetic resonance (NMR)50 and molecular dynamics simulations51, have identified dynamic conformational states in the intracellular regions of the receptor
related to transducer couplings in the presence of GDP or GTP. Different GPCR–G-protein interfaces have been proposed to contribute to the differential kinetics of G proteins during
association and dissociation with the receptor52,53. Our pharmacological characterization of the KOR–G-protein interface identified key residues that have different roles in G-protein
coupling. The complexes that we targeted in this study displayed varying interface areas dependent on the receptors and G proteins. However, time-resolved studies are needed in the future
for the direct measurement of G-protein association and dissociation rates, especially for different Gi/o subtypes, as the strength of the receptor–G-protein interface may be another factor
that affects the coupling efficiency. In the structures of KOR in complex with different G-protein subtypes, we also revealed the binding poses of two selective KOR agonists—momSalB and
GR89,696. Although they occupied the same binding pocket, they adopted different conformations and interacting patterns, probably due to their unique chemical structures. GR89,696 displays
stronger interactions in KOR–Gz than in KOR–Gg, which may contribute to its higher potency observed in the BRET2 assay. Owing to the unique scaffold and pharmacology of salvinorin ligands,
extensive studies have been conducted to elucidate their binding and function13,25,26. Several residues or motifs in KOR (for example, Val1082.53, His2916.52, Ile3167.39) have been
identified that are important for salvinorin’s agonism, which can now be explained by their direct interactions with momSalB. Notably, using multiple structural templates, previous
salvinorin docking suggested different binding poses13,25,26, and our structures now provide direct evidence of how momSalB sits in the binding pocket of KOR. We also observed allosteric
differences among these highly conserved G-protein subtypes. As positive allosteric modulators, the four representative G proteins display distinct allosteric activity in potentiating
agonist binding (Gi1 > GoA > Gz > Gg) (Fig. 4g). This is consistent with measurements of the binding affinity of different G proteins (Gi1 > GoA > Gz > Gg) at KOR. These
intrinsic differences in G proteins, including the binding affinity and coupling efficiency, add pharmacological evidence to determinants for G-protein-subtype selectivity. Our structural
observations from different G proteins in complex with KOR show that the Gi/o family subtypes share a highly conserved mechanism in interacting with KOR, but that each maintains
pharmacological differences. Considering that many GPCRs can couple to different G-protein families, such as the β2AR coupling with both Gs and Gi/o (refs. 38,54), whether β2AR displays
differential binding affinities with Gs and Gi/o may help to explain its G-protein coupling specificity. Furthermore, the allosteric activity of G proteins can be regulated by GDPs or GTPs
that decouple G proteins from the receptor. It is known that nucleotide-specific conformations exist between nucleotide-free G proteins and GDP- or GTP-bound G proteins. When comparing the
crystal structure of the uncoupled GDP-bound Gi1 heterotrimer55 and nucleotide-free (KOR–Gi1, GoA, Gz and Gg) heterotrimers (Extended Data Fig. 11a), several conformational displacements are
noted (Extended Data Fig. 11b). The activated receptor engages the C terminus of the α5 helix of Gαi1, which undergoes an upward helical extension (8.6 Å) into the receptor core (Extended
Data Fig. 11c) compared with the uncoupled G protein structure. The insertion of the α5 helix into the transmembrane helical bundle of the receptor has the following two consequences. First,
the loop connecting the α5 helix and β6 strand moves outward 5 Å. Second, the movement of the α5 helix disrupts the original hydrophobic interactions between the α5 and α1 helices, leading
to a displacement of the P loop. Both the P loop translocation and loss of coordination with GDPs are necessary for GDP release44,45. In agreement with the ternary complex model, our
saturation binding data show that GDPs or GTPs act as negative regulators of agonist binding kinetics in the presence of G proteins. One limitation of this study is that we used an in vitro
overexpressed system with engineered receptors and G proteins to measure ligand activity, which cannot be extended to in vivo without further experiments. In summary, we have elucidated the
molecular interaction details between highly conserved Gi/o subtypes and KOR using cryo-EM-derived atomic models. We have also examined the structural determinants of ligand selectivity and
efficacy at KOR. Using structural pharmacology analysis, we revealed the intrinsic differences between these previously under-represented Gi/o subtypes and demonstrated that subtype
selectivity is probably a combinational result of receptor conformational dynamics, the binding affinity of G proteins and cooperativity between agonist binding and G-protein coupling. Such
findings are important both in understanding GPCR-mediated signalling and in the generation of new research tools and therapeutics based on the potential of G-protein-selective agonists.
METHODS GENERATION OF CONSTRUCTS FOR CRYO-EM For the human KOR, we used a construct the same as the previously determined active-state KOR13. In brief, the construct (1) lacks N-terminal
residues 1–53; (2) lacks C-terminal residues 359–380; (3) contains Met1–Leu106 of the thermostabilized apocytochrome b562 RIL (BRIL) from _E. coli_ (M7W, H102I, R106L) in place of receptor N
terminus residues Met1–His53. This N-terminal Bril will be removed using a PreScission cleavage site in the end. The single chain Fab scFv16 has the same sequence as previously reported56.
A 6×His tag was added to the C-terminal scFv16 sequence with a PreScission cleavage site inserted between. For the G-protein heterotrimers, individual G-protein constructs (Gi1, GoA, Gz and
Gg) were engineered (labelled as dominant negative, DN)47 for the binding of scFv16, and then subcloned into a designed vector that co-expresses the Gβ1 and Gγ2. Further modifications were
made to enable a stable complex between KOR, G-protein heterotrimer and scFv16. Specifically, Gi1(DN) includes S47N, E245A, G203A and A326S. GoA(DN) includes C3S, S47N, G204A, E246A, A326S
and M249K. For Gz(DN), the N-terminal sequence was replaced with the Gi2 sequence to allow for better interaction with scFv16; other mutations include S47N, G204A, E246A, R249K, N262D and
A327S. For Gg(DN), the N-terminal sequence was replaced with the Gi2 sequence; other mutations include S47A, G203A, E245A, H248K, T261D, A326S and N251D. EXPRESSION OF KOR–G-PROTEIN–SCFV16
COMPLEX The Bac-to-Bac Baculovirus Expression System was applied to generate high-quality recombinant baculovirus (>10−9 viral particles per ml) for protein expression (KOR, G-protein
heterotrimers and scFv16). For the expression of KOR–G–scFv16 protein complex, each heterotrimeric G protein, including Gα (Gi1, GoA, Gz or Gg), Gβ1 and Gγ2 was coexpressed with KOR and
scFv16, respectively, by infection of _Spodoptera frugiperda_ Sf9 cells at a cell density of 2.5 × 106 cells per ml in ESF921 medium (Expression System) with the P1 baculovirus at a
multiplicity of infection (MOI) ratio of 2:2:0.5. Cells were collected by centrifugation (125 rpm at 27 °C) for 48 h after infection, washed with HN buffer (25 mM HEPES pH 7.4, 100 mM NaCl),
and stored at −80 °C for future purification. PURIFICATION OF THE KOR–G-PROTEIN–SCFV16 COMPLEX The compounds used in this study—(−)-U50,488 (0496) and GR89,696 (1483)—were purchased from
Tocris. momSalB was synthesized by a method described previously57. After purification by silica gel column chromatography, momSalB was a single spot on TLC (silica, 20% ethyl acetate,
dichloromethane) with an _R_f of 0.49. An NMR spectrum of momSalB was collected to confirm the chemical identity (Supplementary Fig. 6), which is consistent with the expected spectrum
reported previously58. We thawed the cell pellet and incubated it in buffer containing 20 mM HEPES pH 7.5, 50 mM NaCl, 1 mM MgCl2, 2.5 units Apyrase (NEB), 10 μM agonist (final
concentration) and protease inhibitors (500 mM AEBSF, 1 mM E-64, 1 mM leupeptin, 150 nM aprotinin) for 1.5 h at room temperature. We then collected the membrane by centrifugation at 25,000
rpm for 30 min at 4 °C. The membrane was solubilized in buffer (40 mM HEPES pH 7.5, 100 mM NaCl, 5% (w/v) glycerol, 0.6% (w/v) lauryl maltose neopentyl glycol (LMNG), 0.06% (w/v) cholesteryl
hemisuccinate (CHS), 10 μM agonist and protease inhibitors) with 200 μg scFv16 in the cold room. After 5 h, the supernatant was collected by centrifugation at 30,000 rpm for 30 min at 4 °C
and incubated with 1 ml TALON IMAC resin (Clontech) and 20 mM imidazole overnight in the cold room. The next day, the resin was collected and washed with 10 ml buffer containing 20 mM HEPES
pH 7.5, 100 mM NaCl, 30 mM imidazole, 0.01% (w/v) LMNG, 0.001% (w/v) CHS, 5% glycerol and 5 μM agonist. The protein was then eluted with the same buffer supplemented with 300 mM imidazole,
concentrated and further purified by size-exclusion chromatography on the Superdex 200 increase 10/300 column (GE healthcare), which was pre-equilibrated with 20 mM HEPES pH 7.5, 100 mM
NaCl, 100 μM TCEP, 0.00075% (w/v) LMNG, 0.00025% (w/v) glyco-diosgenin (GDN) and 0.00075% (w/v) CHS, 1 μM agonist. Peak fractions were collected, concentrated and incubated with PNGase F
(NEB), PreScission protease (GenScript) to remove the potential glycosylation and N-terminal His–BRIL, respectively, and 100 μg scFv16 at 4 °C overnight. The next day, cleaved His–BRIL and
protein, uncleaved protein and proteases were separated by the same procedure as described above. Peak fractions were concentrated to 3–5 mg ml−1 for electron microscopy analysis. Four
KOR–G-protein–scFv16 complexes were purified according to the same procedure except that different agonists were used. EXPRESSION AND PURIFICATION OF SCFV16 PROTEIN The scFv16 protein was
expressed by infection of Sf9 cells at a cell density of 2.5 × 106 cells per ml in ESF921 medium (Expression System) with the P1 baculovirus at an MOI of 2. After 96 h, the cell culture
medium containing secreted scFv16 protein was collected by centrifugation at 4,000 rpm for 15 min. The pH of the supernatant was adjusted to 7.5 by addition of Tris-base power. Chelating
agents were quenched by the addition of 1 mM nickel chloride and 5 mM calcium chloride and incubation with stirring for 1 h at room temperature and 5 h in the cold room. We removed the
precipitates by centrifugation and the resultant supernatant was further cleaned with 0.45 μm filter paper, and incubated with 2 ml Ni-NTA resin and 10 mM imidazole overnight in the cold
room. The Ni-NTA resin was washed the next day with 20 ml buffer (20 mM HEPES pH 7.5, 100 mM NaCl, 0.00075% (w/v) LMNG, 0.000075% (w/v) CHS, 0.00025% (w/v) GDN, 20 mM imidazole). The protein
was eluted with the same buffer supplemented with 300 mM imidazole, concentrated and further purified on the Superdex 200 increase 10/300 column. Monomeric fractions were pooled,
concentrated, flash-frozen in liquid nitrogen and stored at −80 °C until future use. EXPRESSION AND PURIFICATION OF HETEROTRIMERIC G PROTEINS The expression of heterotrimeric G protein was
achieved by infection of Sf9 cells at a cell density of 2.5 × 106 cells per ml in ESF921 medium (Expression System) with the P1 baculovirus at an MOI of 2. After 48 h, cells were collected
and lysed in buffer containing 200 mM NaCl, 40 mM HEPES pH 7.5, 0.2% Triton X-100, 5% glycerol, 3 mM β-me and protease inhibitors. The supernatant was isolated by centrifugation at 40,000
rpm for 50 min and incubated with 1 ml Ni-NTA resin and 20 mM imidazole overnight at 4 °C. The resin was collected the next day and washed with 20 ml buffer containing 100 mM NaCl, 20 mM
HEPES pH 7.5, 5% glycerol, 20 mM imidazole and 3 mM β-me. The protein was then eluted with elution buffer (300 mM NaCl, 20 mM HEPES pH 7.5, 5% glycerol, 3 mM β-me and 300 mM imidazole),
concentrated and further purified on the Superdex 200 increase 10/300 column, which was pre-equilibrated with buffer the same as the elution buffer except without the imidazole. The peak
fractions were concentrated, flash-frozen in liquid nitrogen and stored at −80 °C for future binding assays. CRYO-EM DATA COLLECTION AND 3D RECONSTRUCTION The purified samples (3–4 μl) were
applied to glow-discharged 300-mesh Au grids (Quantifoil R1.2/1.3) individually and vitrified using a Vitrobot mark IV (Thermo Fisher Scientific). Cryo-EM imaging was performed on the Talos
Artica system operated at 200 kV at a nominal magnification of ×45,000 using a Gatan K3 direct electron detector at a physical pixel size of 0.88 Å. Each stack video was recorded for 2 to
2.7 s in 60 frames at a dose rate of about 15 e− px−1 s−1, leading to a total exposure dose indicated in Extended Data Table 1. Videos were collected automatically with SerialEM59 using an
optimized multishot array procedure60. Dose-fractioned image stacks were processed for beam-induced motion correction followed by contrast transfer function estimation. Particles were
selected using Blob particle picker, extracted from the micrograph and then used for 2D classification and 3D classification followed by non-uniform refinement. All of these steps were
performed in cryoSPARC61,62. MODEL BUILDING AND REFINEMENT Maps from cryoSPARC were used for map building, refinement and subsequent structural interpretation. The dominant-negative Gi1
trimer model and scFv16 model were adapted from the cryo-EM structure of the MRGPRX2–Gi1 complex (Protein Data Bank (PDB): 7S8M)63. GoA, Gz and Gg trimer models were built from the Gi1
trimer model, followed by mutating the non-conserved residues back to the wild-type GoA, Gz and Gg. The receptor KOR model was taken from the active-state KOR–Nb39 structure (PDB: 6B73)13.
The receptor, G proteins and scFv16 were docked into the cryo-EM map using Chimera64. The complex models (KOR–G-protein–scFv16) were manually built in Coot65, followed by several rounds of
real-space refinement using Phenix66. The model statistics were validated using Molprobity67. Structural figures were prepared using Chimera or PyMol (https://pymol.org/2/). CAMP INHIBITION
ASSAY For the KOR–Gαi-mediated cAMP inhibition assay, HEK293T (ATCC CRL-11268) cells were co-transfected with human KOR or various mutants along with a split-luciferase-based cAMP biosensor
(GloSensor, Promega) at a 1:1 ratio (KOR:GloSensor). After 16 h, the transfected cells were plated into poly-l-lysine-coated 96-well white clear-bottom cell culture plates with DMEM + 1%
dialysed FBS at a density of 40,000–50,000 cells per 200 μl per well and incubated at 37 °C with 5% CO2 overnight. The next day, 3× drug solutions were prepared in fresh drug buffer (20 mM
HEPES, 1× HBSS, 0.3% bovine serum albumin (BSA), pH 7.4). The plates were decanted the next day and received 40 μl per well of drug buffer (20 mM HEPES, 1× HBSS, pH 7.4) followed by addition
of 20 μl of 3× drug solutions for 15 min in the dark at room temperature. Cells then received 20 μl luciferin (4 mM final concentration) supplemented with isoproterenol (300 nM final
concentration), stimulating the production of endogenous cAMP through β2 adrenergic Gs activation, and incubated in the dark at room temperature. After 15 min, luminescence intensity was
quantified using the Mithras LB 940 multimode microplate reader (Berthold Technologies). Data were plotted as a function of drug concentration, normalized to percentage U50,488 stimulation,
and analysed using log (agonist) versus response in GraphPad Prism (v.9.3.1). BRET2 ASSAY To measure the agonist-stimulated G-protein (wild type and mutants) activation by KOR and various
mutants, a BRET2-based cell assay was used. Specifically, four plasmids (KOR, Gα, Gβ, Gγ) were used, in which each Gα is tagged with a luciferase (Rluc8) and Gγ is tagged with an N-terminal
GFP. Specifically, the Gαi1/Gβ3/Gγ9, GαoA/Gβ3/Gγ8, Gαz/Gβ3/Gγ1 and Gαg/Gβ3/Gγ1 combinations were used for BRET2 Gi1, GoA, Gz and Gg experiments, respectively. Detailed information of the
GFP-Gγ and Gα-Rluc8 constructs was described previously30. HEK293T cells were then transfected with the four plasmids (KOR, Gα-Rluc8, Gβ, Gγ–GFP) using a 1:5:5:5 DNA ratio of
receptor:Gα-RLuc8:Gβ:Gγ-GFP2 (100 ng receptor, 500 ng Gα–RLuc8, Gβ and Gγ–GFP2 for 10 cm dishes). Transit 2020 (Mirus Biosciences) was used to complex the DNA at a ratio of 2 μl Transit per
μg DNA in Opti-MEM (Gibco-Thermo Fisher Scientific). Then, 16 h after transfection, cells were plated in poly-l-lysine-coated 96-well white clear-bottom plates in plating medium (DMEM + 1%
dialysed FBS) at a density of 40,000–50,000 cells in 200 μl per well and incubated overnight. The next day, the plates were decanted and washed once with 60 μl drug buffer (20 mM HEPES, 1×
HBSS, pH 7.4) and then 60 μl drug buffer containing coelenterazine 400a (Nanolight Technology) at a final concentration of 5 μM was added to each well. After 5 min for substrate diffusion,
30 μl 3× drug solutions in fresh drug buffer (20 mM HEPES, 1× HBSS, 0.3% BSA, pH 7.4) was added to each well and incubated for an additional 5 min. Finally, the plates were read on the
Mithras LB 940 multimode microplate reader (Berthold Technologies) with 400 nm (RLuc8-coelenterazine 400a) and 510 nm (GFP2) emission filters for 1 s per well. The GFP to Rluc8 ratio was
calculated, plotted as a function of drug concentration, normalized to percentage U50,488 stimulation and analysed using log (agonist) vs response in GraphPad Prism (v.9.3.1).
RADIOLIGAND-BINDING ASSAY Saturation binding assays were performed using the construct BRIL-wt-KOR(54–368) reconstituted into nanodiscs comprised of KOR, spMSP1D1 and lipid mixture
(POPC:POPE:POPG = 3:1:1) at a molar ratio of 1:3:100. Binding assays were set-up in 96-well plates in standard binding buffer (50 mM Tris-HCl, 10 mM MgCl2, 0.1 mM EDTA, 0.1% BSA, pH 7.4) at
room temperature. Saturation binding assays with 0.1–20 nM 3H-U69,593 in the standard binding buffer were performed to determine the equilibrium dissociation constant (_K_d) and _B_max. To
determine the effects of G proteins on 3H-U69,593 binding, each G protein (final concentration 1 μM) was incubated with 3H-U69,593 and homogenous membrane fractions for 3.5 h at room
temperature. Data were analysed using GraphPad Prism (v.9.3.1) using a one-site model. For the competitive binding assay, 3H-JDTic (0.68 nM), homogenous membrane fractions expressing KOR and
3× GR89,696 solutions were incubated in 96-well plates in standard binding buffer in the absence or presence of four G proteins in various concentrations (final concentration: 1,900 nM, 190
nM, 19 nM, 1.9 nM, 0 nM) for 3.5 h at room temperature in the dark, and then terminated by rapid vacuum filtration onto chilled 0.3% PEI-soaked GF/A filters followed by three quick washes
with cold wash buffer (50 mM Tris-HCl, pH 7.4) and read. Results (with or without normalization) were analysed using GraphPad Prism (v.9.3.1) using one-site or allosteric IC50 shift models.
CELL-SURFACE EXPRESSION STUDIES The cell-surface expression levels of wild-type KOR and its mutants were measured using an enzyme-linked immunosorbent assay (ELISA). In brief, HEK293T (ATCC
CRL-11268) cells were transiently transfected with wild-type KOR and KOR mutant DNA at the same quantity. After 24 h, cells were plated in poly-l-lysine-coated 96-well white clear-bottom
plates in plating medium (DMEM + 1% dialysed FBS) at a density of 40,000–50,000 cells in 200 μl per well and incubated overnight. The next day, plates were decanted and fixed with 4% (w/v)
paraformaldehyde for 10 min at room temperature. Cells were then washed twice with 1× phosphate-buffered saline (PBS) (pH 7.4) and blocked by 1× PBS containing 0.5% (w/v) non-fat milk for at
least 30 min at room temperature followed by incubation with anti-Flag (M2)–horseradish peroxidase-conjugated antibodies (Sigma-Aldrich, A8592) diluted 1:20,000 in the same buffer for 1 h
at room temperature. After washing three times with 1× PBS, 1-Step Ultra-TMB ELISA substrate (Thermo Fisher Scientific, 34028) was added to the plates and the plates were incubated at 37 °C
for 15–30 min and terminated by addition of 1 M sulfuric acid (H2SO4) stop solution. Finally, the plates were read at a wavelength of 450 nm using the BioTek Luminescence reader. The data
were analysed using GraphPad Prism (v.9.3.1). G-PROTEIN EXPRESSION STUDIES To measure the expression levels of four wild-type G proteins and their mutants, HEK293T (ATCC CRL-11268) cells
were transiently transfected with the same quantity of wild-type and mutant G proteins DNA. After 16 h, cells were plated in poly-l-lysine-coated 96-well white clear-bottom plates in plating
medium (DMEM + 1% dialysed FBS) at a density of 40,000–50,000 cells in 200 μl per well and incubated overnight. The next day, the plates were decanted and washed once with 60 μl drug buffer
(20 mM HEPES, 1× HBSS, pH 7.4), then 60 μl drug buffer containing coelenterazine 400a (Nanolight Technology) at a final concentration of 5 μM was added to each well. After 5 min for
substrate diffusion, plates were read in a Mithras LB 940 multimode microplate reader (Berthold Technologies) with 400 nm (RLuc8-coelenterazine 400a) and 510 nm (GFP2) emission filters for 1
s per well. The Rluc8 values represented the G-protein expression levels and were plotted in the GraphPad Prism (v.9.3.1). GTP TURNOVER ASSAY Analysis of GTPase activity of G proteins (Gi1,
GoA, Gz, Gg) was performed by using a modified protocol of the GTPase-Glo assay (Promega). G proteins were serially (1:1) diluted into various concentrations with a buffer of 300 mM NaCl,
20 mM HEPES pH 7.5 and 1 mM DTT, and 5 μl was dispensed into each well of a 384-well plate. The reaction was initiated by adding 5 μl 1 μM GTP solution to 5 μl G proteins. After incubation
for 90 min at room temperature, 10 μl reconstituted GTPase-Glo reagent was added to the sample and incubated for 30 min at room temperature. Luminescence was measured after addition of 20 μl
detection reagent and incubation for 10 min at room temperature using the Mithras LB 940 multimode microplate reader (Berthold Technologies). The data were analysed using GraphPad Prism
(v.9.3.1). MOLECULAR DYNAMICS SIMULATIONS The Gromacs simulation engine (v.2020.3)68 was used to run all molecular dynamics simulations under the Charmm36 force-field topologies and
parameters69,70. Charmm force-field parameters and topologies for the ligands momSalB and GR89,696 were generated using Charmm-GUI’s Ligand Reader & Modeller tool70. The loop grafting
and optimization for modelling missing side chains and loops was performed in the ICM-Pro (v.3.9-2b) molecular modelling and drug discovery suite (Molsoft)71. The structurally conserved
helix-8 (Hx8) amphipathic helical motifs in KOR were modelled using human antagonist-bound KOR (PDB: 4DJH)26 as the template structure. The lobe in Gi1, GoA, Gz and Gg proteins was modelled
using a human agonist-bound CB2–Gi structure (PDB: 6PT0)72. Structure regularization and torsion profile scanning were performed using ICMFF force field73. The GR89,696-bound structures of
KOR complexes with Gz and Gg proteins as well as momSalB-bound KOR with Gi1 and GoA proteins were then uploaded to the Charmm-GUI webserver69, where the starting membrane coordinates were
determined by the PPM74 server using the Charmm-GUI interface. The complexes were then embedded in a lipid bilayer composed of 1,2-dipalmitoyl-sn-glycero-3-phosphatidylcholine (DPPC),
1,2-dioleoyl-sn-glycero-3-phosphatidylcholine (DOPC) and cholesterol (CHL1) following the recommended ratio of 0.55:0.15:0.30, respectively75. The GR89,696-bound KOR complex with Gz
contained 330 DPPC, 90 DOPC and 180 CHL1 lipids, 64,400 water molecules, and 178 sodium and 176 chloride ions. The GR89,696-bound KOR complex with Gg contained 330 DPPC, 90 DOPC and 180 CHL1
lipids, 64,227 water molecules, and 184 sodium and 175 chloride ions. The momSalB-bound KOR complex with Gi1 contained 220 DPPC, 60 DOPC and 120 CHL1 lipids, 43,172 water molecules, 124
sodium and 116 chloride ions. The momSalB-bound KOR complex with GoA contained 220 DPPC, 60 DOPC and 120 CHL1 lipids, 41,663 water molecules, and 126 sodium and 113 chloride ions. All of the
systems were first processed for 50,000 steps of initial energy minimizations, then 60 ns of equilibration, followed by production runs of up to 750 ns for the KOR–Gg based system and 550
ns for the rest (Gi1, GoA and Gz-bound KOR systems). The simulations were carried out on GPU clusters at the University of Southern California’s High-Performance Computing Center. The
temperature of 310 K and _v_-rescale thermostat algorithm were used during the production run76. The analyses of molecular dynamics trajectories were performed using the GROMACS software
package68. DATA STATISTICAL ANALYSIS For BRET2 and cAMP-inhibition assays, in the case of more than two groups, log-transformed EC50 values were first analysed using one-way ANOVA. If
significant, the Dunnett’s multiple-comparison test was used to compare each mutant with the wild-type one, and the Tukey’s multiple-comparison test was used to compare log-transformed EC50
values between each group. In the case of two groups, log-transformed EC50 values were analysed using unpaired two-tailed Student’s _t_-tests to compare each mutant with a wild-type
receptor. For the cell-surface expression studies, the optical density at 450 nm values of each mutant were normalized to the wild-type KOR receptor (normalized as 100%), and the resultant
values were then first analysed using one-way ANOVA. If significant, a Dunnett’s multiple-comparison test was used to compare each mutant with the wild-type receptor. For G-protein
expression studies, the Rluc values of each mutant were normalized to the wild-type G protein (normalized as 100%), and the resultant values were then first analysed using one-way ANOVA. If
significant, a Dunnett’s multiple-comparison test was used to compare each mutant with the wild-type G protein. For radioligand binding and GTP turnover assays, data were analysed using
unpaired two-tailed Student’s _t_-tests. In one-way ANOVA and unpaired two-tailed Student’s _t_-test analysis, the significance threshold was set at _α_ = 0.05. Asterisks denote statistical
significance; *_P_ < 0.05, **_P_ < 0.01, ***_P_ < 0.001; ****_P_ < 0.0001; NS represents not significant. REPORTING SUMMARY Further information on research design is available in
the Nature Portfolio Reporting Summary linked to this article. DATA AVAILABILITY The coordinate and cryo-EM map of KOR–Gi1–momSalB, KOR–GoA–momSalB, KOR–Gz–GR89,696 and KOR–Gg–GR89,696 have
been deposited at the PDB and Electron Microscopy Data Bank under accession codes 8DZP (EMD-27804), 8DZQ (EMD-27805), 8DZS (EMD-27807) and 8DZR (EMD-27806), respectively. All data
supporting the findings of this study are available within the Article and its Supplementary Information. REFERENCES * Chavkin, C. The therapeutic potential of κ-opioids for treatment of
pain and addiction. _Neuropsychopharmacology_ 36, 369–370 (2011). Article PubMed Google Scholar * Pfeiffer, A., Brantl, V., Herz, A. & Emrich, H. M. Psychotomimesis mediated by kappa
opiate receptors. _Science_ 233, 774–776 (1986). Article ADS CAS PubMed Google Scholar * Bruchas, M. R. & Roth, B. L. New technologies for elucidating opioid receptor function.
_Trends Pharmacol. Sci._ 37, 279–289 (2016). Article CAS PubMed PubMed Central Google Scholar * Che, T. & Roth, B. L. Structural insights accelerate the discovery of opioid
alternatives. _Annu. Rev. Biochem_. https://doi.org/10.1146/annurev-biochem-061620-044044 (2021). * Ivanina, T. et al. Gαi1 and Gαi3 differentially interact with, and regulate, the G
protein-activated K+ channel. _J. Biol. Chem._ 279, 17260–17268 (2004). Article CAS PubMed Google Scholar * Jeong, S. W. & Ikeda, S. R. G protein alpha subunit Gαz couples
neurotransmitter receptors to ion channels in sympathetic neurons. _Neuron_ 21, 1201–1212 (1998). Article CAS PubMed Google Scholar * Wong, G. T., Gannon, K. S. & Margolskee, R. F.
Transduction of bitter and sweet taste by gustducin. _Nature_ 381, 796–800 (1996). Article ADS CAS PubMed Google Scholar * Wall, M. J. et al. Selective activation of Galphaob by an
adenosine A1 receptor agonist elicits analgesia without cardiorespiratory depression. _Nat. Commun._ 13, 4150 (2022). Article ADS CAS PubMed PubMed Central Google Scholar * Beck, T.
C., Hapstack, M. A., Beck, K. R. & Dix, T. A. Therapeutic potential of kappa opioid agonists. _Pharmaceuticals_ https://doi.org/10.3390/ph12020095 (2019). * Peet, M. M. & Baker, L.
E. Salvinorin B derivatives, EOM-Sal B and MOM-Sal B, produce stimulus generalization in male Sprague–Dawley rats trained to discriminate salvinorin A. _Behav. Pharmacol._ 22, 450–457
(2011). Article CAS PubMed Google Scholar * Hayes, A. G. et al. A series of novel, highly potent and selective agonists for the κ-opioid receptor. _Br. J. Pharmacol._ 101, 944–948
(1990). Article CAS PubMed PubMed Central Google Scholar * Koehl, A. et al. Structure of the micro-opioid receptor–Gi protein complex. _Nature_ 558, 547–552 (2018). Article ADS CAS
PubMed PubMed Central Google Scholar * Che, T. et al. Structure of the nanobody-stabilized active state of the kappa opioid receptor. _Cell_ 172, 55–67 (2018). Article CAS PubMed
PubMed Central Google Scholar * Rasmussen, S. G. et al. Crystal structure of the β2 adrenergic receptor–Gs protein complex. _Nature_ 477, 549–555 (2011). Article ADS CAS PubMed PubMed
Central Google Scholar * Huang, W. J. et al. Structural insights into mu-opioid receptor activation. _Nature_ 524, 315–321 (2015). Article ADS CAS PubMed PubMed Central Google Scholar
* Rasmussen, S. G. et al. Structure of a nanobody-stabilized active state of the β2 adrenoceptor. _Nature_ 469, 175–180 (2011). Article ADS CAS PubMed PubMed Central Google Scholar *
Roth, B. L. et al. Salvinorin A: a potent naturally occurring nonnitrogenous kappa opioid selective agonist. _Proc. Natl Acad. Sci. USA_ 99, 11934–11939 (2002). Article ADS CAS PubMed
PubMed Central Google Scholar * Giroud, C. et al. Salvia divinorum: an hallucinogenic mint which might become a new recreational drug in Switzerland. _Forensic Sci. Int._ 112, 143–150
(2000). Article CAS PubMed Google Scholar * Kivell, B. M., Ewald, A. W. & Prisinzano, T. E. Salvinorin A analogs and other κ-opioid receptor compounds as treatments for cocaine
abuse. _Adv. Pharmacol._ 69, 481–511 (2014). Article CAS PubMed PubMed Central Google Scholar * Wang, Y. et al. 2-Methoxymethyl-salvinorin B is a potent kappa opioid receptor agonist
with longer lasting action in vivo than salvinorin A. _J. Pharmacol. Exp. Ther._ 324, 1073–1083 (2008). Article CAS PubMed Google Scholar * Butelman, E. R. et al. GR89,696: a potent
kappa-opioid agonist with subtype selectivity in rhesus monkeys. _J. Pharmacol. Exp. Ther._ 298, 1049–1059 (2001). CAS PubMed Google Scholar * Wang, Y. et al. Structures of the entire
human opioid receptor family. _Cell_ 186, 413–427 (2023). Article CAS PubMed Google Scholar * Ballesteros, J. A. & Weinstein, H. Integrated methods for the construction of
three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. _Methods Neurosci._ 25, 366–428 (1995). Article CAS Google Scholar *
Vardy, E. et al. A new DREADD facilitates the multiplexed chemogenetic interrogation of behavior. _Neuron_ 86, 936–946 (2015). Article CAS PubMed PubMed Central Google Scholar * Vardy,
E. et al. Chemotype-selective modes of action of κ-opioid receptor agonists. _J. Biol. Chem._ 288, 34470–34483 (2013). Article CAS PubMed PubMed Central Google Scholar * Wu, H. et al.
Structure of the human κ-opioid receptor in complex with JDTic. _Nature_ 485, 327–332 (2012). Article ADS CAS PubMed PubMed Central Google Scholar * Vortherms, T. A., Mosier, P. D.,
Westkaemper, R. B. & Roth, B. L. Differential helical orientations among related G protein-coupled receptors provide a novel mechanism for selectivity. _J. Biol. Chem._ 282, 3146–3156
(2007). Article CAS PubMed Google Scholar * Hauser, A. S. et al. GPCR activation mechanisms across classes and macro/microscales. _Nat. Struct. Mol. Biol._ 28, 879–888 (2021). Article
CAS PubMed PubMed Central Google Scholar * Zarzycka, B., Zaidi, S. A., Roth, B. L. & Katritch, V. Harnessing ion-binding sites for GPCR pharmacology. _Pharmacol. Rev._ 71, 571–595
(2019). Article CAS PubMed PubMed Central Google Scholar * Olsen, R. H. J. et al. TRUPATH, an open-source biosensor platform for interrogating the GPCR transducerome. _Nat. Chem. Biol._
16, 841–849 (2020). Article CAS PubMed PubMed Central Google Scholar * Mueller, K. L. et al. The receptors and coding logic for bitter taste. _Nature_ 434, 225–229 (2005). Article ADS
CAS PubMed Google Scholar * Che, T. et al. Nanobody-enabled monitoring of kappa opioid receptor states. _Nat. Commun._ 11, 1145 (2020). Article ADS CAS PubMed PubMed Central Google
Scholar * Flock, T. et al. Universal allosteric mechanism for Galpha activation by GPCRs. _Nature_ 524, 173–179 (2015). Article ADS CAS PubMed PubMed Central Google Scholar * Mafi,
A., Kim, S. K. & Goddard III, W. A. The mechanism for ligand activation of the GPCR-G protein complex. _Proc. Natl Acad. Sci. USA_ 119, e2110085119 (2022). Article CAS PubMed PubMed
Central Google Scholar * Rose, A. S. et al. Position of transmembrane helix 6 determines receptor G protein coupling specificity. _J. Am. Chem. Soc._ 136, 11244–11247 (2014). Article CAS
PubMed Google Scholar * Glukhova, A. et al. Rules of engagement: GPCRs and G proteins. _ACS Pharmacol. Transl. Sci._ 1, 73–83 (2018). Article CAS PubMed PubMed Central Google Scholar
* Flock, T. et al. Selectivity determinants of GPCR-G-protein binding. _Nature_ 545, 317–322 (2017). Article ADS CAS PubMed PubMed Central Google Scholar * Inoue, A. et al.
Illuminating G-protein-coupling selectivity of GPCRs. _Cell_ 177, 1933–1947 (2019). Article CAS PubMed PubMed Central Google Scholar * Garcia-Nafria, J., Nehme, R., Edwards, P. C. &
Tate, C. G. Cryo-EM structure of the serotonin 5-HT1B receptor coupled to heterotrimeric Go. _Nature_ 558, 620–623 (2018). Article ADS CAS PubMed PubMed Central Google Scholar * De
Lean, A., Stadel, J. M. & Lefkowitz, R. J. A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled beta-adrenergic receptor. _J. Biol.
Chem._ 255, 7108–7117 (1980). Article PubMed Google Scholar * Samama, P., Cotecchia, S., Costa, T. & Lefkowitz, R. J. A mutation-induced activated state of the β2-adrenergic receptor.
Extending the ternary complex model. _J. Biol. Chem._ 268, 4625–4636 (1993). Article CAS PubMed Google Scholar * Staus, D. P. et al. Allosteric nanobodies reveal the dynamic range and
diverse mechanisms of G-protein-coupled receptor activation. _Nature_ 535, 448–452 (2016). Article ADS CAS PubMed PubMed Central Google Scholar * Christopoulos, A. & Kenakin, T. G
protein-coupled receptor allosterism and complexing. _Pharmacol. Rev._ 54, 323–374 (2002). Article CAS PubMed Google Scholar * Dror, R. O. et al. Structural basis for nucleotide exchange
in heterotrimeric G proteins. _Science_ 348, 1361–1365 (2015). Article ADS CAS PubMed PubMed Central Google Scholar * Gregorio, G. G. et al. Single-molecule analysis of ligand
efficacy in β2AR–G-protein activation. _Nature_ 547, 68–73 (2017). Article ADS CAS PubMed PubMed Central Google Scholar * Chung, K. Y. et al. Conformational changes in the G protein Gs
induced by the β2 adrenergic receptor. _Nature_ 477, 611–615 (2011). Article ADS CAS PubMed PubMed Central Google Scholar * Liang, Y. L. et al. Dominant negative G proteins enhance
formation and purification of agonist-GPCR-G protein complexes for structure determination. _ACS Pharmacol. Transl. Sci._ 1, 12–20 (2018). Article CAS PubMed PubMed Central Google
Scholar * Gilman, A. G. G proteins: transducers of receptor-generated signals. _Annu. Rev. Biochem._ 56, 615–649 (1987). Article CAS PubMed Google Scholar * Seyedabadi, M., Gharghabi,
M., Gurevich, E. V. & Gurevich, V. V. Structural basis of GPCR coupling to distinct signal transducers: implications for biased signaling. _Trends Biochem. Sci._
https://doi.org/10.1016/j.tibs.2022.03.009 (2022). * Cong, X. et al. Molecular insights into the biased signaling mechanism of the mu-opioid receptor. _Mol. Cell_ 81, 4165–4175 (2021).
Article CAS PubMed PubMed Central Google Scholar * Suomivuori, C. M. et al. Molecular mechanism of biased signaling in a prototypical G protein-coupled receptor. _Science_ 367, 881–887
(2020). Article ADS CAS PubMed PubMed Central Google Scholar * Lane, J. R., May, L. T., Parton, R. G., Sexton, P. M. & Christopoulos, A. A kinetic view of GPCR allostery and biased
agonism. _Nat. Chem. Biol._ 13, 929–937 (2017). Article CAS PubMed Google Scholar * Grundmann, M. & Kostenis, E. Temporal bias: time-encoded dynamic GPCR signaling. _Trends
Pharmacol. Sci._ 38, 1110–1124 (2017). Article CAS PubMed Google Scholar * Wenzel-Seifert, K. & Seifert, R. Molecular analysis of β2-adrenoceptor coupling to Gs-, Gi-, and
Gq-proteins. _Mol. Pharmacol._ 58, 954–966 (2000). Article CAS PubMed Google Scholar * Wall, M. A. et al. The structure of the G protein heterotrimer Gi_α_1_β_1_γ_2. _Cell_ 83, 1047–1058
(1995). Article CAS PubMed Google Scholar * Maeda, S. et al. Development of an antibody fragment that stabilizes GPCR/G-protein complexes. _Nat. Commun._ 9, 3712 (2018). Article ADS
PubMed PubMed Central Google Scholar * Lee, D. Y. et al. Synthesis and in vitro pharmacological studies of new C(2) modified salvinorin A analogues. _Bioorg. Med. Chem. Lett._ 15,
3744–3747 (2005). Article CAS PubMed Google Scholar * Munro, T. A. et al. Standard protecting groups create potent and selective kappa opioids: salvinorin B alkoxymethyl ethers. _Bioorg.
Med. Chem._ 16, 1279–1286 (2008). Article CAS PubMed Google Scholar * Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. _J.
Struct. Biol._ 152, 36–51 (2005). Article PubMed Google Scholar * Peck, J. V., Fay, J. F. & Strauss, J. D. High-speed high-resolution data collection on a 200 keV cryo-TEM. _IUCrJ_ 9,
243–252 (2022). Article CAS PubMed PubMed Central Google Scholar * Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised
cryo-EM structure determination. _Nat. Methods_ 14, 290–296 (2017). Article CAS PubMed Google Scholar * Punjani, A., Zhang, H. & Fleet, D. J. Non-uniform refinement: adaptive
regularization improves single-particle cryo-EM reconstruction. _Nat. Methods_ 17, 1214–1221 (2020). Article CAS PubMed Google Scholar * Cao, C. et al. Structure, function and
pharmacology of human itch GPCRs. _Nature_ 600, 170–175 (2021). Article ADS CAS PubMed PubMed Central Google Scholar * Pettersen, E. F. et al. UCSF Chimera—a visualization system for
exploratory research and analysis. _J. Comput. Chem._ 25, 1605–1612 (2004). Article CAS PubMed Google Scholar * Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and
development of Coot. _Acta Crystallogr. D_ 66, 486–501 (2010). Article CAS PubMed PubMed Central Google Scholar * Adams, P. D. et al. PHENIX: a comprehensive Python-based system for
macromolecular structure solution. _Acta Crystallogr. D_ 66, 213–221 (2010). Article CAS PubMed PubMed Central Google Scholar * Chen, V. B. et al. MolProbity: all-atom structure
validation for macromolecular crystallography. _Acta Crystallogr. D_ 66, 12–21 (2010). Article CAS PubMed Google Scholar * Abraham, M. J. et al. GROMACS: high performance molecular
simulations through multi-level parallelism from laptops to supercomputers. _SoftwareX_ 1, 19–25 (2015). Article ADS Google Scholar * Jo, S., Kim, T., Iyer, V. G. & Im, W. CHARMM-GUI:
a web-based graphical user interface for CHARMM. _J. Comput. Chem._ 29, 1859–1865 (2008). Article CAS PubMed Google Scholar * Kim, S. et al. CHARMM-GUI ligand reader and modeler for
CHARMM force field generation of small molecules. _J. Comput. Chem._ 38, 1879–1886 (2017). Article CAS PubMed PubMed Central Google Scholar * Abagyan, R., Totrov, M. & Kuznetsov, D.
ICM—a new method for protein modeling and design: applications to docking and structure prediction from the distorted native conformation. _J. Comput. Chem._ 15, 488–506 (1994). Article
CAS Google Scholar * Xing, C. et al. Cryo-EM structure of the human cannabinoid receptor CB2–Gi signaling complex. _Cell_ 180, 645–654 (2020). Article CAS PubMed PubMed Central Google
Scholar * Arnautova, Y. A., Abagyan, R. A. & Totrov, M. Development of a new physics-based internal coordinate mechanics force field and its application to protein loop modeling.
_Proteins Struct. Funct. Bioinform._ 79, 477–498 (2011). Article CAS Google Scholar * Lomize, M. A., Pogozheva, I. D., Joo, H., Mosberg, H. I. & Lomize, A. L. OPM database and PPM web
server: resources for positioning of proteins in membranes. _Nucleic Acids Res._ 40, D370–D376 (2012). Article CAS PubMed Google Scholar * Leonard, A. N. & Lyman, E. Activation of
G-protein-coupled receptors is thermodynamically linked to lipid solvation. _Biophys. J._ 120, 1777–1787 (2021). Article ADS CAS PubMed PubMed Central Google Scholar * Bussi, G.,
Donadio, D. & Parrinello, M. Canonical sampling through velocity rescaling. _J. Chem. Phys._ https://doi.org/10.1063/1.2408420 (2007). * Krissinel, E. & Henrick, K. Inference of
macromolecular assemblies from crystalline state. _J. Mol. Biol._ 372, 774–797 (2007). Article CAS PubMed Google Scholar Download references ACKNOWLEDGEMENTS We thank J. Peck and J.
Strauss for technical assistance in this project; the staff at the Washington University Center for Cellular Imaging for sample screening; the staff of the NIDA Drug Supply Program for
providing the 3H-JDTic; X.-P. Huang and the members of the Psychoactive Drug Screening Program (PDSP) at UNC-Chapel Hill for the GPCRome screening analysis; and staff at the Center for
Advanced Research Computing (CARC) at the University of Southern California for providing computing resources that have contributed to the research results reported within this publication.
The work is supported by NIH grants R35GM143061 (to T.C.) and R01NS099341 (to P.Y.). The Titan X Pascal used for this research was donated to J.F.F. by NVIDIA. AUTHOR INFORMATION Author
notes * Jingying Zhang & Peng Yuan Present address: Department of Pharmacological Sciences, Icahn School of Medicine at Mount Sinai, New York, NY, USA * Jingying Zhang & Peng Yuan
Present address: Department of Neuroscience, Icahn School of Medicine at Mount Sinai, New York, NY, USA AUTHORS AND AFFILIATIONS * Department of Anesthesiology, Washington University in St
Louis, St Louis, MO, USA Jianming Han, Sarah M. Bernhard, Lei Zhao, Susruta Majumdar & Tao Che * Center for Clinical Pharmacology, University of Health Sciences and Pharmacy in St Louis
and Washington University School of Medicine, St Louis, MO, USA Jianming Han, Sarah M. Bernhard, Vipin A. Rangari, Susruta Majumdar & Tao Che * Department of Cell Biology and Physiology,
Washington University School of Medicine, St Louis, MO, USA Jingying Zhang & Peng Yuan * Center for the Investigation of Membrane Excitability Diseases, Washington University School of
Medicine, St Louis, MO, USA Jingying Zhang & Peng Yuan * Department of Quantitative and Computational Biology, University of Southern California, Los Angeles, CA, USA Antonina L.
Nazarova, Jordy Homing Lam & Vsevolod Katritch * Department of Chemistry, University of Southern California, Los Angeles, CA, USA Antonina L. Nazarova, Jordy Homing Lam & Vsevolod
Katritch * Center for New Technologies in Drug Discovery and Development, Bridge Institute, Michelson Center for Convergent Biosciences, University of Southern California, Los Angeles, CA,
USA Antonina L. Nazarova, Jordy Homing Lam & Vsevolod Katritch * Department of Pharmacology, University of North Carolina School of Medicine, Chapel Hill, NC, USA Brian E. Krumm *
Washington University Pain Center, Washington University in St Louis, St Louis, MO, USA Susruta Majumdar & Tao Che * Division of Chemical Biology and Medicinal Chemistry, Eshelman School
of Pharmacy, University of North Carolina, Chapel Hill, NC, USA David E. Nichols * Department of Biochemistry and Molecular Biology, University of Maryland Baltimore, Baltimore, MD, USA
Jonathan F. Fay Authors * Jianming Han View author publications You can also search for this author inPubMed Google Scholar * Jingying Zhang View author publications You can also search for
this author inPubMed Google Scholar * Antonina L. Nazarova View author publications You can also search for this author inPubMed Google Scholar * Sarah M. Bernhard View author publications
You can also search for this author inPubMed Google Scholar * Brian E. Krumm View author publications You can also search for this author inPubMed Google Scholar * Lei Zhao View author
publications You can also search for this author inPubMed Google Scholar * Jordy Homing Lam View author publications You can also search for this author inPubMed Google Scholar * Vipin A.
Rangari View author publications You can also search for this author inPubMed Google Scholar * Susruta Majumdar View author publications You can also search for this author inPubMed Google
Scholar * David E. Nichols View author publications You can also search for this author inPubMed Google Scholar * Vsevolod Katritch View author publications You can also search for this
author inPubMed Google Scholar * Peng Yuan View author publications You can also search for this author inPubMed Google Scholar * Jonathan F. Fay View author publications You can also search
for this author inPubMed Google Scholar * Tao Che View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS J.H. prepared the four KOR–G protein
complexes, performed the functional validation assays and prepared the manuscript. J.Z. helped with the cryo-EM data processing. A.L.N. and J.H.L. performed the molecular dynamics
simulations. S.M.B. helped with construct optimization, sample preparation and saturation binding experiments. B.E.K. provided the expression plasmid for Gg. L.Z. helped with protein
expression. V.A.R. and S.M. helped to collect the NMR spectrum of momSalB. D.E.N. synthesized the compound momSalB. V.K. supervised the molecular dynamics simulations and manuscript
discussion. P.Y. helped with the map discussion. J.F.F. prepared grids, collected the cryo-EM data and resolved the cryo-EM density map. P.Y., J.F.F. and T.C. prepared the manuscript and
supervised the project. CORRESPONDING AUTHORS Correspondence to Jonathan F. Fay or Tao Che. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. PEER REVIEW
PEER REVIEW INFORMATION _Nature_ thanks Graham Ladds and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. EXTENDED DATA FIGURES AND
TABLES EXTENDED DATA FIG. 1 COMPARISON OF KOR-GI1 COMPLEX STRUCTURE WITH MOR-GI1 AND ACTIVE-STATE KOR-NB39 STRUCTURES. A. Opioid receptors primarily couple to Gi/o family subtypes upon
activation. B. Sequence alignment (αN and α5 helices) of Gαi/o family subtypes. The percentage represents the sequence similarity related to Gi1 (set as 100%). C. Overall alignment of
KOR-Gi1 and MOR-Gi1 structures. The two structures are globally similar to each other, including TM6 of the receptor and α5 helix of the Gα subunit, but different in the αN helix of Gα. D.
The Gi1-bound KOR differs from the Nb39-bound KOR in the degrees of TM6 outward movement (by 2 Å). E. Both Gi1 and Nb39 act as positive allosteric modulators of KOR, but differentially
increase the binding affinity of KOR agonists. Data are grouped data ± s.e.m. from n = 3 biological replicates. Full quantitative parameters from this experiment are listed in Supplementary
Table 8. EXTENDED DATA FIG. 2 THE BINDING PHARMACOLOGY OF MOMSALB AND GR89,696. A. GPCRome screening at 320 GPCRs that measures agonist activity of tested ligands shows that momSalB and
GR89,696 are selective at KOR. B. The binding poses of momSalB and GR89,696 show that they adopt different planes in the orthosteric pocket. C. A cartoon model of G protein-mediated cAMP
reporter assays. The Gαi/o here represents all subtypes expressed in the cells. D. Mutagenesis screening of key binding-pocket residues using G protein-mediated cAMP reporter assay. Data are
grouped data ± s.e.m. of n = 3 biological replicates. Full quantitative parameters from this experiment are listed in Supplementary Table 2. E. Measurement of cell surface expression of KOR
binding pocket mutants by ELISA. In general, these mutants maintained robust cell surface expression. Although some mutations significantly altered surface expression compared to the wild
type, the increased or decreased expression appeared to minimally affect the potency in agonist-mediated cAMP inhibition. Bar-graphs are OD450 ± s.e.m. from n = 3 biological replicates.
Statistical significance for each mutant is compared in a one-way analysis of variance (ANOVA) with Dunnett’s multiple comparisons test to the wild type (* = _p_ < 0.05, ** = _p_ <
0.01, *** = _p_ < 0.001, **** = _p_ < 0.0001, “ns” represents no significance; V108A: _p_ = 0.3919, W124A: _p_ = 0.0893, V134A: _p_ = 0.0308, M142A: _p_ = 0.0398, V230A: _p_ = 0.0004,
H291A: _p_ = 0.0002, Y320L: _p_ = 0.0008). Signalling curves are grouped data ± s.e.m. of n = 3 biological replicates. Full quantitative parameters from this experiment are listed in
Supplementary Table 9. EXTENDED DATA FIG. 3 MOLECULAR DETERMINANTS OF MOMSALB AGONISM. A. The positive effect of additional D1383.32N mutation on the momSalB-mediated cAMP inhibition through
KOR. The additional D1383.32N mutation does not rescue U50,488 or GR89,696-mediated cAMP inhibition. Data are grouped data ± s.e.m. from n = 3 biological replicates. Full quantitative
parameters from this experiment are listed in Supplementary Table 10. B. Effects of mutations in the hydrophobic pocket on the binding affinity of momSalB. Data are grouped data ± s.e.m.
from n = 3 independent biological replicates. Full quantitative parameters from this experiment are listed in Supplementary Table 11. C and D. Chemical structures of SalB, momSalB, and
EOM-SalB. Differences are highlighted by red colour. The agonist activity of each analogue is shown in the parentheses. Data for EOM-SalB was taken from ref. 19. Binding poses of SalB and
EOM-SalB at KOR were revealed by molecular docking performed in the Schrodinger Maestro v12.9. The three ligands occupy a similar binding pocket with different extents toward the hydrophobic
pocket. EXTENDED DATA FIG. 4 COMPARISON OF INTERFACE DETAILS AND AREAS BETWEEN KOR AND EACH G PROTEIN SUBTYPES. Specific interactions between the receptor and individual Gα subunits, Gi1
(A), GoA (B), Gz (C) and Gg (D). (Cartoon) Key residues from the intracellular side of KOR, and residues in the αN and α5 helix were mapped out. The H-bond or salt-bridge interactions are
shown as red dashed lines. The closest distances between the intracellular KOR residues and the Gα residues were labelled. The distance cutoff is 4 Å. The interface area was calculated by
the online server PDBePISA77. EXTENDED DATA FIG. 5 THE EFFECT OF KOR-G-PROTEIN INTERFACE RESIDUES ON THE G PROTEIN COUPLING. A. Mutagenesis screening of KOR-G protein interface (KOR side)
residues by cAMP inhibition assays. Data are grouped data ± s.e.m. from n = 3 biological replicates. B. Mutagenesis analysis of intracellular KOR residues by cAMP inhibition assays. Data are
mean LogEC50 ± s.e.m. from n = 3 biological replicates. Statistical significance for each mutant is compared in a one-way analysis of variance (ANOVA) with Dunnett’s multiple comparisons
test to the wild type (* = _p_ < 0.05, ** = _p_ < 0.01, *** = _p_ < 0.001, **** = _p_ < 0.0001, “ns” represents no significance, GR89,696: V160A: _p_ = 0.0001, P163A: _p_ =
0.0020, R252A: _p_ = 0.0014, L253A: _p_ = 0.0003, R257A: _p_ = 0.0003, R271A: _p_ = 0.0004; momSalB: L167A: _p_ = 0.0003, R257A: _p_ = 0.1514, R271A: _p_ = 0.0004, I272A: _p_ = 0.0002). Full
quantitative parameters from this experiment are listed in Supplementary Table 12. C. Measurement of cell surface expression of KOR-G protein interface mutants by ELISA. The BRET results
suggest a minor effect with different concentrations of KOR plasmids. Bar-graphs are OD450 ± s.e.m. from n = 3 independent biological replicates. Statistical significance for each mutant is
compared in a one-way analysis of variance (ANOVA) with the Dunnett’s multiple comparisons test to the wild type (* = _p_ < 0.05, ** = _p_<0.01, *** = _p_ < 0.001, **** = _p_ <
0.0001, “ns” represents no significance; V160A: _p_ = 0.2732, P163A: _p_ = 0.9992, R252A: _p_ = 0.9998, L253A: _p_ = 0.8943, R257A: _p_ = 0.9997, N336A: _p_ = 0.9992). Signalling curves are
grouped data ± s.e.m. of n = 3 biological replicates. Full quantitative parameters from this experiment are listed in Supplementary Table 13. D. The KOR-R156A mediated decrease of efficacy
was confirmed by the BRET2-based kinetic measurement. Data are net BRET ± s.e.m. from n = 3 independent biological replicates. EXTENDED DATA FIG. 6 MOLECULAR DYNAMICS (MD) SIMULATIONS REVEAL
DISTANCE TRACES OF INTERACTIONS BETWEEN THE KOR RESIDUES (R1563.50) WITH GΑ PROTEIN RESIDUES. MD simulations revealed the distance traces and frequency between KOR-R1563.50 and
L353/354H5.25 in the α5 of each Gα subunit. The position of KOR-R1563.50 is also supported by the stable interaction with KOR-Y2465.58. Five independent simulations of KOR-G complex (coral,
orange, green, cyan, blue) are shown, spanning 0.55-0.75 μs of cumulative time per system, with the sampling rate of 10 frames per ns, solid lines and same-colour shadows representing moving
average values and one standard deviation respectively from 50 frames in all cases. EXTENDED DATA FIG. 7 MOLECULAR DYNAMICS (MD) SIMULATIONS REVEAL DISTANCE TRACES OF INTERACTIONS BETWEEN
THE KOR RESIDUES (N3368.49) WITH GΑ PROTEIN RESIDUES. The closest distances of polar residues N3368.49 in KOR with residues in Gi1, GoA, Gz, or Gg are shown. Distance histograms for each
plot are also shown in parallel. Five independent simulations of KOR-G complex (coral, orange, green, cyan, blue) are shown, spanning 0.55 μs (KOR-Gi1, GoA, Gz) and 0.75 μs (KOR-Gg) of
cumulative time per system, with the sampling rate of 10 frames per ns, solid lines and same-colour shadows representing moving average values and one standard deviation respectively from 50
frames in all cases. EXTENDED DATA FIG. 8 THE ROLE OF G PROTEIN INTERFACE RESIDUES ON KOR-G PROTEIN COUPLING. A. The nonconserved residues in the G protein α5 and αN helices. B-E. The
effects of individual mutations in each G protein subtype were screened using the BRET2 KOR-G protein assays. Data are grouped data ± s.e.m. from n = 3 biological replicates. Full
quantitative parameters from this experiment are listed in Supplementary Table 14. F. The expression levels of G protein mutants were quantified based on the luminescence counts from the
Gα-Rluc excitation. Data are BRET ratio ± s.e.m. from n = 3 biological replicates. Statistical significance for each mutant is compared in a one-way analysis of variance (ANOVA) with
Dunnett’s multiple comparisons test to the wild type (* = _p_ < 0.05, ** = _p_<0.01, *** = _p_ < 0.001, **** = _p_ < 0.0001, “ns” represents no significance; Gi1-K345A: _p_ =
0.0015, Gi1-C351A: _p_ = 0.7984; GoA-A345Q: _p_ = 0.0210, GoA-R349A: _p_ = 0.0019, GoA-G350A: _p_ = 0.0284, GoA-C351A: _p_ = 0.1102, GoA-Y354A: _p_ = 0.6955; Gz-R31A: _p_ > 0.9999,
Gz-Y351A: _p_ = 0.9947, Gz-I352A: _p_ = 0.5286, Gz-C355A: _p_ = 0.0161; Gg-K345A: _p_ = 0.5849, Gg-E346A: _p_ = 0.0050, Gg-D350A: _p_ = 0.9936, Gg-C351A: _p_ = 0.9808, Gg-F354A: _p_ =
0.0091). EXTENDED DATA FIG. 9 COMPARISON OF KOR-COUPLED GΑ SUBUNIT WITH OTHER G PROTEIN FAMILIES. A. The KOR-GoA displays a different conformation from the 5-HT1BR-miniGo structure,
including both TM6 of the receptor and α5 helix of the Gα subunit. B. Comparison of receptor-G protein interface among Gs, Gi1, Gq bound complexes. The TM6 of the receptor, αN and α5 helices
of the Gα subunits adopt different conformations. C. The receptor-Gα interface of β2AR-Gαs and 5-HT2AR-Gαq. The dashed lines represent the closest distance between the intracellular
receptor residues and the Gα residues. The distance cutoff is 4 Å. The interface area shown in the brackets was calculated by the online server PDBePISA. EXTENDED DATA FIG. 10 THE PRESENCE
OF GDP OR GTP REDUCES THE ALLOSTERIC POTENTIATION OF GOA, GZ, OR GG, RESPECTIVELY. A. Comparison of functional activity between wild-type G proteins and engineered G proteins by GTP turnover
assay. Data are luminescence ± s.e.m. of n = 3 biological replicates with each performed in triplicate. Significance analyses were performed using unpaired two-tailed student’s t-test to
compare each point in engineered G protein (G protein-DN) to the corresponding point in wild-type G proteins. (* = _p_ < 0.05, ** = _p_ < 0.01, *** = _p_ < 0.001, **** = _p_ <
0.0001). B. Measurement of allosteric potentiation of engineered G proteins on KOR agonist binding. The concentration of 3H-U69,593 was 8 nM, and the G protein was 250 nM. DN, dominant
negative. Data are cpm ± s.e.m. from n = 4 biological replicates. Significance analyses were performed using the unpaired two-tailed student’s t-test to compare each group to the KOR-only
group (* = _p_ < 0.05, ** = _p_ < 0.01, *** = _p_ < 0.001, **** = _p_ < 0.0001, “ns” represents no significance; (KOR+GoAwt) vs KOR: _p_ = 0.0002, (KOR+Ggwt) vs KOR: _p_ =
0.2469, (KOR+Gi1DN) vs KOR: _p_ = 0.0012, (KOR+GoADN) vs KOR: _p_ = 0.0007, (KOR+GzDN) vs KOR: _p_ = 0.0029, (KOR+GgDN) vs KOR: _p_ = 0.0719). C. The saturation binding assays were conducted
with or without 50 μM GDP/GTP. Data are Bmax ± s.e.m. from n = 3 biological replicates with each performed in duplicate. Statistical significance analyses between groups (with GTP/GDP and
without GTP/GDP) are compared in the unpaired two-tailed student’s t-test (* = _p_ < 0.05, ** = _p_ < 0.01, *** = _p_ < 0.001, **** = _p_ < 0.0001; (KOR+GoA) vs (KOR+GoA+GTP):
_p_ = 0.0001, (KOR+Gg) vs (KOR+Gg+GDP): _p_ = 0.0008, (KOR+Gg) vs (KOR+Gg+GTP): _p_ = 0.0010). Full quantitative parameters from this experiment are listed in Supplementary Table 15.
EXTENDED DATA FIG. 11 COMPARISON OF UNCOUPLED GDP-BOUND GI1 AND KOR-COUPLED NUCLEOTIDE-FREE GI1. A. Comparison of GDP-bound Gi1 with KOR bound Gi1, GoA, Gz, and Gg. B. Conformational
translocation of α1 and α5 helices upon GPCR engagement. C. α5 helix shows an 8.6 Å upward movement into the KOR intracellular core. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION
Supplementary Methods (allosteric IC50 shift model equation), Supplementary Figs. 1–6 and Supplementary Tables 1–17. REPORTING SUMMARY PEER REVIEW FILE RIGHTS AND PERMISSIONS OPEN ACCESS
This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as
long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third
party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the
article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright
holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Han, J., Zhang, J., Nazarova, A.L.
_et al._ Ligand and G-protein selectivity in the κ-opioid receptor. _Nature_ 617, 417–425 (2023). https://doi.org/10.1038/s41586-023-06030-7 Download citation * Received: 14 August 2022 *
Accepted: 29 March 2023 * Published: 03 May 2023 * Issue Date: 11 May 2023 * DOI: https://doi.org/10.1038/s41586-023-06030-7 SHARE THIS ARTICLE Anyone you share the following link with will
be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt
content-sharing initiative