Play all audios:
ABSTRACT Selfish genetic elements contribute to hybrid incompatibility and bias or ‘drive’ their own transmission1,2. Chromosomal drive typically functions in asymmetric female meiosis,
whereas gene drive is normally post-meiotic and typically found in males. Here, using single-molecule and single-pollen genome sequencing, we describe _Teosinte Pollen Drive_, an instance of
gene drive in hybrids between maize (_Zea mays_ ssp. _mays_) and teosinte _mexicana_ (_Z. mays_ ssp. _mexicana_) that depends on RNA interference (RNAi). 22-nucleotide small RNAs from a
non-coding RNA hairpin in _mexicana_ depend on _Dicer-like 2_ (_Dcl2_) and target _Teosinte Drive Responder 1_ (_Tdr1_), which encodes a lipase required for pollen viability. _Dcl2_, _Tdr1_
and the hairpin are in tight pseudolinkage on chromosome 5, but only when transmitted through the male. Introgression of _mexicana_ into early cultivated maize is thought to have been
critical to its geographical dispersal throughout the Americas3, and a tightly linked inversion in _mexicana_ spans a major domestication sweep in modern maize4. A survey of maize
traditional varieties and sympatric populations of teosinte _mexicana_ reveals correlated patterns of admixture among unlinked genes required for RNAi on at least four chromosomes that are
also subject to gene drive in pollen from synthetic hybrids. _Teosinte Pollen Drive_ probably had a major role in maize domestication and diversification, and offers an explanation for the
widespread abundance of ‘self’ small RNAs in the germ lines of plants and animals. SIMILAR CONTENT BEING VIEWED BY OTHERS TWO COMPLEMENTARY GENES IN A PRESENCE-ABSENCE VARIATION CONTRIBUTE
TO _INDICA-JAPONICA_ REPRODUCTIVE ISOLATION IN RICE Article Open access 28 July 2023 PERICENTROMERIC RECOMBINATION SUPPRESSION AND THE ‘LARGE X EFFECT’ IN PLANTS Article Open access 07
December 2023 THREE TYPES OF GENES UNDERLYING THE _GAMETOPHYTE FACTOR1_ LOCUS CAUSE UNILATERAL CROSS INCOMPATIBILITY IN MAIZE Article Open access 03 August 2022 MAIN The introduction of
novel genetic variation through hybridization is an important evolutionary catalyst5, as adaptive introgression in hybrid individuals can increase fitness under new environmental conditions
and lead to geographical expansion and diversification6. Modern maize, for example, was first domesticated from a close relative of _Z. mays_ ssp. _parviglumis_ (teosinte _parviglumis_) in
the lowlands of southwest Mexico approximately 9000 bp, but admixture from a second teosinte, _Z. mays_ ssp. _mexicana_, 4,000 years later, appears to have catalysed rapid expansion across
the Americas3. The combination of divergent genomes, however, can also result in hybrid sterility, inviability and necrosis7,8,9. The Bateson–Dobzhansky–Muller (BDM) model accounts for such
scenarios, via the interaction of deleterious mutations in distinct populations and at least some of these incompatibilities stem from intragenomic conflict triggered by selfish genetic
elements2,10. Meiotic drive depends on selfish elements that actively manipulate reproductive development to facilitate their own preferential transmission11. Chromosomal drive refers to the
manipulation of chromosome segregation during asymmetric female meiosis, as centromeres, heterochromatic knobs and telomeres exert mechanical advantages that favour their inclusion in the
egg cell1,12,13,14. Examples include _Abnormal 10_ (_Ab10_) in both maize and teosinte populations15,16. Conversely, gene drive occurs preferentially in males and is achieved via disruption
of post-meiotic reproductive development resulting in segregation distortion17,18. These systems tend to occur in sperm or haploid spores and involve toxin–antidote (or distorter–responder)
pairs in close genetic linkage. Gametes that do not inherit the drive locus are selectively killed, resulting in overrepresentation of the driver11. The mouse _t_-complex19,20, _Drosophila
Segregation Distorter_ (_SD_) complex21,22 and _Schizosaccharomyces pombe/kombucha wtf_ spore killers23,24 are all autosomal drivers that selectively kill competing wild-type gametes in
heterozygotes. Because selfish genetic elements often impose fitness and fertility penalties, tremendous selective pressure is placed on regions of the genome that can evolve suppressors25.
As a consequence, drive systems undergo recurrent cycles of suppression and counter-suppression; although drive is predicted to be widespread, most systems exist in a cryptic state, either
through suppression or fixation11,26. It is through hybridization with naive individuals that suppression is lost and drive is once again apparent27, reinforcing species barriers and
influencing patterns of introgression in hybrid individuals via genetic linkage28,29. Here we characterize a male-specific segregation distortion system in introgression lines between maize
(_Z. mays_ ssp. _Mays_) and teosinte _mexicana_ (_Z. mays_ ssp. _mexicana_), hereafter referred to as _Teosinte Pollen Drive_ (_TPD_). We implicate small interfering RNAs (siRNAs) from a
_mexicana-_specific long non-coding hairpin RNA in close genetic linkage with the centromere of chromosome 5 as the primary factor mediating pollen killing. Co-segregation of a genetically
linked hypomorphic (partially functional) _Dcl2_ allele suppresses this effect via the reduction of secondary 22-nucleotide (nt) siRNAs and is reinforced by a second unlinked antidote
(_Tpd2_) on chromosome 6. Survey sequencing of modern and traditional varieties of maize from Mexico and sympatric populations of teosinte implicate _TPD_ in patterns of _mexicana_
introgression, and in maize dispersal and domestication. _TPD_ IN MAIZE HYBRIDS Hybridization between maize and teosinte is subject to unilateral cross-incompatibility30,31, but pollination
of maize by _mexicana_ pollen is frequent32. Consistently, genome-wide assessments of introgression in sympatric collections have provided evidence for asymmetric gene flow from _mexicana_
to maize32,33. To further explore the reproductive consequences of hybridization, multiple sympatric collections of _mexicana_ were crossed to the Midwestern US dent inbred W22, resulting in
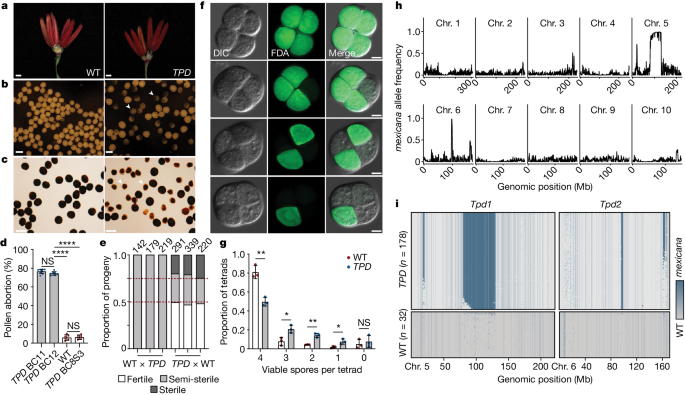
variable rates of pollen abortion that typically decreased in subsequent generations. However, a subset of late backcross (BC) lines (hereafter _TPD_) displayed an unusually consistent rate
of pollen abortion (75.5 ± 2.48%) relative to W22 (6.02 ± 2.95%; _P_ < 0.0001, Welch’s _t_-test) despite normal vegetative and reproductive development (Fig. 1a–c and Extended Data Fig.
1a). The pollen abortion phenotype was absent after three rounds of selfing in _TPD_ BC8S3 plants (6.40 ± 2.26%; _P_ < 0.0001, Welch’s _t_-test), suggesting that heterozygosity was
required (Fig. 1d). In reciprocal crosses, pollination of _TPD_ ears with W22 pollen resulted in the independent assortment of fertile, semi-sterile and fully male sterile progeny in a 2:1:1
ratio (Fig. 1e and Supplementary Table 1). These results indicated the presence of two unlinked loci responsible for pollen survival that were transmitted to all individuals in the next
generation, but only through pollen. Because this phenotype was observed only in heterozygotes, we reasoned that it stemmed from an incompatibility between the W22 genome and regions of
_mexicana_ introgression after meiosis, reminiscent of genic drivers that distort patterns of inheritance via selective gamete killing20,24. Consistently, meiotic progression in _TPD_ plants
was normal until the tetrad stage, following the separation of each haploid complement (Fig. 1f). This phenotype, although strictly post-meiotic, appeared to progress gradually, ultimately
resulting in arrested pollen grains with a heterogenous overall diameter and varying degrees of starch accumulation (Fig. 1c,g). Genetic mapping revealed that _brittle endosperm 1_ (_bt1_)
on chromosome 5 and _yellow endosperm 1_ (_y1_) on chromosome 6 were linked with the pollen abortion phenotype (Extended Data Fig. 1b,c). Backcrosses to _y1_;_bt1_ yielded 100% _Bt1_ kernels
instead of 50%, but only when _TPD_ was used as a pollen parent (Extended Data Fig. 1b). The frequency of white kernels (_y1_) was in agreement with recombination estimates (21–22%). This
bias was strongly indicative of gene drive resembling similar incompatibility systems in rice34, although we could not formally exclude other forms of incompatibility that also result in
segregation distortion. To exclude such possibilities, we sequenced the genomes of two homozygous _TPD_ lines (BC8S3 and BC5S2) to define 408,031 high-confidence single-nucleotide
polymorphisms (SNPs) corresponding to regions of _mexicana_ introgression. Next, we sequenced the genomes of individual surviving pollen grains from _TPD_ plants, rationalizing that if
segregation distortion was occurring in pollen, the causative regions would be overrepresented. We found that several intervals occurred at much higher frequencies than expected after eight
backcrosses (Fig. 1h). Of note, introgression intervals on chromosomes 5 and 6 were consistently observed in all surviving pollen (Fig. 1i), strongly indicative of post-meiotic gene drive.
We designated these loci as _Tpd1_ and _Tpd2_, respectively. A _DICER-LIKE 2_ TOXIN–ANTIDOTE COMPLEX To determine the relative contributions of _Tpd1_ and _Tpd2_ to pollen abortion and
survival, we separated the components by maternal transmission into fertile, semi-sterile (‘drive’) and fully sterile classes (Fig. 2a). Each progeny class had distinct rates of pollen
abortion (Fig. 2b) and showed significant differences in flowering time (Fig. 2c). Fertile segregants were phenotypically wild type and showed no transmission defects, whereas drive plants
recapitulated the canonical _TPD_ pollen abortion phenotype. By contrast, male reproductive development in sterile plants was developmentally retarded, displaying severely delayed anthesis
and reduced overall shed (Fig. 2a,c). Consequently, crosses performed with this pollen showed minimal seed set and often failed entirely. We collected pools of plants from the fertile and
sterile phenotypic classes (Fig. 2d) for bulk segregant analysis, and found that _Tpd1_ was differentially enriched in sterile plants, whereas _Tpd2_ was enriched in fertile plants (Fig.
2e). This indicated that _Tpd1_ alone was sufficient to ‘poison’ the male germ line and that this most likely occurred pre-meiotically, as only a single copy of _Tpd1_ was required. Genetic
mapping placed _Tpd1_ in a large interval surrounding the centromere of chromosome 5, whereas _Tpd2_ was placed in a 1.5-Mb interval on chromosome 6L (Extended Data Fig. 1c,d). The unusual
transmission of _TPD_ led us to liken it to previously described selfish genetic elements that operate via post-meiotic gamete killing20,22,24. These systems generally encode a toxin (or
distorter) that acts in _trans_ to disrupt proper reproductive development. Only gametes containing a cell-autonomous antidote (or resistant responder allele) can suppress these effects in a
gametophytic manner. Although the toxin was clearly encoded by _Tpd1_, the _TPD_ system was unusual in that it featured a genetically unlinked antidote, namely, _Tpd2_. However, the absence
of _tpd1;Tpd2_ recombinants in the progeny of W22 × _TPD_ crosses argued that _Tpd2_ alone was insufficient for suppression of pollen abortion (Fig. 2d and Supplementary Table 2). We
reasoned that this might reflect the additional requirement for another antidote, linked to _Tpd1_, that could explain the observed rate of pollen abortion (approximately 75%). Linked
modifiers in drive systems are common and generally ascribed to the co-evolutionary struggle between distorters and rapidly accumulating suppressors11,22. SNP genotyping of the two
homozygous lines identified 13 _mexicana_ introgression intervals, 7 of which were shared between backcross generations (Extended Data Fig. 2a). As predicted from the single-pollen
sequencing data, the highest regions of SNP density were present on chromosome 5 (_Tpd1_) and chromosome 6 (_Tpd2_), coinciding with _Bt1_ and close to _Y1_, respectively (Extended Data Fig.
2a). However, other regions strongly overrepresented in homozygous progeny were only partially overrepresented in _TPD_ pollen, including additional peaks on chromosomes 5S, 6S and 6L
(Extended Data Fig. 2b). This probably reflected the presence of recombinant pollen grains that competed poorly during pollination. To determine gene content in these and other introgression
intervals, we performed de novo genome assembly from homozygous _Tpd1;Tpd2_ BC8S3 seedlings (see Methods; Supplementary Table 3) with fully scaffolded _mexicana_ introgression intervals on
chromosomes 5 and 6 (Fig. 2f,g). We noted the presence of a 1.9-Mb _mexicana_ introgression interval on chromosome 5S linked to the _Tpd1_ haplotype and strongly overrepresented in both our
bulk sequencing and single-pollen grain data (Figs. 1h,i and 2f). Within this interval, we identified ten genes with expression in pollen, one of which, _Dcl2_, had excess nonsynonymous
substitutions within conserved domains (Fig. 3a), suggesting the possibility of adaptive evolutionary change35. Absolute genetic linkage (_n_ = 214) between this locus (hereafter _dcl2__T_)
and _Tpd1_ was conditioned on passage through the male germ line from heterozygous _TPD_ plants, whereas recombination between _dcl2__T_ and _Tpd1_ occurred at the expected frequency
(approximately 12%) when crossed as female (Fig. 3b). This was very strong evidence for a linked antidote and probably explained the maintenance of this interval across 13 backcross
generations. _Dcl2_ encodes a Dicer-like protein responsible for the production of 22-nt siRNAs from hairpins, as well as secondary small RNAs from double-stranded RNA templates produced by
the coordinated action of RNA-DEPENDENT RNA POLYMERASE 6 (RDR6) and SUPPRESSOR OF GENE SILENCING 3 (SGS3)36. In _Arabidopsis thaliana_, DCL2 function is superseded by DCL4 and endogenous
levels of 22-nt siRNAs are low37. However, DCL2 can fulfill roles in silencing and antiviral immunity when DCL4 function is lost37,38, sometimes resulting in ‘toxic’ pleiotropic defects
associated with gene targets of 22-nt siRNAs37,39,40. These observations stem from the unique biological properties of 22-nt siRNAs, which are responsible for propagation of systemic
silencing signals that move between cells41 and transitive amplification of silencing in both _cis_ and _trans_42. In _dcl2__T_, nonsynonymous changes were clustered within the DExD/H RNA
helicase domain of Dicer (Fig. 3a), which has been shown to alter substrate preference and processing efficiency of double-stranded RNA, but not hairpin RNA, in both plants and
invertebrates43,44,45. To explore the role of 22-nt siRNAs in the _TPD_ phenotype, we tested mutants in 22-nt siRNA biogenesis for their ability to act as antidotes. We isolated maternal
_dcl2__T_ recombinants and compared them with the _dcl2-mu1_ allele in the W22 inbred background, which has a _Mu_ transposon insertion in the 5′ untranslated region, 200 bp upstream of the
start codon. In _dcl2__T_/_dcl2-mu1_ _Tpd1_, pollen abortion was partially suppressed, whereas pollen from _dcl2-mu1/dcl2-mu1_ _Tpd1_ plants were almost fully viable (Fig. 3c). This meant
that stacking over the _dcl2__T_ allele had a synergistic effect, strongly supporting its role as a partial antidote, and indicating that the sporophytic production of 22-nt siRNAs in
diploid meiotic cells was responsible for the _TPD_ phenotype. To test the idea that 22-nt siRNAs might be responsible for _TPD_, we sequenced pollen small RNAs from _TPD_ and wild-type
siblings and found that although small RNA composition was similar overall, the _Tpd1_ haplotype triggered a strong, 22-nt-specific response (Fig. 3d). Consistent with these 22-nt small RNAs
being responsible for the _TPD_ phenotype, we observed almost complete rescue of sterility in _dcl2-mu1/dcl2-mu1_ _Tpd1/_ + pollen parents (Fig. 3e). Several other introgression intervals
observed in one or the other backcross individual also included genes encoding components of the small RNA biogenesis pathway, including _ago1a_, _ago1b_ and _rgd1_, the homologue of SGS3
(Fig. 3a and Extended Data Fig. 2a). These intervals were also observed in single-pollen grain sequencing along with _dcl2__T_ (Extended Data Fig. 2b). To determine whether these genes were
also capable of acting as an antidote, we crossed mutants in _rgd1_ to _TPD_ plants. Segregation of _rgd1_ in the germ line of heterozygotes resulted in close to 50% viable pollen (Extended
Data Fig. 2c), suggesting that it functions as a cell-autonomous gametophytic suppressor in a manner similar to _Tpd2_. We concluded that mutants in primary 22-nt small RNA synthesis
(_dcl2-mu1_) blocked production of the toxin, whereas mutants in secondary 22-nt small RNA synthesis (_dcl2__T_ and _rgd1_), and potentially in small RNA function (_ago1a_ and _ago1b_),
acted as antidotes. 22-NT SMALL RNAS TARGET A POLLEN LIPASE To identify the origin and the targets of DCL2-dependent small RNAs, we performed small RNA sequencing from wild-type, _dcl2__T_
and _dcl2-mu1_ plants. Analysis revealed that 22-nt siRNAs were the dominant species in wild-type pollen (Extended Data Fig. 3a,b) and defined 804 high-confidence 22-nt siRNA pollen-specific
clusters (log2 fold change ≥ 2, false discovery rate (FDR) ≤ 0.01; Supplementary Table 4). As expected, these clusters depended on _Dcl2_ (_P_ < 0.0001, determined by analysis of
variance (ANOVA)) and there were even fewer 22-nt siRNAs in _dcl2-mu1_ than in _dcl2__T_ (Extended Data Fig. 3c). Over half (54.6%) of all pollen-specific 22-nt species were derived from
endogenous hairpin precursors (hpRNAs; Extended Data Fig. 3d,e,g). Hairpin short interfering RNAs (hp-siRNAs) were disproportionately 22 nt long, derived from a single strand (Extended Data
Fig. 4a,b) with high thermodynamic stability (Extended Data Fig. 4c,d). On the basis of these criteria (and a minimum expression cut-off), we identified 28 hp-siRNA-producing loci in the
genome, with at least one hairpin on every chromosome except chromosome 4 (average 2.1 ± 1.3 per chromosome). hp-siRNAs can serve as a powerful means to silence transposons46, and 22-nt
siRNAs targeting _Gypsy_ and _Copia_ LTR retrotransposons were abundant in pollen, as were those targeting _Mutator_ and _CACTA_ elements (Extended Data Fig. 3d). We also found evidence for
pollen-specific silencing of at least 30 protein-coding genes (Extended Data Fig. 3d,f,g). Germline specificity is a common feature in _SD_ systems, as such factors can avoid the
evolutionary conflicts imposed by pleiotropic fitness defects in the diploid stage of the life cycle47. In _TPD_ pollen, we observed the accumulation of 158 ectopic 22-nt siRNA clusters
across the genome (log2 fold change ≥ 2, FDR ≤ 0.01; Supplementary Table 5), and a general upregulation of genes associated with 22-nt siRNA biogenesis and function (Extended Data Fig. 5a).
Nearly 60% of all ectopic 22-nt siRNAs in _TPD_ pollen targeted transposable elements of the _P Instability Factor_ (PIF)/Harbinger superfamily (Extended Data Fig. 5b), whose expression was
_TPD_ specific (Extended Data Fig. 5c–e). This superfamily is also activated following intraspecific hybridization and anther culture in rice48. However, a subset of protein-coding genes was
also targeted in _TPD_ pollen specifically (Extended Data Fig. 5b). Given that a reduction in 22-nt siRNAs suppressed the _TPD_ phenotype, we hypothesized that inappropriate silencing of
these genes might disrupt male reproductive development. In total, we identified four genes that gained ectopic 22-nt siRNAs in _TPD_ pollen, approximately 62% of which came from a single
gene (Zm00004b012122) that is also located on chromosome 5S (Extended Data Fig. 6a). Relative to other targets, this gene exhibited highly specific expression in pollen (Extended Data Fig.
6b,c). Zm00004b012122 encodes a GDSL triacylglycerol lipase/esterase, defined by a core catalytic sequence motif (GDSxxDxG), with roles in lipid metabolism, host immunity and reproductive
development49. In maize, both _male sterile 30_ (_ms30_) and _irregular pollen exine 1_ (_ipe1_) mutants disrupt genes encoding a GDSL lipase and are completely male sterile50,51. Similar
functions have been reported in rice52 and _Arabidopsis_53. DCL2-dependent 22-nt siRNAs engage primarily in translational repression of their targets54, and consistently all four target
genes had similar or higher levels of mRNA in _TPD_ pollen (Extended Data Fig. 6c). We raised antiserum to the GDSL lipase for immunoblotting, choosing a surface-exposed peptide located
between putative pro-peptide-processing sites reflecting endoplasmic reticulum localization51. The GDSL lipase protein accumulated strongly in both 5-mm anthers and mature pollen from
wild-type plants, but was absent from leaf and from _TPD_ anthers and pollen, supporting the conclusion that 22-nt siRNAs mediate translational repression (Extended Data Fig. 6d).
Furthermore, whole-protein extracts from _TPD_ anthers had reduced esterase activity, which was ameliorated in pollen containing _Tpd2_ but not in pollen with _Tpd1_ alone (Extended Data
Fig. 6e). Gene ontology analysis of genes upregulated in wild-type and _TPD_ pollen strongly supported translational suppression of the GDSL lipase as the primary cause of developmental
arrest and abortion of pollen in _TPD_ plants (Extended Data Fig. 6f,g). Finally, mRNA expression began post-meiotically at the 3-mm (tetrad) stage, peaking in 5-mm anthers and mature pollen
(Extended Data Fig. 7a). This expression pattern was conspicuously similar to the developmental window in which _TPD_ pollen abortion begins (Fig. 1f), suggesting that this gene might act
as a ‘responder’ to _Tpd1-_driven distortion. On the basis of all these observations, we defined Zm00004b012122 as the primary candidate for targeting by _Tpd1_ toxin activity, renaming it
_Teosinte drive responder 1 (Tdr1)_. HAIRPIN SIRNAS TRIGGER POLLEN ABORTION As ectopic silencing at protein-coding genes only occurred in the presence of the _Tpd1_ haplotype, we reasoned
that the distorter must generate small RNAs capable of triggering silencing in _trans_. In plants, microRNAs, secondary siRNAs and hp-siRNAs all have this capacity55. Processed small RNA
duplexes are loaded into ARGONAUTE (AGO) proteins, passenger strands are released and RNase H-like slicing activity is targeted by guide strand homology, as is translational repression56.
Silencing can be amplified via the coordinated action of RDR6 and SGS3 (ref. 42). RNase H-mediated slicing results in an exposed 5′-phosphate that allows for ligation of 3′ cleavage
products. Using an improved degradome sequencing technique in _TPD_ pollen, iPARE-seq (see Methods), we identified putative cleavage sites responsible for triggering silencing at the _Tdr1_
locus (Fig. 4a,b). We simultaneously searched for non-coding RNA within the _Tpd1_ haplotype that produced 22-nt sRNAs capable of triggering silencing. This approach yielded only one
candidate: a large hpRNA similar to those identified previously in wild-type pollen (Fig. 4c). This hairpin was uninterrupted in the _mexicana_-derived _Tpd1_ interval and produced high
levels of _TPD_-specific 22-nt hp-siRNAs (Fig. 4d,e). In the W22 genome, we identified two large transposon insertions that interrupted this locus, which produced no small RNA, indicating
that it was non-functional in maize, consistent with being responsible for _TPD_ (Fig. 4c). By comparison with centromere placement in other maize inbreds4, the hairpin is on the short arm
of chromosome 5, 5 Mb from the centromere. Target site prediction uncovered four abundant hp-siRNA species predicted to target the _Tdr1_ transcript in _trans_ (Fig. 4f) resembling
‘proto-microRNA’57. Three of these began with 5′-C, indicating loading into Ago5, and had iPARE-seq support, indicating cleavage of _Tdr1_ (Fig. 4g). However, the most abundant hp-siRNA,
_Tpd1-_siRNAb, was 22 nt in length and began with 5′-A, indicating loading into Ago2 (Fig. 4g). _Tpd1-_siRNAb has an asymmetric bulge predicted to enhance silencing transitivity and systemic
spread between cells58, and had only limited iPARE-seq support, indicating translational repression (Fig. 4b). To confirm that silencing of _Tdr1_ was responsible for the _TPD_ phenotype,
we generated two independent frameshift alleles within the catalytic domain using CRISPR–Cas9 (Fig. 4h). Homozygotes for _tdr1-1_ and _tdr1-2_ had identical male sterile phenotypes, with
extensive pollen abortion that phenocopied _Tpd1_ (Fig. 4i–k). Expression of the _Tpd1_ hairpin was observed pre-meiotically in 1–3-mm anthers, as well as in microspores (4-mm anthers) where
expression of _Tdr1_ was first detected, but not in mature pollen (Extended Data Fig. 7b,c). According to published single-cell RNA sequencing data from developing maize pollen59, _Dcl2_ is
also expressed pre-meiotically, consistent with its role in generating 22-nt hp-siRNA from _Tpd1_ (Extended Data Fig. 7d). _Dcl2_ is not expressed in bicellular microspores, but is
expressed in mature pollen consistent with an additional function in production of secondary small RNAs from _Tdr1_ (Extended Data Fig. 7d). These results indicate a sequential order of
events, in which expression of _Tpd1_ pre-meiotically deposits small RNAs in microspores where they target _Tdr1_. Subsequent expression of _Dcl2_ in mature pollen then promotes secondary
small RNA production and translational suppression. Identification of _Tdr1_ provided insight into the function of _Tpd2_. _Tpd1_ hp-siRNAs were unaffected by _Tpd2_, which was instead
required to suppress secondary small RNA biogenesis from _Tdr1_, along with the _mexicana_ allele of _Dcl2_, namely, _dcl2__T_ (Extended Data Fig. 8a). This indicates that _Tpd2_ and
_dcl2__T_ have additive effects on suppressing secondary small RNAs, consistent with their role as partial antidotes (Extended Data Fig. 8b). Although the molecular identity of _Tpd2_
remains unknown, the 1.5-Mb _Tpd2_ interval contains six pollen-expressed genes in W22 (Extended Data Fig. 8c). One of these genes encodes the maize homologue of _Arabidopsis_ RNA-DIRECTED
DNA METHYLATION (RDM1), a critical component of the RNA-directed DNA methylation pathway60. This gene is significantly overexpressed in _TPD_ pollen (Extended Data Fig. 8c), and it is
possible that increased activity of RNA-directed DNA methylation might compete with the production of secondary small RNAs61,62, although further experimentation is required to support this
idea. _TPD_, RNAI AND THE ORIGIN OF MODERN MAIZE Population-level studies of maize traditional varieties identified an uninterrupted _mexicana_-derived haplotype surrounding the centromere
of chromosome 5 (refs. 32,63) with high rates of linkage disequilibrium63. Consistent with reduced recombination, fine-mapping of _Tpd1_ yielded very few informative recombinants (21 of
7,549) and none proximal to the hairpin (Extended Data Fig. 1c). Comparative analysis of the _TPD_ and W22 genomes revealed three megabase-scale inversions, one of which corresponded to a
13-Mb event within the _Tpd1_ haplotype and including _Bt1_ on chromosome 5L (Fig. 2f,g). The presence of this inversion, along with its physical proximity to the centromere, explained our
mapping data (Extended Data Fig. 1c) and strongly suggested that the _Tpd1_ haplotype behaves as a single genetic unit. The 13-Mb paracentric inversion in the _Tpd1_ haplotype (W22
chromosome 5: 115,316,812–124,884,039) almost entirely encompasses ‘region D’ adjacent to centromere 5 (W22 chromosome 5: 118,213,716–126,309,970), which has undergone a dramatic
domestication sweep in all maize inbreds relative to teosinte4. This region includes _Bt1_, which undergoes drive in the TPD system (Extended Data Fig. 1c). Our synthetic hybrids with maize
inbred W22 retained approximately 13 intervals of the _mexicana_ genome that persisted in serial backcrosses (Extended Data Fig. 2a,b). Four of these intervals are tightly linked to genes
encoding AGO proteins, specifically _Ago1a_, _Ago1b_, _Ago2b_ and _Ago5b_, all of which are expressed in the male germ line (Extended Data Fig. 2). According to 5′ nucleotide analysis, these
AGO proteins are predicted to bind to _Tpd1_-hp-siRNAa–d (Ago2 and Ago5), as well as secondary _Tdr1_ 22-nt siRNAs (Ago1), and it is conceivable that hypomorphic alleles could also act as
partial antidotes in combination with _Tpd2_. In addition to intervals encoding _Dcl2_, _Rdm1_ and _Rgd1/Sgs3_, this means that 7 of the 13 intervals are tightly linked to genes required for
RNAi. These correlations suggest that there has been strong selection on all of these modifiers to ameliorate the toxic effects of _Tpd1_, resulting in apparent gene drive. In traditional
maize varieties, but not in sympatric _mexicana_, significant correlations were observed in _mexicana_ ancestry between 11 of the 13 intervals (Extended Data Fig. 9a, Supplementary Tables 6
and 7 and Supplementary Discussion). By contrast, variation at _Tdr1_ displays no such correlation with the co-inherited intervals in traditional maize varieties (Extended Data Fig. 9a). In
fact, _Tdr1_ is strongly monomorphic in traditional maize varieties, whereas in _mexicana_, _Tdr1_ displays extreme polymorphism (Extended Data Fig. 9b). We considered the possibility that
this locus has evolved to become immune to silencing in modern maize, a predicted outcome of selfish genetic systems11. A recent survey of maize and teosinte genome sequences64 has revealed
that three of the four _Tpd1_-hp-siRNA target sites in _Tdr1_ exhibit extensive polymorphism in maize and teosinte, including an in-frame deletion of the target site seed region for
_Tpd1_-hp-siRNAa and a SNP at position 11 in target sites for _Tpd1-_hp-siRNAb, which are predicted to reduce or abolish cleavage and translational inhibition, respectively (Fig. 5a). _TPD_
pollinations of the temperate inbred B73, which carries the deletion haplotype, resulted in 50% partially sterile (44 of 83) and fully fertile (35 of 83) offspring in advanced backcrosses,
as well as rare fully sterile presumptive recombinants (4 of 83), consistent with these expectations. Surveys of the frequency of the deletion haplotype across _Zea_ found it widespread,
suggesting an origin before speciation of _Z. mays_ from _Zea luxurians_ and _Zea diploperennis_ (Fig. 5b), whereas it is absent from _Zea_ _nicaraguagensis_ and _Tripsacum dactyloides_. The
frequency of the deletion haplotype is relatively low in _mexicana_ (12%) compared with _parviglumis_ (46%), and increases in tropical maize, traditional maize varieties, popcorn and
inbreds, where it is nearly fixed in several modern inbred groups (98%), suggesting a trajectory of spread to North and South America. DISCUSSION _T__PD_ is a toxin–antidote system that
defies Mendelian inheritance and may have a history of selfish evolution, like other hybrid incompatibilities that cause gamete killing. Unlike teosinte crossing barriers _tcb-1_, _Ga-1_ and
_Ga-2_ (ref. 65), which prevent fertilization, _TPD_ resembles BDM incompatibility (also known as Dobzhansky–Muller incompatibility or DMI) in that it acts post-zygotically, resulting in
sterile progeny. In canonical BDM, however, hybrid sterility is due to the unmasking of deleterious alleles, so that fertility eventually recovers in recurrent backcrosses to either parent.
In _TPD_, backcrosses to maize result in pollen abortion no matter how many backcross generations are observed. This is because _TPD_ is a special case of BDM that is consistent with meiotic
drive. For gamete killers to spread via meiotic drive, they must compensate somehow for loss of fertility66. Loss of fertility may have posed a challenge for the spread of _TPD_ in
populations of teosinte. Therefore, establishing the evolutionary origin of _TPD_ by meiotic drive will require additional population-level data and modelling, so that other explanations for
gamete killing can be excluded67. In practice, maize–teosinte hybrids are extremely vigorous with numerous tassels, so that these wind-pollinated species may be less sensitive to reductions
in male fertility. This is especially true during domestication, when early domesticates are typically less prolific than wild relatives, and at lower population size. In such
circumstances, segregation distortion in hybrids could affect patterns of introgression between maize and teosinte. _Tpd1_ encodes a long non-coding hpRNA that produces specific 22-nt
hp-siRNAs in the male germline and kill pollen grains by targeting the genetically linked responder gene _Tdr1_ (Extended Data Fig. 10a,b). This effect is countered by at least two
gametophytic antidotes: a linked hypomorphic allele of _Dcl2_ and the unlinked _Tpd2_ locus on chromosome 6 (Extended Data Fig. 10c). The genetic architecture of this system, consisting of
multiple linked and unlinked loci, deviates from previously established toxin–antidote systems. In rice, for instance, the _qHMS7_ quantitative trait locus is a selfish genetic element
composed of two tightly linked open reading frames34. Similarly, the _wtf4_ driver in _S. pombe_ features two alternatively spliced transcripts derived from the same locus24. By contrast,
the _Tpd1_ haplotype results from tight pseudolinkage between _Tpd1_, _Tdr1_ and _dcl2__T_ on chromosome 5, but only when transmitted through the male (Extended Data Fig. 8b). Although
recombinants occur in single-pollen grains, they are not transmitted to the next generation (Fig. 1), and maternal recombinants between _dcl2__T_ and _Tpd1_ are completely male sterile (Fig.
2). These recombinants produce far more secondary 22-nt small RNAs at _Tdr1_ (Extended Data Fig. 8a), providing an explanation for the failure to transmit recombinants through pollen.
_Tpd2_ is unlinked but acts cell autonomously, so that independent assortment of _Tpd1_ and _Tpd2_ occurs in female gametes, but never in male, implying that gametophytic suppression of
pollen killing requires co-segregation of _Tpd2_ with _Tpd1_. Although unlinked suppressors are relatively rare, a similar system has been reported in fission yeast68. In both cases, the
selective suppression of drive can be interpreted as selfish behaviour on the part of the antidote. Ultimately, cycles of suppression and counter-suppression can be expected to result in
complex, polygenic drivers that exist in a continuum of cryptic states (Extended Data Fig. 11), and the conspicuous maintenance of _mexicana_ introgression intervals containing RNAi factors
supports this idea (Extended Data Figs. 2a and 11). Genome scans of sympatric maize and _mexicana_ have identified multiple regions of introgression associated with adaptive variation, some
of which overlap with the genomic interval corresponding to the _Tpd1_ haplotype32 and other intervals undergoing distortion69, and we found that intervals associated with drive in pollen
are significantly correlated with each other in maize traditional varieties, but not in sympatric _mexicana_ populations (Extended Data Fig. 9). We postulated that the most powerful
suppressor of all would be an ‘immune’ target gene, in which hp-siRNA target sites in _Tdr1_ had been mutated. Such in-frame immune haplotypes were found in wild taxa in _Zea_ and have been
progressively fixed from tropical to temperate stiff-stalk maize inbreds (Fig. 5), suggesting that _TPD_ may be an ancient system that has influenced admixture throughout the history of the
genus, reaching fixation in modern maize. _TPD_ complements the hypothesized role of _Ab10_, a chromosomal driver of female meiosis that simulations suggest may have been responsible for the
redistribution of heterochromatic knobs in maize, _parviglumis_ and _mexicana_15,70, potentially along with thousands of linked genes16. Our results suggest that DCL2-dependent 22-nt small
RNAs stemming from long hpRNAs function as selfish genetic elements in pollen. In _Arabidopsis_, 22-nt siRNA biogenesis is carefully regulated due to ectopic silencing of host
genes37,40,42,54, and 21–22-nt siRNAs from pollen mediate triploid seed abortion71,72 and can block self-fertilization73. In _Drosophila melanogaster_74,75, silencing of protein-coding genes
by recently evolved hairpins is important for male reproductive development75, whereas in _Drosophila_ _simulans_, the Winters sex-ratio distortion system is actually suppressed by two
hpRNAs, _Not much yang_ (_Nmy_) and _Too much yin_ (_Tmy_), which act as antidotes and are essential for male fertility and sex balance76,77. In mammals, endo-siRNAs in the oocyte are
generated from hairpin and antisense precursors by an oocyte-specific Dicer isoform (_Dcr-O_) and have an essential function in global translational suppression78,79,80. The remarkable
parallels between all of these systems, and between Dcr-O and _dcl2__T_, which both have potential defects in the helicase domain, invites speculation that selection for selfish behaviour is
an efficient means by which germline small RNAs can propagate within a population. Such propagation provides a plausible origin for ‘self’-targeting small RNAs in the germlines of plants
and animals. METHODS PLANT MATERIAL AND GROWTH CONDITIONS The _TPD_ lineage traces to teosinte _mexicana_ collected near Copándaro, Michoacán, Mexico in December 1993. Gamete a, plant 4 of
collection 107 was used in an initial outcross to the Midwestern US dent inbred W22 and subsequently backcrossed. _Tpd1;Tpd2_ (BC8S3) homozygous lines were used for whole-genome sequencing
and de novo genome assembly. All additional experiments were performed using _Tpd1/tpd1; Tpd2/tpd2_ (BC11–BC13) plants or populations derived from maternal segregation of these lines. The
_lbl1-rgd1_ and _dcl2-mu1_ alleles were backcrossed to W22 four or more times. _dcl2-mu1_ was isolated from the Uniform-Mu line UFMu-12288. All genetic experiments used segregating wild-type
progeny as experimental controls. Plants were grown under greenhouse and field conditions. PHENOTYPING AND MICROSCOPY All pollen phenotyping was performed using mature 5-mm anthers before
anthesis. Individual anthers were suspended in PBS and dissected using forceps and an insulin syringe. Starch viability staining was performed using Lugol solution (L6146-1L, Sigma).
Measurements for days to anthesis were taken for three replicate crosses (_Tpd1/tpd1;Tpd2/tpd2_ × W22) with staggered planting dates in three different field positions. The leaf collar
method81 was combined with routine manual palpation of the topmost internode to track reproductive stages. Meiotic anthers were dissected, fixed in 4% paraformaldehyde plus MBA buffer82, and
stained with DAPI for visualization. For tetrad viability assays, anthers from the upper floret of an individual spikelet were dissected and stored in MBA. One anther was used for staging
and the others were dissected to release the tetrads. FDA viability staining was performed as previously described83. To control for artefacts associated with sample handling, only intact
tetrads (four physically attached spores) were considered. GENOTYPING AND MARKER DESIGN For routine genotyping, tissue discs were collected with a leaf punch and stored in 96-well plates. To
extract genomic DNA, 20 μl of extraction solution (0.1 M NaOH) was added to each well and samples were heated to 95 °C for 10 min and then placed immediately on ice. To neutralize this
solution, 90 μl of dilution solution (10 mM Tris + 1 mM EDTA, pH to 1.5 with HCl) was added. PCRs, using 1–2 μl of this solution as template, were performed using GoTaq G2 Green Master Mix
(M7822, Promega). Secondary validation of genotyping reactions was performed as needed using the Quick-DNA Plant/Seed Miniprep kit (D6020, Zymo Research). Bulk Illumina and Nanopore data
from _Tpd1;Tpd2_ seedlings was used for co-dominant molecular marker design (Supplementary Table 8). When possible, markers based on simple sequence length polymorphisms were prioritized,
but a number of restriction fragment length polymorphisms were also designed. W22, _Tpd1/tpd1;Tpd2/tpd2_ and _Tpd1;Tpd2_ genomic DNA was used to validate marker segregation before use. The
_dcl2-mu1_ insertion was amplified by combining gene-specific forward and reverse primers with a degenerate terminal inverted repeat primer cocktail. The insertion was subsequently validated
by Sanger sequencing. HIGH-MOLECULAR-WEIGHT GENOMIC DNA EXTRACTION High-molecular-weight (HMW) genomic DNA was used as input for all Nanopore and bulk Illumina sequencing experiments. For
extraction, bulked seedlings were dark treated for 1 week before tissue collection. Four grams of frozen tissue was ground under liquid N2 and pre-washed twice with 1.0 M sorbital. The
tissue was then transferred to 20 ml pre-warmed lysis buffer (100 mM Tris-HCl (pH 9.0), 2% w/v CTAB, 1.4 M NaCl, 20 mM EDTA, 2% PVP-10, 1% 2-mercaptoethanol, 0.1% sarkosyl and 100 μg ml−1
proteinase K), mixed gently and incubated for 1 h at 65 °C. Organic extraction in phase-lock tubes was performed using 1 vol phenol:chloroform:isoamyl alcohol (25:24:1) followed by 1 vol
chloroform:isoamyl alcohol. DNA was precipitated by adding 0.1 vol 3 M NaOAc (pH 5.2) followed by 0.7 vol isopropanol. HMW DNA was hooked out with a pasteur pipette and washed with 70% EtOH,
air dried for 2 min and resuspended in 200 μl Tris-HCl (pH 8.5; EB). The solution was treated with 2 μl 20 mg ml−1 RNase A at 37 °C for 20 min followed by 2 µl 50 mg ml−1 proteinase K at 50
°C for 30 min. 194 μl EB, 100 µl NaCl and 2 μl 0.5 M EDTA were added, and organic extractions were performed as before. DNA was precipitated with 1.7 vol EtOH, hooked out of solution with a
pasteur pipette, washed with 70% EtOH and resuspended in 50 μl EB. NANOPORE AND HI-C SEQUENCING, _TPD_ GENOME ASSEMBLY AND ANNOTATION HMW DNA from _Tpd1;Tpd2_ BC8S3 was gently sheared by
passage through a P1000 pipette 20 times before library preparation with the Oxford Nanopore Technologies Ligation Sequencing gDNA (SQK-LSK109) protocol with the following modifications: (1)
DNA repair, end-prep and ligation incubation times were extended to 20 min each; (2) 0.8× vol of a custom SPRI bead solution was used for reaction cleanups84,85; and (3) bead elutions were
carried out at 50 °C for 5 min. Libraries were sequenced on the MinION device with R9.4.1 flow cells. Offline base calling of Oxford Nanopore Technologies reads was performed with Guppy
5.0.7 and the R9.4.1 450-bp super accuracy model. Reads longer than 1 kb were assembled into contigs using Flye 2.9-b1768 (ref. 86) with options ‘--extra-params max_bubble_length=2000000 -m
20000 -t 48 --nano-raw’. The same long reads were aligned to the Flye contigs (filtered to keep only the longest alternatives) using minimap2 2.22-r1101 (ref. 87), and these alignments were
passed to the PEPPER-Margin-DeepVariant 0.4 pipeline88 to polish the initial consensus. To correct remaining single-nucleotide variants and small indels, two Illumina PCR-free genomic DNA
PE150 libraries were mapped to the long read polished consensus with bwa-mem2 2.2.1 (ref. 89) for further polishing with NextPolish 1.3.1 (ref. 90) followed by Hapo-G 1.2 (ref. 91), both
with default options. Two biological replicate samples of BC8S3 leaf tissue were used to prepare Dovetail Omni-C Kit libraries following the manufacturer’s protocol, and sequenced as a PE150
run on a NextSeq500. These Hi-C reads were mapped to the polished contigs with the Juicer pipeline release 1.6 UGER scripts with options ‘enzyme=none’92. The resulting ‘merged_nodups.txt’
alignments were passed to the 3D DNA pipeline to iteratively order and orient the input contigs and correct misjoins93. This initial automatic scaffolding resulted in 11 superscaffolds
longer than 10 Mb. Correcting a single centromeric break during manual review with JBAT94 resulted in the expected 10 pseudomolecules. One 6-Mb contig was identified as bacterial with no
contacts and was discarded. The remaining unscaffolded contigs were of organelle origin (_n_ = 9, 625 kb) or aligned to the pseudomolecules (_n_ = 116, 12 Mb). Coding gene predictions from
the NRGene 2.0 W22 (ref. 95) were projected onto the _TPD_ genome assembly using Liftoff 1.6.2 (ref. 96) with options ‘-polish -copies -chroms <chrom_map>’. An average Phred quality
value (QV) score for the assembly was estimated from a 20-mer database of the Illumina reads using merqury 1.4.1 (ref. 97) with default options. Assembly completeness was also assessed with
BUSCO 5.5.0 (ref. 98) with options ‘-m genome --miniprot’. See Supplementary Table 3 for assembly metrics. RNA EXTRACTION Tissue was collected, snap frozen in liquid nitrogen and stored at
−80 °C. Samples were ground into a fine powder using a mortar and pestle on liquid nitrogen. Of pre-extraction buffer (100 mM Tris-HCl (pH 8.0), 150 mM LiCl, 50 mM EDTA (pH 8.0), 1.5% v/v
SDS and 1.5% 2-mercaptoethanol), 800 µl was added and mixed by vortexing. Of acid phenol:chloroform (pH 4.7–5.0), 500 µl was added and samples were mixed then spun down at 13,000_g_ for 15
min at 4 °C. The aqueous layer was extracted, and 1 ml TRIzol per 200 mg input tissue was added. Samples were mixed by vortex and incubated at room temperature for 10 min. Chloroform (200
µl) per 1 ml TRIzol was added and samples were mixed by vortexing and then incubated at room temperature for 2 min. Samples were then spun down at 13,000_g_ for 15 min at 4 °C. The aqueous
phase was extracted and cleaned up using the Zymo RNA Clean and Concentrator-5 kit (R1013, Zymo Research). Only samples with RNA integrity scores of 9 or more were used for quantitative PCR
(qPCR) and sequencing. REVERSE TRANSCRIPTION AND RT–QPCR For reverse transcription, 1 µg of total RNA was treated with ezDNase (11766051, Thermo Fisher) according to the manufacturer’s
instructions. Reverse transcription was performed with SuperScript IV VILO Master Mix (11756050, Thermo Fisher). Following reverse transcription, complementary DNA (cDNA) was diluted 1:20 in
dH20 to be used as template in qPCR with reverse transcription (RT–qPCR). All RT–qPCR experiments were performed on an Applied Biosystems QuantStudio 6 system in 96-well plate format using
PowerUp SYBR Green Master Mix (A25741, Thermo Fisher). Before use in experiments, primer efficiency was tested for each primer set using a standard curve generated from serial dilutions of
cDNA template. Only primer sets with efficiencies between 90% and 110% were used (Supplementary Table 9). For experiments, three or more biological replicates (independent cDNA samples from
discrete plants) were assayed per genotype, and two or more technical replicates were set up for each reaction condition. Raw Ct (cycle threshold) from technical replicates were averaged,
and ∆Ct (mean Ctexp – mean Ctref) was calculated using _Elfa9_ as a housekeeping reference. ∆∆Ct values (∆Ctcond1 – ∆Ctcond2) were calculated between genotypes and converted to fold change
(2(–∆∆Ct)). WHOLE-GENOME SEQUENCING AND SNP CALLING For HMW DNA from separately maintained _Tpd1;Tpd2_ lineages (BC8S3 and BC5S2) and from bulk segregation analysis maternal pools,
extractions were as detailed above. Libraries were prepared using the Illumina TruSeq DNA PCR-Free kit (20015962, Illumina) with 2 μg of DNA input. Samples were sequenced on a NextSeq500
platform using 2 × 150-bp high-output run. Adapter trimming was performed with Cutadapt (v3.1)99. Paired-end reads were aligned to the W22 reference genome95 with BWA-MEM (v0.7.17)100.
Alignments were filtered by mapping quality (mapQ ≥ 30), and PCR duplicates were removed using SAMtools (v1.10)101. SNP calling was performed using Freebayes (v1.3.2)102. Putative SNP calls
were filtered by quality, depth and allele frequency (allele frequency = 1) to obtain a high-confidence _mexicana_ marker set that was subsequently validated against the _TPD_ assembly. For
bulk segregation analysis103, SNP calls were filtered against the gold-standard _TPD_ marker set. Reference and alternate allele frequencies at each marker were calculated and the average
signal was consolidated into 100-kb bins. The ∆SNP index was then calculated for each bin in a sliding window. SINGLE-POLLEN GRAIN SEQUENCING Pollen grains from _Tpd1/tpd1;Tpd2/tpd2_ plants
were suspended in ice-cold PBS on a microscope slide under a dissecting scope. Individual plump, viable pollen grains were deposited into the 0.2-ml wells of a 96-well plate using a p20
pipette. Lysis and whole-genome amplification were performed using the REPLI-g single-cell kit (150345, Qiagen) with the following modifications: one-fourth of the specified volume of
amplification mix was deposited in each well and isothermal amplification was limited to 5 h. All steps before amplification were performed in a UV-decontaminated PCR hood. Whole-genome
analysis products were cleaned up using a Genomic DNA Clean & Concentrator kit (D4067, Zymo Research), and yields were quantified using with the QuantiFluor dsDNA system (E2670, Promega)
in a 96-well microplate format. Libraries were prepared using the TruSeq Nano DNA High Throughput kit (20015965, Illumina) with 200 ng input. Samples were sequenced on a NextSeq500 platform
using 2 × 101-bp high-output runs. Quality control, adapter trimming, alignment and SNP calling were performed as above. BCFtools 1.14 (ref. 104) was used to derive genotype calls from
single-pollen grains at the predefined marker positions and then passed to GLIMPSE 1.1.1 (ref. 105) for imputation. All calls at validated marker sites were extracted and encoded in a sparse
matrix format (rows = markers, columns = samples) and encoded (1 = alt allele, −1 = ref allele, 0 = missing). To assess _mexicana_ introgression in individual pollen grains, mean SNP signal
was calculated in 100-kb bins across the genome. A sliding window (1-Mb window, 200-kb step) was applied to smooth the data and identify regions with _mexicana_ SNP density. To identify
genomic intervals overrepresented in surviving _TPD_ pollen grains, aggregate allele frequency was calculated across all pollen grains at each marker site. RNA SEQUENCING AND ANALYSIS Five
biological replicates were prepared for each biological condition (_Tpd1/tpd1;Tpd2/tpd2_ and _tpd1;tpd2_ siblings). Of total RNA, 5 µg was ribosome depleted using the RiboMinus Plant Kit
(A1083808, Thermo Fisher), and libraries were prepared using the NEXTFLEX Rapid Directional RNA-seq kit (NOVA-5138-08, PerkinElmer). The size distribution of completed libraries was assessed
using an Agilent Bioanalyzer, and quantification was performed using a KAPA Library Quantification kit (KK4824, Roche). Libraries were sequenced on a NextSeq500 platform using a 2 × 150-bp
high-output run. Trimmed reads were aligned to the W22 reference with STAR in two-pass alignment mode106. Read counts were assigned to annotated features using featureCounts107. For
transposable element expression, multi-mapping reads were assigned fractional counts based on the number of identical alignments. Differential expression analysis was performed using
edgeR108. To avoid false positives, a stringent cut-off (log2 fold change ≥ 2, FDR ≤ 0.001) was used to call differentially expressed genes. Gene ontology analysis (Fisher’s exact test, _P_
< 0.01) was performed using topGO109, and the results were visualized using rrvgo110. For data visualization, alignment files were converted to a strand-specific bigwig format using
deepTools111. SMALL RNA SEQUENCING AND ANALYSIS For comparisons between _Tpd1/tpd1;Tpd2/tpd2_ and _tpd1;tpd2_ pollen, three biological replicates were used. Two biological replicates were
used for _dcl2__T−/−_ and _dcl2-mu1__−/−_ pollen samples. Libraries were constructed with the NEXTFLEX Small RNA-Seq V3 kit (NOVA-5132-06, PerkinElmer) using 2 μg of total RNA input per
library and the gel-free size selection protocol. The size distribution of completed libraries was assessed using an Agilent Bioanalyzer, and quantification was performed using a KAPA
Library Quantification kit (KK4824, Roche). Libraries were sequenced on a NextSeq500 platform using a 1 × 76-bp run. Adapters were trimmed using cutadapt99, and the 4-bp unique molecular
identifier sequences on either side of each read were removed. Reads were filtered using pre-alignment to a maize structural RNA consensus database using bowtie2 (ref. 112). Alignment and de
novo identification of small RNA loci were performed with ShortStack113, using a minimum CPM cut-off of 5, and only clusters with clear size bias (21, 22 or 24 nt) were retained in
downstream analysis. Differential sRNA accumulation was performed with edgeR108 (log2 fold change ≥ 2, FDR ≤ 0.01). The accumulation of size and strand-biased hp-siRNAs was used to identify
hairpin loci throughout the genome. For each locus, the underlying primary sequence was tested for reverse complementarity, and RNA secondary structure prediction was performed using
RNAfold114. Non-hp-siRNA targets were only retained if they showed negligible strand bias (that is, evidence of a double-stranded RNA template for processing by a Dicer-like enzyme).
IPARE-SEQ AND ANALYSIS iPARE-seq is an improvement on degradome sequencing by PARE-seq115. For iPARE-seq libraries, 40 μg of total RNA was poly(A) selected using a Dynabeads mRNA
Purification Kit (61006, Thermo Fisher). Of poly(A) RNA, 1 µg was ligated to the 5′ PARE adapter (100 pmol) in 10% DMSO, 1 mM ATP, 1X T4 RNA ligase 1 buffer (B0216L, New England Biolabs),
25% PEG8000 with 1 μl (40U) of RNaseOUT (10777019, Thermo Fisher) and 1 μl T4 RNA ligase 1 (M0204S, New England Biolabs) in a reaction volume of 100 μl. Ligation reactions were performed for
2 h at 25 °C followed by overnight incubation at 16 °C. Samples were then purified using RNA Clean XP beads (A63987, Beckman Coulter) and eluted in 18 μl dH20. Chemical fragmentation of
ligated RNA to 200 nt or fewer was performed using the Magnesium RNA fragmentation kit (E6150S, New England Biolabs). Of RNA fragmentation buffer, 2 µl was added and samples were incubated
at 94 °C for 5 min followed by a transfer to ice and the addition of 2 μl of RNA Stop solution. Samples were purified using the RNA Clean & Concentrator-5 kit (R1013, Zymo Research) and
eluted in 11 μl H20. Reverse transcription was performed as follows: 10 μl of RNA, 1 μl of 10 mM dNTP and 2 μl of random primer mix (S1330S New England Biolabs) were mixed and incubated for
10 min at 23 °C, and then put on ice for 1 min. The following was then added: 4 μl of 5X SuperScript IV buffer, 1 μl of 100 mM DTT, 1 μl of RNaseOUT and 1 μl of Superscript IV (200U). The
reaction was incubated for 10 min at 23 °C, followed by 10 min at 50 °C. Of Tris-EDTA, 80 µl was then added to this mixture. Target indirect capture was performed with 100 μl Dynabeads MyOne
Streptavidin T1 beads (65601, Thermo Fisher) as per the manufacturer's instructions. Of the reverse transcription reaction, 100 µl was used as input, and captured cDNA molecules were
eluted in 50 μl. Second-strand synthesis was performed using 5U Klenow fragment (M0210S, New England Biolabs) with 100 µM dNTPs and 1 μM of iPARE adapter primer
(5′-NNNNTCTAGAATGCATGGGCCCTCCAAG-3′) for 1 h at 37 °C and incubation at 75 °C for 20 min. Samples were purified with a 1:1 ratio of AMPure XP SPRI beads (A63880, Beckman Coulter) and
resuspended in 51 μl EB. Of sample, 50 µl was used for library preparation with the NEB Ultra DNA library kit (E7370S, New England Biolabs). Barcoded samples were sequenced with a NextSeq500
2 × 150-bp high-output run. Use of the directional iPARE adapter allows for the retention of directionality even when using a non-directional DNA-seq kit. Cutadapt99 was used to search and
recover the adapter sequence in both 5′ and 3′ orientation (forward in read1 or read2, respectively). Read1 adapter reads were trimmed for the 3′ adapter if present, and the 5′ iPARE adapter
was subsequently removed. Potential polyA tails were also removed, and only reads of 20 nt or more were retained. Read2 adapter reads were processed in an identical manner. Filtered reads
were mapped using Bowtie2 (ref. 112) and the 5′ position of each read (the cloned 5′-monophosphate corresponding to the position of AGO-mediated cleavage) was extracted using BEDtools116
with CPM normalization. Small RNA target prediction was performed using psRNATarget117. PROTEIN EXTRACTION AND WESTERN BLOTTING Fresh anthers or pollen were collected and snap frozen in
liquid nitrogen. Samples were then ground to a fine powder in a mortar and pestle over liquid nitrogen and resuspended in freshly prepared extraction buffer (2 mM Tris-HCl (pH 7.4), 150 mM
NaCl, 1 mM EDTA, 1% v/v NP-40, 5% v/v glycerol, 1 mM PMSF and 1 ml Roche protease inhibitor cocktail per 30 g input tissue) and vortexed thoroughly. Samples were then centrifuged at 14,000
rpm at 4 °C for 5 min to pellet cellular debris, and the aqueous fraction was transferred to another tube. This step was then repeated twice more. Protein extracts were quantified using the
Pierce Detergent Compatible Bradford Assay Kit (23246, Thermo Fisher) on a Promega Glomax-Multi+ plate reader. To assess the role of 22-nt siRNAs in translational repression, antiserum was
raised to a peptide (SRKGAPPSSPPLSPPKLGA) from the Zm00004b012122 protein in collaboration with PhytoAB. Specificity was determined as follows: (1) blots using pollen protein extracts showed
a single band at roughly the expected size, and (2) blots using leaf protein extracts showed no band in concordance with expected pollen/anther specificity. A rabbit polyclonal HSP90-2
antibody (AS11 1629, Agrisera), a constitutive isoform with high expression, was used as loading control in all western blot experiments. For comparisons of protein abundance between
wild-type and _TPD_ pollen/anthers, 2 µg of protein was denatured at 95 °C for 5 min in an appropriate volume of 2X Laemmli buffer (120 nM Tris-Cl (pH 6.8), 4% v/v SDS, 0.004% bromophenol
blue, 20% v/v glycerol, 0.02% w/v bromophenol blue and 350 mM DTT). Samples were run on a 4–20% Mini-PROTEAN TGX Precast Gel (4561094, Bio-Rad) with a Precision Plus Protein Dual Xtra
Prestained standard (1610377, Bio-Rad). Transfer to a PVDF membrane was performed using a Bio-Rad Trans-Blot Turbo Transfer system. Membranes were blocked using 5% w/v powdered milk in 1X
TBS-T (20 mM Tris, 150 mM NaCl and 0.1% Tween-20) for 1 h at room temperature. Subsequently, the membrane was cut and incubated with primary antibody (1:3,000 dilution in blocking solution)
at 4 °C overnight with gentle agitation. Three 15-min membrane washes were performed with 1X TBS-T at room temperature. Membranes were then incubated with a 1:3,000 goat anti-rabbit IgG
H&L (PHY6000, PhytoAB) secondary antibody for 1 h at room temperature. Following three more washes with 1X TBS-T, membranes were incubated for 5 min with ECL Prime detection reagent
(RPN2236, Amersham) and visualized using a Bio-Rad ChemiDoc Touch Imaging System. ESTERASE ENZYMATIC ACTIVITY ASSAY Esterase activity assays were performed using the colorimetric substrate
_p_-nitrophenyl butyrate (N9876, Sigma) at a final concentration of 1 mM in 0.5 M HEPES (pH 6.5). For assays using whole 5-mm anthers, 100 μg of total protein was used as input for each
sample, whereas 50 μg was used for pollen. Individual samples were prepared in cuvettes at a volume of 1.5 ml. Upon addition of the total protein extract, samples were gently mixed, and an
initial 410-nm absorbance reading was taken to serve as a per sample baseline. Samples were then incubated at 30 °C, and absorbance readings were taken every 5 min for a total of 12
timepoints. This experiment was replicated three times for each genotype. All absorbance readings were taken using a Thermo Scientific Genesys 20 spectrophotometer. DETECTION OF SELECTIVE
SWEEPS IN CANDIDATE REGIONS ASSOCIATED WITH _TPD_ We investigated signals of selection in genomic regions associated with _TPD_ using selscan (v1.2.0a)118 to calculate the genome-wide
normalized absolute integrated haplotype score (iHS) statistics for individual SNPs and in 10-kb windows. iHS is suitable for identifying selection in a single population and relies on the
presence of ongoing sweeps and a signal of selection from unusually long-range linkage disequilibrium. We also used VCFtools (v0.1.16)119 to calculate Weir and Cockerham’s _F_ST in 10-kb
windows to assess signals of selection based on changes in allele frequency between populations. Phased SNPs for modern temperate maize lines, teosinte and _T. dactyloides_ were obtained
from Grzybowski et al.120, and SNPs for 265 CIMMYT traditional varieties were obtained from Yang et al.121 and phased with Beagle (v5.4)122. A phased and imputed set of 42,387,706
genome-wide concatenated SNPs was used for the analysis of selection. The _T_. _dactyloide_s allele was set to be the ancestral allele. A consensus genetic map curated by Ed Coe was obtained
from MaizeGDB123, and SNP positions were interpolated to genetic positions. Weighted _F_ST was calculated for each unique population pair. For iHS, 10-kb windows were binned into 10
quantiles based on the number of SNPs they contained, and empirical _P_ values for each window were calculated within each quantile. The statistic calculated was the number of extreme (top
5%) |iHS| scores per window. Empirical _P_ values for iHS and _F_ST were then calculated from the rank of each window based on the respective statistics. We adjusted these _P_ values for
multiple testing of different populations using the Bonferroni method. _TPD_-linked regions (_dcl2_, _rdm1_, _tdr1_ and hairpin region) and their 1-kb upstream and downstream regions were
intersected with the 10-kb windows using bedtools (v2.30)116 and assigned the lowest _P_ value of all intersecting windows. To validate our selection scan, we also investigated windows
intersecting with a set of four known domestication genes124. REPORTING SUMMARY Further information on research design is available in the Nature Portfolio Reporting Summary linked to this
article. DATA AVAILABILITY Sequencing datasets generated during the current study are available at the NCBI (Gene Expression Omnibus SuperSeries: GSE234925). Datasets used for genome
assembly are available at the Sequence Read Archive (BioProject: PRJNA937229). This Whole-Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession JARBIH000000000.
The version described in this paper is version JARBIH010000000. All materials are available on request. CODE AVAILABILITY All code is available on Github
(https://github.com/martienssenlab/TPD-manuscript). REFERENCES * Sandler, L. & Novitski, E. Meiotic drive as an evolutionary force. _Am. Nat._ 91, 105–110 (1957). Article Google Scholar
* Presgraves, D. C. The molecular evolutionary basis of species formation. _Nat. Rev. Genet._ 11, 175–180 (2010). Article CAS PubMed Google Scholar * Kistler, L. et al. Multiproxy
evidence highlights a complex evolutionary legacy of maize in South America. _Science_ 362, 1309–1313 (2018). Article ADS CAS PubMed Google Scholar * Schneider, K. L., Xie, Z.,
Wolfgruber, T. K. & Presting, G. G. Inbreeding drives maize centromere evolution. _Proc. Natl Acad. Sci. USA_ 113, E987–E996 (2016). Article ADS CAS PubMed PubMed Central Google
Scholar * Anderson, E. & Stebbins, G. L. Hybridization as an evolutionary stimulus. _Evolution_ 8, 378–388 (1954). Article Google Scholar * Arnold, M. L. Transfer and origin of
adaptations through natural hybridization: were Anderson and Stebbins right? _Plant Cell_ 16, 562–570 (2004). Article CAS PubMed PubMed Central Google Scholar * Bayes, J. J. &
Malik, H. S. Altered heterochromatin binding by a hybrid sterility protein in _Drosophila_ sibling species. _Science_ 326, 1538–1541 (2009). Article ADS CAS PubMed PubMed Central Google
Scholar * Tang, S. & Presgraves, D. C. Evolution of the _Drosophila_ nuclear pore complex results in multiple hybrid incompatibilities. _Science_ 323, 779–782 (2009). Article ADS CAS
PubMed PubMed Central Google Scholar * Bomblies, K. et al. Autoimmune response as a mechanism for a Dobzhansky–Muller-type incompatibility syndrome in plants. _PLoS Biol._ 5, e236
(2007). Article PubMed PubMed Central Google Scholar * McLaughlin, R. N. Jr & Malik, H. S. Genetic conflicts: the usual suspects and beyond. _J. Exp. Biol._ 220, 6–17 (2017). Article
PubMed PubMed Central Google Scholar * Lindholm, A. K. et al. The ecology and evolutionary dynamics of meiotic drive. _Trends Ecol. Evol._ 31, 315–326 (2016). Article PubMed Google
Scholar * Fishman, L. & Saunders, A. Centromere-associated female meiotic drive entails male fitness costs in monkeyflowers. _Science_ 322, 1559–1562 (2008). Article ADS CAS PubMed
Google Scholar * Chmátal, L. et al. Centromere strength provides the cell biological basis for meiotic drive and karyotype evolution in mice. _Curr. Biol._ 24, 2295–2300 (2014). Article
PubMed PubMed Central Google Scholar * Fishman, L. & McIntosh, M. Standard deviations: the biological bases of transmission ratio distortion. _Annu. Rev. Genet._ 53, 347–372 (2019).
Article CAS PubMed Google Scholar * Buckler, E. S. 4th et al. Meiotic drive of chromosomal knobs reshaped the maize genome. _Genetics_ 153, 415–426 (1999). Article CAS PubMed PubMed
Central Google Scholar * Dawe, R. K. et al. A kinesin-14 motor activates neocentromeres to promote meiotic drive in maize. _Cell_ 173, 839–850.e18 (2018). Article CAS PubMed Google
Scholar * Lyon, M. F. Transmission ratio distortion in mice. _Annu. Rev. Genet._ 37, 393–408 (2003). Article CAS PubMed Google Scholar * McDermott, S. R. & Noor, M. A. F. The role
of meiotic drive in hybrid male sterility. _Phil. Trans. R. Soc. B_ 365, 1265–1272 (2010). Article PubMed PubMed Central Google Scholar * Herrmann, B. G., Koschorz, B., Wertz, K.,
McLaughlin, K. J. & Kispert, A. A protein kinase encoded by the t complex responder gene causes non-Mendelian inheritance. _Nature_ 402, 141–146 (1999). Article ADS CAS PubMed Google
Scholar * Bauer, H., Willert, J., Koschorz, B. & Herrmann, B. G. The t complex-encoded GTPase-activating protein Tagap1 acts as a transmission ratio distorter in mice. _Nat. Genet._
37, 969–973 (2005). Article CAS PubMed Google Scholar * Hartl, D. L. Genetic dissection of segregation distortion. I. Suicide combinations of SD genes. _Genetics_ 76, 477–486 (1974).
Article CAS PubMed PubMed Central Google Scholar * Larracuente, A. M. & Presgraves, D. C. The selfish segregation distorter gene complex of _Drosophila melanogaster_. _Genetics_
192, 33–53 (2012). Article CAS PubMed PubMed Central Google Scholar * Zanders, S. E. et al. Genome rearrangements and pervasive meiotic drive cause hybrid infertility in fission yeast.
_eLife_ 3, e02630 (2014). Article PubMed PubMed Central Google Scholar * Nuckolls, N. L. et al. wtf Genes are prolific dual poison–antidote meiotic drivers. _eLife_ 6, e26033 (2017).
Article PubMed PubMed Central Google Scholar * Lewontin, R. C. & Dunn, L. C. The evolutionary dynamics of a polymorphism in the house mouse. _Genetics_ 45, 705–722 (1960). Article
CAS PubMed PubMed Central Google Scholar * Hurst, L. D. & Pomiankowski, A. Causes of sex ratio bias may account for unisexual sterility in hybrids: a new explanation of Haldane’s
rule and related phenomena. _Genetics_ 128, 841–858 (1991). Article CAS PubMed PubMed Central Google Scholar * Coughlan, J. M. The role of conflict in shaping plant biodiversity. _New
Phytol._ https://doi.org/10.1111/nph.19233 (2023). * Phadnis, N. & Orr, H. A. A single gene causes both male sterility and segregation distortion in _Drosophila_ hybrids. _Science_ 323,
376–379 (2009). Article ADS CAS PubMed Google Scholar * Zhang, L., Sun, T., Woldesellassie, F., Xiao, H. & Tao, Y. Sex ratio meiotic drive as a plausible evolutionary mechanism for
hybrid male sterility. _PLoS Genet._ 11, e1005073 (2015). Article PubMed PubMed Central Google Scholar * Kermicle, J. L. & Allen, J. P. Cross-incompatibility between maize and
teosinte. _Maydica_ 35, 399–408 (1990). Google Scholar * Lu, Y., Hokin, S. A., Kermicle, J. L., Hartwig, T. & Evans, M. M. S. A pistil-expressed pectin methylesterase confers
cross-incompatibility between strains of _Zea mays_. _Nat. Commun._ 10, 2304 (2019). Article ADS PubMed PubMed Central Google Scholar * Hufford, M. B. et al. The genomic signature of
crop-wild introgression in maize. _PLoS Genet._ 9, e1003477 (2013). Article CAS PubMed PubMed Central Google Scholar * Rojas-Barrera, I. C. et al. Contemporary evolution of maize
landraces and their wild relatives influenced by gene flow with modern maize varieties. _Proc. Natl Acad. Sci. USA_ 116, 21302–21311 (2019). Article ADS CAS PubMed PubMed Central Google
Scholar * Wang, C. et al. A natural gene drive system confers reproductive isolation in rice. _Cell_ 186, 3577–3592.e18 (2023). Article CAS PubMed Google Scholar * Yang, Z. &
Bielawski, J. P. Statistical methods for detecting molecular adaptation. _Trends Ecol. Evol._ 15, 496–503 (2000). Article CAS PubMed PubMed Central Google Scholar * Yoshikawa, M.,
Peragine, A., Park, M. Y. & Poethig, R. S. A pathway for the biogenesis of trans-acting siRNAs in _Arabidopsis_. _Genes Dev_ 19, 2164–2175 (2005). Article CAS PubMed PubMed Central
Google Scholar * Parent, J.-S., Bouteiller, N., Elmayan, T. & Vaucheret, H. Respective contributions of _Arabidopsis_ DCL2 and DCL4 to RNA silencing. _Plant J._ 81, 223–232 (2015).
Article CAS PubMed Google Scholar * Deleris, A. et al. Hierarchical action and inhibition of plant Dicer-like proteins in antiviral defense. _Science_ 313, 68–71 (2006). Article ADS
CAS PubMed Google Scholar * Bouché, N., Lauressergues, D., Gasciolli, V. & Vaucheret, H. An antagonistic function for _Arabidopsis_ DCL2 in development and a new function for DCL4 in
generating viral siRNAs. _EMBO J._ 25, 3347–3356 (2006). Article PubMed PubMed Central Google Scholar * Wu, Y.-Y. et al. DCL2- and RDR6-dependent transitive silencing of SMXL4 and SMXL5
in _Arabidopsis_ dcl4 mutants causes defective phloem transport and carbohydrate over-accumulation. _Plant J._ 90, 1064–1078 (2017). Article CAS PubMed Google Scholar * Taochy, C. et al.
A genetic screen for impaired systemic RNAi highlights the crucial role of DICER-LIKE 2. _Plant Physiol._ 175, 1424–1437 (2017). Article CAS PubMed PubMed Central Google Scholar *
Mlotshwa, S. et al. DICER-LIKE2 plays a primary role in transitive silencing of transgenes in _Arabidopsis_. _PLoS ONE_ 3, e1755 (2008). Article ADS PubMed PubMed Central Google Scholar
* Tagami, Y., Motose, H. & Watanabe, Y. A dominant mutation in DCL1 suppresses the hyl1 mutant phenotype by promoting the processing of miRNA. _RNA_ 15, 450–458 (2009). Article CAS
PubMed PubMed Central Google Scholar * Welker, N. C. et al. Dicer’s helicase domain discriminates dsRNA termini to promote an altered reaction mode. _Mol. Cell_ 41, 589–599 (2011).
Article MathSciNet CAS PubMed PubMed Central Google Scholar * Aderounmu, A. M., Aruscavage, P. J., Kolaczkowski, B. & Bass, B. L. Ancestral protein reconstruction reveals
evolutionary events governing variation in Dicer helicase function. _eLife_ 12, e85120 (2023). Article CAS PubMed PubMed Central Google Scholar * Slotkin, R. K., Freeling, M. &
Lisch, D. Heritable transposon silencing initiated by a naturally occurring transposon inverted duplication. _Nat. Genet._ 37, 641–644 (2005). Article CAS PubMed Google Scholar *
Bhutani, K. et al. Widespread haploid-biased gene expression enables sperm-level natural selection. _Science_ 371, eabb1723 (2021). Article CAS PubMed Google Scholar * Shan, X. et al.
Mobilization of the active MITE transposons mPing and Pong in rice by introgression from wild rice (_Zizania latifolia_ Griseb.). _Mol. Biol. Evol._ 22, 976–990 (2005). Article CAS PubMed
Google Scholar * Ding, L.-N. et al. Advances in plant GDSL lipases: from sequences to functional mechanisms. _Acta Physiol. Plant_ 41, 151 (2019). Article Google Scholar * An, X. et al.
ZmMs30 encoding a novel GDSL lipase is essential for male fertility and valuable for hybrid breeding in maize. _Mol. Plant_ 12, 343–359 (2019). Article CAS PubMed Google Scholar * Huo,
Y. et al. IRREGULAR POLLEN EXINE2 encodes a GDSL lipase essential for male fertility in maize. _Plant Physiol._ 184, 1438–1454 (2020). Article CAS PubMed PubMed Central Google Scholar *
Zhao, J. et al. RMS2 encoding a GDSL lipase mediates lipid homeostasis in anthers to determine rice male fertility. _Plant Physiol._ 182, 2047–2064 (2020). Article CAS PubMed PubMed
Central Google Scholar * Tsugama, D., Fujino, K., Liu, S. & Takano, T. A GDSL-type esterase/lipase gene, GELP77, is necessary for pollen dissociation and fertility in _Arabidopsis_.
_Biochem. Biophys. Res. Commun._ 526, 1036–1041 (2020). Article CAS PubMed Google Scholar * Wu, H. et al. Plant 22-nt siRNAs mediate translational repression and stress adaptation.
_Nature_ 581, 89–93 (2020). Article ADS CAS PubMed Google Scholar * Borges, F. & Martienssen, R. A. The expanding world of small RNAs in plants. _Nat. Rev. Mol. Cell Biol._ 16,
727–741 (2015). Article CAS PubMed PubMed Central Google Scholar * Fang, X. & Qi, Y. RNAi in plants: an Argonaute-centered view. _Plant Cell_ 28, 272–285 (2016). Article CAS
PubMed PubMed Central Google Scholar * Axtell, M. J., Westholm, J. O. & Lai, E. C. Vive la différence: biogenesis and evolution of microRNAs in plants and animals. _Genome Biol._ 12,
221 (2011). Article CAS PubMed PubMed Central Google Scholar * Manavella, P. A., Koenig, D. & Weigel, D. Plant secondary siRNA production determined by microRNA-duplex structure.
_Proc. Natl Acad. Sci. USA_ 109, 2461–2466 (2012). Article ADS CAS PubMed PubMed Central Google Scholar * Nelms, B. & Walbot, V. Gametophyte genome activation occurs at pollen
mitosis I in maize. _Science_ 375, 424–429 (2022). Article ADS CAS PubMed Google Scholar * Wongpalee, S. P. et al. CryoEM structures of _Arabidopsis_ DDR complexes involved in
RNA-directed DNA methylation. _Nat. Commun._ 10, 3916 (2019). Article ADS PubMed PubMed Central Google Scholar * Jauvion, V., Rivard, M., Bouteiller, N., Elmayan, T. & Vaucheret, H.
RDR2 partially antagonizes the production of RDR6-dependent siRNA in sense transgene-mediated PTGS. _PLoS ONE_ 7, e29785 (2012). Article ADS CAS PubMed PubMed Central Google Scholar *
Creasey, K. M. et al. miRNAs trigger widespread epigenetically activated siRNAs from transposons in _Arabidopsis_. _Nature_ 508, 411–415 (2014). Article ADS CAS PubMed PubMed Central
Google Scholar * Romero Navarro, J. A. et al. A study of allelic diversity underlying flowering-time adaptation in maize landraces. _Nat. Genet._ 49, 476–480 (2017). Article CAS PubMed
Google Scholar * Chen, L. et al. Genome sequencing reveals evidence of adaptive variation in the genus _Zea_. _Nat. Genet._ 54, 1736–1745 (2022). Article ADS CAS PubMed Google Scholar
* Lu, Y., Kermicle, J. L. & Evans, M. M. S. Genetic and cellular analysis of cross-incompatibility in _Zea mays_. _Plant Reprod._ 27, 19–29 (2014). Article CAS PubMed Google Scholar
* Hartl, D. L. Population dynamics of sperm and pollen killers. _Theor. Appl. Genet._ 42, 81–88 (1972). Article CAS PubMed Google Scholar * Sweigart, A. L., Brandvain, Y. & Fishman,
L. Making a murderer: the evolutionary framing of hybrid gamete-killers. _Trends Genet._ 35, 245–252 (2019). Article CAS PubMed Google Scholar * Bravo Núñez, M. A., Lange, J. J. &
Zanders, S. E. A suppressor of a wtf poison–antidote meiotic driver acts via mimicry of the driver’s antidote. _PLoS Genet._ 14, e1007836 (2018). Article PubMed PubMed Central Google
Scholar * Barnes, A. C. et al. An adaptive teosinte _mexicana_ introgression modulates phosphatidylcholine levels and is associated with maize flowering time. _Proc. Natl Acad. Sci. USA_
119, e2100036119 (2022). Article CAS PubMed PubMed Central Google Scholar * McClintock, B., Kato Yamakake, T. A., Blumenschein, A. & Escuela Nacional de Agricultura (Mexico).
_Chromosome Constitution of Races of Maize: Its Significance in the Interpretation of Relationships between Races and Varieties in the Americas_ (Colegio de Postgraduados, 1981). * Borges,
F. et al. Transposon-derived small RNAs triggered by miR845 mediate genome dosage response in _Arabidopsis_. _Nat. Genet._ 50, 186–192 (2018). Article CAS PubMed PubMed Central Google
Scholar * Martinez, G. et al. Paternal easiRNAs regulate parental genome dosage in _Arabidopsis_. _Nat. Genet._ 50, 193–198 (2018). Article CAS PubMed Google Scholar * Durand, E. et al.
Dominance hierarchy arising from the evolution of a complex small RNA regulatory network. _Science_ 346, 1200–1205 (2014). Article ADS CAS PubMed Google Scholar * Czech, B. et al. An
endogenous small interfering RNA pathway in _Drosophila_. _Nature_ 453, 798–802 (2008). Article ADS CAS PubMed PubMed Central Google Scholar * Wen, J. et al. Adaptive regulation of
testis gene expression and control of male fertility by the _Drosophila_ hairpin RNA pathway. _Mol. Cell_ 57, 165–178 (2015). Article CAS PubMed Google Scholar * Tao, Y. et al. A
sex-ratio meiotic drive system in _Drosophila simulans_. II: an X-linked distorter. _PLoS Biol._ 5, e293 (2007). Article PubMed PubMed Central Google Scholar * Lin, C.-J. et al. The
hpRNA/RNAi pathway is essential to resolve intragenomic conflict in the _Drosophila_ male germline. _Dev. Cell_ 46, 316–326.e5 (2018). Article CAS PubMed PubMed Central Google Scholar *
Flemr, M. et al. A retrotransposon-driven Dicer isoform directs endogenous small interfering RNA production in mouse oocytes. _Cell_ 155, 807–816 (2013). Article CAS PubMed Google
Scholar * Tam, O. H. et al. Pseudogene-derived small interfering RNAs regulate gene expression in mouse oocytes. _Nature_ 453, 534–538 (2008). Article ADS CAS PubMed PubMed Central
Google Scholar * Su, R. et al. Global profiling of RNA-binding protein target sites by LACE-seq. _Nat. Cell Biol._ 23, 664–675 (2021). Article CAS PubMed Google Scholar * Begcy, K.
& Dresselhaus, T. Tracking maize pollen development by the leaf collar method. _Plant Reprod._ 30, 171–178 (2017). Article CAS PubMed PubMed Central Google Scholar * Bass, H. W. et
al. A maize root tip system to study DNA replication programmes in somatic and endocycling nuclei during plant development. _J. Exp. Bot._ 65, 2747–2756 (2014). Article CAS PubMed Google
Scholar * Kalkar, S. A. & Neha, K. Evaluation of FDA staining technique in stored maize pollen. _Middle East J. Sci. Res._ 12, 560–562 (2012). * Nagar, R. & Schwessinger, B. DNA
size selection (>3–4 kb) and purification of DNA using an improved homemade SPRIbeads solution. _Protocols.io_ https://doi.org/10.17504/protocols.io.n7hdhj6 (2018). * Schalamun, M.,
Nagar, R. & Kainer, D. Harnessing the MinION: an example of how to establish long‐read sequencing in a laboratory using challenging plant tissue from _Eucalyptus pauciflora_. _Mol.
Ecol._ https://doi.org/10.1111/1755-0998.12938 (2018). * Kolmogorov, M., Yuan, J., Lin, Y. & Pevzner, P. A. Assembly of long, error-prone reads using repeat graphs. _Nat. Biotechnol._
37, 540–546 (2019). Article CAS PubMed Google Scholar * Li, H. Minimap2: pairwise alignment for nucleotide sequences. _Bioinformatics_ 34, 3094–3100 (2018). Article CAS PubMed PubMed
Central Google Scholar * Shafin, K. et al. Haplotype-aware variant calling with PEPPER-Margin-DeepVariant enables high accuracy in nanopore long-reads. _Nat. Methods_ 18, 1322–1332 (2021).
Article CAS PubMed PubMed Central Google Scholar * Vasimuddin, M., Misra, S., Li, H. & Aluru, S. Efficient architecture-aware acceleration of BWA-MEM for multicore systems. In
_2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS)_ 314–324 (IEEE, 2019). * Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome
polishing tool for long read assembly. _Bioinformatics_ https://doi.org/10.1093/bioinformatics/btz891 (2019). * Aury, J.-M. & Istace, B. Hapo-G, haplotype-aware polishing of genome
assemblies with accurate reads. _NAR Genom. Bioinform._ 3, lqab034 (2021). Article PubMed PubMed Central Google Scholar * Durand, N. C. et al. Juicer provides a one-click system for
analyzing loop-resolution Hi-C experiments. _Cell Syst._ 3, 95–98 (2016). Article CAS PubMed PubMed Central Google Scholar * Dudchenko, O. et al. De novo assembly of the _Aedes aegypti_
genome using Hi-C yields chromosome-length scaffolds. _Science_ 356, 92–95 (2017). Article ADS CAS PubMed PubMed Central Google Scholar * Durand, N. C. et al. Juicebox provides a
visualization system for Hi-C contact maps with unlimited zoom. _Cell Syst._ 3, 99–101 (2016). Article CAS PubMed PubMed Central Google Scholar * Springer, N. M. et al. The maize W22
genome provides a foundation for functional genomics and transposon biology. _Nat. Genet._ 50, 1282–1288 (2018). Article CAS PubMed Google Scholar * Shumate, A. & Salzberg, S. L.
Liftoff: accurate mapping of gene annotations. _Bioinformatics_ 37, 1639–1643 (2021). Article CAS PubMed PubMed Central Google Scholar * Rhie, A., Walenz, B. P., Koren, S. &
Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. _Genome Biol._ 21, 245 (2020). Article CAS PubMed PubMed Central Google
Scholar * Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for
scoring of eukaryotic, prokaryotic, and viral genomes. _Mol. Biol. Evol._ 38, 4647–4654 (2021). Article CAS PubMed PubMed Central Google Scholar * Martin, M. Cutadapt removes adapter
sequences from high-throughput sequencing reads. _EMBnet.journal_ 17, 10 (2011). Article Google Scholar * Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM.
Preprint at https://arxiv.org/abs/1303.3997 (2013). * Li, H. et al. The Sequence Alignment/Map format and SAMtools. _Bioinformatics_ 25, 2078–2079 (2009). Article PubMed PubMed Central
Google Scholar * Garrison, E. & Marth, G. Haplotype-based variant detection from short-read sequencing. Preprint at https://arxiv.org/abs/1207.3907 (2012). * Takagi, H. et al. QTL-seq:
rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. _Plant J._ 74, 174–183 (2013). Article CAS PubMed Google Scholar *
Danecek, P. et al. Twelve years of SAMtools and BCFtools. _Gigascience_ 10, giab008 (2021). Article PubMed PubMed Central Google Scholar * Rubinacci, S., Ribeiro, D. M., Hofmeister, R.
J. & Delaneau, O. Efficient phasing and imputation of low-coverage sequencing data using large reference panels. _Nat. Genet._ 53, 120–126 (2021). Article CAS PubMed Google Scholar *
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. _Bioinformatics_ 29, 15–21 (2013). Article CAS PubMed Google Scholar * Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an
efficient general purpose program for assigning sequence reads to genomic features. _Bioinformatics_ 30, 923–930 (2014). Article CAS PubMed Google Scholar * Robinson, M. D., McCarthy,
D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. _Bioinformatics_ 26, 139–140 (2009). Article PubMed PubMed
Central Google Scholar * Alexa, A. & Rahnenfuhrer, J. topGO: enrichment analysis for gene ontology. _R package_ version 2.42.0 (2023). * Sayols, S. rrvgo: a Bioconductor package for
interpreting lists of Gene Ontology terms. _MicroPubl. Biol._ https://doi.org/10.17912/micropub.biology.000811 (2023). * Ramirez, F. et al. deepTools2: a next generation web server for
deep-sequencing data analysis. _Nucleic Acids Res._ 44, 160–165 (2016). Article Google Scholar * Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. _Nat. Methods_
9, 357–359 (2012). Article CAS PubMed PubMed Central Google Scholar * Axtell, M. J. ShortStack: comprehensive annotation and quantification of small RNA genes. _RNA_ 19, 740–751
(2013). Article CAS PubMed PubMed Central Google Scholar * Gruber, A. R., Lorenz, R., Bernhart, S. H., Neuböck, R. & Hofacker, I. L. The Vienna RNA websuite. _Nucleic Acids Res._
36, W70–W74 (2008). Article CAS PubMed PubMed Central Google Scholar * German, M. A., Luo, S., Schroth, G., Meyers, B. C. & Green, P. J. Construction of parallel analysis of RNA
ends (PARE) libraries for the study of cleaved miRNA targets and the RNA degradome. _Nat. Protoc._ 4, 356–362 (2009). Article CAS PubMed Google Scholar * Quinlan, A. R. & Hall, I. M.
BEDTools: a flexible suite of utilities for comparing genomic features. _Bioinformatics_ 26, 841–842 (2010). Article CAS PubMed PubMed Central Google Scholar * Dai, X., Zhuang, Z.
& Zhao, P. X. psRNATarget: a plant small RNA target analysis server (2017 release). _Nucleic Acids Res._ 46, W49–W54 (2018). Article CAS PubMed PubMed Central Google Scholar *
Szpiech, Z. A. selscan 2.0: scanning for sweeps in unphased data. _Bioinformatics_ 40, btae006 (2024). * Danecek, P. et al. The variant call format and VCFtools. _Bioinformatics_ 27,
2156–2158 (2011). Article CAS PubMed PubMed Central Google Scholar * Grzybowski, M. W. et al. A common resequencing-based genetic marker data set for global maize diversity. _Plant J._
113, 1109–1121 (2023). Article CAS PubMed Google Scholar * Yang, N. et al. Two teosintes made modern maize. _Science_ 382, eadg8940 (2023). * Browning, B. L., Tian, X., Zhou, Y. &
Browning, S. R. Fast two-stage phasing of large-scale sequence data. _Am. J. Hum. Genet._ 108, 1880–1890 (2021). Article CAS PubMed PubMed Central Google Scholar * Portwood, J. L. II et
al. MaizeGDB 2018: the maize multi-genome genetics and genomics database. _Nucleic Acids Res._ 47, D1146–D1154 (2019). Article PubMed Google Scholar * Stitzer, M. C. & Ross-Ibarra,
J. Maize domestication and gene interaction. _New Phytol._ 220, 395–408 (2018). Article PubMed Google Scholar * Walley, J. W. et al. Integration of omic networks in a developmental atlas
of maize. _Science_ 353, 814–818 (2016). Article ADS CAS PubMed PubMed Central Google Scholar * Liu, L. & Li, J. Communications between the endoplasmic reticulum and other
organelles during abiotic stress response in plants. _Front. Plant Sci._ 10, 749 (2019). Article PubMed PubMed Central Google Scholar * Taurino, M. et al. SEIPIN proteins mediate lipid
droplet biogenesis to promote pollen transmission and reduce seed dormancy. _Plant Physiol._ 176, 1531–1546 (2018). Article CAS PubMed Google Scholar * Beissinger, T. M. et al. Recent
demography drives changes in linked selection across the maize genome. _Nat. Plants_ 2, 16084 (2016). Article PubMed Google Scholar Download references ACKNOWLEDGEMENTS We thank K. Dawe
and J. Birchler for discussions and for sharing cytogenetic data. Research in the Martienssen laboratory is supported by the US National Institutes of Health (NIH) grant R35GM144206, the
National Science Foundation Plant Genome Research Program and the Howard Hughes Medical Institute. The authors acknowledge assistance from the Cold Spring Harbor Laboratory Shared Resources,
which are funded in part by a Cancer Center Support grant (5PP30CA045508). B.B. was supported by a predoctoral fellowship from the National Science Foundation. AUTHOR INFORMATION AUTHORS
AND AFFILIATIONS * Howard Hughes Medical Institute, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY, USA Benjamin Berube, Evan Ernst, Jonathan Cahn, Benjamin Roche, Cristiane de Santis
Alves, Jason Lynn & Robert A. Martienssen * Simons Center for Quantitative Biology, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY, USA Armin Scheben & Adam Siepel * DIADE,
Université de Montpellier, Montpellier, France Daniel Grimanelli * Department of Evolution and Ecology, Center for Population Biology and Genome Center, University of California at Davis,
Davis, CA, USA Jeffrey Ross-Ibarra * Laboratory of Genetics, University of Wisconsin, Madison, WI, USA Jerry Kermicle Authors * Benjamin Berube View author publications You can also search
for this author inPubMed Google Scholar * Evan Ernst View author publications You can also search for this author inPubMed Google Scholar * Jonathan Cahn View author publications You can
also search for this author inPubMed Google Scholar * Benjamin Roche View author publications You can also search for this author inPubMed Google Scholar * Cristiane de Santis Alves View
author publications You can also search for this author inPubMed Google Scholar * Jason Lynn View author publications You can also search for this author inPubMed Google Scholar * Armin
Scheben View author publications You can also search for this author inPubMed Google Scholar * Daniel Grimanelli View author publications You can also search for this author inPubMed Google
Scholar * Adam Siepel View author publications You can also search for this author inPubMed Google Scholar * Jeffrey Ross-Ibarra View author publications You can also search for this author
inPubMed Google Scholar * Jerry Kermicle View author publications You can also search for this author inPubMed Google Scholar * Robert A. Martienssen View author publications You can also
search for this author inPubMed Google Scholar CONTRIBUTIONS B.B., J.K. and R.A.M. designed the study. B.B., E.E., B.R., C.d.S.A. and J.L. performed the experiments with advice from D.G.
B.B., E.E., J.C., A.Scheben, J.R.-I. and R.A.M. analysed the data and/or its significance. B.B. and R.A.M. wrote the manuscript with contributions from J.C., J.R.-I. and A.Scheben, A.Siepel
and R.A.M. acquired funding. CORRESPONDING AUTHOR Correspondence to Robert A. Martienssen. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. PEER REVIEW
PEER REVIEW INFORMATION _Nature_ thanks the anonymous reviewers for their contribution to the peer review of this work. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains
neutral with regard to jurisdictional claims in published maps and institutional affiliations. EXTENDED DATA FIGURES AND TABLES EXTENDED DATA FIG. 1 _TEOSINTE POLLEN DRIVE_ AND GENETIC
MAPPING OF _TPD1_ AND _TPD2._ A, Crossing scheme of the _TPD_ phenotype. When back-crossed as male, all the progeny of semi-sterile _TPD_ plants display the semi-sterile pollen phenotype
instead of the expected 1:1 fertile:semi-sterile ratio. Graphics in panel A were created using BioRender (https://biorender.com). B, Representative ears from _Tpd1 Bt1/bt1_; _Tpd2 Y1/y1_
reciprocal crosses with _bt1; y1_ testers, demonstrating severe segregation distortion (“drive” of _Bt1_), but only through the male. _bt1 (brittle1_, collapsed kernels); _y1_ (_yellow1_,
white kernels). C, Summary of molecular and morphological mapping of the _Tpd1_ interval. Molecular mapping was performed using _Tpd/++_ x W22 segregating progeny, whereas morphological
mapping was performed by crossing _Tpd1 Bt1/tpd1 bt1_ plants to _bt1_ testers. D, Molecular mapping of the _Tpd2_ interval. SNP markers are shown in blue with recombination frequencies in
red. EXTENDED DATA FIG. 2 _MEXICANA_ INTERVALS INTROGRESSED INTO MAIZE CARRY RNAI GENES. A, Whole genome plots of homozygous _mexicana_ SNP density present within _Tpd1; Tpd2_ lines. The
upper plot corresponds to data from bulked seedlings after 8 backcrosses and 3 self-pollinations (BC8S3) whereas the lower plot is from BC5S2 plants. SNP density is consolidated in 250 kb
genomic bins. Physical locations for morphological markers _Bt1_ and _Y1_, as well as _mexicana_ derived RNAi genes, are labelled in red. 7/13 introgression intervals overlap in both
independently maintained homozygous lines. B, Allele frequency at _mexicana_ markers in 96 pollen grains from four different _TPD_ plants subjected to single pollen grain sequencing. Regions
highlighted in red were over-represented in viable pollen grains. C, Quantification of pollen viability in _Tpd1_ + _/−; Tpd2_ + _/−; rgd1/+_ and _Tpd1_ + _/−; Tpd2_ + _/−; Rgd1_ pollen
demonstrating gametophytic suppression via germline segregation of the _rgd1_ null allele. n ≥ 9 plants per genotype, ≥ 200 pollen grains per plant. **** p < 0.0001 (Welch’s t-test).
EXTENDED DATA FIG. 3 _DCL2_-DEPENDENT 22NT SIRNAS FROM HAIRPINS ARE PREVALENT IN MAIZE POLLEN. A, Distribution and relative abundances of small RNA size classes in WT pollen libraries. Bars
indicate mean ± SD. n = 3 biological replicates. B, Comparison of relative abundances for 21-nt, 22-nt, and 24-nt sRNA size classes in WT leaf and pollen samples. Both 22-nt and 24-nt sRNAs
show significant increases in pollen. Bars indicate mean ± SD. n = 3 biological replicates. **** p < 0.0001 (Welch’s t-test). C, Comparison of 22-nt sRNA levels in _Dcl2_, _dcl2__T_, and
_dcl2-mu1_ pollen at 804 pollen-specific loci. Values shown are log2 transformed counts per million (CPM) averaged across replicates. n = 3 replicates per genotype. **** p < 0.0001 (ANOVA
test). D, Summary of relative contributions for 22-nt sRNA producing loci in WT pollen. Hairpin/inverted repeat (IR) hp-siRNAs represent the largest fraction of 22-nt species. E, Heatmap
showing 22-nt hp-siRNA levels at hpRNA loci in leaf and pollen. F, Heatmap showing 22-nt siRNA levels at protein-coding genes in leaf and pollen. G, Browser shots showing 22-nt hp-siRNA
accumulation at a hpRNA locus on chromosome 1 (left) and 22 nt siRNA silencing at a representative protein-coding gene. Scale is CPM. EXTENDED DATA FIG. 4 VALIDATION OF HIGHLY ABUNDANT
POLLEN HAIRPIN PRECURSORS. A, hp-siRNAs are expected to show strand bias. Measurement of strand score (min[plus, minus]/max[plus, minus]) at 28 putative hairpin precursors and randomly
selected siRNA clusters from wild-type (W22) maize. A value of zero indicates complete strand bias, whereas a value of 1 indicates unbiased accumulation from both strands. n = 28. **** p
< 0.0001 (Welch’s t-test). B, log2 read count at hairpin precursors indicate 22 nt size bias. n = 28. **** p < 0.0001 (ANOVA). C, Example of a 73 nt stretch from a 4,480 bp hairpin
precursor demonstrating near-complete reverse complementarity. D, Mountain plots measuring thermodynamic stability of the _Tpd1_ hairpin from _mexicana_ and another randomly selected hairpin
structure. EXTENDED DATA FIG. 5 ORIGINS AND TARGETS OF 22 NT SMALL RNAS IN _TPD_ POLLEN. A, RNAi genes (_Sgs3_/_Rgd1_, _Rdr6_, _Ago1e_ and _Dcl2_) associated with 22 nt biogenesis and
function are upregulated in _TPD_ pollen. Expression is shown in TMM normalized counts. Bars show mean ± SD. n = 5 replicates per condition. **** p < 0.0001, *** p < 0.001, ** p <
0.01 (FDR). B, Relative abundances of _TPD_-dependent 22 nt siRNAs mapping to annotated elements. Pie chart inset shows proportions of 22 nt siRNAs targeting genes in CPM. C, Browser shot
showing transcriptional activation at PIF/Harbinger elements in _TPD_ pollen as well as 22 nt siRNA accumulation. D, Quantification of mRNA expression at 258 PIF/Harbinger superfamily
elements in WT and _TPD_ pollen. **** p < 0.0001 (Mann-Whitney test). E, 22 nt siRNA levels at 42 PIF/Harbinger elements in WT and _TPD_ pollen. **** p < 0.0001 (Mann-Whitney test).
EXTENDED DATA FIG. 6 _TPD_-DEPENDENT SILENCING OF A GDSL LIPASE DISRUPTS LIPID METABOLISM. A, Browser shots showing ectopic accumulation of 22-nt siRNAs at protein-coding genes in _TPD_
pollen. Scale in counts per million (CPM). B, RNA-seq tissue expression of 22-nt siRNA targets specific to _TPD_ pollen, data from ref. 125. Bars show mean ± SD. n = 3 replicates per tissue.
C, RNA-seq expression of 22-nt siRNA targets in WT and _TPD_ pollen. Bars show mean ± SD. n = 5 replicates per condition. D, Western blot comparing TDR1 protein levels in WT and _TPD_
pollen, anthers, and leaf. Protein levels were normalized using Heat Shock Protein 90 (HSP90). E, p-nitrophenyl butyrate esterase activity assay in 5 mm anthers and pollen from WT, _TPD_,
and _Tpd1_ + _/−_ plants. F, G, GO term biological processes up-regulated in F, _TPD_ and G, WT pollen (FDR ≤ 0.001). Upregulated genes in _TPD_ pollen were associated with RNA metabolism,
ribosome assembly, and cytoplasmic translation as well as G2 mitotic arrest. This could reflect translational repression via 22-nt siRNAs. Interestingly, a subset of genes associated with
endoplasmic reticulum (ER)-nucleus signalling was also up-regulated126, while genes associated with glycerol metabolism, the primary backbone for TAG synthesis, were downregulated. In
pollen, the accumulation of TAGs in lipid droplets (LDs) is critical for proper membrane expansion and pollen tube growth127. EXTENDED DATA FIG. 7 _TPD1_ AND _DCL2_ ARE EXPRESSED
PRE-MEIOTICALLY, WHEREAS _TDR1_ IS EXPRESSED IN MICROSPORE AND POLLEN. A, RT-qPCR of the _Tdr1_ transcript throughout anther development and in mature pollen. Bars show mean ± SD. n = 2
replicates per condition. B, RT-qPCR of the _Tpd1_ transcript during anther development and in mature pollen in WT and _Tpd_. Bars show mean ± SD. n = 2 replicates per condition. C, D,
Single-cell expression at different stages of meiosis of C, _Tdr1_ and D, _Dcl2_, using single cell RNAseq data59. Early and late expression of _Dcl2_ coincides with _Tpd1_ and _Tdr1_,
respectively. EXTENDED DATA FIG. 8 _TPD2_ SUPPRESSES 22NT SECONDARY SMALL RNAS. A, Browser shots showing ectopic accumulation of 22nt siRNAs at _Tdr1_ (left) and _Tpd1_ hairpin (right) in
fertile (_tpd1; tpd2_, grey), drive (_Dcl2__T_ _Tpd1/Dcl2 tpd1; Tpd2/tpd2_, blue) and sterile (_Dcl2 Tpd1/Dcl2 tpd1; tpd2_, red) pollen from maternal segregants. Scale in counts per million
(CPM). _Tpd2_ and _Dcl2__T_ reduce small RNAs from _Tdr1_ (left) but not from the _Tpd1_ hairpin (right), consistent with a cell autonomous role in secondary small RNA biogenesis and
silencing. B, Table summarizing genotypes transmitted when _TPD_ is backcrossed to W22 as female (left column) or male (right column). Only the combination of _dcl2__T_ _Tpd1_ (linked) and
_Tpd2_ is transmitted through pollen. Recombinants between _dcl2__T_ and _Tpd1_ occur at the expected frequency but are not transmitted through pollen (Fig. 3b), presumably because of higher
siRNA production during and after meiosis. C, _Rdm1_ (Zm00004b029511) is one of six genes in the _Tpd2_ interval expressed in pollen, and is overexpressed in _TPD_ pollen. EXTENDED DATA
FIG. 9 SIGNATURES OF _TEOSINTE POLLEN DRIVE_ IN MODERN MAIZE, MAIZE TRADITIONAL VARIETIES AND SYMPATRIC MEXICANA POPULATIONS. A, Frequency of mexicana-derived alleles were calculated for 1
Mb intervals associated with _TPD_ on chromosomes 1,2,3,4,5,6 and 10. Correlations are shown between population means from each of 14 maize traditional varieties (left) and sympatric
_mexicana_ populations (right). Intervals on chromosomes 5, 6 and 10 include _Dcl2_ (5.19), _Tdr1_ (5.40), _Tpd1_ (5.79), _Rgd1/Sgs3_ (6.3) and candidate genes _Ago1a_ (6.3), _Tpd2/Rdm1_
(6.98), _Ago1b_ and _Ago2b_ (10.134). Correlations were observed for most of the intervals in maize traditional varieties, except for _Tdr1_ (green arrow), but only for intervals including
_Tpd1_, _Rgd1_ and _Tpd2_ in _mexicana_. Spearman correlation coefficients are displayed as a heatmap. B, _mexicana_-derived ancestry in each of 14 maize traditional varieties (above) and
sympatric _mexicana_ populations (below) in _Dcl2_, _Tdr1, Tpd1_ and _Tpd2_ intervals. The _Tdr1_ interval (green) is monomorphic in most of the maize traditional varieties, but shows
extreme dimorphism in 7 out of 14 sympatric _mexicana_ populations. EXTENDED DATA FIG. 10 MECHANISTIC MODEL OF _TEOSINTE POLLEN DRIVE._ A, The _TPD_ system is defined by _mexicana_
introgression intervals on chromosomes 5 and 6. _Tpd1_ encodes a pre-meiotically expressed _mexicana_-specific hairpin that produces abundant 22nt hp-siRNAs. B, These hp-siRNAs trigger
secondary siRNAs amplification by RDR6 and SGS3/RGD1 at the _Tdr1_ gene when it starts being transcribed at the late tetrad stage, which in turn target _Tdr1_ for translational repression
(red ribosomes). In surviving microspores (dark yellow background) _Tpd2_ and _dcl2__T_ repress secondary siRNAs processing, restoring translation and fertility (green ribosome). C, Only
pollen grains of the genotype _dcl2__T_ _Tpd1_; _Tpd2_ are viable, and all other competing gametes are eliminated. Other RNAi genes (_Sgs3_/_Rgd1_, _Ago1_, _Ago2_, _Ago5_) can act as partial
suppressors by affecting levels of siRNAs. EXTENDED DATA FIG. 11 EVOLUTIONARY MODEL OF _TEOSINTE POLLEN DRIVE._ After the antidotes arise in an ancestral teosinte population, the _Tpd1_
toxin arises and gains a transmission advantage when linked to the antidote genes. In extant populations of _Z. mexicana_ and _Z. mays_, some antidotes are fixed, while others are
polymorphic or lost. The demographic model was based on ref. 128 and the conceptual framework of selfish evolution was adapted from ref. 67. Graphics were created using BioRender
(https://biorender.com). SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION Supplementary Discussion, Supplementary Tables 1–3 and Supplementary Tables 6–9 REPORTING SUMMARY SUPPLEMENTARY
TABLES SUPPLEMENTARY TABLE 4 22nt clusters enriched in WT pollen vs WT leaf SUPPLEMENTARY TABLE 5 22nt clusters enriched in _TPD_ pollen vs WT pollen RIGHTS AND PERMISSIONS OPEN ACCESS This
article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as
you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party
material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s
Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Berube, B., Ernst, E., Cahn, J. _et al._
_Teosinte Pollen Drive_ guides maize diversification and domestication by RNAi. _Nature_ 633, 380–388 (2024). https://doi.org/10.1038/s41586-024-07788-0 Download citation * Received: 14 June
2023 * Accepted: 04 July 2024 * Published: 07 August 2024 * Issue Date: 12 September 2024 * DOI: https://doi.org/10.1038/s41586-024-07788-0 SHARE THIS ARTICLE Anyone you share the following
link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature
SharedIt content-sharing initiative