Play all audios:
ABSTRACT Chaperonins are large barrel-shaped complexes that mediate ATP-dependent protein folding1,2,3. The bacterial chaperonin GroEL forms juxtaposed rings that bind unfolded protein and
the lid-shaped cofactor GroES at their apertures. In vitro analyses of the chaperonin reaction have shown that substrate protein folds, unimpaired by aggregation, while transiently
encapsulated in the GroEL central cavity by GroES4,5,6. To determine the functional stoichiometry of GroEL, GroES and client protein in situ, here we visualized chaperonin complexes in their
natural cellular environment using cryo-electron tomography. We find that, under various growth conditions, around 55–70% of GroEL binds GroES asymmetrically on one ring, with the remainder
populating symmetrical complexes. Bound substrate protein is detected on the free ring of the asymmetrical complex, defining the substrate acceptor state. In situ analysis of GroEL–GroES
chambers, validated by high-resolution structures obtained in vitro, showed the presence of encapsulated substrate protein in a folded state before release into the cytosol. Based on a
comprehensive quantification and conformational analysis of chaperonin complexes, we propose a GroEL–GroES reaction cycle that consists of linked asymmetrical and symmetrical subreactions
mediating protein folding. Our findings illuminate the native conformational and functional chaperonin cycle directly within cells. SIMILAR CONTENT BEING VIEWED BY OTHERS CRYOEM REVEALS THE
STOCHASTIC NATURE OF INDIVIDUAL ATP BINDING EVENTS IN A GROUP II CHAPERONIN Article Open access 06 August 2021 IN SITU ANALYSIS REVEALS THE TRIC DUTY CYCLE AND PDCD5 AS AN OPEN-STATE
COFACTOR Article Open access 11 December 2024 SNAPSHOTS OF ACTIN AND TUBULIN FOLDING INSIDE THE TRIC CHAPERONIN Article Open access 21 April 2022 MAIN The bacterial chaperonin GroEL
cooperates with its cofactor GroES in assisting the folding of roughly 10% of newly synthesized proteins, including proteins with α/β topology that fail to fold spontaneously1,2,7,8. GroEL
is a cylindrical complex of around 800 kDa containing two heptameric rings of 57 kDa subunits stacked back to back. The subunits consist of apical, intermediate and equatorial domains and a
flexible C-terminal tail protruding into the ring cavity9 (Fig. 1a, top left inset). The apical domains mediate substrate protein (SP) binding and the equatorial domains mediate ATP binding
and hydrolysis. Hydrophobic residues at the apical domains recruit unfolded SP. ATP-dependent binding of the lid-shaped GroES (a heptamer of 10 kDa subunits), capping the SP-containing ring
(the _cis_-ring), results in the burial of hydrophobic surfaces on GroEL and displaces the bound protein into an enclosed chamber. SP folds inside this chamber during ATP hydrolysis on the
GroEL _cis_-ring, and a second SP can bind to the _trans_-ring. The _cis_-chamber opens following ATP binding to the _trans_-ring, dissociating GroES through negative inter-ring allostery to
allow SP release1,2,10,11,12. Thus, the two rings of GroEL are sequentially folding active. However, in vitro studies1,2,13 showed that GroES not only binds asymmetrically with GroEL
(‘bullet’ complexes, EL–ES1), but can also associate symmetrically with both rings (‘football’ complexes, EL–ES2). Some reports have suggested that SP binding shifts GroEL entirely from an
asymmetrical cycle to a symmetrical mode14. The cell cytosol is characterized by a high degree of macromolecular crowding, which profoundly affects protein–protein interactions15. To
investigate how the available in vitro data apply to the situation in the intact cell, here we explored the chaperonin mechanism within its natural cellular context by cryo-electron
tomography (cryo-ET)—a technique enabling in situ visualization of macromolecular assemblies at subnanometre resolution16,17,18,19,20,21,22,23. We find that the native chaperonin cycle
consists of linked asymmetrical and symmetrical subreactions mediating protein folding. GROEL–GROES COMPLEXES IN SITU For visualization of GroEL by cryo-ET in situ, _Escherichia coli_
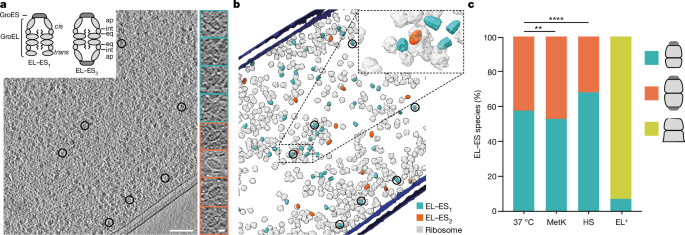
BL21(DE3) cells were vitrified on electron microscopy grids and thinned by cryogenic focused ion beam milling before imaging (Fig. 1a,b and Extended Data Fig. 1a). EL–ES1 and EL–ES2
complexes were readily observed in raw tomograms (Fig. 1a, right insets), whereas GroEL alone was undetectable. We used template matching with reference structures for systematic
identification and classification (Extended Data Fig. 1b), showing the relative proportions and cellular distribution of these complexes. In cells growing at 37 °C, EL–ES1 and EL–ES2
complexes occurred at an approximate ratio of 60:40% (Fig. 1c). To validate the accuracy of the template-matching results we compared the numbers of identified chaperonin complexes with
those of ribosomes, which can be readily identified in cryo-ET23,24. We localized essentially all cellular ribosomes (Extended Data Fig. 2a,b), and determined a median ratio of GroEL to
ribosomes of 1:23 during growth at 37 °C (Extended Data Fig. 2c). Quantification by mass spectrometry (MS) confirmed these results (Extended Data Fig. 2c, blue crosses), indicating that our
cryo-ET analysis had identified most GroEL complexes. However, owing to the inherent limitations of template matching, we cannot rule out a small fraction of false-positive or false-negative
particles. To load GroEL with chaperonin-dependent SP, we first increased the level of both GroEL and GroES by around sixfold (Extended Data Fig. 3a–c), to reduce occupancy with endogenous
SP, and then strongly overexpressed the obligate GroEL substrate _S_-adenosylmethionine synthase (MetK)25,26 (Extended Data Fig. 3d,e). Biochemical analysis by GroEL immunoprecipitation and
MS demonstrated that, on average, about 1.3 MetK molecules bound per GroEL complex, corresponding to over 50% of GroEL rings containing MetK (Extended Data Fig. 3f,g). The relative abundance
of EL–ES1 and EL–ES2 complexes in tomograms was about 55% and 45%, respectively, similar to growth without MetK overexpression (Fig. 1c). To explore changes in chaperonin function under
stress, we exposed cells to heat stress (HS) at 46 °C for 2 h. Note that _E. coli_ grows efficiently under HS in full medium (Extended Data Fig. 3d) although numerous proteins are
destabilized27, increasing the demand for chaperonin. HS induced a roughly threefold increase in GroEL and GroES abundance (Extended Data Fig. 3a,b), with a ratio of GroEL to ribosomes of
about 1:10 in MS and cryo-ET data (Extended Data Fig. 2c). Notably, the level of EL–ES1 complexes increased to 70% of total (Fig. 1c); GroEL alone remained undetectable. Thus, HS promotes
the formation of asymmetric chaperonin complexes. We next investigated whether EL–ES2 complexes form as a consequence of GroES:GroEL concentration ratio. Expression of the _groES_ and
_groEL_ genes (_groESL_), organized in an operon28, resulted in an approximate 1:1 GroES:GroEL ratio29, equivalent to around a twofold excess of GroES (7-mer) over GroEL (14-mer)30, with
both proteins being essential28. To reverse the physiological ratio of GroES and GroEL we selectively overexpressed GroEL (EL+ cells) at 37 °C, resulting in a roughly 4.5-fold increase in
GroEL (Extended Data Fig. 3h). EL+ cells grew essentially as wild-type (WT) (Extended Data Fig. 3d) but contained only free GroEL (EL complex) and EL–ES1 (around 90% and 10% of total GroEL,
respectively) and no EL–ES2 complexes (Fig. 1c). Notably, because EL–ES1 complexes were of similar abundance relative to ribosomes as in WT cells (Extended Data Fig. 3i), the absence of
EL–ES2 resulted in a reduction in the overall level of GroEL–GroES complexes. Nevertheless, overexpression of MetK did not impair the growth of EL+ cells (Extended Data Fig. 3e). In summary,
asymmetrical and symmetrical chaperonin complexes coexist in vivo, with EL–ES1 predominating under all growth conditions tested, including high SP load and HS. Cells grew efficiently when
EL–ES2 complexes were not populated, indicating that EL–ES1 complexes are sufficient for function. IN SITU STRUCTURES OF CHAPERONIN COMPLEXES Subtomogram averaging (STA) produced structural
models for EL–ES1, EL–ES2 and EL complexes at around 10–12 Å resolution following the application of symmetry (Fig. 2a–d, Extended Data Fig. 4a–d and Extended Data Table 1). Molecular models
were derived, starting from rigid-body fitting of high-resolution GroEL structures. EL–ES1 complexes were further classified based on the positioning of the apical domains of the GroEL
_trans_-ring, resulting in two conformations referred to as ‘narrow’ and ‘wide’ (Fig. 2a,b,e,f). In the narrow state the opening of the _trans_-ring has a diameter of around 45 Å (Fig. 2f),
similar to the EL–ADP7–ES1 crystal structure (PDB 1AON31) (Extended Data Fig. 4e). By contrast, the wide conformation shows a significant reorientation of the apical domains, extending the
ring opening to around 65 Å (Fig. 2f), which would facilitate the exit of larger SPs such as folded MetK (approximately 70 × 60 × 30 Å3 in size). Consistent with this interpretation, a
similar conformation was observed in a cryo-electron microscopy (cryo-EM) structure of EL–ES1 with bound ADP (PDB 7PBJ32) (Extended Data Fig. 4f). Under all conditions analysed, the wide
_trans_-ring conformation was more abundant than the narrow state, especially following HS (Extended Data Fig. 4g). The in situ structure of EL–ES2 at the given resolution showed no major
deviations from the crystal structure of the non-cycling symmetrical complex with bound ADP–BeFx (PDB 4PKO33) (Extended Data Fig. 4h). Interestingly, in the in situ structure of GroEL alone
at a resolution of about 9.8 Å (Extended Data Fig. 4d)—attained following GroEL overexpression (EL+)—one ring mirrored the wide _trans_-ring conformation of EL–ES1 whereas the other was in a
more narrow state (Fig. 2d) with continuous additional density at the apical domains (Extended Data Fig. 4i,j). This density probably resulted from symmetry-averaged, unfolded SP that had
accumulated on GroEL at substoichiometric GroES. Thus the GroEL complex shows intrinsic inter-ring asymmetry in vivo, reflecting the negative allosteric coupling between rings and leading to
preferential substrate binding to one ring. VISUALIZATION OF SUBSTRATE IN THE GROEL–GROES CYCLE Similar to GroEL alone, the _trans_-ring of EL–ES1 in the narrow state also contained central
density at the apical domains (indicated by arrowheads in Fig. 3a), presumably representing bound SP before encapsulation by GroES. No SP density was observed in the wide _trans_-ring, nor
was a narrow state without bound SP resolved (Fig. 3a). Indeed, the apical domains in the narrow state expose the functionally critical hydrophobic residues in helices αI and αH, forming a
continuous furrow for SP binding34, whereas in the wide state the coherent binding surface was disrupted (Fig. 2f). Thus, following GroES dissociation, the _trans_-ring in its wide
conformation would allow SP release whereas binding of new SP presumably occurs following conversion to the narrow conformation. Interestingly, the ratio of EL–ES1 with wide _trans_-ring to
EL–ES1 with narrow _trans_-ring (Extended Data Fig. 4g) correlated closely with the overall ratio of EL–ES1 to EL–ES2 (Fig. 1c). This suggests that binding of SP to the _trans_-ring may
facilitate the formation of symmetrical complexes by lowering negative inter-ring allostery14. Furthermore, because EL–ES1 species with SP bound in _trans_ are populated, association of the
second GroES must be a relatively slow step. Next, for visualization of encapsulated SP we extracted and pooled GroEL–GroES chambers from all EL–ES1 and EL–ES2 complexes and analysed them by
averaging and three-dimensional classification of the chamber interior (Extended Data Fig. 4k). For each growth condition we identified two distinct classes of complex (Fig. 3b): the
GroEL–GroES chambers of class I contained a well-defined globular density close to the bottom of the cavity, consistent with structured SP. The chambers of class II showed only a weak and
fuzzy density, representing empty cavities and/or the presence of dynamic, non-native SP conformations that would be obscured by averaging. Sorting the EL–ES1 and EL–ES2 complexes in the in
situ datasets according to the presence of encapsulated and/or bound SP allowed us to quantify a total of seven different states of EL–ES1 and EL–ES2 (Fig. 3c). At 37 °C growth, the relative
proportions of these species were largely independent of MetK overexpression, with a subset of EL–ES2 complexes containing structured SP in both chambers. Interestingly, following HS,
EL–ES1 complexes with wide _trans_-ring conformation (no bound SP) were enriched (Fig. 3c(i,ii)) and EL–ES2 complexes reduced (Fig. 3c(v–vii)), perhaps due to changes in the ATP:ADP ratio
during HS35. This is consistent with SP binding to the _trans_-ring facilitating EL–ES2 formation. These results define the chaperonin species that are populated in vivo and demonstrate that
complexes EL–ES1 and EL–ES2 are both functionally active. STRUCTURE OF METK INSIDE CHAPERONIN To what extent does SP fold inside the chaperonin chamber during the functional GroEL–GroES
cycle in vivo? Previous in vitro cryo-EM analyses of encapsulated client protein under non-cycling conditions had shown a distinct density in the equatorial half of the chamber, representing
SP folding intermediates at low resolution36,37,38,39. We performed a similar in vitro analysis on encapsulated MetK, by both cryo-EM and cryo-ET, for comparison with the in situ cryo-ET
structures. We prepared SP-bound GroEL by heat denaturation of MetK in the presence of GroEL40. Encapsulation occurred following the addition of GroES and ATP-BeFx (Extended Data Fig. 5a,b).
BeFx favours the formation of stable (non-cycling) EL–ES2 complexes with bound ADP–BeFx (ref. 41). MS analysis indicated a stoichiometry of MetK to GroEL 14-mer of roughly 1.2 (Extended
Data Fig. 5c), similar to MetK overexpression (Extended Data Fig. 3f,g). Reference-free, two-dimensional classification demonstrated the presence of EL–ES2 as well as some EL–ES1 complexes
(Extended Data Figs. 5d and 6a–d). The latter exhibited subpopulations with wide and narrow _trans_-ring conformations resembling those observed in situ (Extended Data Fig. 6e,f), with
density for bound SP in the narrow state (Extended Data Fig. 6f). For visualization of encapsulated SP, GroEL–GroES chambers were processed for cryo-EM structure determination (Extended Data
Figs. 5d and 6c,d). Alignment and classification showed that around 40% of GroEL–GroES units contained density for an ordered MetK molecule close to the equatorial region of the chamber
(Fig. 4a–d, Extended Data Fig. 7 and Extended Data Table 1). The remainder contained only a faint, smeared-out density, representing empty chambers and chambers with incompletely folded or
misaligned MetK. The substructure of the ordered MetK molecules was solved at a resolution of approximately 3.7 Å, showing side-chain density in its hydrophobic core (Extended Data Fig.
7d–f). The encapsulated MetK was native-like, with a root mean squared deviation relative to the crystal structure (PDB 7LOO42) of 1.4 Å for 366 of the 379 Cα atoms (Fig. 4e). The main
difference was in the conformation of residues 97–111, the so-called core loop. This region packs against bound _S_-adenosylmethionine and an adjacent subunit in the MetK tetramer42,43
(Extended Data Fig. 8a), but in the encapsulated MetK subunit adopted a more extended conformation that was not well resolved (Fig. 4e). The core loop apparently remains unstructured until
tetramer assembly following release from chaperonin. The encapsulated MetK makes multiple contacts with the GroEL cavity wall, contacting two subunits at Phe44 in the equatorial GroEL domain
as well as five subunits at Phe281 and three at Tyr360, both protruding from the apical GroEL domains (Fig. 4b–d). These residues appear to interact with MetK via van der Waals contacts.
However, the side chains of the interacting residues are poorly defined, indicating heterogeneity in these regions of the structure (Fig. 4f). The GroEL subunits contacting MetK show only
minor conformational rearrangements, with root mean squared deviation values of 0.5–1.0 Å compared with a new 2.5 Å cryo-EM structure of empty GroEL–(ADP–BeFx)7–ES chambers (Extended Data
Fig. 8b–g and Extended Data Table 1). Of note, the GroEL cavity wall does not contact the interface regions of the MetK subunit that become buried following assembly. These regions
apparently remain solvent exposed in the chamber (Extended Data Fig. 8a) but could be reached by flexible C-terminal Gly–Gly–Met repeat sequences (23 residues) of the GroEL subunits not
resolved in the cryo-EM structure. To further rationalize our in situ cryo-ET analysis of encapsulated SP (Fig. 3b), we next performed cryo-ET on isolated GroEL–GroES–MetK complexes using
the same imaging parameters as for in situ tomography (Extended Data Table 1). In agreement with the single-particle data, the classification of chambers within these complexes again yielded
two classes. Class I (around 40% of particles) contained a strong density in the chaperonin cavity, corresponding well with symmetry-averaged folded MetK (Fig. 4g, left), whereas class II
chambers (roughly 60% of particles) showed a weak, diffuse density (Fig. 4h, left). The location of the structured MetK near the equatorial region of the GroEL–GroES chamber and its density
relative to the GroEL wall (Fig. 4g, left) coincided with that of the folded SP in situ (Fig. 4g, right). Specifically, the position of the SP centre of mass following MetK overexpression,
in which MetK is highly enriched on GroEL (Extended Data Fig. 3f,g), was in almost perfect agreement with the position of the folded MetK in the in vitro tomograms (Extended Data Fig. 9).
Although other SPs besides MetK may be present within GroEL in situ, our data suggest that these proteins occupy a similar location within the chamber. Thus, encapsulation in vivo resulted
in SP folding to a native or native-like, compact state. CONCLUSIONS Our analysis of GroEL–GroES complexes in situ using cryo-ET allows us to define the intermediate steps of the bacterial
chaperonin cycle in vivo. We find that both asymmetric and symmetric chaperonin complexes operate in linked subreactions (Fig. 5). GroEL without bound GroES is below detectability and may
exist only transiently (Fig. 5(i)). By contrast, asymmetric EL–ES1 with and without bound SP on the _trans_-ring is abundant, defining the main SP acceptor state (Fig. 5(ii)). In the
asymmetric reaction the GroEL rings alternate between folding active and binding active. Following GroES dissociation, SP exits the folding chamber (Fig. 5(iii–i)), facilitated by a wide
conformation of the apical GroEL domains, possibly generating a short-lived GroEL-only intermediate (Fig. 5(i)). Alternatively, rather than completing the asymmetric cycle, GroES binding to
the _trans_-ring gives rise to EL–ES2 (Fig. 5(iii–iv)), in which both rings can be folding active. Because folding begins in the _cis_-chamber of EL–ES1 and can continue in the EL–ES2
complex, the symmetric cycle may benefit SPs with slow folding kinetics13. How is the partitioning between asymmetric and symmetric chaperonin reactions regulated? In the canonical
asymmetric cycle in vitro the GroEL rings are coupled by negative allostery, with ATP binding to the _trans_-ring causing ADP and GroES release from the _cis_-ring (Fig. 5 (species
ii/iii–i))1,44,45. Negative inter-ring allostery also operates in vivo, favouring EL–ES1 formation, because exclusively EL–ES1 complexes mediate protein folding at GroEL excess over GroES.
In WT cells, EL–ES2 complexes are also functional. Conversion of the EL–ES1 _trans_-ring from a wide conformation to the narrow, SP-binding state (Fig. 5(ii–iii)) appears to be limiting for
EL–ES2 formation (Fig. 5(iii–iv)), because the ratio of EL–ES1 wide to EL–ES1 narrow correlates closely with the overall EL–ES1 to EL–ES2 ratio. Our cryo-ET analysis also demonstrated that,
before release into bulk cytosol, SP reaches a folded state in the GroEL–GroES chamber. To validate this finding we solved as a reference the structure of stably encapsulated MetK, an
obligate GroEL substrate26, in vitro. The MetK subunit is natively folded and is located close to the equatorial region of the GroEL–GroES cavity36,38,39. Encapsulated MetK makes weak
contacts with specific GroEL residues (Fig. 4) and is in close proximity to flexible C-terminal GGM repeat sequences of the equatorial GroEL domains, which may promote efficient
folding46,47. The position and density of folded, encapsulated MetK closely resemble those of structured SP in the GroEL–GroES chamber in situ. In summary, our analysis provides a detailed
view of the chaperonin reaction cycle in vivo, in which asymmetric and symmetric GroEL–GroES complexes are functionally linked. SP accumulates inside the chaperonin chamber in a folded state
before release into cytosol. METHODS PLASMIDS AND STRAINS _Escherichia coli_ BL21(DE3) Gold cells (Stratagene) were used for growth analysis, electron tomography and protein expression. For
tomography and biochemical experiments, GroEL was expressed from a pBAD33 plasmid containing the _groEL_ gene under the control of an araBAD promotor (EL+ cells)26. For overexpression of
GroEL and GroES, a pBAD33 plasmid containing both _groEL_ and _groES_ genes under the control of an araBAD promotor was used48. MetK was expressed from a pET22b plasmid previously
described26. ANTIBODIES Polyclonal antisera used against GroEL, GroES, MetK and GAPDH were previously described26, and the rabbit antiserum against α-lactalbumin was a product of East Acres
Biologicals immunization service. _E. COLI_ GROWTH _E. coli_ cells were grown in lysogeny broth (LB) medium that contained, depending on the plasmids used, the antibiotics ampicillin (200 μg
ml−1, pET22b-MetK) and chloramphenicol (32 μg ml−1, pBAD33 variants). For overexpression of GroEL, GroES and MetK (MetK cells), transformed _E. coli_ Bl21 (DE3) pBAD33-GroEL:ES pET22b-MetK
cells were grown to early exponential phase at 37 °C, and GroEL–GroES expression using the pBAD33 promoter was induced for 90 min by supplementation of LB medium with arabinose to a final
concentration of 0.2% (w/v). Cells were subsequently harvested by centrifugation at 8,000_g_ (4 °C for 10 min) and resuspended to an optical density (600 nm, OD600) of 0.1–0.2 in fresh LB
medium containing both antibiotics and 1 mM isopropyl β-d-thiogalactopyranoside (IPTG), to induce MetK expression under control of the T7 promoter for 40 min. GroEL expression (EL+) was
induced in _E. coli_ Bl21 (DE3) pBAD33-GroEL by supplementation of LB medium with arabinose to a final concentration of 0.1% (w/v) and growth of the culture at 37 °C. To expose _E. coli_
Bl21 (DE3) cells to HS, cells were first cultured to early exponential phase at 37 °C and then incubated in a shaking water bath at 46 °C for 2 h. _E. COLI_ GROWTH CURVES Cells were cultured
as described above. Aliquots were removed at the time points indicated for optical density measurement at OD600. To ensure exponential growth conditions, growing cultures were diluted to an
OD600 of 0.1 with prewarmed LB medium containing the necessary antibiotics and arabinose when OD600 just exceeded 0.4. Growth curves for MetK and EL+/MetK cells were measured following
termination of GroEL induction by transfer of cells into arabinose-free medium containing 1 mM IPTG for MetK overexpression. The first sample was taken 5 min after changing the medium. Data
were processed for fitting in R. PROTEIN EXPRESSION AND PURIFICATION GroEL, GroES and MetK proteins were expressed and purified as previously described26,49. MEASUREMENT OF PROTEIN
CONCENTRATION Concentrations of purified proteins were determined by measurement of absorbance at 280 nm using absorbance coefficients calculated from the protein sequence with the program
ProtParam50. Protein concentrations of cell lysates were determined with the Pierce Coomassie Plus (Bradford) Assay Kit (Thermo Fisher Scientific) as described by the manufacturer.
PREPARATION OF CELL LYSATES Cultures were prepared as described above, harvested by centrifugation and the cell pellet flash-frozen in liquid nitrogen before further processing. Spheroplasts
were prepared at 4 °C as previously described51. In brief, cells were resuspended in 100 mM Tris-HCl pH 8.0 and washed twice with 2 ml of buffer. The pellet was then resuspended in HMK
buffer (50 mM HEPES-KOH pH 7.2, 20 mM Mg acetate, 50 mM K acetate) supplemented with 20% (w/v) sucrose and 0.25 mg ml−1 lysozyme. Cells were then incubated on ice for 7 min and transferred
to 37 °C for 10 min. The resulting suspension was supplemented with Complete EDTA-free protease inhibitor cocktail (Roche), and spheroplasts were lysed by the addition of 0.1% (v/v) Triton
X-100 and subsequent sonication. MASS SPECTROMETRY Cell lysates were reduced by the addition of dithiothreitol (DTT) to a final concentration of 10 mM and heated to 56 °C for 45 min.
Acylation of thiol groups was performed by the addition of chloroacetamide to a final concentration of 55 mM and incubation for 45 min in the dark, followed by a first digestion step with
Lys-C (Wako) at a w/w ratio of 1:20 for 2 h at 37 °C. This was followed by a second digestion step overnight with trypsin (Roche) at a 1:20 (w/w) ratio at 37 °C. The reaction was stopped by
the addition of trifluoroacetic acid to a final volume of 1%. Peptides were desalted using OMIX C18 (100 μl) tips (Agilent Technologies, no. A57003100) according to the manufacturer’s
instructions. Desalted peptides were dissolved in 12 µl of 5% formic acid, sonicated in an ultrasonic bath, centrifuged and transferred to autosampler vials (Waters). Samples were analysed
on an Easy nLC-1200 nanoHPLC system (Thermo) coupled to a Q-Exactive Orbitrap HF mass spectrometer (Thermo). Peptides were separated on pulled-spray columns (ID 75 μm, length 30 cm, tip
opening 8 μm, NewObjective) packed with 1.9 μm C18 particles (Reprosil-Pur C18-AQ, Dr Maisch) using either a stepwise 196 min gradient (comparison of 37 °C, HS and MetK) or a stepwise 67 min
gradient (all other samples) between buffer A (0.2% formic acid in water) and buffer B (0.2% formic acid in 80% acetonitrile). Samples were loaded on the column by the nanoHPLC autosampler
at a pressure of 900 bar. The high-performance liquid chromatography flow rate was set to 0.25 μl min−1 during analysis. No trap column was used. The following parameters were used for
comparison of growth conditions 37 °C, HS and MetK: MS, resolution 60,000 (full-width at half-maximum (FWHM) setting); MS mass range 300–1,650 _m_/_z_; MS-AGC-setting 3 × 106; MS-MaxIT 50
ms; MS/MS fragmentation of the 15 most intense ions (charge state 2 or higher) from the MS scan; MS/MS resolution 15,000 (FWHM setting); MS/MS-AGC-setting 105; MS/MS-MaxIT 50 ms; MS/MS
isolation width 1.8 _m_/_z_; collision-energy setting 29 (NCE). All other samples were analysed with the following parameters: MS resolution 120,000 (FWHM setting); MS mass range 300–1,650
_m_/_z_; MS-AGC-setting 3 × 106; MS-MaxIT 100 ms; MS/MS fragmentation of the ten most intense ions (charge state 2 or higher) from the MS scan; MS/MS resolution 15,000 (FWHM setting);
MS/MS-AGC-setting 105; MS/MS-MaxIT 50 ms; MS/MS isolation width 1.2 _m_/_z_; collision-energy setting 29 (NCE). MS DATA ANALYSIS Protein identification was performed using MaxQuant with
default settings. The _E. coli_ K12 strain sequences of UNIPROT (v.2023-03-01) were used as the database for protein identification (Supplementary Information). MaxQuant uses a decoy version
of the specified UNIPROT database to adjust false discovery rates for proteins and peptides below 1%. QUANTIFICATION OF METK BINDING TO GROEL To quantify the fraction of GroEL with bound
MetK in MetK-overexpressing cells, we immunoprecipitated GroEL with GroEL antibody followed by GroEL and MetK immunoblotting and liquid chromatography–tandem mass spectrometry. Cells were
prepared and lysed as described above, but with the addition of apyrase (25 U ml−1 final concentration) to rapidly deplete the ATP pool in the lysate and arrest the GroEL reaction cycle26.
The lysate was clarified by centrifugation at 16,000_g_ (4 °C for 10 min). Either 20 μl of a non-specific antibody (against α-lactalbumin) or a GroEL-specific antibody was coupled to 100 μl
of recombinant protein A Sepharose 4B beads (Thermo Fisher Scientific) as described by the manufacturer. The beads were loaded with sample (180 μg of protein) and incubated in 650 μl of HMK
buffer for 1 h. The beads were washed twice with 600 μl of HMK buffer and then twice more with HMK containing 0.1% Triton X-100. For immunoblotting, elution was performed with 50 μl of 2×
lithium dodecyl sulfate (Pierce) containing β-mercaptoethanol 5% (v/v) as prescribed by the manufacturer. For liquid chromatography–tandem mass spectrometry analysis, elution and digestion
were performed with the IST MS sample preparation kit (Preomics) using the manufacturer’s on-bead digestion protocol. Mass spectrometry was performed as described above. SDS–PAGE AND
IMMUNOBLOTTING Before SDS–polyacrylamide gel electrophoresis (SDS–PAGE) analysis, cells were resuspended in HMK buffer supplemented with 2 mM DTT, 1 mM EDTA and 5% glycerol and subsequently
sonicated, followed by centrifugation (20 min, 16,000_g_ at 4 °C). Protein samples were separated by electrophoresis on NuPAGE 10% Bis-Tris SDS gels (Invitrogen) using NuPAGE MES SDS running
buffer (Invitrogen) at 150 V. Proteins were transferred to polyvinylidene difluoride membranes in blotting buffer (25 mM Tris, 192 mM glycine, 20% methanol) at 150 mA. Membranes were first
incubated with primary antibodies in TBST buffer overnight at 4 °C and subsequently with horseradish peroxidase-conjugated secondary antibody for chemiluminescence detection. Uncropped
immunoblots are provided in the Source Data file to Extended Data Fig. 3. IN SITU CRYO-ET ANALYSIS Cell cultures were grown as described above. For cryo-ET analysis, cells in exponential
growth (approximate OD600 0.4) were rapidly (for about 2 min) concentrated to an approximate OD600 of 10 by centrifugation at 8,000_g_ and subsequently applied to R 2/1 100 Holey carbon film
Cu 200 mesh grids (Quantifoil) that were previously plasma cleaned for 30 s. The sample was blotted for 9 s at force 10 and then plunge-frozen in a mixture of liquid ethane and propane
cooled by liquid nitrogen using a Vitrobot Mark IV (Thermo Fisher Scientific) at 70% humidity and 22 °C. Frozen grids were transferred to a dual-beam, cryo-focused ion beam (FIB)/scanning
electron microscope (Thermo Fisher Scientific; either Scios, Quanta, Aquilos or Aquilos 2). Cells were coated with a layer of inorganic platinum, if available in the system used, followed by
the deposition of organometallic platinum using an in situ gas injection system (working distance, 10 mm; heating, 27 °C; time, 8 s). Removal of bulk material was done at a stage angle of
20–25° using gallium ions at 30 kV, 0.5 nA. Fine milling of lamellae was done at 11–13° stage tilt with successively lower currents between 0.3 nA and 30 pA, aiming for a final thickness of
100–200 nm (ref. 52). Lamellae for the selective GroEL overexpression dataset were prepared using Serial FIB53, and an additional layer of inorganic platinum was added following fine milling
to avoid charging during image acquisition54. The resulting lamellae were transferred to a TEM (Titan Krios, field emission gun 300 kV, Thermo Fisher Scientific) equipped with an energy
filter (Quantum K2, Gatan), a direct detection camera (K2 Summit, Gatan), and tomograms were acquired at a magnification of ×42,000 (pixel size 3.52 Å), defocus ranging from −5.0 to −3.0 μm
and the energy filter slit set to 20 eV using SerialEM 3.9.0 (ref. 55). Tomograms were recorded in dose-fractionated super-resolution mode, with a total dose of roughly 120 e−/Å2 per tilt
series. A dose-symmetric tilt scheme was used with an increment of 2–3° in a total range of ±60° from a starting angle of approximately 10° to compensate for lamellar pretilt (mostly around
11°)56. Frames were aligned using MotionCor2 (v.1.4.0, https://emcore.ucsf.edu/ucsf-software)57. The reconstruction was performed in IMOD using patch tracking (v.4.11.1, RRID:SCR_003297,
https://bio3d.colorado.edu/imod/)58 using the TOMOgram MANager (TOMOMAN) wrapper scripts59. Tilt-series images were dose filtered using TomoMAN’s implementation of the Grant and Grigorieff
exposure filter60. Defocus was estimated using CTFFIND4 (ref. 61). Tomograms of the EL+ dataset were acquired on a Krios G4 equipped with a Selectris X energy filter and Falcon 4 direct
electron detector (Thermo Fisher Scientific). Tilt series were collected with a dose-symmetric tilt scheme using TEM Tomography 5 software (Thermo Fisher Scientific). A tilt span of ±60° was
used with 2° steps, starting at ±10°, to compensate for lamellar pretilt. Target focus was changed for each tilt series in steps of 0.5 µm over a range of −2.5 µm to +5 µm. Data were
acquired in EER mode of Falcon 4 with a calibrated physical pixel size of 3.02 Å and a total dose of 3e−/Å2 per tilt over ten frames. A 10 eV slit was used for the entire data collection.
Data were preprocessed using TOMOMAN59. EER images were motion corrected using RELION’s implementation of MotionCor2 (ref. 62). Defocus was estimated using CTFFIND4 (ref. 61). Reconstruction
was performed with IMOD using local deposits of the inorganic platinum that was applied by sputtering following milling as fiducials. All tomograms were reconstructed using NovaCTF63. _E.
coli_ membranes were segmented for visualization using TomoSegMemTV 1.0. CRYO-ET ANALYSIS OF IN VITRO RECONSTITUTED GROEL–GROES COMPLEXES For generation of a GroEL–GroES reference for in
situ tomographic analysis containing a defined substrate protein in a folded state and in a known topology, we imaged in vitro reconstituted GroEL–GroES–MetK complexes using the same data
collection strategy and parameters as above for WT cells. SUBTOMOGRAM AVERAGING For subtomogram averaging, all datasets acquired on the same microscope (37 °C, HS, MetK) were combined and
processed together; the EL+ dataset was processed separately. The overall processing workflow is depicted in Extended Data Fig. 1b. For template matching, PDB entry 1AON was used for EL–ES1,
4PKO for EL–ES2 and 5MDZ for 70S ribosomes to generate templates at a resolution of 40 Å using the molmap64 command in Chimera65. Initial positions for a subset of EL–ES1 and EL–ES2
complexes and ribosomes were determined using the noise correlation template-matching approach implemented in STOPGAP, by fourfold binning to a pixel size of 14.08 Å (ref. 66). This subset
of the data was subsequently aligned and classified in STOPGAP to generate a reference from the tomographic data with a Fourier shell correlation (FSC) value close to 1 at 40 Å
template-matching resolution. Template matching with various GroEL14 species was attempted, but never yielded an average of GroEL14 with a resolution better than the template resolution. The
data-derived references of all three different structures were used for an additional round of template matching on the complete dataset. Cross-correlation cut-off was chosen separately for
every tomogram by visual inspection of the generated hits and comparison with the tomogram. To reduce the level of false-positive detection, a mask for the cytosol of the cell was first
created using AMIRA (Thermo Fisher Scientific) and subsequently used to filter out hits outside of the cytosol. Putative particles were deliberately overpicked with low-resolution templates
in the initial stage to avoid false-negative assignments. This procedure yielded 176,408 initial subtomograms for the EL–ES1 reference and 125,860 for the EL–ES2 reference. These were then
further aligned and classified separately in STOPGAP, each yielding classes containing both EL–ES1 and EL–ES2 particles. The combined number of particles contained in classes with emergent
high-resolution features (Supplementary Fig. 1a) for the EL–ES1 reference was 19,239, and 17,614 for the EL–ES2 reference (Extended Data Fig. 1 and Supplementary Fig. 1b). Because both
references pick up a subset of the other particles, the particles were then combined and duplicates removed. The resulting combined dataset was split by reference-free, three-dimensional
classification in STOPGAP, resulting in a set of 17,598 EL–ES1 and 11,213 EL–ES2 complexes that were then independently refined. This resulted in a resolution at the FSC cut-off of 0.143
following the application of symmetry at 11.6 Å for the EL–ES1 complex (_C_7 symmetry) and 11.9 Å for the EL–ES2 complex (_D_7 symmetry). Classification was performed using simulated
annealing stochastic hill-climbing multireference alignment as previously described67. All classifications were done repeatedly with different, random initial starting sets of 250–500
subtomograms to generate the initial references. Only particles that ended up in the same class for all independent rounds of classifications were retained67. Further refinements with the
established WARP, RELION, M pipeline were attempted but did not yield any further improvements. EL–ES1 wide and narrow complexes were separated by classification with a focused, disk-shaped
mask on the apical domains of the EL–ES1 _trans_-ring. This resulted in 6,681 narrow complexes that were refined to a resolution of 13.5 Å, and 10,130 wide EL–ES1 complexes refined to a
resolution of 12.0 Å. The EL+ dataset was processed in the same way, but starting with the structures from the other datasets, low-pass filtered to 40 Å, as initial references for template
matching. Template matching was then repeated once with structures generated by averaging a subset of particles from this dataset. To improve the resolution for model building, the dataset
was exported to WARP68 and angles and positions refined using RELION v.3.0.8 (ref. 69). This yielded a GroEL14 structure at a global resolution of 13 Å. GroEL 14-mer particles were corefined
for geometric distortions with ribosomes in M. The resulting GroEL 14-mer particles were exported for further alignment and classification in RELION. Classification was performed with a
regularization parameter T of four and six classes for 25 iterations without angular search, resulting in a more homogeneous subset of 12,421 particles. These particles were again corefined
in M for geometric distortions and per-particle defocus for contrast transfer function (CTF) estimation, resulting in a final structure with nominal resolution of 9.8 Å at 0.143 FSC cut-off.
Owing to their high molecular weight and density, ribosome template matching achieves a higher precision and recall. During initial rounds of classification in STOPGAP, because no
false-positive particles were detected, all ribosomal hits from template matching were aligned first in STOPGAP at progressively lower binnings (bin4, bin2, bin1). The resulting particles
were then exported to WARP using TOMOMAN. Subtomograms were reconstructed for RELION v.3.0.8 using WARP at a pixel size of 3.52 Å per pixel. An iterative approach with subtomogram alignment
in RELION and tilt-series refinement in M70 were performed until no further improvement in gold-standard FSC was obtained. This resulted in a final structure of the ribosome at a resolution
of 8.6 Å for the combined 37 °C, HS and MetK datasets, and 6.3 Å for the EL+ dataset, which was processed separately. In vitro cryo-ET data for GroEL–GroES complexes were processed analogous
to the in situ data, resulting in 39,518 initial hits for the EL–ES2 template and 46,093 for the EL–ES1 template, with both sets having a significant overlap. These were then further
aligned and classified separately in STOPGAP, yielding 5,832 and 13,688 particles, respectively, following duplicate removal. CLASSIFICATION OF SP OCCUPANCY OF GROEL–GROES COMPLEXES IN SITU
For the resolution of densities corresponding to substrate proteins in the GroEL–GroES chamber we first performed symmetry expansion around the _C_2 axis of the EL–ES2 complexes and aligned
the new set of GroEL–GroES chambers with the _cis_-ring of the EL–ES1 complexes. The resulting subtomograms of the chambers were then denoised using TOPAZ’s three-dimensional pretrained
denoising function71. Because initial attempts to classify the interior of the chamber using STOPGAP multireference-based alignment showed only separation by missing wedge, the subtomograms
were combined into 5,000 random bootstraps containing 250 random subtomograms each. These averages were then used to perform _k_-means clustering with two classes. Bootstraps from the
resulting clusters were averaged and used as initial start structures for multireference alignment in STOPGAP. For this, stochastic hill climbing was performed with a temperature factor of
10 for simulated annealing, followed by 40 iterations of multireference alignment with two classes and a mask around the interior of the chamber. This process was repeated five times. Only
particles consistently assigned to the same classes were used for a final round of subtomogram averaging, resulting in one class showing weak diffuse density inside the chamber and a second
showing strong density near the bottom. Attempts to further subdivide these two classes resulted only in separation based on missing wedge. Because it was not possible to resolve the _C_7
symmetry mismatch of the substrate and enclosing chamber, final averages were produced for all different biological conditions with _C_7 symmetry applied to increase the signal-to-noise
ratio. The class showing a strong density near the bottom contained 12,255 subtomograms, the one showing only a weak diffuse density with 24,435 subtomograms for the combined 37 °C, HS and
MetK datasets. In vitro data were processed analogously. The resulting classes were then again split into EL–ES1 and EL–ES2 complexes corresponding to their substrate state and exported to
WARP68. An additional round of alignments was performed in RELION for all different classes and complexes. A prior was set for all angles. Local search was performed with a sigma of 0.5 and
search angle of 0.9°. The resulting particles were separately refined in M, correcting for geometrical distortions. Particles were again exported from M70 and signal subtraction preformed in
RELION of the _trans_-ring for EL–ES1 and the opposing chambers for EL–ES2. Based on their previous classification results in STOPGAP, the refined signal-subtracted, single-chamber
complexes were combined in two groups resulting in 7,087 GroEL–GroES chambers containing an ordered SP and 14,371 that either contained a disordered SP or were empty. The resulting chambers
were again locally refined in RELION using priors and a sigma on all angles, yielding a resolution of 9.4 Å for GroEL–GroES chambers containing ordered SP and 8.8 Å for the remaining
chambers. CRYO-EM SINGLE-PARTICLE ANALYSIS OF GROEL–GROES–METK COMPLEXES For generation of substrate-bound GroEL–GroES complexes, 4 μM MetK was denatured in the presence of 1 μM GroEL
(14-mer) in buffer A (20 mM MOPS-NaOH pH 7.4, 200 mM KCl, 10 mM MgCl2, 5 mM DTT) containing 30 mM NaF and 5 mM BeSO4 by first incubation of the mixture at 60 °C for 15 min and then cooling
to 25 °C in a thermomixer (Eppendorf). The addition of 2 μM GroES (7-mer) and 1 mM ATP (pH 7.0) resulted in stable chaperonin complexes with encapsulated MetK40. Biochemical analysis of this
preparation was performed by size exclusion chromatography on a Superdex 200 3.2/300 GL column. Fractions were analysed by SDS–PAGE electrophoresis (NuPAGE, Bis-Tris 4–12% gels), and MetK
loading of GroEL–GroES complexes was estimated by mass spectrometry using intensity-based absolute quantification values72. For analysis by mass spectrometry, fractions F1 and F2 (Extended
Data Fig. 5a) were analysed separately but intensities pooled for the determination of intensity-based absolute quantification ratios. GroEL–GroES–MetK samples were concentrated tenfold by
ultrafiltration using a 100 kDa Amicon centrifugal concentrator (Millipore) at room temperature. As a control, GroEL and GroES were treated identically in the absence of MetK. Before
freezing, 1 μl of a _n_-octyl-β-d-glucopyranoside stock solution (87.5 mg ml−1 in buffer A) was added per 50 μl of sample. For single-particle analysis and in vitro cryo-ET experiments, 4 μl
of the sample was applied onto R 2/1 100 Holey carbon film Cu 200 mesh grids (Quantifoil) previously plasma cleaned for 30 s. This grid was blotted for 3.5 s at force 4 and plunge-frozen in
a mixture of liquid ethane and propane cooled by liquid nitrogen using a Vitrobot Mark IV (Thermo Fisher Scientific) at 100% humidity and 4 °C. Cryo-EM data for the EL–ES–MetK dataset were
acquired using a FEI Titan Krios transmission electron microscope and SerialEM software55. Video frames were recorded at a nominal magnification of ×22,500 using a K3 direct electron
detector (Gatan), with a total electron dose of around 55 electrons per Å2 distributed over 30 frames at a calibrated physical pixel size of 1.09 Å. Micrographs were recorded within a
defocus range of −0.5 to −3.0 μm. On-the-fly image processing and CTF refinement of cryo-EM micrographs were carried out using the Focus software package73. Only micrographs that met the
selection criteria (ice thickness under 1.05, drift 0.4 Å < _x_ < 70 Å, refined defocus 0.5 μm < _x_ < 5.5 μm, estimated CTF resolution under 6 Å) were retained. Micrograph
frames were aligned using MotionCor2 (ref. 57), and the CTF for aligned frames was determined using GCTF74. The control dataset of GroEL–GroES complexes without MetK was acquired similarly
but with a nominal magnification of ×29,000, resulting in a calibrated pixel size of 0.84 Å. IMAGE PROCESSING, CLASSIFICATION AND REFINEMENT FOR SINGLE-PARTICLE ANALYSIS From the resulting
8,945 micrographs of the GroEL–GroES–MetK dataset, 1,561,482 particles were picked using a trained crYOLO network75 and extracted with RELION v.3.1.3 (ref. 69). An initial round of
two-dimensional classification was performed and the remaining particles were passed into CryoSPARC76 for further two-dimensional classification, ab initio model building, alignment and
initial three-dimensional classification to separate EL–ES1 from EL–ES2 complexes. The remaining EL–ES2 (659,866 particles) and EL–ES1 (294,250 particles) complexes were then exported
separately to RELION for additional alignment with imposed symmetry, CTF refinement and Bayesian polishing. For the EL–ES2 complexes, symmetry expansion around the _C_2 axis was performed
and the opposing half removed using RELION’s signal subtraction. The resulting asymmetric EL–ES1 complexes were then classified further with CryoDRGN77, resulting in a clean subset of
242,276 particles. The _trans-_rings of the EL–ES1 complexes were classified in CryoSPARC using a focused mask on the apical domains of the _trans_-ring, resulting in 169,454 particles in
the narrow conformation and 34,755 in the wide conformation. The resulting structures were refined in CryoSPARC under the application of _C7_ symmetry to a nominal resolution of 2.9 and 3.1
Å, respectively. For the analysis of the _cis_-chamber, all EL–ES1 particles were pooled and the _trans_-ring was removed by signal subtraction in RELION. The resulting GroES-bound,
single-ring particles (1,562,002 particles) were then aligned to a common reference in RELION and exported to CryoSPARC for further alignment without imposed symmetry. The resulting mask and
reference were reimported into RELION and used for an additional alignment step with the goal of aligning the asymmetric MetK substrate contained inside the chamber (Extended Data Fig. 5d).
Subsequently a second round of signal subtraction was performed and the resulting particles, comprising only MetK density, were further subjected to three-dimensional classification without
angular search in RELION. A subset of the resulting classes showed visible secondary structure elements in different orientations (Extended Data Fig. 5d). These classes were then combined
and aligned into a single frame of reference in Matlab 2015b by manual rotation with the respective multiple of 360°/7 around the sevenfold symmetry axis. This was done by adding the
corresponding increment to particle rotation angles in the particle table (.star file). These folded MetK (fMetK) particles were then further locally aligned in CryoSPARC. An additional
round of three-dimensional classification was performed followed by a final round of local alignment (322,800 particles), resulting in density for MetK at a resolution of 3.7 Å. For the
study of MetK contacts with the inner wall of GroEL–GroES chamber, we reverted the signal subtraction in RELION to generate single-ring GroEL–GroES–MetK particles for both the folded MetK
and mixed population of chambers either containing disordered MetK or empty; both were refined and aligned in CryoSPARC. The subset containing a mixed population was additionally classified
in CryoDRGN between the final alignment steps, resulting in a global resolution of 3.04 Å for the GroEL–GroES–MetK complex containing folded MetK and of 2.94 Å for the complex containing a
mixed population of disordered MetK or empty chambers. GroEL–GroES complexes without MetK were processed analogously but without Bayesian polishing and CTF refinement in RELION. Signal
subtraction was performed in CryoSPARC; using 293,974 particles, this resulted in a map with a global resolution of 2.5 Å following the application of _C_7 symmetry. Densities were
visualized and rendered using ChimeraX78,79. MODEL BUILDING AND REFINEMENT Model building was initiated by rigid-body fitting the GroEL subdomains, GroES and MetK from the crystal structures
PDB 1SX3 (ref. 80), 5OPW12 and 7LOO42, respectively, into cryo-EM density, followed by manual editing using Coot81. The models were subsequently refined in real space with Phenix82. For the
refinement of models against low-resolution data from STA, automatically generated restraints from reference structures such as PDB 8P4M (this study) were used. Residues with disordered
sidechains were truncated at C-beta. REPORTING SUMMARY Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article. DATA AVAILABILITY
The mass spectrometry data have been deposited to the ProteomeXchange Consortium83 via the PRIDE partner repository with the dataset identifier PXD042587. Model coordinates and electron
density maps have been deposited to the wwPDB database under PDB/EMDB accession code nos. 8P4M/EMD-17418 (empty GroEL–GroES chamber), 8P4N/EMD-17420 (GroEL–GroES chamber with no or
disordered MetK), 8P4O/EMD-17421/EMD-17422 (GroEL–GroES chamber with ordered MetK), 8QXS/EMD-18735 (EL–ES1–MetK wide), 8QXT/EMD-18736 (EL–ES1–MetK narrow), 8P4R/EMD-17426 (in situ EL–ES2),
8QXU/EMD-18737 (in situ EL–ES1 wide), 8QXV/EMD-18738 (in situ EL–ES1 narrow) and 8P4P/EMD-17425 (in situ EL). Primary electron density maps have been deposited to the wwPDB database under
EMDB accession code nos. EMD-17423 (in vitro GroEL–GroES chamber with no or disordered MetK), EMD-17424 (in vitro GroEL–GroES chamber with ordered MetK), EMD-17534 (empty EL–ES2), EMD-17535
(empty EL–ES1), EMD-17559 (GroEL–GroES chamber with no or disordered substrate), EMD-17560 (GroEL–GroES chamber with encapsulated, ordered substrate), EMD-17561 (70S ribosomes in 37 °C, HS
and MetK _E. coli_ cells), EMD-17562 (70S ribosomes in EL+ _E. coli_ cells), EMD-17563 (EL–ES1 with encapsulated ordered MetK), EMD-17564 (EL–ES1 with no or encapsulated disordered MetK),
EMD-17565 (EL–ES2 with two chambers with no or disordered MetK), EMD-17566 (EL–ES2 with ordered MetK in one chamber and no or disordered MetK substrate in the other) and
EMD-17567/EMD-17568/EMD-17569/EMD-17570/EMD-17571/EMD-17572/EMD-17573 (conformers 1–7 of EL–ES2 with two encapsulated, ordered MetK). Because of their large file size, original cryo-ET
imaging data are available from the corresponding author on request. Source data are provided with this paper. REFERENCES * Hayer-Hartl, M., Bracher, A. & Hartl, F. U. The GroEL–GroES
chaperonin machine: a nano-cage for protein folding. _Trends Biochem. Sci._ 41, 62–76 (2016). Article CAS PubMed Google Scholar * Horwich, A. L. & Fenton, W. A. Chaperonin-assisted
protein folding: a chronologue. _Q. Rev. Biophys._ 53, e4 (2020). Article PubMed Google Scholar * Gestaut, D., Limatola, A., Joachimiak, L. & Frydman, J. The ATP-powered gymnastics of
TRiC/CCT: an asymmetric protein folding machine with a symmetric origin story. _Curr. Opin. Struct. Biol._ 55, 50–58 (2019). Article CAS PubMed PubMed Central Google Scholar * Brinker,
A. et al. Dual function of protein confinement in chaperonin-assisted protein folding. _Cell_ 107, 223–233 (2001). Article CAS PubMed Google Scholar * Mayhew, M. et al. Protein folding
in the central cavity of the GroEL–GroES chaperonin complex. _Nature_ 379, 420–426 (1996). Article ADS CAS PubMed Google Scholar * Weissman, J. S., Rye, H. S., Fenton, W. A., Beechem,
J. M. & Horwich, A. L. Characterization of the active intermediate of a GroEL–GroES-mediated protein folding reaction. _Cell_ 84, 481–490 (1996). Article CAS PubMed Google Scholar *
Balchin, D., Hayer-Hartl, M. & Hartl, F. U. In vivo aspects of protein folding and quality control. _Science_ 353, aac4354 (2016). Article PubMed Google Scholar * Balchin, D.,
Hayer-Hartl, M. & Hartl, F. U. Recent advances in understanding catalysis of protein folding by molecular chaperones. _FEBS Lett._ 594, 2770–2781 (2020). Article CAS PubMed Google
Scholar * Krishna, K. A., Rao, G. V. & Rao, K. R. Chaperonin GroEL: structure and reaction cycle. _Curr. Protein Pept. Sci._ 8, 418–425 (2007). Article CAS PubMed Google Scholar *
Ranson, N. A. et al. Allosteric signaling of ATP hydrolysis in GroEL-GroES complexes. _Nat. Struct. Mol. Biol._ 13, 147–152 (2006). Article CAS PubMed PubMed Central Google Scholar *
Rye, H. S. et al. GroEL-GroES cycling: ATP and nonnative polypeptide direct alternation of folding-active rings. _Cell_ 97, 325–338 (1999). Article CAS PubMed Google Scholar * Yan, X. et
al. GroEL Ring separation and exchange in the chaperonin reaction. _Cell_ 172, 605–617 (2018). Article CAS PubMed Google Scholar * Horovitz, A., Reingewertz, T. H., Cuellar, J. &
Valpuesta, J. M. Chaperonin mechanisms: multiple and (mis)understood? _Annu. Rev. Biophys._ 51, 115–133 (2022). Article CAS PubMed Google Scholar * Ye, X. & Lorimer, G. H. Substrate
protein switches GroE chaperonins from asymmetric to symmetric cycling by catalyzing nucleotide exchange. _Proc. Natl Acad. Sci. USA_ 110, E4289–E4297 (2013). Article ADS CAS PubMed
PubMed Central Google Scholar * Dutta, P., Roy, P. & Sengupta, N. Effects of external perturbations on protein systems: a microscopic view. _ACS Omega_ 7, 44556–44572 (2022). Article
CAS PubMed PubMed Central Google Scholar * Allegretti, M. et al. In-cell architecture of the nuclear pore and snapshots of its turnover. _Nature_ 586, 796–800 (2020). Article ADS CAS
PubMed Google Scholar * Baumeister, W. Cryo-electron tomography: the power of seeing the whole picture. _Biochem. Biophys. Res. Commun._ 633, 26–28 (2022). Article CAS PubMed Google
Scholar * Berger, C. et al. Cryo-electron tomography on focused ion beam lamellae transforms structural cell biology. _Nat. Methods_ 20, 499–511 (2023). Article CAS PubMed Google Scholar
* Chen, Z. et al. In situ cryo-electron tomography reveals the asymmetric architecture of mammalian sperm axonemes. _Nat. Struct. Mol. Biol._ 30, 360–369 (2023). Article CAS PubMed
PubMed Central Google Scholar * Gemmer, M. et al. Visualization of translation and protein biogenesis at the ER membrane. _Nature_ 614, 160–167 (2023). Article ADS CAS PubMed PubMed
Central Google Scholar * Guo, Q. et al. In situ structure of neuronal C9orf72 poly-GA aggregates reveals proteasome recruitment. _Cell_ 172, 696–705 (2018). Article CAS PubMed PubMed
Central Google Scholar * O’Reilly, F. J. et al. In-cell architecture of an actively transcribing-translating expressome. _Science_ 369, 554–557 (2020). Article ADS PubMed PubMed Central
Google Scholar * Xue, L. et al. Visualizing translation dynamics at atomic detail inside a bacterial cell. _Nature_ 610, 205–211 (2022). Article ADS CAS PubMed PubMed Central Google
Scholar * Hoffmann, P. C. et al. Structures of the eukaryotic ribosome and its translational states in situ. _Nat. Commun._ 13, 7435 (2022). Article ADS CAS PubMed PubMed Central
Google Scholar * Fujiwara, K., Ishihama, Y., Nakahigashi, K., Soga, T. & Taguchi, H. A systematic survey of in vivo obligate chaperonin-dependent substrates. _EMBO J._ 29, 1552–1564
(2010). Article CAS PubMed PubMed Central Google Scholar * Kerner, M. J. et al. Proteome-wide analysis of chaperonin-dependent protein folding in _Escherichia coli_. _Cell_ 122, 209–220
(2005). Article CAS PubMed Google Scholar * Zhao, L. et al. Bacterial RF3 senses chaperone function in co-translational folding. _Mol. Cell_ 81, 2914–2928 (2021). Article CAS PubMed
Google Scholar * Fayet, O., Ziegelhoffer, T. & Georgopoulos, C. The groES and groEL heat shock gene products of _Escherichia coli_ are essential for bacterial growth at all
temperatures. _J. Bacteriol._ 171, 1379–1385 (1989). Article CAS PubMed PubMed Central Google Scholar * PaxDb: Protein Abundance Database. E.coli - Whole organism (Integrated).
https://pax-db.org/dataset/511145/2297923011/ (2023). * Huang, Q., Szklarczyk, D., Wang, M., Simonovic, M. & von Mering, C. PaxDb 5.0: curated protein quantification data suggests
adaptive proteome changes in yeasts. _Mol. Cell. Proteomics_ 22, 100640 (2023). Article CAS PubMed PubMed Central Google Scholar * Xu, Z. H., Horwich, A. L. & Sigler, P. B. The
crystal structure of the asymmetric GroEL-GroES-(ADP)7 chaperonin complex. _Nature_ 388, 741–749 (1997). Article ADS CAS PubMed Google Scholar * Kudryavtseva, S. S. et al. Novel cryo-EM
structure of an ADP-bound GroEL-GroES complex. _Sci. Rep._ 11, 18241 (2021). Article ADS CAS PubMed PubMed Central Google Scholar * Fei, X., Ye, X., LaRonde, N. A. & Lorimer, G.
H. Formation and structures of GroEL:GroES2 chaperonin footballs, the protein-folding functional form. _Proc. Natl Acad. Sci. USA_ 111, 12775–12780 (2014). Article ADS CAS PubMed PubMed
Central Google Scholar * Fenton, W. A., Kashi, Y., Furtak, K. & Horwich, A. L. Residues in chaperonin GroEL required for polypeptide binding and release. _Nature_ 371, 614–619 (1994).
Article ADS CAS PubMed Google Scholar * Soini, J. et al. Transient increase of ATP as a response to temperature up-shift in _Escherichia coli_. _Microb. Cell Fact._ 4, 9 (2005). Article
PubMed PubMed Central Google Scholar * Chen, D. H. et al. Visualizing GroEL/ES in the act of encapsulating a folding protein. _Cell_ 153, 1354–1365 (2013). Article CAS PubMed PubMed
Central Google Scholar * Clare, D. K., Bakkes, P. J., van Heerikhuizen, H., van der Vies, S. M. & Saibil, H. R. Chaperonin complex with a newly folded protein encapsulated in the
folding chamber. _Nature_ 457, 107–113 (2009). Article ADS CAS PubMed PubMed Central Google Scholar * Kim, H. et al. Cryo-EM structures of GroEL:ES(2) with RuBisCO visualize molecular
contacts of encapsulated substrates in a double-cage chaperonin. _iScience_ 25, 103704 (2022). Article ADS CAS PubMed Google Scholar * Gardner, S., Darrow, M. C., Lukoyanova, N.,
Thalassinos, K. & Saibil, H. R. Structural basis of substrate progression through the bacterial chaperonin cycle. _Proc. Natl Acad. Sci. USA_ 120, e2308933120 (2023). Article CAS
PubMed PubMed Central Google Scholar * Koike-Takeshita, A., Yoshida, M. & Taguchi, H. Revisiting the GroEL-GroES reaction cycle via the symmetric intermediate implied by novel aspects
of the GroEL(D398A) mutant. _J. Biol. Chem._ 283, 23774–23781 (2008). Article CAS PubMed PubMed Central Google Scholar * Taguchi, H., Tsukuda, K., Motojima, F., Koike-Takeshita, A.
& Yoshida, M. BeF(x) stops the chaperonin cycle of GroEL-GroES and generates a complex with double folding chambers. _J. Biol. Chem._ 279, 45737–45743 (2004). Article CAS PubMed
Google Scholar * Gade, M. et al. Substrate dynamics contribute to enzymatic specificity in human and bacterial methionine adenosyltransferases. _JACS Au_ 1, 2349–2360 (2021). Article CAS
PubMed PubMed Central Google Scholar * Takusagawa, F., Kamitori, S., Misaki, S. & Markham, G. D. Crystal structure of _S_-adenosylmethionine synthetase. _J. Biol. Chem._ 271, 136–147
(1996). Article CAS PubMed Google Scholar * Horovitz, A. & Willison, K. R. Allosteric regulation of chaperonins. _Curr. Opin. Struct. Biol._ 15, 646–651 (2005). Article CAS PubMed
Google Scholar * Saibil, H. R., Fenton, W. A., Clare, D. K. & Horwich, A. L. Structure and allostery of the chaperonin GroEL. _J. Mol. Biol._ 425, 1476–1487 (2013). Article CAS
PubMed Google Scholar * Tang, Y. C. et al. Structural features of the GroEL-GroES nano-cage required for rapid folding of encapsulated protein. _Cell_ 125, 903–914 (2006). Article CAS
PubMed Google Scholar * Weaver, J. et al. GroEL actively stimulates folding of the endogenous substrate protein PepQ. _Nat. Commun._ 8, 15934 (2017). Article ADS CAS PubMed PubMed
Central Google Scholar * Guzman, L. M., Belin, D., Carson, M. J. & Beckwith, J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD
promoter. _J. Bacteriol._ 177, 4121–4130 (1995). Article CAS PubMed PubMed Central Google Scholar * Hayer-Hartl, M. K., Weber, F. & Hartl, F. U. Mechanism of chaperonin action:
GroES binding and release can drive GroEL-mediated protein folding in the absence of ATP hydrolysis. _EMBO J._ 15, 6111–6121 (1996). Article CAS PubMed PubMed Central Google Scholar *
Gasteiger, E. et al. in _The Proteomics Protocols Handbook_ (ed. Walker, J. M.) 571–607 (Humana, 2005). * Ewalt, K. L., Hendrick, J. P., Houry, W. A. & Hartl, F. U. In vivo observation
of polypeptide flux through the bacterial chaperonin system. _Cell_ 90, 491–500 (1997). Article CAS PubMed Google Scholar * Rigort, A. et al. Focused ion beam micromachining of
eukaryotic cells for cryoelectron tomography. _Proc. Natl Acad. Sci. USA_ 109, 4449–4454 (2012). Article ADS CAS PubMed PubMed Central Google Scholar * Klumpe, S. et al. A modular
platform for automated cryo-FIB workflows. _eLife_ 10, e70506 (2021). Article CAS PubMed PubMed Central Google Scholar * Khavnekar, S. et al. Optimizing cryo-FIB lamellas for sub-5Å in
situ structural biology. Preprint at _bioRxiv_ https://doi.org/10.1101/2022.06.16.496417 (2022). * Mastronarde, D. N. Automated electron microscope tomography using robust prediction of
specimen movements. _J. Struct. Biol._ 152, 36–51 (2005). Article PubMed Google Scholar * Hagen, W. J. H., Wan, W. & Briggs, J. A. G. Implementation of a cryo-electron tomography
tilt-scheme optimized for high resolution subtomogram averaging. _J. Struct. Biol._ 197, 191–198 (2017). Article PubMed PubMed Central Google Scholar * Zheng, S. Q. et al. MotionCor2:
anisotropic correction of beam-induced motion for improved cryo-electron microscopy. _Nat. Methods_ 14, 331–332 (2017). Article CAS PubMed PubMed Central Google Scholar * Mastronarde,
D. N. & Held, S. R. Automated tilt series alignment and tomographic reconstruction in IMOD. _J. Struct. Biol._ 197, 102–113 (2017). Article PubMed Google Scholar * Wan, W.
williamnwan/TOMOMAN: TOMOMAN 08042020. Zenodo https://doi.org/10.5281/zenodo.4110737 (2020). * Grant, T. & Grigorieff, N. Measuring the optimal exposure for single particle cryo-EM using
a 2.6 A reconstruction of rotavirus VP6. _eLife_ 4, e06980 (2015). Article PubMed PubMed Central Google Scholar * Rohou, A. & Grigorieff, N. CTFFIND4: fast and accurate defocus
estimation from electron micrographs. _J. Struct. Biol._ 192, 216–221 (2015). Article PubMed PubMed Central Google Scholar * Zivanov, J. et al. A Bayesian approach to single-particle
electron cryo-tomography in RELION-4.0. _eLife_ 11, e83724 (2022). Article CAS PubMed PubMed Central Google Scholar * Turonova, B., Schur, F. K. M., Wan, W. & Briggs, J. A. G.
Efficient 3D-CTF correction for cryo-electron tomography using NovaCTF improves subtomogram averaging resolution to 3.4 A. _J. Struct. Biol._ 199, 187–195 (2017). Article CAS PubMed
PubMed Central Google Scholar * Baker, M. L., Zhang, J., Ludtke, S. J. & Chiu, W. Cryo-EM of macromolecular assemblies at near-atomic resolution. _Nat. Protoc._ 5, 1697–1708 (2010).
Article CAS PubMed PubMed Central Google Scholar * Pettersen, E. F. et al. UCSF Chimera–a visualization system for exploratory research and analysis. _J. Comput. Chem._ 25, 1605–1612
(2004). Article CAS PubMed Google Scholar * Wan, W., Khavnekar, S. & Wagner, J. STOPGAP: an open-source package for template matching, subtomogram alignment and classification. _Acta
Crystallogr. D Struct. Biol._ 80, 336–349 (2024). Article ADS CAS PubMed PubMed Central Google Scholar * Erdmann, P. S. et al. In situ cryo-electron tomography reveals gradient
organization of ribosome biogenesis in intact nucleoli. _Nat. Commun._ 12, 5364 (2021). Article ADS CAS PubMed PubMed Central Google Scholar * Tegunov, D. & Cramer, P. Real-time
cryo-electron microscopy data preprocessing with Warp. _Nat. Methods_ 16, 1146–1152 (2019). Article CAS PubMed PubMed Central Google Scholar * Zivanov, J. et al. New tools for automated
high-resolution cryo-EM structure determination in RELION-3. _eLife_ 7, e42166 (2018). Article PubMed PubMed Central Google Scholar * Tegunov, D., Xue, L., Dienemann, C., Cramer, P.
& Mahamid, J. Multi-particle cryo-EM refinement with M visualizes ribosome-antibiotic complex at 3.5 A in cells. _Nat. Methods_ 18, 186–193 (2021). Article CAS PubMed PubMed Central
Google Scholar * Bepler, T. et al. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. _Nat. Methods_ 16, 1153–1160 (2019). Article CAS
PubMed PubMed Central Google Scholar * Schwanhausser, B. et al. Global quantification of mammalian gene expression control. _Nature_ 473, 337–342 (2011). Article ADS PubMed Google
Scholar * Biyani, N. et al. Focus: the interface between data collection and data processing in cryo-EM. _J. Struct. Biol._ 198, 124–133 (2017). Article CAS PubMed Google Scholar *
Zhang, K. Gctf: real-time determination and correction. _J. Struct. Biol._ 193, 1–12 (2016). Article ADS CAS PubMed PubMed Central Google Scholar * Wagner, T. et al. SPHIRE-crYOLO is a
fast and accurate fully automated particle picker for cryo-EM. _Commun. Biol._ 2, 218 (2019). Article PubMed PubMed Central Google Scholar * Punjani, A., Rubinstein, J. L., Fleet, D. J.
& Brubaker, M. A. CryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. _Nat. Methods_ 14, 290–296 (2017). Article CAS PubMed Google Scholar * Zhong, E. D.,
Bepler, T., Berger, B. & Davis, J. H. CryoDRGN: reconstruction of heterogeneous cryo-EM structures using neural networks. _Nat. Methods_ 18, 176–185 (2021). Article CAS PubMed PubMed
Central Google Scholar * Goddard, T. D. et al. UCSF ChimeraX: meeting modern challenges in visualization and analysis. _Protein Sci._ 27, 14–25 (2018). Article CAS PubMed Google Scholar
* Pettersen, E. F. et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. _Protein Sci._ 30, 70–82 (2021). Article CAS PubMed Google Scholar *
Chaudhry, C., Horwich, A. L., Brunger, A. T. & Adams, P. D. Exploring the structural dynamics of the _E. coli_ chaperonin GroEL using translation-libration-screw crystallographic
refinement of intermediate states. _J. Mol. Biol._ 342, 229–245 (2004). Article CAS PubMed Google Scholar * Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics.
_Acta Crystallogr. D Biol. Crystallogr._ 60, 2126–2132 (2004). Article ADS PubMed Google Scholar * Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons
and electrons: recent developments in Phenix. _Acta Crystallogr. D Struct. Biol._ 75, 861–877 (2019). Article ADS CAS PubMed PubMed Central Google Scholar * Deutsch, E. W. et al. The
ProteomeXchange consortium in 2020: enabling ‘big data’ approaches in proteomics. _Nucleic Acids Res._ 48, D1145–D1152 (2020). CAS PubMed Google Scholar * Vila-Sanjurjo, A. et al. X-ray
crystal structures of the WT and a hyper-accurate ribosome from _Escherichia coli_. _Proc. Natl Acad. Sci. USA_ 100, 8682–8687 (2003). Article ADS CAS PubMed PubMed Central Google
Scholar * Baldwin, P. R. & Lyumkis, D. Non-uniformity of projection distributions attenuates resolution in Cryo-EM. _Prog. Biophys. Mol. Biol._ 150, 160–183 (2020). Article CAS PubMed
Google Scholar * Baldwin, P. R. & Lyumkis, D. Tools for visualizing and analyzing Fourier space sampling in Cryo-EM. _Prog. Biophys. Mol. Biol._ 160, 53–65 (2021). Article PubMed
Google Scholar Download references ACKNOWLEDGEMENTS We thank S. Gärtner, R. Lange and N. Wischnewski for expert technical assistance. We thank L. Zhang and C. Sitron for help in developing
the immunoprecipitation protocol and improving the text, respectively. This study used the infrastructure of the Department of Cell and Virus Structure at the MPI of Biochemistry. We thank
J. Plitzko for valuable technical advice. Funding was provided by the German Research Foundation (Deutsche Forschungsgemeinschaft) under Germany’s Excellence Strategy (EXC 2067/1-390729940)
and SFB 1035, as well as the European Research Council (ERC Advanced Grant no. 101052783-INSITUFOLD) and the Ministry of Science and Culture of the State of Lower Saxony (74ZN1949). FUNDING
Open access funding provided by Max Planck Society. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Cellular Biochemistry, Max Planck Institute of Biochemistry, Martinsried,
Germany Jonathan Wagner, Alonso I. Carvajal, Andreas Bracher, Roman Körner & F. Ulrich Hartl * Research Group Molecular Structural Biology, Max Planck Institute of Biochemistry,
Martinsried, Germany Jonathan Wagner & Wolfgang Baumeister * Institute of Neuropathology, University Medical Center Göttingen, Göttingen, Germany Jonathan Wagner & Ruben
Fernandez-Busnadiego * Cluster of Excellence “Multiscale Bioimaging: from Molecular Machines to Networks of Excitable Cells” (MBExC), University of Göttingen, Göttingen, Germany Jonathan
Wagner & Ruben Fernandez-Busnadiego * Research Group CryoEM Technology, Max Planck Institute of Biochemistry, Martinsried, Germany Florian Beck & Stefan Bohn * Vanderbilt University
Center for Structural Biology, Nashville, TN, USA William Wan * Institute of Structural Biology, Helmholtz Center Munich, Oberschleissheim, Germany Stefan Bohn * Faculty of Physics,
University of Göttingen, Göttingen, Germany Ruben Fernandez-Busnadiego Authors * Jonathan Wagner View author publications You can also search for this author inPubMed Google Scholar * Alonso
I. Carvajal View author publications You can also search for this author inPubMed Google Scholar * Andreas Bracher View author publications You can also search for this author inPubMed
Google Scholar * Florian Beck View author publications You can also search for this author inPubMed Google Scholar * William Wan View author publications You can also search for this author
inPubMed Google Scholar * Stefan Bohn View author publications You can also search for this author inPubMed Google Scholar * Roman Körner View author publications You can also search for
this author inPubMed Google Scholar * Wolfgang Baumeister View author publications You can also search for this author inPubMed Google Scholar * Ruben Fernandez-Busnadiego View author
publications You can also search for this author inPubMed Google Scholar * F. Ulrich Hartl View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS
J.W. performed cryo-ET and cryo-EM single-particle analyses with help from W.W., F.B. and S.B. A.I.C. performed biochemical experiments and MS analysis together with J.W. and R.K. A.B. built
the structural models and helped with data interpretation. F.U.H., W.B. and R.F.-B. designed the project and wrote the manuscript together with the other coauthors. CORRESPONDING AUTHORS
Correspondence to Wolfgang Baumeister, Ruben Fernandez-Busnadiego or F. Ulrich Hartl. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. PEER REVIEW PEER
REVIEW INFORMATION _Nature_ thanks Han Remaut and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available. ADDITIONAL
INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. EXTENDED DATA FIGURES AND TABLES EXTENDED
DATA FIG. 1 CRYO-ET AND SUBTOMOGRAM AVERAGING. (A) Sample preparation for cryo-ET. _E. coli_ cells were vitrified, thinned by cryo focussed ion beam (FIB) milling and tomograms aquired in a
cryo-transmission electron microscope (TEM). Representative scanning electron micrograph of a sample before and after FIB milling is shown along with an overview of a lamella from a cryo-TEM
(a total of 166 tomograms were acquired for 37 °C, HS and MetK combined). (B) Processing flowchart used for EL–ES1 and EL–ES2 subtomogram averaging in situ. The color of the box indicates
whether the respective step was performed in STOPGAP (blue) or with the indicated program (white). See Methods for details. EXTENDED DATA FIG. 2 IN SITU STRUCTURAL ANALYSIS OF 70 S
RIBOSOMES. (A-B) Subtomogram averaging of ribosomes. Ribosomes from three datasets (37 °C, HS, MetK) were averaged and refined to a global resolution of 8.7 Å. The resulting subtomogram
structure with the superposed molecular model (PDB code 4V4A84) in ribbon representation (a) and the corresponding FSC curve (b) are shown. (C) Analysis of ribosome to GroEL 14-mer ratio in
tomograms (box plots; 37 °C n = 48, HS n = 58, MetK n = 60 tomograms) and by MS using intensity-based absolute quantification (iBAQ) (blue crosses; n = 3 independent experiments). Box plots
show median (center line), interquartile range (IQR) (box edges) and 1.5 × IQR (whiskers). The MS measurements fall mainly within the range of the first to third quartile of the tomography
data, indicating that most EL complexes were identified in situ. Source data EXTENDED DATA FIG. 3 BIOCHEMICAL ANALYSIS OF GROEL/GROES LEVELS AND METK BINDING. (A) Representative immunoblot
of GroEL and GroES for the different growth conditions analyzed (37 °C, HS, MetK and EL+). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as loading control. (B) Quantification of
GroEL and GroES levels by label-free mass spectrometry of cell lysates. The total amount of GroEL was quantified by label-free mass spectrometry using iBAQ. iBAQ values of GroEL and GroES
of cells grown at 37 °C cells were set to 1 and used for normalization (n = 3 independent experiments). The horizontal line in the boxplots indicates the median value; boxes indicate upper
and lower quartile and whisker caps the largest or smallest value within 1.5 times the interquartile range above the 75th percentile or below the 25th percentile, respectively. (C) Ratio of
GroEL 14-mer and GroES 7-mer, based on the iBAQ values from (b). The GroEL:GroES ratio in wild-type cells at 37 °C was set to 1 and used for normalization. The differences between the groups
were not statistically significant when compared with a 1-way ANOVA test. (D) Growth of _E. coli_ BL21(DE3) at 37 °C, upon exposure to HS at 46 °C or upon ~4,5-fold overexpression of GroEL
(EL+) at 37 °C. Data points are averages ± SD (n = 3, independent repeats). Growth curves were standardized to start at a log2(OD600) value of 0. (E) Growth of transformed _E. coli_
BL21(DE3) at 37 °C, upon sequential overexpression of GroEL/GroES and MetK (MetK cells) or upon ~4,5-fold overexpression of GroEL with subsequent overexpression of MetK (EL+/MetK) at 37 °C
(see Methods). For comparison, the growth of the latter strain without induction (n.i.) of MetK (EL+/MetK(n.i.)) is shown. Data points are averages ± SD (n = 3, independent repeats). Growth
curves were standardized to start at a log2(OD600) value of 0. (F) Quantification of MetK bound to GroEL complexes in 37 °C and MetK overexpressing cells. Apyrase treatment was performed
upon cell lysis to stop GroEL cycling. GroEL was immunoprecipitated (IP), followed by immunoblotting with antibodies against GroEL and MetK. Anti-lactalbumin antibodies were used as
non-specific control. (G) Quantification of MetK:GroEL stoichiometry by MS in GroEL IPs from (d). The fraction of MetK per GroEL 14-mer was calculated based on iBAQ values (n = 3 independent
experiments). Box plots show median (center line), interquartile range (IQR) (box edges) and 1.5 × IQR (whiskers). (H) Cellular abundance of GroEL in 37 °C and EL+ cells. The data was
normalized to a median of 1 for 37 °C (n = 3 independent experiments). Boxplots are defined as in (g). (I) Cellular abundance of EL–ES1 in 37 °C and EL+ cells. The abundance of EL–ES1
relative to ribosomes in tomograms was calculated as a proxy for its cytosolic concentration and normalized to a median of 1 for 37°C. Source data EXTENDED DATA FIG. 4 IN SITU STRUCTURAL
ANALYSIS OF GROEL COMPLEXES. (A-D) FSC curves for EL–ES1 narrow (a), EL–ES1 wide (b) and EL–ES2 complexes (c), as well as EL (d) from Fig. 2a–d. The resolution at the 0.143 FSC cut-off is
indicated. Note that free GroEL (EL) was only observed upon GroEL overexpression (EL+) and thus this structure was obtained from a separate data set. (E) Comparison of GroEL subunits in the
_trans_-ring of the in situ structure of EL–ES1 narrow (dark blue) and the crystal structure of GroEL·ADP7–GroES7 (yellow) (PDB 1AON31). Two orthogonal views of the superimposed models are
shown. The models are depicted in ribbon representation. (F) Comparison of GroEL subunits in the _trans_-ring of the in situ structure of EL–ES1 wide (light blue) and the cryoEM structure of
EL–ES1 in complex with 14 ADP molecules (orange) (PDB 7PBJ32), using the same representation as in (e). (G) Distribution of narrow and wide EL–ES1 complexes at 37 °C, upon overexpression of
GroEL, GroES and MetK at 37 °C (MetK cells) or upon exposure to HS at 46 °C (37 °C, n = 48; MetK, n = 60; HS, n = 58 tomograms). (H) Comparison of GroEL–GroES units of the in situ structure
of EL–ES2 (orange) and the crystal structure of EL–ES2 in complex with 14 ADP·BeFx ligands (teal) (PDB 4PKO33). The models are depicted in ribbon representation. (I) Overlay of the rings in
the wide conformation in the in situ structures of EL–ES1 (teal) and the EL complex (yellow). The models are depicted in ribbon representation. (J) Cross section through the EL complex
density. Additional density not accounted for by the molecular model is present in the more narrow ring at the SP binding sites, as shown in side and top view. There is no additional density
at the given contour level in the opposing ring in the wide conformation. (K) Processing workflow of tomograms for analysis of encapsulated SP. To discern the SP states in GroEL–GroES
chambers of EL–ES1 and EL–ES2 complexes, isolated chambers were aligned. After denoising the resulting subtomograms, initial structures for subsequent 3D classification were produced by
bootstrapping and k means clustering. The resulting averages were used as starting structures for 3D classification (see Methods). Source data EXTENDED DATA FIG. 5 PREPARATION AND CRYO-EM
ANALYSIS OF THE GROEL–GROES–METK COMPLEX. (A-B) Size exclusion chromatography of the stable GroEL–GroES–MetK complex prepared in the presence of ATP·BeFx (see Material and Methods). A
representative chromatogram is shown in (a). Eluate fractions F1−F5 were analyzed by SDS-PAGE and Coomassie staining (b). Purified GroEL, MetK and GroES were analyzed for comparison.
Fractions F1 and F2 contain the GroEL:ES-encapsulated MetK. (C) The complex was analyzed by MS and the ratio of MetK to the GroEL 14-mer calculated based on iBAQ values (n = 2 independent
samples, each 3 technical repeats). Box plots show median (center line), interquartile range (IQR) (box edges) and 1.5 × IQR (whiskers). (D) Data processing workflow for single particle
analysis of GroEL–GroES–MetK complexes. A flow diagram is shown. The color of the box borders indicate that the respective step was performed in CryoSPARC (green), RELION 3.1.3 (blue) or
with the indicated program (black). After data collection, particle picking and initial 2D classification (I), EL–ES1 and EL–ES2 complexes were processed separately (II). GroEL–GroES
chambers were extracted and combined for further processing. Subsequently, the GroEL–GroES density was subtracted and the chamber interiors separated by 3D classification without alignment
(III). The picture row shows central slices of the resulting 3D class averages, which were used to separate folded MetK from disordered MetK or empty chambers (IV). Red arrows mark the MetK
densities differing by 2π/7 rotation in the GroEL–GroES chambers. The other 3D class averages had no visible secondary structure elements. The GroEL–GroES–MetK chambers containing folded
MetK were aligned and refined to a resolution of 3.0 Å. Local refinement of MetK after signal subtraction resulted in a 3.7 Å resolution map. The final resolution for GroEL–GroES chambers
with disordered MetK or empty chambers was 2.9 Å. See Methods for details. Source data EXTENDED DATA FIG. 6 SINGLE PARTICLE ANALYSIS OF GROEL–GROES–METK COMPLEXES. (A) A representative
micrograph of the GroEL–GroES–MetK sample at a magnification of 22,500-fold. (8,945 micrographs were used after on-the-fly preselection) (B) Corresponding 2D classes of particles selected
for further refinement. (C, D) Surface representation of densities for MetK-containing EL–ES2 (c) and EL–ES1 (narrow conformation) complexes (d). The red boxes indicate the GroEL–GroES
chambers that were processed further to solve the GroEL–GroES–MetK complex structure. (E, F) Bottom views of the densities and molecular models for the apical domains of the _trans_-ring in
EL–ES1 complexes with wide (e) and narrow (f) conformation. Surface views of the density are shown. The models are depicted in ribbon representation, with the substrate binding helices αH
and αI highlighted in orange and yellow, respectively. Additional density in the narrow _trans_-ring not accounted for by the model – presumably from the substrate MetK – is high-lighted in
teal (f). The insert shows one apical domain of the narrow _trans_-ring in detail. EXTENDED DATA FIG. 7 RESOLUTION AND DENSITY FIT ANALYSIS OF GROEL–GROES CHAMBERS AND METK. (A-D) FSC curves
(top) and local resolution maps (bottom) of the empty GroEL–GroES chamber (a), the GroEL–GroES chamber with disordered MetK or without substrate (b), the GroEL–GroES chamber with ordered
MetK (c) and isolated MetK (d), respectively. The rainbow color gradient indicates the local resolution scale. (E, F) Cryo-EM density of MetK with superposed molecular model in ribbon
representation. Two views for the entire protein are shown (e). Exemplary portions of the structure are shown below with side chains in stick representation (f). The respective residue
ranges are indicated. (G) Angular sampling of MetK. Planar and spherical representation of the Fourier sampling of MetK depicted in (d). The color gradient from dark blue to pale yellow
corresponds to the number of images in each bin. The sampling compensation factor was calculated to be 0.987 with values over 0.81, generally indicating adequate sampling85,86. Image was
generated using the CryoSPARC Orientation Diagnostics job76. EXTENDED DATA FIG. 8 COMPARISON OF ISOLATED TETRAMERIC METK WITH GROEL–GROES-ENCAPSULATED METK AND THE EFFECT OF METK
ENCAPSULATION ON GROEL–GROES CHAMBERS. (A) Overview of the crystal structure of the MetK tetramer (left; PDB 7LOO42). One subunit is shown in ribbon representation in gold, the other three
as molecular surfaces in violet, blue and yellow, respectively. The insert highlights the location of the core loop, which is marked by a red asterisk. Bound ligands pyrophosphate and
S-adenosylmethionine are shown as stick models in green. Cut-away view of GroEL–GroES encapsulated, folded MetK in the GroEL–GroES chamber in the same orientation (right). MetK is shown in
ribbon representation (teal). The GroEL and GroES subunits are shown as molecular surfaces. The core loop of MetK is indicated by a red asterisk, and the last resolved residue Pro525 in the
GroEL subunits is indicated with the letter C. The disordered C-terminal GGM repeats, GroEL residues 536−548, could easily reach the exposed MetK interface regions. (B-D) Overlay of the
_C_7-symmetric model of the empty GroEL–GroES chamber (green) with the chamber containing either disordered MetK or no substrate (blue) at the level of the equatorial GroEL domains (b), the
intermediate domains and the hinge regions between equatorial and intermediate domains (c and d). (E-G) Overlay of the _C_7-symmetric model of the empty GroEL–GroES chamber (green) with the
chamber containing folded MetK (purple) at the level of the equatorial GroEL domains (e), the intermediate domains and the hinge regions between equatorial and intermediate domains (f and
g). EXTENDED DATA FIG. 9 ANALYSIS OF THE SP DENSITY IN GROEL–GROES CHAMBER SUBTOMOGRAMS. Central xy slices of the subtomogram averages of in vitro assembled GroEL–GroES chambers with ordered
MetK (left) and class I in situ chambers (containing structured substrate; see Fig. 3b) from cells overexpressing GroEL/GroES and MetK (right), with gray values normalized to 2 standard
deviations. The center of mass for the density within a spherical volume in the chamber indicated by a circle is depicted by a red asterisk. Within error, the centers of mass were identical
(x, y, z in voxels: 65, 65, 51). SUPPLEMENTARY INFORMATION SUPPLEMENTARY FIG. 1 Classification of GroEL–GroES complexes following template matching. REPORTING SUMMARY SUPPLEMENTARY DATA
Zipped folder containing Uniprot_Ecoli_20230301.fasta file and UniProt sequences used for mass spectrometry analysis. SUPPLEMENTARY DATA Validation reports for wwPDB and emDB deposition.
PEER REVIEW FILE SOURCE DATA SOURCE DATA FIG. 1 SOURCE DATA FIG. 3 SOURCE DATA EXTENDED DATA FIG. 2 SOURCE DATA EXTENDED DATA FIG. 3 SOURCE DATA EXTENDED DATA FIG. 4 SOURCE DATA EXTENDED
DATA FIG. 5 RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution
and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if
changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the
material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to
obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS
ARTICLE Wagner, J., Carvajal, A.I., Bracher, A. _et al._ Visualizing chaperonin function in situ by cryo-electron tomography. _Nature_ 633, 459–464 (2024).
https://doi.org/10.1038/s41586-024-07843-w Download citation * Received: 11 January 2024 * Accepted: 18 July 2024 * Published: 21 August 2024 * Issue Date: 12 September 2024 * DOI:
https://doi.org/10.1038/s41586-024-07843-w SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative