Play all audios:
ABSTRACT It remains unclear why acute depletion of CTCF (CCCTC-binding factor) and cohesin only marginally affects expression of most genes despite substantially perturbing three-dimensional
(3D) genome folding at the level of domains and structural loops. To address this conundrum, we used high-resolution Micro-C and nascent transcript profiling in mouse embryonic stem cells.
We find that enhancer–promoter (E–P) interactions are largely insensitive to acute (3-h) depletion of CTCF, cohesin or WAPL. YY1 has been proposed as a structural regulator of E–P loops, but
acute YY1 depletion also had minimal effects on E–P loops, transcription and 3D genome folding. Strikingly, live-cell, single-molecule imaging revealed that cohesin depletion reduced
transcription factor (TF) binding to chromatin. Thus, although CTCF, cohesin, WAPL or YY1 is not required for the short-term maintenance of most E–P interactions and gene expression, our
results suggest that cohesin may facilitate TFs to search for and bind their targets more efficiently. SIMILAR CONTENT BEING VIEWED BY OTHERS COHESIN PREVENTS CROSS-DOMAIN GENE COACTIVATION
Article Open access 24 July 2024 CTCF AND TRANSCRIPTION INFLUENCE CHROMATIN STRUCTURE RE-CONFIGURATION AFTER MITOSIS Article Open access 27 August 2021 TRANSCRIPTION REGULATES THE
SPATIO-TEMPORAL DYNAMICS OF GENES THROUGH MICRO-COMPARTMENTALIZATION Article Open access 25 June 2024 MAIN High-throughput chromosomal conformation capture (Hi-C)-based assays have
transformed our understanding of 3D genome folding1,2. Based on such studies, we can distinguish at least three levels of 3D genome folding. First, the genome is segregated into A and B
compartments, which largely correspond to active and inactive chromatin segments, respectively, and appear as a plaid-like pattern in Hi-C contact maps3. Second, the proteins CTCF and
cohesin help fold the genome into topologically associating domains (TADs)4,5 and structural chromatin loops6, probably through DNA loop extrusion7,8. Third, at a much finer scale,
transcriptional elements engage in long-range chromatin interactions such as E–P and promoter–promoter (P–P) interactions to form local domains9,10,11. Elegant experiments combining acute
protein depletion of CTCF, cohesin and cohesin-regulatory proteins with Hi-C or imaging approaches have revealed the role of CTCF and cohesin in regulating the first two levels: TADs and
compartments12,13,14,15,16. However, Hi-C is ineffective for capturing the third level of 3D genome folding: the fine-scale transcriptionally important E–P/P–P interactions9,17,18. Our
understanding of the role of CTCF and cohesin in regulating gene expression has mainly come from genetic experiments focusing on a few developmental loci19,20,21. Thus, it remained unclear
whether, when, where and how CTCF/cohesin regulates E–P/P–P interactions and gene expression. We recently reported that Micro-C can effectively resolve ultra-fine 3D genome folding at
nucleosome resolution22,23, including E–P/P–P interactions9,17. In the present study, we used Micro-C, chromatin immunoprecipitation sequencing (ChIP-seq), total RNA-sequencing (RNA-seq) and
nascent RNA-seq24 to systematically investigate how acutely depleting CTCF, RAD21 (cohesin subunit), WAPL (cohesin unloader) or YY1 (a putative structural protein25) affects gene regulatory
chromatin interactions and transcription in mouse embryonic stem cells (mESCs). Finally, focusing on the dynamics of YY1 uncovered an unexpected role for cohesin in facilitating TF binding.
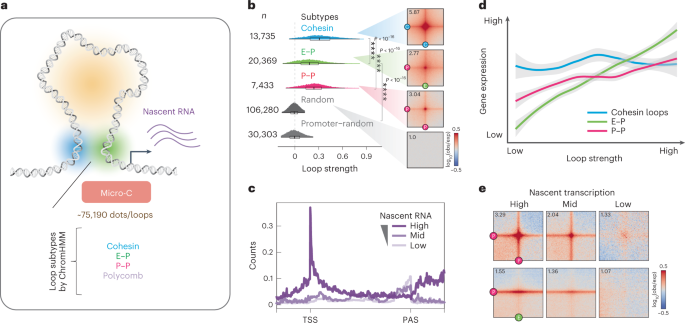
RESULTS GENOME-WIDE IDENTIFICATION OF TRANSCRIPTION-LINKED CHROMATIN LOOPS Our previous study used Micro-C to reveal that fine-scale 3D genome structure correlates well with transcriptional
activity, forming ‘dots’ or ‘loops’ (see Methods for terminology) at E–P and P–P intersections9. In the present study, we identified over 75,000 statistically significant loops in mESCs
using the newly developed loop caller Mustache26 (Fig. 1a) or Chromosight27 (Extended Data Fig. 1a), approximately 2.5× more than in our previous report9,26 and about 4× more than Hi-C26,28
(Extended Data Fig. 1b). Through analysis of local chromatin state at loop anchors (Extended Data Fig. 1c,d), we subclassified these loops into cohesin loops (~13,735), E–P loops (~20,369),
P–P loops (~7,433) and polycomb-associated contacts (~700) (Fig. 1a,b), with a median size of ~160 kb for cohesin loops and ~100 kb for E–P/P–P loops (Extended Data Fig. 1e). We profiled
nascent transcription by mammalian native elongating transcript sequencing (mNET-seq)24 in mESCs to better understand the relationship between active transcription and chromatin loops (Fig.
1c and Extended Data Fig. 1f). Newly transcribed RNAs generally have a higher correlation with E–P contacts than with compartments and TADs (Extended Data Fig. 1g). Specifically, the
strength of E–P/P–P loops positively correlates with the level of gene expression, whereas cohesin loops show no such correlation (Fig. 1d,e). Thus, by coupling Micro-C with nascent RNA-seq,
we can more precisely delineate which chromatin loops are associated with active transcription in a cell type of interest. We note that Micro-C assay is superior to Hi-C at detecting
E–P/P–P contacts (Extended Data Fig. 1h)26,27, as illustrated by the region around the _Klf2_ gene (Fig. 2a and Extended Data Fig. 1i), providing a less biased method for studying genome
organization relevant to transcription regulation29 (see Supplementary Note). E–P/P–P LOOPS CAN CROSS TAD BOUNDARIES TAD boundaries formed by CTCF and cohesin are thought to regulate E–P/P–P
interactions in two ways: by increasing interactions inside the TAD and by blocking interactions across TADs2. Nevertheless, it remains debatable whether TAD boundaries can absolutely
prevent an enhancer from interacting with and activating a gene in another TAD30,31,32,33,34. Our genome-wide analysis uncovered that, although loop interactions largely decay across
distance (Extended Data Fig. 1j), ~22.5% of E–P and ~33.2% of P–P loops that cross TAD boundaries retain a comparable level of contact intensity to equidistant loops within a TAD (Fig. 2b).
Genes located at the anchors of these inter-TAD loops also show similar or even higher expression levels in nascent or total RNA analysis (Fig. 2c). We postulated two possibilities that
could lead to this observation: the TAD boundaries that are crossed by E–P/P–P loops have either lower CTCF/cohesin occupancy or weaker insulation propensity. We first split the TAD
boundaries into two groups: ‘crossed’ or ‘not crossed’ by loops. Strikingly, CTCF and RAD21 occupancy at the boundaries is almost the same regardless of whether the boundaries are crossed by
loops (Extended Data Fig. 1k). The TAD boundaries crossed by either E–P or P–P loops show only slightly weaker insulation strength than the noncrossed boundaries (Fig. 2d). In contrast, the
boundaries that insulate the cohesin loops are substantially stronger than those that allow their crossing (Fig. 2d). Together, these results indicate that TAD boundaries are much more
effective at insulating cohesin loops than insulating E–P/P–P loops and that strong E–P interactions can overcome structural barriers18,35,36,37. ACUTE DEPLETION OF CTCF, COHESIN OR WAPL
ALTER CTCF AND COHESIN BINDING ON CHROMATIN To test whether active loop extrusion is essential for maintaining various types of chromatin loops and transcription, we endogenously and
homozygously tagged each of the three primary loop extrusion factors (CTCF, RAD21 or WAPL) with an auxin-inducible degron (AID) by clustered regularly repeating interspaced short palindromic
repeats (CRISPR)–Cas9-mediated genome editing in mESC lines expressing the F-box protein OsTir1 (Fig. 3a and Extended Data Fig. 2a)38. Despite CTCF-AID and RAD21–AID cell lines showing some
basal degradation (Extended Data Fig. 2b,c), we found no substantial change in their chromatin association, 3D genome organization or transcriptome compared with wild-type cells (see
Supplementary Note). Previous studies employing acute CTCF/cohesin depletion used prolonged degradation (6–48 h (refs. 12,13,39), which may confound the primary molecular response with
potential secondary effects40. To minimize indirect effects, we used a shorter degradation time and achieved almost-complete degradation of AID-tagged proteins after 3 h of iodoacetamide
(IAA) treatment, confirmed by western blotting (Fig. 3b and Extended Data Fig. 2d) and biochemical fractionation experiments (Extended Data Fig. 2e,f, red box). We then asked how the loss of
each loop extrusion factor affects the binding of the remaining factors. We obtained high-quality and high-reproducibility ChIP-seq data for CTCF, RAD21, SMC1A and SMC3 in the AID-tagged
lines treated with either ethanol (untreated (UT)) or IAA to degrade the tagged protein (Extended Data Fig. 3a–d). Consistent with previous studies12,41, both CTCF and cohesin lose their
occupancy after CTCF depletion (Fig. 3c,d and Extended Data Fig. 3e,f). Differential peak analysis42 confirmed that >90% of CTCF peaks and 60% of cohesin peaks are significantly decreased
on loss of CTCF (_P_adj < 0.05; Fig. 3e and Extended Data Fig. 3g). Despite the substantial loss of cohesin peaks, biochemical fractionation experiments show that the fraction of RAD21
associated with chromatin remains fairly constant 3 h after CTCF degradation (Extended Data Fig. 2f, green box). Thus, our results are in line with the widely accepted conclusion that CTCF
positions cohesin43. On the other hand, loss of cohesin affects a subset of CTCF binding (Fig. 3c,d)13, resulting in ~20% reduction in the number of CTCF peaks (Fig. 3e) and a slight
decrease in its global chromatin association (Extended Data Fig. 2f, blue box). ACUTE DEPLETION OF CTCF, COHESIN AND WAPL PERTURBS STRUCTURAL LOOPS Next, we used Micro-C to analyze the
effect of CTCF, RAD21 and WAPL depletion on fine-scale 3D genome structures. We pooled the highly reproducible replicates to achieve ~1–2 billion unique reads for each sample (Extended Data
Fig. 4a–c). At the levels of compartments and TADs, our findings largely agree with previous studies12,13,14,16 (Extended Data Fig. 4d,e). In addition, loop-strength analysis revealed that
nearly 90% of cohesin loops were lost after depletion of CTCF or RAD21 (Fig. 3f,g), whereas most loops were retained in a similar or slightly higher strength after WAPL depletion (Fig. 3g
and Extended Data Fig. 4f). Indeed, after WAPL depletion, an additional ~6,000 loops extended over longer distances (median size = 570 kb) were sufficiently strengthened to meet our
detection threshold (Extended Data Fig. 4g,h). In summary, cohesin-mediated DNA extrusion operates in a more unrestricted manner after depletion of CTCF (loss of well-positioned loops) or
WAPL (gain of longer-range loops). ACUTE LOSS OF CTCF, COHESIN AND WAPL DOES NOT AFFECT EXPRESSION OF MOST GENES We next asked whether acute disruption of active loop extrusion impacts the
maintenance of gene expression. To capture the immediate and temporal effects of depleting loop extrusion factors on transcription, we profiled nascent transcription by mNET-seq24 and
messenger RNA by ribosomal RNA-depleted RNA-seq for untreated and IAA-treated degron lines at 0, 3, 12 and 24 h after depletion. After validating the reproducibility and sensitivity of the
methods44 (Extended Data Fig. 5a,b), we performed differential expression tests of ~30,000 genes and identified ~50 transcripts changed in CTCF depletion, ~5 changed in RAD21 depletion and
only 2 changed in WAPL depletion after 3 h of IAA treatment (Fig. 3h,i and Extended Data Fig. 5c,d). Differentially expressed genes (DEGs) became more numerous with longer degradation times,
in line with previous findings12,39,45 (Extended Data Fig. 5c–e). We noticed that the early deregulated genes after loss of CTCF and cohesin include many cell-type-specific TFs (for
example, _Sox21_, _Myc_ and _Klf4_; Fig. 3i and Extended Data Fig. 5f). Chromatin structures around the DEGs were strongly disrupted, often featuring loss of a boundary or domain and gain of
de novo chromatin interactions (Fig. 3j and Extended Data Fig. 5g). Indeed, the early DEGs are associated with loop anchors and TAD boundaries, whereas the DEGs detected at the later time
points are not (Fig. 3k). This finding highlights the importance of distinguishing between primary and indirect effects of perturbations in the study of 3D genome and gene expression40. In
summary, despite CTCF, cohesin and WAPL probably regulating some gene expression in mESCs, their acute depletion affects the transcription of only a handful of genes that mostly encode
pluripotency and differentiation factors. E–P AND P–P INTERACTIONS ARE LARGELY MAINTAINED AFTER DEGRADATION OF LOOP EXTRUSION FACTORS The very modest transcriptional changes seen after CTCF
and cohesin degradation suggest that transcription-linked interactions may persist for at least 3 h after the depletion of CTCF, cohesin or WAPL. To test this hypothesis, we quantified the
loop strength at all 75,000 dots identified in wild-type mESCs in both control and depletion conditions. About 20% of loops are significantly decreased, but the remaining 60,000 loops are
largely unaltered (Fig. 4a,b). Consistent with our previous results, the disrupted loops are CTCF or cohesin dependent, whereas the persistent and upregulated loops are mostly anchored by
promoters and enhancers (Fig. 4c and Extended Data Fig. 6a,b). To further validate this, we specifically quantified the strength of loops that are anchored by E–P/P–P. Remarkably, acute
depletion of CTCF and cohesin has only a limited impact on the E–P/P–P loops, with ~80% of E–P contacts and 90% of P–P contacts remaining unaltered (Fig. 4d). Despite being less drastic than
for cohesin loops (Fig. 3f), E–P interactions appear to be slightly weakened globally, deviating from the midpoint line, but P–P loops remain largely insensitive to CTCF/cohesin depletion
(Fig. 4d,e and Extended Data Fig. 6c,d). WAPL depletion also has a negligible impact on E–P/P–P interactions (Fig. 4e and Extended Data Fig. 6e,f). What are the CTCF-/cohesin-sensitive
E–P/P–P loops? We found that these loops span a longer distance (Extended Data Fig. 6g) and have higher CTCF and cohesin occupancy at their anchors (Extended Data Fig. 6h), but these anchors
are not specifically associated with TAD boundaries (Fig. 4f,g and Extended Data Fig. 6i). We further tested whether the affected E–P/P–P interactions were associated with the DEGs in
nascent RNA-seq. Indeed, E–P/P–P interactions showed a greater decrease when their associated genes were deregulated on loss of CTCF/cohesin (Fig. 4h). Together, most E–P/P–P contacts and
fine-scale gene folding largely persist and remain transcriptionally functional even after almost-complete depletion of CTCF, cohesin or WAPL, suggesting that, in mESCs, these proteins are
not strictly required to maintain E–P/P–P interactions and transcription at least within a 3-h degradation, despite a broad but weak reduction in E–P interactions after cohesin depletion.
ACUTE YY1 DEPLETION HAS LITTLE EFFECT ON GLOBAL GENE EXPRESSION AND E–P/P–P INTERACTIONS A multifunctional zinc finger-containing TF, YY1 (ref. 46) (Extended Data Fig. 7a), has been
implicated as a master structural regulator of chromatin looping25, particularly during early neural lineage commitment47. To investigate the function of YY1 in genome organization and
transcriptional regulation in mESCs, we fused the mini-IAA7 tag48 to the endogenous _Yy1_ locus to allow for rapid protein degradation within 3 h (Fig. 5a and Extended Data Fig. 7b).
ChIP-seq analysis showed a clear depletion of YY1 at its cognate sites (Fig. 5b and Extended Data Fig. 7c), with ~90% of peaks (_n_ = 34,342) being called significantly changed by
differential peak analysis42 (Fig. 5b and Extended Data Fig. 7d,e). These peaks are primarily enriched at promoters, enhancers and bivalent loop anchors (Fig. 5c and Extended Data Fig. 7f),
consistent with its reported role in E–P interactions. We also noticed a modest decrease in cohesin occupancy after loss of YY1 (Fig. 5b and Extended Data Fig. 7g), which may be associated
with YY1’s potential to position or halt cohesin25. Similarly, biochemical fractionation analysis shows a decrease of ~87% in the chromatin-associated YY1 fraction and a reduction of ~7% in
the chromatin-associated cohesin fraction (Extended Data Fig. 7h, orange box). To characterize YY1’s role in 3D genome organization, we acquired ~850 × 106 unique Micro-C reads after pooling
high-quality replicates from untreated or YY1-depleted cells (Extended Data Fig. 7i). We found that YY1 depletion has no strong effect on chromatin compartments, TADs and cohesin loops
(Fig. 5d and Extended Data Fig. 7j). YY1 was proposed to be a causally required structural regulator of transcription and E–P interactions in a study conducted with 24-h depletion in
mESCs25. Surprisingly, acute removal of YY1 only mildly affected ~1% of loops (Fig. 5e) and ~11 and ~34 genes in the RNA-seq and mNET-seq profiling, respectively (Fig. 5f and Extended Data
Fig. 7k). Genome-wide pileup analysis for YY1, E–P and P–P loops showed only a very minor change in loop intensity after YY1 depletion (Fig. 5g and Extended Data Fig. 7l). Nevertheless, a
specific set of loci appears to require the presence of YY1 to interact with their _cis_-regulatory elements (for example, the _Ifnar2_, _Ikzf2_ and _NES_ gene loci) (Fig. 5h and Extended
Data Fig. 7m). Taken together, although YY1 may be required for a limited set of E–P/P–P interactions, YY1 is generally dispensable for maintaining genome organization and transcription in
mESCs, at least within a 3-h depletion window. SINGLE-MOLECULE IMAGING REVEALS YY1 BINDING DYNAMICS AND NUCLEAR ORGANIZATION The surprisingly modest effects of YY1 on chromatin looping might
result from YY1 DNA binding being very transient and/or due to only a small fraction of YY1 proteins being bound to DNA. To better understand the dynamics and mechanisms underlying YY1
function in living cells, we homozygously tagged YY1 with HaloTag49 (designated YN11 and YN31 clones) for live-cell, single-molecule imaging using CRISPR–Cas9-mediated genome editing (Fig.
6a and Extended Data Fig. 8a–c). Live-cell confocal imaging validated that HaloTag-YY1 was predominantly localized within the nucleus and appeared to be nonhomogeneously distributed
throughout the nucleoplasm, with noticeable puncta sporadically clustered within nucleoplasm and nucleoli (Fig. 5b,c and Extended Data Fig. 8d). We then visualized the nuclear distribution
of YY1 at single-molecule resolution by using photoactivated localization microscopy (PALM) (Fig. 6d), confirming its high-density punctate clusters. Furthermore, YY1 has been thought to be
evicted from chromosomes during mitosis in fixed-cell imaging experiments50. However, our live-cell imaging showed continued YY1 residency on mitotic chromosomes, suggesting that YY1 may be
involved in mitotic bookmarking (Fig. 6b and Extended Data Fig. 8d)51. Together, these results validate our homozygous HaloTag-YY1 knock-in cell lines and reveal that YY1 binds mitotic
chromosomes and forms local high concentration hubs in the nucleus. Having characterized our cell lines, we next interrogated YY1 protein dynamics and target search mechanisms. We took
advantage of the stroboscopic photoactivation, single-particle-tracking technique (spaSPT)52,53 to minimize motion blur and tracking errors to unambiguously trace the movement of individual
YY1 molecules at a frame rate of ~133 Hz (Fig. 6e and Extended Data Fig. 8e). YY1 molecules were then subclassified into bound and freely diffusing populations using a Bayesian-based
approach54 (Extended Data Fig. 8e,f). We found that ~31% of YY1 is in an immobile state, presumably bound to chromatin, with the remaining population exhibiting either slow diffusion (~26%)
or fast diffusion in the nucleoplasm (~43%) (Fig. 6f). These measurements largely agree with kinetic modeling of displacements obtained with the Spot-On algorithm (Extended Data Fig. 8g)53
and biochemical fractionation experiments (Fig. 6g). We note that the fraction of YY1 stably associating with chromatin is substantially lower than CTCF (~43%) and cohesin (~65%). The
residence times of TFs bound at their targets often correlate with their functional outcomes55,56,57. To estimate the overall residence time of the bound fraction of YY1, we used
fluorescence recovery after photobleaching (FRAP) to measure in vivo protein-binding kinetics by fitting the fluorescence recovery curve to a kinetic model58,59. Using a reaction-dominant
FRAP model, we estimated a residence time of ~13 s for most of the YY1 molecules (Fig. 6h and Extended Data Fig. 8h,i). We also employed slow-SPT60 as an orthogonal approach to measure YY1
residence times and obtained a residence time of ~13 s for YY1 at an exposure time of 100 ms (Fig. 6i). Slow-SPT with exposure times from 50 ms to 250 ms (ref. 61) further revealed a
subpopulation of YY1 that binds to chromatin for 40–60 s (Fig. 6j), consistent with the FRAP results showing that ~15% of YY1 recovers slowly (Fig. 6h and Extended Data Fig. 8h,i). Thus, YY1
proteins appear to have two distinct binding modes with apparent residence times of ~13 s and ~1 min. Taken together, our imaging experiments suggest that a smaller fraction of YY1 (~31%)
is bound to chromatin and that YY1 binding is more dynamic (average residence time of ~13 to 60 s) than CTCF (~50% bound for ~1 to 4 min) and cohesin (~40 to 50% bound for ~20 to 25 min),
which may help explain why YY1 protein depletion has a much weaker effect on looping and 3D genome folding. COHESIN DEPLETION ALTERS TF CHROMATIN-BINDING KINETICS We recently showed that
CTCF clusters enrich diffusive CTCF proteins near their binding sites, thereby accelerating their target search62. To test whether CTCF and cohesin may similarly affect YY1’s target search,
we endogenously fused an AID to CTCF or RAD21 in the HaloTag-YY1 parental line and confirmed >90% depletion after 3 h of IAA treatment (Fig. 7a and Extended Data Fig. 9a). Despite the
high degradation efficiency, neither YY1’s nuclear distribution nor its clustering was strongly affected after acute loss of CTCF and cohesin in either live or fixed cells (Fig. 7b,c and
Extended Data Fig. 9b). This suggests that the maintenance of YY1 hubs is independent of CTCF and cohesin. We next examined YY1 nuclear target search efficiency in the absence of CTCF and
cohesin using spaSPT. Although CTCF depletion had no major effect, cohesin depletion resulted in a modest but reproducible decrease from ~33% to 22% (~31% drop; _P_ < 0.01) in the bound
fraction of YY1 (Fig. 7d and Extended Data Fig. 9c). A lower bound fraction could either result from a shorter residence time (_k_off) or slower target search (_k_on). To distinguish between
these possibilities, we analyzed the FRAP data and found YY1 residence times (_k_off) to be only weakly affected by CTCF and cohesin depletion (Fig. 7e). We therefore conclude that cohesin
loss may affect the YY1 target search (_k_on). Specifically, we estimated a ~54% decrease in _k_on after cohesin depletion, resulting in a ~2.2-fold longer YY1 search time (UT = 28 s; IAA =
61 s), the time it takes YY1 on average to find and bind a cognate binding site after dissociating from DNA. To independently test this SPT finding, we analyzed our ChIP-seq data. We found
that ~3,504 YY1 peaks (total peaks = ~41,989 (~8.3%)) were lost after RAD21 degradation and >82% of these loci were associated with promoter regions (Fig. 7f and Extended Data Fig. 9d,e).
In contrast, both CTCF and WAPL depletion had a negligible effect on YY1 occupancy (Fig. 7f and Extended Data Fig. 9d,e). In biochemical fractionation analysis, we also observed a similar,
though less pronounced, reduction in YY1 chromatin association after RAD21 depletion (Extended Data Fig. 9f). To test whether cohesin facilitates the target search of TFs in general, we
performed spaSPT on additional TFs. We thus generated RAD21–AID cell lines stably expressing either HaloTag-conjugated SOX2 or KLF4 and found that the bound fraction of both TFs was reduced
by ~20% after 3-h cohesin degradation (Extended Data Fig. 9g). These results suggest that cohesin probably facilitates chromatin binding of TFs in general. Taken together, our results reveal
a role for cohesin in accelerating the target search of TFs, resulting in increased YY1 chromatin binding as measured by SPT, FRAP and ChIP-seq. Cohesin or cohesin-mediated genome structure
is likely to facilitate transcriptional establishment via more efficient target sampling of TFs (Fig. 7g). These findings also suggest that long-term cohesin depletion experiments must be
interpreted with caution because cohesin depletion results in both direct and indirect effects, including diminished general TF binding to DNA. DISCUSSION Both the extent and mechanism by
which CTCF- and cohesin-mediated loop extrusion regulates transcription have remained puzzling and hotly debated12,13,32,39,43,63,64,65,66,67. In the present study we applied high-resolution
Micro-C to overcome this limitation. Surprisingly, we found that CTCF, cohesin, WAPL or YY1 is not required for the maintenance of most E–P/P–P loops or transcription at least within a 3-h
depletion in mESCs. When affected, the altered E–P/P–P interactions only result in moderate expression changes of the underlying genes. Our findings, together with other evidence63,68, allow
us to distinguish and/or eliminate several models of E–P interactions previously assigned to these ubiquitous structural proteins (Fig. 8). First, CTCF and cohesin have been proposed to
either directly bridge E–P interactions69 or indirectly mediate E–P interactions by increasing contact frequency inside TADs (Fig. 8, Model 1)70. Our findings that acute CTCF, cohesin and
WAPL depletion minimally affect gene expression (Fig. 3h–j) and E–P interactions (Fig. 4) disfavor this model for short-term maintenance of E–P interactions, although CTCF and cohesin may
still help establish E–P interactions indirectly. We propose that loop extrusion may often be a separable mechanism from most E–P interactions and transcription, which is further supported
by the following observations: (1) >20% of E–P/P–P loops can cross TAD boundaries and retain high contact probability and transcriptional activity (Fig. 2)18,35; (2) only a very small
handful of genes showed altered expression levels after CTCF, cohesin or WAPL depletion (Fig. 3)12,13,14,15,16; (3) CTCF and cohesin loops are both rare (~5% of the time) and dynamic (median
lifetime ~10–30 min)34; (4) most of the E–P/P–P loops persist after depletion of these structural proteins (Fig. 4)39,63; (5) CTCF/cohesin generally does not colocalize with transcription
loci67; and (6) E–P loops and transcription can be established before CTCF/cohesin interactions on mitotic exit71, in some cases even with no CTCF/cohesin expression36,65,66. Second, YY1 was
proposed to be a master structural regulator of E–P interactions25 (Fig. 8, Model 2). However, our Micro-C data are inconsistent with this model, because acute YY1 depletion has little
effect on E–P/P–P interactions or gene expression. It is still possible that YY1 specifically connects development-related chromatin loops during neural lineage commitment47, but is less
important in the pluripotent state. In summary, we conclude that, in mESCs, CTCF, cohesin, WAPL or YY1 is not generally required for the short-term maintenance of most E–P interactions and
the subsequent expression of most genes after acute depletion and loss of function. The evidence that CTCF and cohesin can directly or indirectly regulate E–P interactions and affect gene
expression in many cases is overwhelming72,73,74,75,76,77,78,79. To reconcile these studies with our observations, we propose a ‘time-buffering’ model (Fig. 8, Model 3). In this model, CTCF,
cohesin and architectural factors contribute to the establishment of E–P interactions, but not to their maintenance. Instead, once established, a molecular memory (for example, histone
modifications80, chromatin remodeling81,82,83, DNA modification84,85,86, long noncoding RNAs87,88) may be sufficient to maintain E–P interactions and gene expression for several hours
without the contribution of these architectural factors. We propose that this time-buffering model and its variants89,90 reconcile our observations with the unambiguous genetic evidence that
CTCF and cohesin regulate some E–P interactions. An alternative, more conservative, interpretation of our data and the evidence cited above is that CTCF and cohesin only regulate a very
small, unique set of genes in specific biological processes and cell types and their effect on a handful of loci simply cannot be generalized as a universal rule. In the present study, we
also provide the first comprehensive study, to our knowledge, of YY1 dynamics and nuclear organization (Fig. 6). Surprisingly, we found that cohesin depletion, but not CTCF depletion,
significantly reduces YY1 chromatin binding and slows down its target search time from 28 s to 61 s. A similar effect was also observed in SOX2 and KLF4 in the present study, as well as
independently in glucocorticoid receptors by another group91. Furthermore, a study using high-throughput ChIP-seq analysis suggested that cohesin is critical to promote TF rebinding after
mitosis92. We therefore propose that cohesin could facilitate TF binding to chromatin in general (Fig. 8, Model 3). After cohesin depletion, TFs take a longer time to find their targets,
which may decrease transcription activation efficiency and eventually lead to changes in gene expression. It is interesting that the subunits of cohesin, as well as its loading and unloading
complexes, are composed of multiple segments of intrinsically disordered regions, which may facilitate TF binding to chromatin via establishing weak multivalent interactions93,94. Although
more quantitative works will be necessary to unveil these mechanisms, in addition to its roles in loop extrusion, DNA repair, replication and chromosome segregation, cohesin might also
facilitate TF binding to chromatin and could be critical for ensuring the precise timing of gene activation and silencing during embryonic development and cell-state transitions36. In
summary, we have comprehensively investigated the role of CTCF, RAD21, WAPL and YY1 in finer-scale chromatin structure, nascent transcription, as well as YY1 dynamics and nuclear
organization in mESCs. We propose a time-buffering model, where architectural proteins generally contribute to the establishment, but not the short-term maintenance, of E–P interactions and
gene expression, and we also propose that cohesin plays an underappreciated role to facilitate TF binding to chromatin. The connection linking protein dynamics to chromatin structure opens a
new avenue to rethink the mechanism of transcriptional regulation in the context of 3D genome organization. METHODS NOMENCLATURE FOR CHROMATIN ‘LOOPS’ OR ‘DOTS’ The focal contact enrichment
in Hi-C maps has historically been described as a ‘loop’ based on the assumption that motor proteins (that is, cohesin complex or RNA polymerase II) or TFs bridge long-range genomic loci
together, forming a ‘loop-shaped’ structure in vitro and in vivo. Unlike cohesin, which is likely to form loops through loop extrusion, E–P or P–P interactions may occur by a variety of
mechanisms without looping. Their interactions are typically detected as ‘dots’ in contact matrices. We agree that the term ‘dot’ is ideal for describing these enhanced focal contacts
without making any assumptions about their folding mechanisms or actual 3D structures. However, we chose to use ‘loop’ over ‘dots’ (that is, cohesin loops or E–P loops) to make the
manuscript more accessible to the general audience and to match the terms that are commonly used in the field. CELL CULTURE, STABLE CELL-LINE CONSTRUCTION AND DYE LABELING JM8.N4 mESCs95
(Research Resource Identifier: RRID:CVCL_J962; obtained from the KOMP Repository at University of California (UC), Davis) were used for all experiments. Cells were cultured on plates
precoated with 0.1% gelatin (Sigma-Aldrich, catalog no. G9291) in knock-out Dulbecco’s modified Eagle’s medium (DMEM; Thermo Fisher Scientific, catalog no. 10829018) supplemented with 15%
fetal bovine serum (HyClone FBS SH30910.03 lot no. AXJ47554), 0.1 mM minimal essential medium nonessential amino acids (Thermo Fisher Scientific, catalog no. 11140050), 2 mM GlutaMAX (Thermo
Fisher Scientific, catalog no. 35050061), 0.1 mM 2-mercaptoethanol (Sigma-Aldrich, catalog no. M3148), 1% penicillin–streptomycin (Thermo Fisher Scientific, catalog no. 15140122) and 1,000
units of leukemia inhibitory factor (Millipore). Medium was replaced daily and cells were passaged every 2 d by trypsinization. Cells were grown at 37 °C and 5.5% CO2 in a Sanyo copper alloy
IncuSafe humidified incubator (MCO-18AIC(UV)). For imaging, the medium was identical except that knock-out DMEM lacking phenol red (Thermo Fisher Scientific, catalog no. 31053028) was used
to minimize background fluorescence. Cell lines stably expressing 3× FLAG-HaloTag-YY1 and YY1-HaloTag-3×FLAG were generated using PiggyBac transposition and drug selection. Full details are
given in Supplementary Methods. For PALM experiments, cells were grown overnight on Matrigel-coated (Corning, catalog no. 354277; purchased from Thermo Fisher Scientific, catalog no.
08-774-552), 25-mm circular no. 1.5H cover glasses (High-Precision, catalog no. 0117650). Before all experiments, the cover glasses were plasma cleaned and then stored in isopropanol until
use. Cells were labeled with 500 nM PA-JFX549 HaloTag ligand for 30 min, washed twice with fresh medium for 5 min and then washed once with phosphate-buffered saline (PBS), pH 7.4. Labeled
cells were fixed with 4% paraformaldehyde and 2% glutaraldehyde in PBS for 20 min at 37 °C, washed once with PBS and imaged in PBS with 0.01% (w:v) NaN3. For FRAP experiments, cells were
grown overnight on Matrigel-coated glass-bottomed 35-mm dishes (MatTek P35G-1.5-14C). Cells were labeled with 500 nM HaloTag tetramethylrhodamine (TMR) ligand (Promega, catalog no. G8251)
for 30 min and washed twice with PBS. GENERATION OF CRISPR–CAS9-MEDIATED KNOCK-IN CELL LINES Endogenously tagged mESC lines were generated by CRISPR–Cas9-mediated genome editing as
previously described96 with modifications. Full details are given in Supplementary Methods. WESTERN BLOTTING See Supplementary Methods. CHIP AND CHIP-SEQ ChIP was performed as described with
a few modifications97(see Supplementary Methods for details). ChIP-seq libraries were prepared using the NEBNext Ultra II DNA Library Prep Kit for Illumina (New England Biolabs (NEB),
catalog no. E7645) according to the manufacturer’s instructions with a few modifications (see Supplementary Methods for details). Library concentration, quality and fragment size were
assessed by Qubit fluorometric quantification (Qubit dsDNA HS Assay Kit, Invitrogen, catalog no. Q32851), quantitative PCR and Fragment analyzer. Twelve multiplexed libraries were pooled and
sequenced in one lane on the Illumina HiSeq4000 sequencing platform (50-bp, single-end reads) at the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley, supported by National
Institutes of Health (NIH, grant no. S10 OD018174) instrumentation grant. See Supplementary Methods for the details on the ChIP-seq analysis. BIOCHEMICAL FRACTIONATION Wild-type JM8.N4 mESCs
were seeded on to 15-cm plates, washed with ice-cold PBS, scraped in PBS and pelleted at 135_g_ for 10 min at 4 °C. Pellets were resuspended in 350 μl of cell lysis buffer A (10 mM Hepes,
pH 7.9, 10 mM KCl, 3 mM MgCl2, 340 mM sucrose, 10% glycerol, v:v, 1 mM dithiothreitol (DTT) and freshly added 0.1% Triton X-100, v:v, and protease inhibitors) and rocked for 8 min at 4 °C.
Nuclei were pelleted at 3,000_g_ for 3 min at 4 °C and the supernatant containing the cytoplasmic fraction was saved. Nuclei were resuspended in 350 μl of buffer B with 75 mM NaCl (9 mM
EDTA, 0.2 mM (ethylenebis(oxonitrilo))tetra-acetate, 1 mM DTT, freshly added 0.1% Triton X-100, v:v, and protease inhibitors) and rocked at 4 °C for 15 min. Nuclei were pelleted again as
above (supernatant saved as the 75 mM wash fraction) and washed with 350 μl of buffer B with increasing NaCl concentrations (150 mM, 300 mM, 500 mM and 1 M; see Extended Data Fig. 2f for a
step-by-step procedure). After collecting the 1 M wash, the pellet was resuspended to 350 μl of 1 M buffer B and sonicated (Covaris S220 sonicator, 20% Duty factor, 200 cycles per burst, 100
peak incident power, 8 cycles of 20 s on and 40 s off). The sonicated lysate was spun down and the insoluble pellet boiled in sodium dodecylsulfate (SDS)-loading buffer. Then, 10 μl of each
fraction was added to 2 μl of 4× SDS-loading buffer and subjected to western blotting as detailed above. Band intensities were quantified with the ImageJ ‘Analyze Gels’ function98. MICRO-C
ASSAY FOR MAMMALIAN CELLS We briefly summarize the Micro-C experiment in Supplementary Methods. The detailed protocol and technical discussion are available in our previous study9.
Micro-C-seq libraries were generated using the NEBNext Ultra II DNA Library Prep Kit for Illumina (NEB, catalog no. E7645) with some minor modifications (detailed in Supplementary Methods).
We used Illumina 100-bp paired-end sequencing (PE100) to obtain ~400 M reads for each replicate in the present study. MICRO-C DATA PROCESSING AND ANALYSES Valid Micro-C contact read pairs
were obtained from the HiC-Pro analysis pipeline (v.2.11.3)99 and the detailed description and code can be found at https://github.com/nservant/HiC-Pro (see Supplementary Methods for a brief
description). Valid Micro-C contacts were assigned to the corresponding ‘pseudo’ nucleosome bin. The bin file was pregenerated from the mouse mm10 genome by a 100-bp window that virtually
resembles the nucleosome resolution. The binned matrix can be stored in HDF5 format as a COOL file using the COOLER package (v.0.8.10) (https://github.com/mirnylab/cooler)100 or in HIC file
format using the JUICER package (v.1.22.01) (https://github.com/aidenlab/juicer)101. Contact matrices were then normalized by using iterative correction in COOL files102 or Knight–Ruiz in
HIC files103. Regions with low mappability and high noise were blocked before matrix normalization. We expect that matrix-balancing normalization corrects systematic biases such as
nucleosome occupancy, sequence uniqueness, GC content or crosslinking effects102. We notice that both normalization methods produce qualitatively equal contact maps. To visualize the contact
matrices, we generated a compilation of COOL files with multiple resolutions (100-bp to 12,800-bp bins) that can be browsed on the HiGlass 3D genome server (http://higlass.io)104. In the
present study, all snapshots of Micro-C or Hi-C contact maps and the one-dimensional (1D) browser tracks (for example, ChIP-seq) were generated by the HiGlass browser (v.1.11.7) unless
otherwise stated. We evaluated the reproducibility and data quality for the Micro-C replicates using two published methods independently
(https://github.com/kundajelab/3DChromatin_ReplicateQC)105 (see Supplementary Methods for details). To analyze the genome-wide, contact-decaying _P_-value curve, we used intrachromosomal
contact pairs to calculate the contact probability in bins with exponentially increasing widths from 100 bp to 100 Mb. Contacts shorter than 100 bp were removed from the analysis to minimize
noise introduced by self-ligation or undigested DNA products. The orientations of ligated DNA are parsed into ‘IN-IN (+/−)’, ‘IN-OUT (+/+)’, ‘OUT-IN (−/−)’ and ‘OUT-OUT (−/+)’ according to
the readouts of Illumina sequencing22,23. ‘UNI’ pairs combine ‘IN-OUT’ and ‘OUT-IN’ because both orientations are theoretically interchangeable. In the present study, we plotted the contact
decaying curves with the ‘UNI’ pairs and then normalized to the total number of valid contact pairs. Slopes of contact decay curves were obtained by measuring slopes in a fixed-width window
searching across the entire range of decaying curves. We then plotted the derivative slope in each window against the corresponding genomic distance. To identify chromosome compartments, we
first transformed the observed:expected Micro-C matrices at the 200-kb resolution to Pearson’s correlation matrices and then obtained the eigenvector of the first principal component of
Pearson’s matrix by principal component analysis. The sign of the eigenvector was corrected using active histone marks (H3K27ac and H3K4me3), because positive values are the A compartment
(gene-rich or active chromatin) and negative values are the B compartment (gene-poor or inactive chromatin). The detailed description can be found in Lieberman-Aiden et al.3. The genome-wide
compartment strength analysis shown as a saddle plot represents the rearrangement and aggregation of the genome-wide, distance-normalized contact matrix with the order of increasing
eigenvector values. The chromosome arm is first divided into quantiles based on the compartment score. All combinations of quantile bins are averaged and rearranged in the saddle plot. The
Cooltools package (v.0.3.2; https://github.com/mirnylab/cooltools) has implemented the ‘call-compartments’ and ‘compute-saddle’ functions with the COOL files. To identify chromatin domains
(TADs) along the diagonal, we used insulation score analysis from the Cooltools package (v.0.3.2; https://github.com/mirnylab/cooltools) or arrowhead transformation analysis from the JUICER
package (v.1.22.01; https://github.com/aidenlab/juicer)101 (see Supplementary Methods for more details). Details of loops/dots identification and related analyses are in Supplementary
Methods. DEFINITION OF CHROMATIN STATES AND STRUCTURE OBSERVED BY MICRO-C We first used the published ChromHMM (http://compbio.mit.edu/ChromHMM)106,107 to define the chromatin states in
mESCs, which subclassifies chromatin into 12 states including: (1) CTCF/insulator, (2) active promoter (designated as ‘P’), (3) strong enhancer, (4) medium enhancer, (5) weak enhancer, (6)
mix of promoter and enhancer, (7) bivalent promoter, (8) gene body, (9) polycomb repressor, (10) intergenic regions, (11) heterochromatin and (12) repeats. To simplify the analysis, we
further combined the groups of strong, medium and weak enhancers and mix of promoter and enhancer into ‘enhancer’ (designated as ‘E’). In the present study, we use the terms that are widely
accepted in the field to describe the chromatin structures in Micro-C contact maps as well as avoid any ambiguous description that implicates their biological functions if they have not been
well characterized, including: (1) TAD: squares along matrix diagonal enriched with self-interactions, which are defined as genomic intervals demarcated by the boundaries characterized by
the insulation score analysis or the arrowhead transformation analysis; (2) cohesin loops: focal enrichment of contacts in contact maps with the coenrichment of CTCF/cohesin ChIP-seq peaks
at loop anchors, which is thought to be formed by active loop extrusion halted by CTCF; and (3) E–P/P–P dots: focal enrichment of contacts in contact maps with the coenrichment of chromatin
states for ‘active promoter (P)’ or ‘enhancer (E)’ at loop anchors. Although not all cohesin loops and E–P/P–P loops are formed through ‘looping’, and some studies suggest using ‘dots’
instead of ‘loops’, to simplify and be consistent with most of the findings, we chose to use ‘loops’ for cohesin-mediated focal contacts and ‘dots’ for other categories of enhanced focal
contacts in this manuscript. RNA-SEQ EXPERIMENTS AND ANALYSIS Total RNA was extracted from ~1 × 107 mESCs (~70% confluent P10 dish) using the standard TRIzol RNA extraction protocol. The
abundant rRNAs were depleted from the sample using the NEBNext rRNA Depletion Kit (NEB, catalog no. E6310). The rRNA-depleted RNAs were then subjected to RNA-seq library construction using
the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (NEB, catalog no. E7765). The final RNA-seq libraries were amplified with seven to eight PCR cycles. For RNA-seq analysis,
we used Kallisto (v.0.46.2)108 to quantify the number of transcripts and performed DEseq2 (v.1.30.1)109 analysis for DEG identification according to the recommended settings in the
walkthrough (http://bioconductor.org/packages/devel/bioc/vignettes/DESeq2/inst/doc/DESeq2.html) with _P_adj < 0.01 and fold-change >2. Full lists of DEGs are available in Supplementary
Table 11. NASCENT RNA-SEQ EXPERIMENT AND ANALYSIS We used the nascent RNA-seq (mNET-seq) protocol described in Nojima et al.110 with minor changes, detailed in Supplementary Methods. RNA
libraries were prepared according to the protocol of the NEBNext Small RNA Library Prep Kit (NEB, catalog no. E7330). The mNET-seq library was obtained by PCR for 12–14 cycles. For mNET-seq
analysis, we wrote a customized pipeline to process raw data as follows: (1) adapter trimming: we used TrimGalore (v.0.6.7) (https://github.com/FelixKrueger/TrimGalore) to remove sequencing
adapters ‘AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC’ and ‘GATCGTCGGACTGTAGAACTCTGAAC’ at each side of the reads; (2) mapping: trimmed reads were mapped to the mouse mm10 reference genome with STAR
RNA-seq aligner (v.2.7.10a)111; (3) identifying the last nucleotide incorporated by Pol II: we used the Python script mNET_snr (https://github.com/tomasgomes/mNET_snr) to locate the
3′-nucleotide of the second read and the strand sign of the first read. The bigWig files were generated using Deeptools (v.3.5.0) as described in Supplementary Methods. To identify DEGs in
mNET-seq, we used either the Nascent RNA Sequencing Analysis (v.2)112 package or FeatureCounts (v.1.22.2)113 and DEseq2 (v.1.30.1)109 to statistically quantify differential changes of the
mNET-seq signal at the gene body between UT- and IAA-treated cells (with _P_adj < 0.01 and fold-change >2). Full lists of DEGs are available in Supplementary Table 12. SINGLE-PARTICLE
IMAGING EXPERIMENTS All single-molecule imaging experiments were performed with a similar setting as described in our previous studies52,53 and detailed in Supplementary Methods. For PALM
experiments, continuous illumination was used for both the main excitation laser (633 nm for PA-JF646 or 561 nm for PA-JF549) and the photoactivation laser (405 nm). The intensity of the
405-nm laser was gradually increased over the course of the illumination sequence to image all molecules and avoid too many molecules being activated at any given frame. The camera was set
for 25-ms exposure time, frame transfer mode and vertical shift speed at 0.9 μs. In total, 40,000–60,000 frames were recorded for each cell (~20–25 min), which was sufficient to image and
bleach all labeled molecules. THE SPASPT ANALYSIS For analysis of spaSPT experiments, we used the QUOT package (v.1; https://github.com/alecheckert/quot) to generate trajectories from raw
spaSPT videos with the steps of spot detection, subpixel localization and tracking. All localization and tracking for this manuscript were performed with the following settings: (1)
detection: generalized log(likelihood ratio test) with a 2D Gaussian kernel (‘llr’ with _k_ = 1.0, pixel window size (_w_) = 15 and a log(ratio threshold (_t_)) = 26.0). (2) Subpixel
localization: Levenberg–Marquardt fitting of a 2D integrated Gaussian point spread function model (‘ls_int_gaussian’ with _w_ = 9, sigma = 1.0, ridge = 0.001, maximal iterations = 20 per
point spread function and damping term = 0.3). (3) Tracking: we chose to use a conservative tracking algorithm with a 1.3-μm search radius (‘conservative’ with maximal blinks = 0). This
setting makes the algorithm search for spot reconnections unambiguously, meaning that no other reconnections are possible within the specified search radius. Jumps were discarded if other
reconnection possibilities given the search radius existed. We next used the SASPT package (v.1; https://github.com/alecheckert/saspt)54 to estimate the likelihood of diffusion coefficients
for each trajectory. The detailed discussion is available in Heckert et al.114 and described in Supplementary Methods. Alternatively, we analyzed the spaSPT data with the kinetic modeling
framework implemented in the Spot-On package (v.1.04)53, briefly described in Supplementary Methods. SLOW-SPT ANALYSIS For analysis of slow-SPT experiments, we used the following tracking
settings for this manuscript: (1) detection: ‘llr’ with _k_ = 1.0, _w_ = 15, _t_ = 18; (2) subpixel localization: ‘ls_int_gaussian’ with _w_ = 9, sigma = 1.0, ridge = 0.001, maximal
iteration = 20 and damping = 0.3; (3) tracking: ‘euclidean’ with search radius = 0.5, maximal blinks = 1 and maximal diffusion constant (μm2 s−1) = 0.08. Details on how we extracted
residence times from slow-SPT are in Supplementary Methods. FRAP IMAGING ANALYSIS FRAP was performed on an inverted Zeiss LSM 900 Axio Observer confocal microscope equipped with Airyscan 2
detector, a motorized stage, a full incubation chamber maintaining 37 °C/5% CO2, a heated stage and an X-Cite 120 illumination source, as well various laser lines. Images were acquired on a
×40 Plan NeoFluar, numerical aperture 1.3, oil-immersion objective at a zoom corresponding to a 76 nm × 76 nm pixel size. The microscope was controlled using the Zeiss Zen imaging software.
In this manuscript, we recorded 60 s of videos for YY1-HaloTag at 1 frame per 250 ms, corresponding to a total of 240 frames. The first 20 frames were acquired before the bleach pulse,
allowing us to accurately measure baseline fluorescence. A circular bleach spot (_r_ = 6 pixels) was chosen in a region of homogeneous fluorescence at a position at least 1 μm from nuclear
or nucleolar boundaries. Alternatively, we bleached a square at one corner of the nucleus, which reduces noise while introducing some uncertainty for our downstream fitting analysis. The
spot was bleached using maximal laser intensity and pixel dwell time corresponding to a total bleach time of ~1 s. We note that, because the bleach duration was relatively long compared with
the timescale of molecular diffusion, it is not possible to accurately estimate the bound and free fractions from our FRAP curves. Details on the analysis of FRAP videos are in
Supplementary Methods. INFERRING PARAMETERS RELATED TO YY1’S TARGET SEARCH MECHANISM We used the parameters inferred from our spaSPT and the residence time measurements from our FRAP or
slow-SPT analysis. The detailed discussion is available in both Hansen et al.52 and Supplementary Methods. PALM ANALYSIS For analysis of PALM experiments, we used the publicly available
ThunderSTORM package (v.1.3; https://github.com/zitmen/thunderstorm)115 with the following setting for this manuscript: (1) image filtering: ‘Wavelet filter (B-Spline)’ with B-Spline order =
3 and B-Spline scale = 2.0; (2) approximate localization: ‘Local maximum’ with peak intensity threshold = 1.5 × std(Wave.F1) and 8-neighbourhood connectivity; (3) subpixel localization:
‘Integrated Gaussian’ with fitting radius = 3 pixels, fitting method = maximum likelihood, initial sigma = 1.6, multi-emitter analysis disabled; and (4) image reconstruction: ‘Averaged
shifted histogram’. After tracking, we further filtered ambiguous emitters with the following setting: (1) filtering: frame > 100 & intensity > 100 & sigma < 220 &
uncertainty_xy < 50; (2) merge: Max distance = 10 & Max frame off = 1 & Max frames = 0; and (3) remove duplicates enabled. This setting combines the blinking molecules into one
and removes the multiple localizations in a frame. ANTIBODIES See Supplementary Table 1 for a complete list of the antibodies used in the present study. STATISTICS AND REPRODUCIBILITY No
statistical method was used to predetermine sample size. No data were excluded from the analyses. The experiments were not randomized. The Investigators were not blinded to allocation during
experiments and outcome assessment. Western blotting, biochemical fractionation and flow cytometry experiments were repeated and confirmed at least twice. REPORTING SUMMARY Further
information on research design is available in the Nature Portfolio Reporting Summary linked to this article. DATA AVAILABILITY The Micro-C, ChIP-seq, nascent RNA-seq and total RNA-seq data
generated in this publication are available in National Center for Biotechnology Information’s Gene Expression Omnibus (GEO) through accession no. GSE178982. We also reanalyzed data that we
previously generated in wild-type mESCs (GEO accession no. GSE130275)9. The spaSPT raw data are accessible through https://doi.org/10.5281/zenodo.5035837. The reference genome mm10 and
sacCer3 are available through UC Santa Cruz genome browser (https://hgdownload.soe.ucsc.edu/downloads.html). Source data are provided with this paper. CODE AVAILABILITY The availability of
the codes used in this manuscript is specified in Methods and Supplementary Methods. REFERENCES * Jerkovic, I. & Cavalli, G. Understanding 3D genome organization by multidisciplinary
methods. _Nat. Rev. Mol. Cell Biol._ https://doi.org/10.1038/s41580-021-00362-w (2021). * Beagan, J. A. & Phillips-Cremins, J. E. On the existence and functionality of topologically
associating domains. _Nat. Genet._ 52, 8–16 (2020). Article CAS Google Scholar * Lieberman-Aiden, E. et al. Comprehensive mapping of long-range interactions reveals folding principles of
the human genome. _Science_ 326, 289–293 (2009). Article CAS Google Scholar * Nora, E. P. et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. _Nature_
485, 381–385 (2012). Article CAS Google Scholar * Dixon, J. R. et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. _Nature_ 485, 376–380
(2012). Article CAS Google Scholar * Rao, S. S. P. et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. _Cell_ 159, 1665–1680 (2014).
Article CAS Google Scholar * Fudenberg, G. et al. Formation of chromosomal domains by loop extrusion. _Cell Rep._ 15, 2038–2049 (2016). Article CAS Google Scholar * Sanborn, A. L. et
al. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. _Proc. Natl Acad. Sci. USA_ 112, E6456–E6465 (2015). Article CAS Google
Scholar * Hsieh, T.-H. S. et al. Resolving the 3D landscape of transcription-linked mammalian chromatin folding. _Mol. Cell_ 78, 539–553.e8 (2020). Article CAS Google Scholar * Mifsud,
B. et al. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. _Nat. Genet._ 47, 598–606 (2015). Article CAS Google Scholar * Schoenfelder, S. et al. The
pluripotent regulatory circuitry connecting promoters to their long-range interacting elements. _Genome Res._ 25, 582–597 (2015). Article CAS Google Scholar * Nora, E. P. et al. Targeted
degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. _Cell_ 169, 930–944.e22 (2017). Article CAS Google Scholar * Rao, S. S. P. et al.
Cohesin loss eliminates all loop domains. _Cell_ 171, 305–320.e24 (2017). Article CAS Google Scholar * Wutz, G. et al. Topologically associating domains and chromatin loops depend on
cohesin and are regulated by CTCF, WAPL, and PDS5 proteins. _EMBO J._ 36, 3573–3599 (2017). Article CAS Google Scholar * Schwarzer, W. et al. Two independent modes of chromatin
organization revealed by cohesin removal. _Nature_ 551, 51–56 (2017). Article Google Scholar * Haarhuis, J. H. I. et al. The cohesin release factor WAPL restricts chromatin loop extension.
_Cell_ 169, 693–707.e14 (2017). Article CAS Google Scholar * Krietenstein, N. et al. Ultrastructural details of mammalian chromosome architecture. _Mol. Cell_ 78, 554–565.e7 (2020).
Article CAS Google Scholar * Gasperini, M. et al. A genome-wide framework for mapping gene regulation via sellular genetic screens. _Cell_ 176, 377–390.e19 (2019). Article CAS Google
Scholar * Robson, M. I., Ringel, A. R. & Mundlos, S. Regulatory landscaping: how enhancer-promoter communication is sculpted in 3D. _Mol. Cell_ 74, 1110–1122 (2019). Article CAS
Google Scholar * Anania, C. et al. In vivo dissection of a clustered-CTCF domain boundary reveals developmental principles of regulatory insulation. _Nat. Genet._ 54, 1026–1036 (2022).
Article CAS Google Scholar * Zheng, H. & Xie, W. The role of 3D genome organization in development and cell differentiation. _Nat. Rev. Mol. Cell Biol._ 20, 535–550 (2019). Article
CAS Google Scholar * Hsieh, T.-H. S. et al. Mapping nucleosome resolution chromosome folding in yeast by Micro-C. _Cell_ 162, 108–119 (2015). Article CAS Google Scholar * Hsieh, T.-H.
S., Fudenberg, G., Goloborodko, A. & Rando, O. J. Micro-C XL: assaying chromosome conformation from the nucleosome to the entire genome. _Nat. Methods_ 13, 1009–1011 (2016). Article CAS
Google Scholar * Nojima, T. et al. Mammalian NET-seq reveals genome-wide nascent transcription coupled to RNA processing. _Cell_ 161, 526–540 (2015). Article CAS Google Scholar *
Weintraub, A. S. et al. YY1 is a structural regulator of enhancer-promoter loops. _Cell_ 171, 1573–1588.e28 (2017). Article CAS Google Scholar * Ardakany, A. R., Gezer, H. T., Lonardi, S.
& Ay, F. Mustache: multi-scale detection of chromatin loops from Hi-C and Micro-C maps using scale-space representation. _Genome Biol._ 21, 256 (2020). Article Google Scholar *
Matthey-Doret, C. et al. Computer vision for pattern detection in chromosome contact maps. _Nat. Commun._ 11, 5795 (2020). Article CAS Google Scholar * Bonev, B. et al. Multiscale 3D
genome rewiring during mouse neural development. _Cell_ 171, 557–572.e24 (2017). Article CAS Google Scholar * Oksuz, B. A. et al. Systematic evaluation of chromosome conformation capture
assays. _Nat. Methods_ 18, 1046–1055 (2021). Article Google Scholar * Bintu, B. et al. Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells.
_Science_ 362, eaau1783 (2018). Article Google Scholar * Flyamer, I. M. et al. Single-nucleus Hi-C reveals unique chromatin reorganization at oocyte-to-zygote transition. _Nature_ 544,
110–114 (2017). Article CAS Google Scholar * Luppino, J. M. et al. Cohesin promotes stochastic domain intermingling to ensure proper regulation of boundary-proximal genes. _Nat. Genet._
52, 840–848 (2020). Article CAS Google Scholar * Szabo, Q. et al. Regulation of single-cell genome organization into TADs and chromatin nanodomains. _Nat. Genet._ 52, 1151–1157 (2020).
Article CAS Google Scholar * Gabriele, M. et al. Dynamics of CTCF- and cohesin-mediated chromatin looping revealed by live-cell imaging. _Science_ 376, 496–501 (2022). Article CAS
Google Scholar * Finn, E. H. et al. Extensive heterogeneity and intrinsic variation in spatial genome organization. _Cell_ 176, 1502–1515.e10 (2019). Article CAS Google Scholar *
Rodríguez-Carballo, E. et al. Chromatin topology and the timing of enhancer function at the HoxD locus. _Proc. Natl Acad. Sci. USA_ 117, 31231–31241 (2020). Article Google Scholar *
Chakraborty, S. et al. High affinity enhancer-promoter interactions can bypass CTCF/cohesin-mediated insulation and contribute to phenotypic robustness. Preprint at _bioRxiv_
https://doi.org/10.1101/2021.12.30.474562 (2022). * Natsume, T., Kiyomitsu, T., Saga, Y. & Kanemaki, M. T. Rapid protein depletion in human cells by auxin-inducible degron tagging with
short homology donors. _Cell Rep._ 15, 210–218 (2016). Article CAS Google Scholar * Kubo, N. et al. Promoter-proximal CTCF binding promotes distal enhancer-dependent gene activation.
_Nat. Struct. Mol. Biol._ 28, 152–161 (2021). Article CAS Google Scholar * Jaeger, M. G. & Winter, G. E. Fast-acting chemical tools to delineate causality in transcriptional control.
_Mol. Cell_ https://doi.org/10.1016/j.molcel.2021.02.015 (2021). * Pugacheva, E. M. et al. CTCF mediates chromatin looping via N-terminal domain-dependent cohesin retention. _Proc. Natl
Acad. Sci. USA_ 117, 2020–2031 (2020). Article CAS Google Scholar * Tu, S. et al. MAnorm2 for quantitatively comparing groups of ChIP-seq samples. _Genome Res._ 31, 131–145 (2020).
Article Google Scholar * Busslinger, G. A. et al. Cohesin is positioned in mammalian genomes by transcription, CTCF and Wapl. _Nature_ 544, 503–507 (2017). Article CAS Google Scholar *
Arnold, M., Bressin, A., Jasnovidova, O., Meierhofer, D. & Mayer, A. A BRD4-mediated elongation control point primes transcribing RNA polymerase II for 3′-processing and termination.
_Mol. Cell_ 81, 3589–3603.e13 (2021). Article CAS Google Scholar * Liu, N. Q. et al. WAPL maintains a cohesin loading cycle to preserve cell-type-specific distal gene regulation. _Nat.
Genet._ 53, 100–109 (2021). Article CAS Google Scholar * Donohoe, M. E. et al. Targeted disruption of mouse Yin Yang 1 transcription factor results in peri-implantation ilethality. _Mol.
Cell Biol._ 19, 7237–7244 (1999). Article CAS Google Scholar * Beagan, J. A. et al. YY1 and CTCF orchestrate a 3D chromatin looping switch during early neural lineage commitment. _Genome
Res._ 27, 1139–1152 (2017). Article CAS Google Scholar * Li, S., Prasanna, X., Salo, V. T., Vattulainen, I. & Ikonen, E. An efficient auxin-inducible degron system with low basal
degradation in human cells. _Nat. Methods_ 16, 866–869 (2019). Article CAS Google Scholar * Los, G. V. et al. HaloTag: a novel protein labeling technology for cell imaging and protein
analysis. _ACS Chem. Biol._ 3, 373–382 (2008). Article CAS Google Scholar * Rizkallah, R. & Hurt, M. M. Regulation of the transcription factor YY1 in mitosis through phosphorylation
of its DNA-binding domain. _Mol. Biol. Cell_ 20, 4766–4776 (2009). Article CAS Google Scholar * Raccaud, M. et al. Mitotic chromosome binding predicts transcription factor properties in
interphase. _Nat. Commun._ 10, 487 (2019). Article CAS Google Scholar * Hansen, A. S., Pustova, I., Cattoglio, C., Tjian, R. & Darzacq, X. CTCF and cohesin regulate chromatin loop
stability with distinct dynamics. _eLife_ 6, e25776 (2017). Article Google Scholar * Hansen, A. S. et al. Robust model-based analysis of single-particle tracking experiments with Spot-On.
_eLife_ 7, e33125 (2018). Article Google Scholar * Heckert, A., Dahal, L., Tijan, R. & Darzacq, X. Recovering mixtures of fast-diffusing states from short single-particle trajectories.
_eLife_ 11, e70169 (2022). Article Google Scholar * Lionnet, T. & Wu, C. Single-molecule tracking of transcription protein dynamics in living cells: seeing is believing, but what are
we seeing? _Curr. Opin. Genet Dev._ 67, 94–102 (2021). Article CAS Google Scholar * Loffreda, A. et al. Live-cell p53 single-molecule binding is modulated by C-terminal acetylation and
correlates with transcriptional activity. _Nat. Commun._ 8, 313 (2017). Article Google Scholar * Trojanowski, J. et al. Transcription activation is enhanced by multivalent interactions
independent of phase separation. _Mol. Cell_ 82, 1878–1893.e10 (2022). Article CAS Google Scholar * Mueller, F., Mazza, D., Stasevich, T. J. & McNally, J. G. FRAP and kinetic modeling
in the analysis of nuclear protein dynamics: what do we really know? _Curr. Opin. Cell Biol._ 22, 403–411 (2010). Article CAS Google Scholar * Sprague, B. L., Pego, R. L., Stavreva, D.
A. & McNally, J. G. Analysis of binding reactions by fluorescence recovery after photobleaching. _Biophys. J._ 86, 3473–3495 (2004). Article CAS Google Scholar * Chen, J. et al.
Single-molecule dynamics of enhanceosome assembly in embryonic stem cells. _Cell_ 156, 1274–1285 (2014). Article CAS Google Scholar * Garcia, D. A. et al. Power-law behavior of
transcription factor dynamics at the single-molecule level implies a continuum affinity model. _Nucleic Acids Res._ 49, 6605–6620 (2021). Article CAS Google Scholar * Hansen, A. S.,
Amitai, A., Cattoglio, C., Tjian, R. & Darzacq, X. Guided nuclear exploration increases CTCF target search efficiency. _Nat. Chem. Biol._ 16, 257–266 (2020). Article CAS Google Scholar
* Thiecke, M. J. et al. Cohesin-dependent and -independent mechanisms mediate chromosomal contacts between promoters and enhancers. _Cell Rep._ 32, 107929 (2020). Article CAS Google
Scholar * Luan, J. et al. Distinct properties and functions of CTCF revealed by a rapidly inducible degron system. _Cell Rep._ 34, 108783 (2021). Article CAS Google Scholar * Stik, G. et
al. CTCF is dispensable for immune cell transdifferentiation but facilitates an acute inflammatory response. _Nat. Genet._ 52, 655–661 (2020). Article CAS Google Scholar * Calderon, L.
et al. Cohesin-dependence of neuronal gene expression relates to chromatin loop length. _eLife_ 11, e76539 (2022). Article CAS Google Scholar * Gu, B. et al. Opposing effects of cohesin
and transcription on CTCF organization revealed by super-resolution imaging. _Mol. Cell_ 80, 699–711.e7 (2020). Article CAS Google Scholar * Aljahani, A. et al. Analysis of sub-kilobase
chromatin topology reveals nano-scale regulatory interactions with variable dependence on cohesin and CTCF. _Nat. Commun._ 13, 2139 (2022). Article CAS Google Scholar * Kagey, M. H. et
al. Mediator and cohesin connect gene expression and chromatin architecture. _Nature_ 467, 430–435 (2010). Article CAS Google Scholar * Symmons, O. et al. Functional and topological
characteristics of mammalian regulatory domains. _Genome Res._ 24, 390–400 (2014). Article CAS Google Scholar * Zhang, H. et al. Chromatin structure dynamics during the mitosis-to-G1
phase transition. _Nature_ 576, 158–162 (2019). Article CAS Google Scholar * Hou, C., Zhao, H., Tanimoto, K. & Dean, A. CTCF-dependent enhancer-blocking by alternative chromatin loop
formation. _Proc. Natl Acad. Sci. USA_ 105, 20398–20403 (2008). Article CAS Google Scholar * Huang, H. et al. CTCF mediates dosage- and sequence-context-dependent transcriptional
insulation by forming local chromatin domains. _Nat. Genet_. https://doi.org/10.1038/s41588-021-00863-6 (2021). * Flavahan, W. A. et al. Insulator dysfunction and oncogene activation in IDH
mutant gliomas. _Nature_ 529, 110–114 (2016). Article CAS Google Scholar * Flavahan, W. A. et al. Altered chromosomal topology drives oncogenic programs in SDH-deficient GISTs. _Nature_
575, 229–233 (2019). Article CAS Google Scholar * Lupiáñez, D. G. et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene–enhancer interactions. _Cell_ 161,
1012–1025 (2015). Article Google Scholar * Ushiki, A. et al. Deletion of CTCF sites in the SHH locus alters enhancer–promoter interactions and leads to acheiropodia. _Nat. Commun._ 12,
2282 (2021). Article CAS Google Scholar * Despang, A. et al. Functional dissection of the Sox9–Kcnj2 locus identifies nonessential and instructive roles of TAD architecture. _Nat. Genet._
51, 1263–1271 (2019). Article CAS Google Scholar * Kraft, K. et al. Serial genomic inversions induce tissue-specific architectural stripes, gene misexpression and congenital
malformations. _Nat. Cell Biol._ 21, 305–310 (2019). Article CAS Google Scholar * Bintu, L. et al. Dynamics of epigenetic regulation at the single-cell level. _Science_ 351, 720–724
(2016). Article CAS Google Scholar * Keung, A. J., Bashor, C. J., Kiriakov, S., Collins, J. J. & Khalil, A. S. Using targeted chromatin regulators to engineer combinatorial and
spatial transcriptional regulation. _Cell_ 158, 110–120 (2014). Article CAS Google Scholar * Stevens, T. J. et al. 3D structures of individual mammalian genomes studied by single-cell
Hi-C. _Nature_ 544, 59–64 (2017). Article CAS Google Scholar * Basu, S. et al. Live-cell 3D single-molecule tracking reveals how NuRD modulates enhancer dynamics. Preprint at _bioRxiv_
https://doi.org/10.1101/2020.04.03.003178 (2020). * Shipony, Z. et al. Dynamic and static maintenance of epigenetic memory in pluripotent and somatic cells. _Nature_ 513, 115–119 (2014).
Article CAS Google Scholar * Meir, Z., Mukamel, Z., Chomsky, E., Lifshitz, A. & Tanay, A. Single-cell analysis of clonal maintenance of transcriptional and epigenetic states in cancer
cells. _Nat. Genet._ 52, 709–718 (2020). Article CAS Google Scholar * Nuñez, J. K. et al. Genome-wide programmable transcriptional memory by CRISPR-based epigenome editing. _Cell_ 184,
2503–2519.e17 (2021). Article Google Scholar * Engreitz, J. M. et al. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. _Science_ 341,
1237973 (2013). Article Google Scholar * Minajigi, A. et al. A comprehensive Xist interactome reveals cohesin repulsion and an RNA-directed chromosome conformation. _Science_ 349, aab2276
(2015). Article Google Scholar * Xiao, J. Y., Hafner, A. & Boettiger, A. N. How subtle changes in 3D structure can create large changes in transcription. _eLife_ 10, e64320 (2021).
Article CAS Google Scholar * Zuin, J. et al. Nonlinear control of transcription through enhancer–promoter interactions. _Nature_ 604, 571–577 (2022). Article CAS Google Scholar *
Rinaldi, L. et al. The glucocorticoid receptor associates with the cohesin loader NIPBL to promote long-range gene regulation. _Sci. Adv._ 8, eabj8360 (2022). Article CAS Google Scholar *
Yan, J. et al. Transcription factor binding in human cells occurs in dense clusters formed around cohesin anchor sites. _Cell_ 154, 801–813 (2013). Article CAS Google Scholar * Brodsky,
S. et al. Intrinsically disordered regions direct transcription factor in vivo binding specificity. _Mol. Cell_ 79, 459–471.e4 (2020). Article CAS Google Scholar * Garcia, D. A. et al. An
intrinsically disordered region-mediated confinement state contributes to the dynamics and function of transcription factors. _Mol. Cell_ 81, 1484–1498.e6 (2021). Article CAS Google
Scholar * Pettitt, S. J. et al. Agouti C57BL/6N embryonic stem cells for mouse genetic resources. _Nat. Methods_ 6, 493–495 (2009). Article CAS Google Scholar * Ran, F. A. et al. Genome
engineering using the CRISPR–Cas9 system. _Nat. Protoc._ 8, 2281–2308 (2013). Article CAS Google Scholar * Testa, A. et al. Chromatin immunoprecipitation (ChIP) on Chip experiments
uncover a widespread distribution of NF-Y binding CCAAT sites outside of core promoters*. _J. Biol. Chem._ 280, 13606–13615 (2005). Article CAS Google Scholar * Schindelin, J. et al.
Fiji: an open-source platform for biological-image analysis. _Nat. Methods_ 9, 676–682 (2012). Article CAS Google Scholar * Servant, N. et al. HiC-Pro: an optimized and flexible pipeline
for Hi-C data processing. _Genome Biol._ 16, 259 (2015). Article Google Scholar * Abdennur, N. & Mirny, L. A. Cooler: scalable storage for Hi-C data and other genomically labeled
arrays. _Bioinformatics_ https://doi.org/10.1093/bioinformatics/btz540 (2019). * Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. _Cell
Syst._ 3, 95–98 (2016). Article CAS Google Scholar * Imakaev, M. et al. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. _Nat. Methods_ 9, 999–1003 (2012).
Article CAS Google Scholar * Knight, P. A. & Ruiz, D. A fast algorithm for matrix balancing. _IMA J. Numer. Anal._ 33, 1029–1047 (2012). Article Google Scholar * Kerpedjiev, P. et
al. HiGlass: web-based visual exploration and analysis of genome interaction maps. _Genome Biol._ 19, 125 (2018). Article Google Scholar * Yardımcı, G. G. et al. Measuring the
reproducibility and quality of Hi-C data. _Genome Biol._ 20, 57 (2019). Article Google Scholar * Pintacuda, G. et al. hnRNPK recruits PCGF3/5-PRC1 to the Xist RNA B-repeat to establish
polycomb-mediated chromosomal silencing. _Mol. Cell_ 68, 955–969.e10 (2017). Article CAS Google Scholar * Ernst, J. & Kellis, M. ChromHMM: automating chromatin-state discovery and
characterization. _Nat. Methods_ 9, 215–216 (2012). Article CAS Google Scholar * Bray, N. L., Pimentel, H., Melsted, P. & Pachter, L. Near-optimal probabilistic RNA-seq
quantification. _Nat. Biotechnol._ 34, 525–527 (2016). Article CAS Google Scholar * Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq
data with DESeq2. _Genome Biol._ 15, 550 (2014). Article Google Scholar * Nojima, T., Gomes, T., Carmo-Fonseca, M. & Proudfoot, N. J. Mammalian NET-seq analysis defines nascent RNA
profiles and associated RNA processing genome-wide. _Nat. Protoc._ 11, 413–428 (2016). Article CAS Google Scholar * Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner.
_Bioinformatics_ 29, 15–21 (2013). Article CAS Google Scholar * Wang, J. et al. Nascent RNA sequencing analysis provides insights into enhancer-mediated gene regulation. _BMC Genom._ 19,
633 (2018). Article Google Scholar * Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features.
_Bioinformatics_ 30, 923–930 (2014). Article CAS Google Scholar * Heckert, A., Dahal, L., Tjian, R. & Darzacq, X. Recovering mixtures of fast diffusing states from short single
particle trajectories. _eLife_ 11, e70169 (2022). Article Google Scholar * Ovesný, M., Křížek, P., Borkovec, J., Švindrych, Z. & Hagen, G. M. ThunderSTORM: a comprehensive ImageJ
plug-in for PALM and STORM data analysis and super-resolution imaging. _Bioinformatics_ 30, 2389–2390 (2014). Article Google Scholar Download references ACKNOWLEDGEMENTS This work was
supported by the California Institute of Regenerative Medicine (grant no. LA1-08013 to X.D.), the Koret UC Berkeley–Tel Aviv University Initiative grant (to T.S.H. and X.D.) and the Howard
Hughes Medical Institute (grant no. 003061 to R.T.). T.S.H. was a postdoctoral fellow of the Koret UC Berkeley–Tel Aviv University Initiative. E.S. was an undergraduate fellow of SURF Rose
Hills Independent at UC Berkeley. A.S.H. acknowledges support from the NIH under grant nos. R00GM130896, DP2GM140938, R33CA257878 and NSF 2036037. We thank Gina M. Dailey for assisting with
cloning and all members of the Tjian and Darzacq laboratory for comments on the manuscript. AUTHOR INFORMATION Author notes * These authors contributed equally: Claudia Cattoglio, Elena
Slobodyanyuk. AUTHORS AND AFFILIATIONS * Department of Molecular and Cell Biology, University of California, Berkeley, Berkeley, CA, USA Tsung-Han S. Hsieh, Claudia Cattoglio, Elena
Slobodyanyuk, Xavier Darzacq & Robert Tjian * Li Ka Shing Center for Biomedical and Health Sciences, University of California, Berkeley, Berkeley, CA, USA Tsung-Han S. Hsieh, Claudia
Cattoglio, Elena Slobodyanyuk, Xavier Darzacq & Robert Tjian * CIRM Center of Excellence, University of California, Berkeley, Berkeley, CA, USA Tsung-Han S. Hsieh, Claudia Cattoglio,
Xavier Darzacq & Robert Tjian * Howard Hughes Medical Institute, University of California, Berkeley, Berkeley, CA, USA Tsung-Han S. Hsieh, Claudia Cattoglio & Robert Tjian * Center
for Computational Biology, University of California, Berkeley, Berkeley, CA, USA Tsung-Han S. Hsieh & Xavier Darzacq * Department of Biological Engineering, Massachussets Institute of
Technology, Cambridge, MA, USA Anders S. Hansen Authors * Tsung-Han S. Hsieh View author publications You can also search for this author inPubMed Google Scholar * Claudia Cattoglio View
author publications You can also search for this author inPubMed Google Scholar * Elena Slobodyanyuk View author publications You can also search for this author inPubMed Google Scholar *
Anders S. Hansen View author publications You can also search for this author inPubMed Google Scholar * Xavier Darzacq View author publications You can also search for this author inPubMed
Google Scholar * Robert Tjian View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS T.S.H., C.C. and E.S. conceived and designed the project.
C.C. performed all biochemical and ChIP-seq experiments and analysis with E.S.’s assistance. E.S. generated all plasmids and cell lines with the assistance of T.S.H. C.C. and A.S.H.
generated the parental degradation cell lines. T.S.H. performed Micro-C assays and analysis. E.S. and T.S.H. performed all imaging experiments and analyses. T.S.H. drafted the manuscript.
C.C., E.S., A.S.H. and R.T. edited the manuscript. R.T. and X.D. supervised the project. CORRESPONDING AUTHORS Correspondence to Xavier Darzacq or Robert Tjian. ETHICS DECLARATIONS COMPETING
INTERESTS The authors declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Nature Genetics_ thanks Charles Danko and the other, anonymous, reviewer(s) for their contribution
to the peer review of this work. Peer reviewer reports are available. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in
published maps and institutional affiliations. EXTENDED DATA EXTENDED DATA FIG. 1 GENOME-WIDE IDENTIFICATION OF CHROMATIN LOOPS. A. Loop strengths. Paired loci quantified using Chromosight27
(results comparable to Mustache26 in Fig. 1b). Loop numbers shown on the left. Box plot: quartiles for the loop strength score distribution as in Fig. 1b. Right: genome-wide averaged
contact signals. Contact map normalized by matrix balancing and distance, shown as a diverging colormap with the gradient of normalized contact enrichment in log10 (red: positive enrichment;
blue: negative signal). Ratio of contact enrichment for the center pixels annotated within each plot. Asterisks: _P_ < 10−16, two-sided Wilcoxon test. n = 37 biological replicates. B.
Comparison of Mustache and Hiccups loop calling algorithms on Micro-C data (top) and Micro-C vs. Hi-C loops called by different algorithms (bottom). C. Enrichment of mESC ChromHMM107 states
at loop anchors. Heatmap: log2 enrichment of each state ± 5-kb around loop anchors. Loops = 75,190; loop anchors = 118,733 after removing duplicates. D. Loop analysis pipeline. E. Loop
length distributions. Colored box: 2-kb resolution Micro-C data; white box: 4-kb resolution data. Box plot: quartiles for the loop length distribution as in Fig. 1b. Median size of loops
annotated on the right. Median lengths are larger than our previous analysis with the insulation score4 due to the high computational expense to quantify the short-range loops with Micro-C
data finer than 1-kb resolution. Asterisks: _P_ < 10−16, two-sided Wilcoxon test. _n_ = 37 biological replicates. F. Left: gel image of the mNET-seq library size (6% PAGE). Right:
resolved bands on a Fragment Analyzer electropherogram. G. Heatmap of Pearson’s correlation between sequencing data and chromatin structures by Micro-C or Hi-C. Compartment, TAD and loop
scores obtained from Cooltools, Arrowhead and Chromosight, respectively. H. Micro-C or Hi-C28 loop numbers. Contacts surrounding the intersections of targets quantified with data at 4-kb
resolution using Chromosight. I. Snapshots of Hi-C data in the same region as Fig. 2a. J. Rank-ordered distribution of loop length against loop strength. Distributions fitted and smoothed by
LOESS regression. Error bands: fitted curve ±SEM with 95% confidence interval. K. CTCF and RAD21 ChIP-seq signal at TAD boundaries grouped by intra-TAD/inter-TAD cohesin, E-P, or P-P loops.
EXTENDED DATA FIG. 2 CELL LINES GENERATION, VALIDATION, AND BIOCHEMICAL FRACTIONATION ASSAY. A. Schematics for endogenously tagging CTCF, RAD21, WAPL with the mAID degron and for
endogenously tagging YY1 with miniIAA7. B. Immunoblots of CTCF, RAD21, WAPL, YY1, and their tags (HaloTag for CTCF, V5 for RAD21, RFP (mScarletI) for YY1, and HA for WAPL) for the protein
expression levels and sizes in wild type mESCs and degron clones C58 (ΔCTCF), F1 (ΔRAD21), YD39 (ΔYY1), and C40 (ΔWAPL). C. Quantification of the levels of WAPL, CTCF, Rad21 and YY1 proteins
in the degron clones C58 (ΔCTCF), F1 (ΔRAD21), C40 (ΔWAPL) and YD39 (ΔYY1) relative to wild type mESCs by immunoblotting (n = 2 independent immunoblots ran on the same cell lysates). Black
asterisks point to the basal degradation level of each degron-tagged factor in the corresponding cell line. D. Immunoblots of CTCF, RAD21 and WAPL proteins across a degradation time course
from 0 (untreated) to 3 hr (IAA treatment) in ΔCTCF, ΔRAD21, and ΔWAPL degron clones. E. Schematic for biochemical salt fractionation experiment in mock-treated (UT) or IAA-treated degron
clones. F. Immunoblots of cytoplasmic (Cyt) and nuclear proteins dissociating from chromatin at increasing salt concentrations (75, 150, 300 and 500 mM NaCl) as schematized in g, probed with
the indicated antibodies (α). Son: sonicated, solubilized chromatin; % of total: signal intensity of each fraction divided by the total signal intensity across all fractions; % of
degradation: 1 – (signal intensity of each fraction in the IAA treated condition divided by the untreated condition), after normalization for total protein amounts (normalizer for ΔWAPL
degron: total CTCF; normalizer for ΔRAD21 degron: total YY1). A blot with anti-histone 2B antibody (almost exclusively found in the solubilized chromatin) controls for chromatin integrity
during the fractionation steps. Source data EXTENDED DATA FIG. 3 CHIP-SEQ ANALYSIS. A. Heatmap of ‘all-by-all’ Spearman’s correlation for all ChIP-seq replicates samples (_n_ = 96). B.
Pairwise correlation of ChIP-seq data for all UT samples. C. Heatmap of Jaccard’s index for the ratio of co-enriched peaks between the ChIP-seq replicates. D. Summary of differential
ChIP-seq peak analysis for all UT degron cell clones. The chart shows the fraction of down-regulated, up-regulated, or unchanged peaks in the UT condition. The total number of peaks for each
protein was summed from all peaks in UT cells. E. Heatmaps of CTCF and cohesin (RAD21, SMC1A, and SMC3 subunits) ChIP-seq signal around WT-CTCF peaks called by MACS2 in the CTCF-, RAD21-,
or WAPL-degron cells. The peaks called by MACS2 are plotted at the center across a ±3-kb region. The colormap shows the maximum signal (log2) in blue and the minimum signal in white. F.
Heatmaps of differential ChIP-seq signals for SMC1A and SMC3 in cells depleted of CTCF, RAD21, or WAPL. The peaks called by MACS2 are plotted at the center across a ±3-kb region. The
colormap shows an increased signal (log2) in orange and a decreased signal in purple after IAA treatment. G. MA plots show the differential ChIP-seq peaks between the UT and IAA-treated
cells. The significantly changed peaks (_P__adj_ < 0.05) are colored in red. X-axis: mean observations of UT and IAA cells. Y-axis: log2 fold-change comparing the UT and IAA-treated
cells. The statistical test for all ChIP-seq in this study are obtained from the statistical model derived from MAnorm2 unless otherwise indicated. EXTENDED DATA FIG. 4 MICRO-C ANALYSIS. A.
Summary of Micro-C experiments in the degron cell lines. Total unique reads annotated for each replicate on the right, consisting of trans-interactions (inter-chromosome), short-range
cis-interactions (<20 kb), and long-range cis-interactions (>20 kb). B. Micro-C reproducibility tests. Top: pairwise similarity scores measured by GenomeDisco between UT vs. IAA and UT
vs. UT samples using 10-kb resolution of Micro-C matrices. Bottom: similarity scores measured by QuASAR between replicates (light lines) or comparing the UT and IAA-treated samples (dark
lines) using Micro-C matrices at 250-kb, 50-kb, 25-kb, and 10-kb resolutions. C. Genome-wide contact decaying P(s) analysis (bottom) and slope distributions of the P(s) curves (top) for UT
cells. D. Micro-C contact maps at specific regions or at genome-wide scale across multiple resolutions in the UT and IAA-treated cells. Left to right: examples of Pearson’s correlation
matrices showing plaid-like chromosome compartments; saddle plots showing overall compartment strength (A-A: bottom-right; B-B: top left); differential saddle plots showing changes in
compartment strength; contact matrices showing TADs along the diagonal; ADA showing all TADs; differential ADA showing TAD strength changes. E. Slope distribution of P(s) curves for UT and
IAA-treated cells. Dashed lines highlight the range of genome distances affected by CTCF, RAD21, or WAPL depletion. CTCF depletion had minimal impact on overall interactions across the
genome. RAD21 depletion reduced contact frequencies in the range of 10–200 kb but increased interactions at 300 kb – 5 Mb. WAPL depletion showed the opposite trend, with increased contacts
at 70–700 kb but reduced contacts at 1–5 Mb. F. Scatter plot of cohesin loops scores in UT and IAA-treated cells. The overlaid heatmap indicates dot density (red: highest, blue: lowest).
Dashed lines along the diagonal delimit unchanged loops. G. Loop numbers called by Mustache for UT and IAA-treated cells. The additional loops (n = 5764) identified after WAPL depletion show
longer lengths, with a 570-kb median. H. APA for loops across multiple ranges of genomic distance in UT and IAA-treated cells. EXTENDED DATA FIG. 5 RNA-SEQ AND MNET-SEQ ANALYSIS. A.
Pairwise correlation of RNA-seq (left) and nascent RNA-seq (right) data for all UT samples. B. MA plots of nascent RNA-seq comparing UT wild type JM8.N4 mESCs with cells treated for 6 hours
with the BRD inhibitor dBET6. Differentially expressed genes (DEGs) with q-value < 0.01 and 2-fold change are highlighted in pink (up) or blue (down). C. MA plot of nascent RNA-seq
comparing wild-type JM8.N4 mESCs with ΔCTCF, ΔRAD21, or ΔWAPL degron cell lines after IAA treatment for 3, 12, and 24 hours. DEGs (q-value < 0.01 and 2-fold change) are highlighted in
pink (up) or blue (down). D. MA plot of total RNA-seq comparing wild-type JM8.N4 mESCs with ΔCTCF, ΔRAD21, or ΔWAPL degron cell lines after IAA treatment for 3, 12, and 24 hours. DEGs
(q-value < 0.01 and 2-fold change) are highlighted in pink (up) or blue (down). Quality of spike-in control (ERCC spike-in) for each condition is plotted in the right panel. E. Bar graph
showing the summary of DEGs identified by nascent RNA-seq with (bottom) or without (top) spike-in calibration. F. Overlap of DEGs between different depletions and assays. Bar graph shows the
odds ratio on the y-axis and is annotated with the corresponding p-value. Many DEGs are consistent between CTCF and cohesin depletion (Odd ratio > 10), suggesting that while CTCF and
cohesin are required for the transcriptional maintenance of only a small subset of genes, those genes tend to require the presence of both factors. Statistical test: Fisher’s exact test. G.
Snapshots of Micro-C maps comparing chromatin interactions in the UT (top-right) and IAA-treated (bottom-left) cells surrounding _Klf4_ locus. Contact maps are annotated with gene boxes and
1D chromatin tracks showing the ChIP-seq signal enrichment in the same region. EXTENDED DATA FIG. 6 ANALYSIS OF E-P/P-P INTERACTIONS. A. Heatmaps of differential ChIP-seq signals for CTCF,
RAD21, SMC1A, and SMC3 comparing UT and IAA-treated degron cell lines. Heatmaps were plotted across a ±3-kb region around four major types of loop anchors. The colormap shows an increased
signal (log2) in orange and a decreased signal in purple after IAA treatment. B. Profiles of differential ChIP-seq signals for CTCF or RAD21 comparing the UT and IAA-treated degron cell
lines across the same regions as in a. C. Bar graph showing the changes in loop intensity quantified from Fig. 4e. D. APA is plotted for E-P (top) or P-P (bottom) loops that are grouped by
up-regulated, down-regulated, or unchanged loops (right) in untreated and IAA-treated cells. E. Scatter plot of loop scores for the called loops in the UT and IAA-treated cells (left). The
violin chart (inset) shows the distribution of loop scores for the UT and IAA-treated conditions. The box plot indicates the quartiles for the loop strength score distribution (see Fig. 1b).
The pile-up contact maps are plotted with loops grouped by up-regulated, down-regulated, or unchanged loops (right). (control = 2; IAA = 3 biological replicates). F. Enrichment of the
ChromHMM states at loop anchors grouped by up-regulated, down-regulated, or unchanged after IAA treatment. G. Scatter plot shows the relationship between loop length (x-axis) and the changes
in loop intensity (y-axis) for E-P (top) and P-P (bottom) loops. H. Profiles of ChIP-seq signals for CTCF (top) or RAD21 (bottom) across a ±3-kb region around the anchors of E-P or P-P
loops that are either unchanged (gray) or reduced after CTCF (blue) or RAD21 (pink) depletion. I. Length distribution of the unchanged or down-regulated E-P/P-P loops relative to TAD
boundaries (left). Ratio of the unchanged (gray) or down-regulated (pink) E-P/P-P loop anchors located within ±10 kb of TAD boundaries (right). EXTENDED DATA FIG. 7 ANALYSIS OF CHIP-SEQ,
RNA-SEQ, MNET-SEQ, AND MICRO-C IN YY1-AID CELLS. A. YY1 protein domains. B. mScarletI signal in HaloTag-YY1 cells (YN11) treated with IAA by flow cytometry (left) and confocal imaging
(right). C. YY1 ChIP-seq signal around YY1 peaks with or without spike-in calibration. Peaks called by MACS2 in the YY1-degron cells with antibodies against RFP or two different YY1
epitopes. Final YY1 peaks used throughout this manuscript summed from all peaks. Colormap: blue: maximum signal (log2), white: minimum signal. D. MA plots showing differential ChIP-seq peaks
between UT and IAA-treated cells. Significantly changed peaks (_P__adj_ < 0.05) in red. X-axis: mean observations of UT and IAA-treated cells. Y-axis: log2 fold-change (UT/IAA-treated).
E. Differential ChIP-seq peak analysis. Fraction of down-regulated, up-regulated, or unchanged peaks after IAA treatment. Total number of peaks for each protein summed from all peaks in UT
and IAA-treated cells. F. Percentage of YY1 peaks enriched with four primary types of ChromHMM states and silent chromatin. G. Heatmaps of differential YY1, SMC1A, and SMC3 ChIP-seq signals
after YY1 depletion. H. Immunoblots of cytoplasmic and nuclear proteins dissociating from chromatin at increasing salt concentrations (Extended Data Fig. 2f), probed with various antibodies
(α). Son: solubilized chromatin; % of total: signal of each fraction / total signal; % of degradation: 1 – (signal of each fraction in IAA-treated / UT), normalized by total CTCF protein. I.
Micro-C of UT and YY1-depleted cells. Right: total unique reads annotated for each replicate, consisting of trans- (inter-chromosome), short-range cis- (<20 kb), and long-range cis-
(>20 kb) interactions. J. Genome-wide contact decaying P(s) analysis (bottom) and slope distributions of the P(s) curves (top) for UT cells. K. MA plot of total RNA-seq and nascent
RNA-seq for YY1 degron 3 to 24 hours after IAA treatment. L. Scatter plots of loop scores (quantified using 2-kb-resolution Micro-C data) plotted for E-P or P-P loops in UT and IAA-treated
cells. APA for YY1, E-P, or P-P anchored loops plotted for the ΔYY1 degron cell line in UT and IAA-treated cells. M. Micro-C maps comparing chromatin interactions in UT and IAA-treated ΔYY1
cells surrounding _Nes_ gene. Source data EXTENDED DATA FIG. 8 DYNAMIC ANALYSIS OF YY1 PROTEIN. A. Schematic for conjugating a fluorescent dye with the HaloTag-YY1 fusion protein, which
emits fluorescence upon excitation by a specific wavelength. B. Schematic for endogenously fusing the N-terminus of YY1 with HaloTag. C. Immunoblots of wild-type (WT), HaloTag-YY1 knock-in
(YN11 and YN31), and stably expressing Halotag-YY1/YY1-HaloTag (PBYN2 and PBYC3) mESC lines for YY1, HaloTag, and FLAG proteins. TBP was used as a loading control. We either added a HaloTag
to the N-terminus of the endogenous YY1 via CRISPR-Cas9-mediated genome editing or ectopically expressed YY1 fused with HaloTag using a minimal L30 promoter via PiggyBac transposition. D.
Confocal or Airyscan-resolved live-cell imaging for HaloTag-YY1 stained with 500 nM TMR Halo ligand. Arrow points to sporadic loci within the nucleolus. Images at the bottom panel are a
z-projection with the mean signal. E. Schematic for the spaSPT experiment and the analysis pipeline with Quot and SASPT54. F. Heatmaps of localization errors obtained by aggregated
likelihood across all trajectories (left) or posterior marginalized localization error (middle) for clones YN11, YN31, PBYN2, PBYC3 and H2B. The distribution of the likelihood of diffusion
coefficients (x-axis) for single cells (each row at the y-axis) is plotted on the right panel. G. spaSPT displacement histograms for YN11, YN31, PBYN2, PBYC3, and H2B. Raw displacement data
for seven different lag times are shown with a three-state Spot-On model53 fit overlaid. The inferred fractions and diffusion coefficients for each cell are shown in the table in the bottom
panel. H. Snapshots of FRAP experiments for multiple time points from ‘before bleach’ to ‘50 sec after bleach’. I. FRAP analysis of YY1 bleached with a small circular spot. Error bars
indicate the standard deviation of each acquired data point. (_n_ = 8 cells examined over 2 independent experiments; error bars: the fitted curve ±SEM with 95% confidence interval). Source
data EXTENDED DATA FIG. 9 YY1 DYNAMICS AFTER CTCF OR COHESIN LOSS. A. Histogram of mNeonGreen intensity of HaloTag-YY1 CTCF or RAD21 degron cells (clones CD1 and RD35) treated with IAA for 0
or 3 hours. B. Airyscan-resolved live-cell imaging of HaloTag-YY1 stained with 500-nM TMR Halo ligand in wild type, CTCF-, or RAD21-depleted cells. C. Localization error heatmaps obtained
by aggregated likelihood across all trajectories (first panel on the left) or posterior marginalized localization error (second panel) for UT or IAA-treated HaloTag-YY1 CTCF or RAD21 degron
cells. Third panel: distribution of the diffusion coefficients likelihood (x-axis) for single cells (each y-axis row). Fourth panel: estimation of YY1 diffusion coefficients by regular
Brownian motion with marginalized localization errors. D. Heatmaps of differential CTCF, RAD21, SMC1A, SMC3, and YY1 ChIP-seq signals in CTCF-, RAD21-, and WAPL-depleted cells. Peaks are
centered on wild-type YY1 peaks across a ±3-kb region. Colormap: increased signal (log2) in orange and decreased signal in purple after IAA treatment. E. MA plot showing the differential
ChIP-seq peaks between UT and IAA-treated cells (left). Significantly changed peaks (_P__adj_ < 0.05) colored in red. X-axis: mean observations of UT and IAA cells. Y-axis: log2
fold-change comparing UT and IAA-treated cells. Pie chart: percentages of downregulated YY1 peaks enriched at promoters and enhancers (right). F. Immunoblots of cytoplasmic (Cyt) and nuclear
YY1 protein dissociating from chromatin at increasing salt concentrations (75-, 150-, 300- and 500-mM NaCl, Extended Data Fig. 2f) in CTCF, RAD21 or WAPL degron lines UT or treated with
auxin (IAA). Son: sonicated, solubilized chromatin. Numbers represent the signal intensity of each fraction divided by the total signal intensity across all fractions (% of total). Red
highlights the percent of YY1 retained on chromatin after all salt washes. G. Stacked bar graph of bound, slow, and fast diffusing KLF4 (left) or SOX2 (right) populations in UT and
IAA-treated RAD21 degron cells, obtained by spaSPT analysis. Source data SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION Supplementary Notes and Methods. REPORTING SUMMARY PEER REVIEW
FILE SUPPLEMENTARY TABLE 1-10 Supplementary Table 1: Plasmids, primers and antibodies used in the present study. Supplementary Table 2: Summary of Micro-C interactions. Supplementary Table
3: TAD arrowhead. Supplementary Table 4: TAD insulation score. Supplementary Table 5: Loop-calling options and filtering parameters for Chromosight. Supplementary Table 6: Mustache loops.
Supplementary Table 7: Chromosight loops. Supplementary Table 8: Cohesin loops called by Chromosight using Micro-C data at 4-kb resolution. Supplementary Table 9: E–P loops called by
Chromosight using Micro-C data at 4-kb resolution. Supplementary Table 10: P–P loops called by Chromosight using Micro-C data at 4-kb resolution. SUPPLEMENTARY TABLE 11 Supplementary Table
11 RNA-seq–DEseq2. SUPPLEMENTARY TABLE 12 Supplementary Table 12 mNET-seq–DEseq2. SUPPLEMENTARY DATA 1 Plasmid sequence. SOURCE DATA SOURCE DATA FIG. 3 Unprocessed western blots. SOURCE DATA
FIG. 5 Unprocessed western blots. SOURCE DATA FIG. 6 Unprocessed western blots. SOURCE DATA FIG. 7 Unprocessed western blots. SOURCE DATA EXTENDED DATA FIG. 2 Unprocessed western blots.
SOURCE DATA EXTENDED DATA FIG. 7 Unprocessed western blots. SOURCE DATA EXTENDED DATA FIG. 8 Unprocessed western blots. SOURCE DATA EXTENDED DATA FIG. 9 Unprocessed western blots. RIGHTS AND
PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any
medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The
images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly
from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Hsieh, TH.S.,
Cattoglio, C., Slobodyanyuk, E. _et al._ Enhancer–promoter interactions and transcription are largely maintained upon acute loss of CTCF, cohesin, WAPL or YY1. _Nat Genet_ 54, 1919–1932
(2022). https://doi.org/10.1038/s41588-022-01223-8 Download citation * Received: 14 July 2021 * Accepted: 11 October 2022 * Published: 05 December 2022 * Issue Date: December 2022 * DOI:
https://doi.org/10.1038/s41588-022-01223-8 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative