Play all audios:
ABSTRACT Polycomb repressive complex 2 (PRC2) interacts with RNA in cells, but there is no consensus on how RNA regulates PRC2 canonical functions, including chromatin modification and the
maintenance of transcription programs in lineage-committed cells. We assayed two separation-of-function mutants of the PRC2 catalytic subunit EZH2, defective in RNA binding but functional in
methyltransferase activity. We find that part of the RNA-binding surface of EZH2 is required for chromatin modification, yet this activity is independent of RNA. Mechanistically, the
RNA-binding surface within EZH2 is required for chromatin modification in vitro and in cells, through interactions with nucleosomal DNA. Contrarily, an RNA-binding-defective mutant exhibited
normal chromatin modification activity in vitro and in lineage-committed cells, accompanied by normal gene repression activity. Collectively, we show that part of the RNA-binding surface of
EZH2, rather than the RNA-binding activity per se, is required for the histone methylation in vitro and in cells, through interactions with the substrate nucleosome. SIMILAR CONTENT BEING
VIEWED BY OTHERS STRUCTURAL INSIGHTS INTO HOW DEK NUCLEOSOME BINDING FACILITATES H3K27 TRIMETHYLATION IN CHROMATIN Article Open access 21 February 2025 AN EVOLVING LANDSCAPE OF PRC2–RNA
INTERACTIONS IN CHROMATIN REGULATION Article 30 April 2025 STRUCTURAL BASIS FOR THE INHIBITION OF PRC2 BY ACTIVE TRANSCRIPTION HISTONE POSTTRANSLATIONAL MODIFICATIONS Article Open access 07
January 2025 MAIN RNA is found in most compartments of the nucleus and is required for their three-dimensional (3D) structural organization1. RNA binds to various chromatin modifiers and is
proposed to regulate them, although the molecular mechanisms and their functional consequences are often not under consensus (reviewed in ref. 2). One chromatin modifier that has been
extensively studied in the context of RNA-mediated regulation is the repressive histone methyltransferase PRC2 (ref. 3). PRC2 is required for normal development and for the maintenance of
transcription programs in lineage-committed cells4. At the molecular level, PRC2 introduces the trimethyl modification to lysine 27 of histone H3 (H3K27me3), which is required for
maintaining genes in a repressed state5. PRC2 interacts with thousands of transcripts in cells with rather broad specificity6,7,8. The functional consequences of PRC2–RNA interactions are
the subject of an ongoing debate (reviewed in refs. 3,9). On one hand, RNA inhibits the histone methyltransferase activity of PRC2 (refs. 10,11,12), competes against nucleosomes for binding
to PRC2 (refs. 13,14) and removes PRC2 from genes15. On the other hand, RNA was previously proposed to recruit PRC2 to chromatin16 and more recently reported to be essential for PRC2
chromatin occupancy17 and polycomb-mediated genome architecture18. Hence, at this time there is no clarity or consensus on how RNA regulates the key canonical functions of PRC2: introducing
the H3K27me3 mark to chromatin and enforcing lineage commitment through maintaining transcription programs4. An inherent challenge in determining how RNA regulates PRC2 was the difficulty in
separating the RNA binding from other biochemical functions of PRC2. Separation-of-function mutations perturb one biochemical function without affecting others, allowing dissection of the
contribution of specific biochemical properties of a given protein. Separation-of-function mutations in PRC2, which reduce its affinity for RNA without affecting its methyltransferase
activity, have already been identified on the basis of in vitro studies19,20. One of these separation-of-function mutations, which includes ten substitutions in two separate patches in the
catalytic subunit EZH2 (ref. 19), has already been tested in human pluripotent stem cells in two recent studies17,18. This mutant—termed mt2 herein (Fig. 1a)—exhibited aberrant targeting of
PRC2 to chromatin, impaired deposition of the H3K27me3 mark17, loss of PRC2-associated DNA looping18 and cardiomyocyte differentiation defects17. These data were used to support a model
where PRC2 requires its RNA-binding activity for its chromatin localization and function17. PRC2 uses multiple faces to contact chromatin. For instance, PRC2 can simultaneously contact the
nucleosomal DNA of two adjacent nucleosomes21 and also interact with the DNA linker that resides between nucleosomes14. This is conceptually similar to the case of RNA binding, which
involves multiple dispersed patches in PRC2 (refs. 19,20,22). Hence, we reasoned that some PRC2 mutants might be bona fide separation-of-function mutants—defective in RNA binding and active
in the methyltransferase of chromatin—while other mutants might be defective in the methylation of only certain substrates. We anticipated that comparing such different RNA-binding-defective
mutants would allow us to determine the molecular function of different RNA-binding sites in PRC2. Herein, we examine two separation-of-function mutations in EZH2 that are defective in RNA
binding but catalytically active against isolated H3 histones. We directly compared these mutants against a catalytically defective EZH2 mutant, which serves as a background for
methyltransferase loss-of-function. In vitro, we show that one of the RNA-binding-defective mutants (mt1; Fig. 1a) is a bona fide separation-of-function mutant, active in the methylation of
H3 histones, nucleosomes and chromatin. The other mutant (mt2; Fig. 1a), which was considered a separation-of-function mutant in earlier studies in vitro17,19 and in cells17,18, is in fact
defective in the modification of H3 histones while they are in the context of chromatin. We then compared these three mutants in cells, aiming to dissect the RNA-binding activity from the
catalytic activity of PRC2. Our data indicate that a portion of the RNA-binding surface of PRC2 is as important as its catalytic activity for the canonical functions of PRC2 in cells:
histone modification, gene repression and the maintenance of cell identity. Yet, these molecular functions of PRC2 were not substantially affected by a separation-of-function mutation in
EZH2, defective in RNA binding and active in chromatin modification. Collectively, our data indicate that part of the RNA-binding surface of PRC2, but not its RNA-binding activity per se, is
required for the modification of chromatin by PRC2. RESULTS A PORTION OF THE RNA-BINDING SURFACE OF EZH2 IS DISPENSABLE FOR THE METHYLATION OF HISTONE TAILS BUT REQUIRED FOR THE
MODIFICATION OF CHROMATIN IN VITRO To identify functions of the RNA-binding surfaces of PRC2, we set out to assay two RNA-binding-defective separation-of-function EZH2 mutants that were
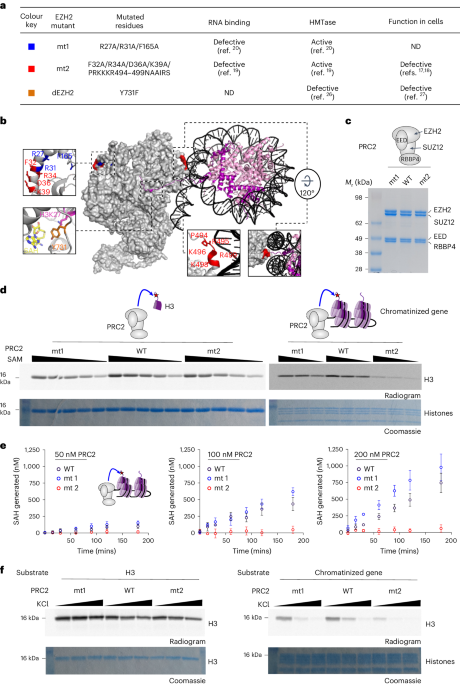
identified in previous studies19,20. These EZH2 mutants, mt1 (ref. 20) and mt2 (ref. 19), include three and ten nonoverlapped amino acids, respectively (Fig. 1a), and are both catalytically
active but defective in RNA binding. The majority of amino acids mutated in mt1 and mt2 are evolutionarily conserved (Supplementary Fig. 1a). We expressed and purified the four-subunit human
PRC2 core complex, including EED, SUZ12, RBBP4 and either the wild-type or mutant EZH2 (Fig. 1c and Supplementary Fig. 1b,c). Quantitative RNA-binding assays confirmed that both PRC2
mutants mt1 and mt2 are defective in RNA binding, although the defect is more obvious in the case of mt1 (Supplementary Fig. 1d–h), which qualitatively in agreement with previous
studies19,20. Nucleosome-binding assays, using electrophoretic mobility shift assay (EMSA), indicated that both mt1 and mt2 bind to nucleosomes with a similar affinity to the wild type
(Supplementary Fig. 1i). This observation persisted whether the nucleosome core particle (NCP) probes included a linker DNA (NCP182) or not (NCP147). Yet, we noticed that a few of the amino
acids that were mutated in mt2 reside in close proximity to the DNA of the substrate nucleosome (marked in red in Fig. 1b). This led us to hypothesize that mt2 might not be able to interact
with chromatin in a conformation required for the efficient modification of histone tails. To test this hypothesis, we assayed the methyltransferase activity of mt1 and mt2 against three
types of substrates that were previously used to characterize mt2 (ref. 17): isolated H3 histones, mononucleosomes and chromatin. We used unmodified human histone proteins in all the
substrates. Unmodified (naïve) chromatin was reconstituted using unmodified human histones and a PCR-amplified 3.6-kilobase pair (kb) DNA from the human _ATOH1_ locus (Supplementary Fig.
1j,k), which is repressed by PRC2 in most lineages23. The wild-type PRC2 and mt1 exhibited similar activities against H3 histones (Fig. 1d, left, and Supplementary Fig. 2a), mononucleosomes
(Supplementary Fig. 2b) and naïve chromatin (Fig. 1d, right, and Supplementary Fig. 2c). This indicates that mt1 is a separation-of-function mutant, defective in RNA binding but active in
the methylation of H3 histones within the context of nucleosomes or chromatin. The histone methyltransferase activity of mt2 resembled that of the wild-type PRC2 when reacted against H3
histones externally to the context of chromatin (Fig. 1d, left, and Supplementary Fig. 2a), in agreement with previous studies17,19. Yet, mt2 was defective in modifying histones while they
are in the context of nucleosomes (Supplementary Fig. 2b) or chromatin (Fig. 1d, right, and Supplementary Fig. 2c). These results indicate that mt2 separates the catalytic activity of PRC2
(Fig. 1d, left, and Supplementary Fig. 2a) from its chromatin modification activity (Fig. 1d, right, and Supplementary Fig. 2c). This also indicates that the nucleosome-interacting surface
of EZH2 is required for the methylation of chromatin, even if this surface is dispensable for the methylation of unchromatinized substrates. A similar observation has been made for another
EZH2 mutant, mutated in the CXC domain24; that mutant was active as the wild-type EZH2 on H3 peptides, exhibited only a modest reduction in affinity for nucleosomes (less than threefold
dissociation constant; _K_d) but substantially reduced methyltransferase activity on nucleosomal substrates. When H3 histones were tested externally to the context of chromatin, the histone
methyltransferase activity of the core PRC2 complex exhibited similar activity across a large range of monovalent salt concentrations (Fig. 1f, left, and Supplementary Fig. 2d). Yet, when
PRC2 was tested in the context of chromatin, monovalent salt inhibits the histone methyltransferase activity of PRC2 (Fig. 1f, right, and Supplementary Fig. 2e). This indicates that
electrostatic interactions are required for the methylation of chromatin by the PRC2 core complex, but not for methyltransferase per se. This strongly implies that interactions between PRC2
and nucleosomal DNA play a pivotal role during the modification of chromatin. This is in agreement with multiple high-resolution structures of PRC2 with nucleosomal constructs that identify
contacts between the nucleosomal DNA and positively charged patches in surfaces of EZH2 external to the catalytic center (Fig. 1b and refs. 21,24,25). While results thus far are internally
consistent, they are not in full agreement with a previous work, which detected no difference between the methyltransferase activity of the wild-type PRC2 and mt2 when tested against a
mononucleosomal construct17. Despite using the experimental conditions that were applied before17, we were unable to reproduce this result. Instead, we found that the difference between the
histone methyltransferase (HMTase) activities of the wild-type PRC2 and mt2 persisted regardless of which nucleosomal construct was used (Supplementary Fig. 3a,b). PRC2 mt2 was less active
than the wild type regardless of whether the nucleosomes were reconstituted using the salt gradient dialysis method, as in other experiments described herein, or whether the nucleosomes were
reconstituted using step dilutions, as in the original work17 (Supplementary Fig. 3a,b). A large difference between the activity of the wild type and mt2 was also observed when PRC2 was
tested on reconstituted human naïve chromatin under initial rate conditions (Fig. 1e). EZH2 mt2 also remained the least active enzyme in the presence of an allosteric-effector H3K27me3
peptide, while we noticed an elevated activity of mt1 (Supplementary Fig. 2f). Collectively, we found that mt2 exhibits lower HMTase activity, in comparison to the wild-type PRC2, against a
variety of nucleosomal substrates and under different experimental conditions. We also noticed a small reduction in the HMTase activity of mt2 with respect to the wild-type PRC2 when tested
on non-nucleosomal H3 substrate at a substrate concentration of 1.2 µM (Supplementary Fig. 3a,b). These differences in activity were not as prominent as in the case of the nucleosomal
substrates and were not visible at the higher concentration of histone H3 (12 µM) that was assayed herein (Fig. 1d and Supplementary Fig. 2a) and elsewhere17. Data thus far indicate that a
portion of the RNA-binding surface of EZH2, mutated in mt2 or a portion of it (marked in red in Fig. 1b), is required for both RNA binding and for the modification of H3 histones in the
context of chromatin (Fig. 1d,e and Supplementary Fig. 2b,c,e,f). Mechanistically, the interactions between the PRC2 core complex and the nucleosomal DNA are largely electrostatic (Fig. 1f)
and probably involve a portion of the RNA-binding surfaces of EZH2 (Fig. 1b). A different portion of the RNA-binding surface of EZH2, represented by mt1 (marked in blue in Fig. 1b), is
required for RNA binding but is dispensable for the methylation of histones, nucleosomes and chromatin (Fig. 1d and Supplementary Fig. 2a–e). A PORTION OF THE RNA-BINDING ACTIVITY OF EZH2 IS
DISPENSABLE FOR MAINTAINING GLOBAL H3K27ME3 IN CELLS While both mt1 and mt2 are defective in RNA binding19,20, they separate different sets of molecular functions: mt1 separates RNA binding
from the modification of chromatin, while mt2 separates the modification of chromatin from catalysis. Given that mt2 is defective in HMTase selectively in the context of chromatin (Fig.
1d,e), we also added to our panel the catalytically defective EZH2 Y731F mutant (dEZH2; Fig. 1a,b)26,27. This mutation is located in the catalytic center of PRC2 (Fig. 1b, in orange)26,27,
away from all the nucleosome-binding surfaces that have been mapped so far21,24,25. The mutant dEZH2 is expected to be catalytically defective against any substrate, while active in RNA
binding (Supplementary Fig. 1e). We reasoned that these mutants would allow us to dissect the function of the RNA-binding activity of EZH2 from its other canonical functions (wild type (WT)
versus mt1); to determine what the function of the nucleosome-interacting site is within the RNA-binding surface of EZH2 (mt1 versus mt2); and to separate the canonical HMTase activity of
EZH2 in the context of chromatin from methyltransferase activity that could, in principle, take place externally to the context of chromatin and may not be limited to H3 histones (mt2 versus
dEZH2). PRC2 is required for the maintenance of transcription programs in cells that are committed to a lineage28. We therefore proceeded to test the different molecular functions of PRC2
in lineage-committed cells. We used the acute myelogenous leukemia (AML) K562 cell line (Fig. 2a), which is dependent on PRC2 for the maintenance of transcription programs29. We developed an
experimental system allowing for knockout with rescue of EZH2 in bulk cell populations, to avoid variations in transcription programs that could otherwise be attributed to clonal selection.
The expression level of the ectopically expressed EZH2 was controlled using flow cytometry (Fig. 2a) and was similar to that of the endogenous EZH2 (Fig. 2b and Supplementary Fig. 4a). A
possible contributing factor for the even expression level of EZH2 across constructs is that the stability of the EZH2 protein is dependent on the expression level of other PRC2 core
subunits30. Coimmunoprecipitation confirmed that all the ectopically expressed EZH2 constructs are effectively incorporated into PRC2 in the cells (Supplementary Fig. 4b). Knockout of EZH2
simultaneously with rescue using the wild-type EZH2 led to the restoration of normal H3K27me3 levels (Fig. 2b), as could be expected. Knockout of EZH2 simultaneously with rescue using dEZH2
led to global depletion of the H3K27me3 mark (Fig. 2b). There were no reproducibly substantial changes in the H3K27me1 or H3K27me2 marks (Supplementary Fig. 4a,d). We noticed an upregulation
of EZH1 after EZH2 knockout, but EZH1 expression was diminished on rescue using all the EZH2 constructs (Supplementary Fig. 4a). Thus far, these results are consistent with the ectopically
expressed EZH2 being the predominant EZH2 protein in PRC2 complexes and being responsible for the bulk of H3K27me3 within these cells. EZH2 mt1 and mt2 exhibited markedly different global
levels of H3K27me3: while mt1 was virtually indistinguishable from the wild type, mt2 phenocopied the catalytically defective dEZH2 (Fig. 2b and Supplementary Fig. 4a). This finding was
further confirmed by repeating the assay after clonal selection of the mt1 and mt2 K562 lines (Supplementary Fig. 4c). The same observation persisted when rescue experiments were carried out
in mouse embryonic stem cells (mouse ES cells), on the background of _Ezh1_ and _Ezh2_ knockout (Ezh1/2 dKO), using two different lentiviral expression systems (Supplementary Fig. 4e). EZH2
mt2 includes mutations in two separate surfaces on EZH2 (Fig. 1b, marked in red). Yet, according to structural data25, only one of the EZH2 mt2-mutated surfaces is responsible for the
interactions with the substrate nucleosome (right red patch in Fig. 1b). Hence, to directly link the loss of methyltransferase activity to the substrate nucleosome-interacting surface, we
generated a new mutant: mt2*. EZH2 mt2* only contains the PRKKKR494-499NAAIRS perturbation, which affects only the substrate nucleosome-interacting surface of EZH2 (right red patch in Fig.
1b). As expected, the mt2* mutant phenocopies its parental mutant mt2 in cells (Supplementary Fig. 4d). These data indicate that the global loss of H3K27me3, as seen for mt2 (Supplementary
Fig. 4a), is attributed to the defective substrate nucleosome-interacting surface of EZH2 (Supplementary Fig. 4d). Results thus far indicate that at least a portion of the RNA-binding
activity of PRC2 is dispensable for the maintenance of global H3K27me3 in cells (that is, WT versus mt1 in Fig. 2b). These results also indicate that a portion of the RNA-binding surface of
PRC2, which is required to modify chromatin in vitro in an RNA-independent manner, is also required to modify chromatin in cells (that is, mt1 versus mt2 in Fig. 2b). The data also imply
that the global levels of H3K27me3 are dependent on direct contacts between nucleosomal DNA to EZH2, externally to the catalytic center and independent of catalysis (that is, mt2 versus
dEZH2 in Fig. 2b). A PORTION OF THE RNA-BINDING ACTIVITY OF EZH2 IS DISPENSABLE FOR DEPOSITING H3K27ME3 AT REPRESSED GENES We next aimed to determine how the different EZH2 mutants affect
the deposition of H3K27me3 to repressed genes. We used the same experimental system (Fig. 2a), but instead of immunoblotting, we carried out CUT&Tag31 using antibodies for H3K27me3 and
H3K27ac (Fig. 2c–e and Supplementary Fig. 5). The level of H3K27me3 at selected target genes was similar between the control cells (‘Ctrl’: no knockout and no rescue) and cells where EZH2
was knocked out and rescued using the wild-type EZH2 (Fig. 2c). This trend was also similar genome wide across all transcription start sites (Fig. 2d,e) and across three biological
replicates that were carried out on three different days (Fig. 2e and Supplementary Fig. 5a–c; compare black to purple enrichment profiles). By contrast, knockout and rescue with a
catalytically defective EZH2 led to a substantial reduction of H3K27me3 with respect to cells expressing the wild-type EZH2 (Fig. 2d,e). Knockout of EZH2 without rescue was less effective in
reducing H3K27me3 than the knockout with dEZH2 rescue (Supplementary Fig. 5c). This was possibly owing to compensation by EZH1, which exhibited elevated protein levels in the absence of
EZH2 rescue (Supplementary Fig. 4a). So far, these data indicate that the knockout and rescue are effective and resemble the global levels of H3K27me3 that were observed using immunoblotting
(Fig. 2b). The two RNA-binding-defective mutants behaved differently from each other: mt1 deposited H3K27me3 at target genes as effectively as the wild-type EZH2, while mt2 resembled the
catalytically defective EZH2 (Fig. 2c). Genome-wide correlation analysis of H3K27me3 levels followed by unsupervised clustering revealed two distinct clusters of cell lines, which we marked
as ‘defective’ and ‘normal’ (Fig. 3a). The defective cluster included the catalytically defective EZH2 and mt2. The normal cluster included mt1, the wild-type EZH2 rescue and the control
cell line that expressed the endogenous EZH2. In contrast to the repressive H3K27me3 mark, the levels of the active H3K27ac mark remained similar between the different mutants (Fig. 2c–e and
Supplementary Figs. 5a–d and 6a). This indicates that the changes in H3K27me3 deposition were directly attributed to the activity of PRC2 at its target genes, rather than to global changes
in transcription or secondary effects. Data thus far indicate that the activities of mt1 and mt2 at H3K27me3-marked genes resemble their in vitro activities: mt1 is active in the
modification of chromatin and mt2 is defective. This indicates that the overall RNA-binding activity of PRC2 can be reduced without compromising its ability to modify chromatin at
H3K27me3-marked genes (Fig. 2; compare WT to mt1). Yet, the data also indicate that a part of the RNA-binding surface of PRC2, which also interacts with the substrate nucleosome (Fig. 1b),
is required for depositing the H3K27me3 mark at target genes (Fig. 2 and Supplementary Fig. 4b, d; compare mt1 to mt2 and mt2*) and is as important as the catalytic center for H3K27me3
deposition to chromatin (Fig. 2; compare mt2 to dEZH2). A PORTION OF THE RNA-BINDING ACTIVITY OF EZH2 IS DISPENSABLE FOR MAINTAINING TRANSCRIPTION PROGRAMS IN LINEAGE-COMMITTED CELLS
H3K27me3 is required for the maintenance of the repressed state of cell type-specific genes32. Maintaining the repressed state of cell type-specific genes enables the maintenance of
transcription programs and, therefore, cell identity. Given that mt2 exhibited a rather similar loss-of-function phenotype to dEZH2 in depositing the H3K27me3 mark, we wished to determine
whether these mutants also mimic each other in transcriptional regulation. For that, we used the same system (Fig. 2a) to carry out RNA-seq. K562 cells are committed to a lineage and are
strongly dependent on PRC2 to maintain gene expression programs29,33. Accordingly, knockout with rescue using catalytically defective EZH2 led to substantial changes in transcription with
respect to the wild-type rescue (Fig. 3b; compare WT to dEZH2). Principal component analysis (PCA) on the gene expression data revealed two distinct clusters that we marked as ‘normal’ and
‘defective’ (Fig. 3b). The normal cluster included the control cells (Fig. 3b, in black), the knockout with rescue using wild-type EZH2 and the knockout with rescue using the
separation-of-function mutant mt1 (Fig. 3b, in purple or blue, respectively). The defective cluster included the knockout with rescue using the catalytic defective EZH2, the knockout without
rescue and the knockout with rescue using mt2 (Fig. 3b, orange, gray and red, respectively). The dEZH2 and mt2 lines led to 1,604 and 708 differentially expressed genes, respectively, in
agreement with a poor maintenance of transcription programs (Supplementary Fig. 6c). Gene ontology (GO) analysis on the differentially expressed genes identified hematopoietic-related GO
terms, in agreement with the hematopoietic origin of K562 cells (Supplementary Fig. 6b). Conversely, only 57 differentially expressed genes were identified in the mt1 line, and no
significant GO terms (Supplementary Fig. 6b,c). This implies that mt1 maintains transcription programs almost as the wild-type EZH2. These results indicate that an RNA-binding-defective EZH2
can maintain transcription programs in lineage-committed cells (compare WT to mt1 in Fig. 3b). Yet, a portion of the RNA-binding surface of PRC2 is required for maintaining transcription
programs (compare mt1 to mt2 in Fig. 3b), probably through interactions with nucleosomes (Fig. 1b), while PRC2 methylates them (Fig. 2). These data also suggest that a surface of EZH2 that
resides externally to the catalytic center and is required for interactions with both RNA17,19 and, independently, nucleosomal DNA25 is as important as the catalytic center for maintaining
transcription programs in lineage-committed cells (compare mt2 to dEZH2 in Fig. 3b). A NUCLEIC ACID-INTERACTING SURFACE IN EZH2 IS REQUIRED FOR METHYLTRANSFERASE, WHICH IS IN TURN REQUIRED
FOR PRC2 CHROMATIN OCCUPANCY We next investigated the impact of the two RNA-binding-defective EZH2 mutants on the chromatin occupancy of PRC2. To this end, we employed the same experimental
system as in Fig. 2a, except that we now carried out quantitative chromatin immunoprecipitation sequencing (ChIP-seq) with exogenous reference genome spike-in (ChIP-Rx), using antibodies for
H3K27me3, SUZ12 and EZH2 (Fig. 4a,b). Consistent with the findings from the CUT&Tag analyses (Fig. 2), ChIP-Rx indicated that mt2 and dEZH2 led to a substantial reduction in H3K27me3
genome wide (Fig. 4a,b). The genome-wide chromatin occupancy of PRC2 followed a similar pattern to H3K27me3, albeit with more subtle changes: EZH2 and SUZ12 occupancy at promoter regions
were slightly higher for EZH2 wild type and mt1, and slightly lower for dEZH2 and mt2 (Fig. 4a,b). These findings are consistent with the reduced PRC2 chromatin occupancy that was previously
observed in the case of another catalytic defective EZH2 mutant (mouse EZH2 Y726D, equivalent to human Y731D)34,35. Accordingly, we also noticed a subtle increment of the soluble fraction
of SUZ12 isolated from the dEZH2 and mt2 cell lines (Supplementary Fig. 7a). Overall, these results further support that mt2 resembles the catalytic defective dEZH2. More importantly, the
resemblance between the chromatin occupancies of mt2 and dEZH2 (Fig. 4) implies that the reduced chromatin occupancy identified for mt2 is more likely to be attributed to defective
methyltransferase rather than to altered interactions with RNA or even nucleosomes. Hence, the substrate nucleosome-interacting surface of EZH2 (Fig. 1b) is essential for correct H3K27me3
deposition (Fig. 2), which is in turn required for transcriptional repression (Fig. 3) and the chromatin occupancy of PRC2 (Fig. 4). DISCUSSION Our data dissect different biochemical
functions of EZH2 through separation-of-function mutagenesis (Fig. 5a). The conclusions are extended beyond RNA-mediated regulation. For instance, mt2 and dEZH2 (Figs. 2–4) phenocopy each
other in cells. This implies that specific contacts between EZH2 and nucleosomes are as critical for the methylation of chromatin as the catalytic center itself is. Hence, although EZH2 is
dispensable for the recruitment of PRC2 to chromatin36, specific contacts between EZH2 to nucleosomes are required for histone methylation (Figs. 1–4). The separation-of-function mutants mt1
and mt2 are not completely inactive in binding to RNA (Supplementary Fig. 1d,e and ref. 20). This is the case for all the RNA-binding-defective PRC2 mutants that were assayed by us and
others19,20. A possible explanation is that PRC2 binds to RNA through dispersed surfaces in multiple subunits, including EZH2, EED and SUZ12 (refs. 19,20,22). Therefore, data herein cannot
in any way exclude the possibility that the RNA-binding activity of PRC2 regulates, or fine tunes, some of its functions. Yet, mt1 reduces the affinity of PRC2 for RNA by over twofold _K_d,
according to data presented here (Supplementary Fig. 1d,e) and elsewhere20. Despite this, mt1 is biochemically (Fig. 1) and phenotypically (Figs. 2–4) nearly indistinguishable from the
wild-type EZH2. This indicates that the full extent of the RNA-binding activity of EZH2 is not required for the canonical functions of PRC2: H3K27 methylation of chromatin and the
maintenance of transcription programs. How does the RNA-binding surface of EZH2 operate? The RNA-binding-defective mt2 modifies H3 histones only when they are isolated, but not in the
context of nucleosomes or naïve chromatin (Fig. 1). These observations have nothing to do with the RNA-binding activity of PRC2, as all the in vitro methyltransferase assays herein (Fig. 1
and Supplementary Figs. 1–3) were carried out in the absence of RNA. In fact, our RNA-binding assays indicate that EZH2 mt2 is quite able to interact with RNA (Supplementary Fig. 1d,e),
despite being defective in chromatin modification (Fig. 1e). Hence, a portion of the RNA-binding surface of PRC2 is required for the methylation of H3 histones in the context of chromatin
and independently of RNA binding. This observation provides a mechanistic explanation to rationalize previous findings, indicating that PRC2 can either bind to nucleosomes or to RNA, but not
to both of them simultaneously13,14. When PRC2 is bound to chromatin, it uses part of its RNA-binding surface to engage with nucleosomes and these interactions are required for the
methylation of chromatin and are independent of RNA (Fig. 5b, left). The interactions between EZH2 and nucleosomes are also required for the chromatin occupancy of PRC2 (Fig. 4), but this
could be attributed to the H3K27me3 deposition (Fig. 4; compare mt2 to dEZH2). When PRC2 is not bound to chromatin, part of its chromatin-interacting surface is required for RNA binding
(Fig. 5b, right). A portion of the RNA-binding surface of EZH2 is required for the function of PRC2 in human pluripotent stem cells17,18. The same RNA-binding surface is also required for
the canonical functions of PRC2 in cells, including H3K27me3 deposition in lineage-committed cells and stem cells and the maintenance of gene expression programs (Figs. 2–4 and Supplementary
Figs. 4–6), but also for the modification of chromatin in vitro (Fig. 1). Given the data herein, the simplest explanation for seemingly contradictory observations in the recent literature,
implicating RNA as either a negative10,11,12,13,14 or a positive17,18 regulator of PRC2, is as follows: a nucleosome-interacting surface of EZH2, not the RNA-binding activity of this
surface, is required for the canonical functions of PRC2 through engaging with chromatin during histone methylation. METHODS PROTEIN EXPRESSION AND PURIFICATION The full-length human EZH2,
SUZ12, RBBP4 and EED (UniProtKB: Q15910-2, Q15022-1, Q09028-1 and O75530-1, respectively) were cloned into the pFastBac1 expression vector containing a PreScission-cleavable N-terminal
hexahistidine-MBP tag, as previously described7,37. Mutations were introduced to EZH2 using Takara PrimeSTAR HS (Clontech, cat. no. R045A) or Pfu DNA polymerase, as previously described20.
Baculovirus production, titration, infection and cell collection, and the purification of PRC2 and the mutants, were carried out as previously described20. All the proteins were snap-frozen
in liquid nitrogen and stored at −80 °C as single-use aliquots. To resolve all PRC2 subunits using SDS–PAGE, 3–8% Tris–acetate gel (Thermo Fisher, cat. no. EA0375BOX) and 1× MES SDS running
buffer (Thermo Fisher, cat. no. NP0002) were used. The 3 µg PRC2 complexes were supplemented to a final concentration of 1× LDS sample buffer (Thermo Fisher, cat. no. NP0007) with 1%
2-mercaptoethanol (Sigma-Aldrich, cat. no. M3148) and heated at 95 °C for 5 min before loading onto a Tris–acetate gel. The gel was run for 30 min at 200 V before staining with InstantBlue
Coomassie protein stain (Expedeon, cat. no. ISB1L). NUCLEOSOME RECONSTITUTION Unlabeled and Cy5-labeled nucleosomes were produced as previously described29. In brief, recombinant human
histones were purified from inclusion bodies and reconstituted into histone octamers as previously described38. Cy5-labeled H2A was produced as previously described39. The 147-Base pair (bp)
or 182-bp DNA including one copy of the 601 Widom sequence was PCR amplified and purified using anion exchange column (Cytiva, cat. no. 17115401) with NaCl gradient from 150 mM to 2 M.
Mononucleosomes were reconstituted by initially titrating across three octamer ratios (from 1:0.8 to 1:1.2 DNA:octamer molar ratio) in a 20-µl mixture of 6 µM DNA, 20 mM Tris pH7.5, 2 M KCl,
1 mM EDTA, 10 mM DTT. For each of these samples, gradient salt dialysis was used at 4 °C in a dialysis device (Thermo Fisher, cat. no. 69572), starting from refolding buffer (20 mM Tris pH
7.5, 2 M KCl, 1 mM EDTA, 1 mM DTT) to a medium salt buffer containing 20 mM Tris pH 7.5, 250 mM KCl, 1 mM EDTA and 1 mM DTT over 18 h, and the final-step dialysis was carried out using a low
salt buffer containing 20 mM Tris pH 7.5, 2.5 mM KCl, 1 mM EDTA, 1 mM DTT. Quality of mononucleosomes was assessed by 5–6% acrylamide TBE gel electrophoresis, and the most appropriate molar
ratio of DNA:octamer was selected for large-scale reconstitution. Large-scale reconstitution was conducted as above, except in a volume of 0.2–2 ml using dialysis tubing (Spectrum, cat. no.
888-11527). Mononucleosomes were stored at 4 °C and the quality was assessed by 5–6% acrylamide TBE gel. For mononucleosomes reconstituted using step dilutions, we followed the procedure as
previously described17. In brief, a 300-µl starting reaction was prepared by mixing 1.95 nmol of DNA with a 1.2 molar ratio of histone octamers (2.34 nmol) in starting buffer (2 M NaCl, 6
mM Tris pH 7.5, 0.3 mM EDTA and 0.3 mM DTT). The mixture was first incubated for 30 min at 37 °C. Dilution buffer (20 mM Tris pH 7.5, 1 mM EDTA, 1 mM DTT) was then added to the mixture in
30-min intervals in the following volumes: 324, 360, 840 and 1,920 µl. Mononucleosomes were then concentrated with Amicon Ultra-0.5 ml 10-kDa cutoff centrifugal filter (Merck, cat. no.
UFC501096) and stored at 4 °C. Chromatinized genes were produced as previously described29. Briefly, _ATOH1_ DNA was amplified using Pfu DNA polymerase and purified by ion exchange
chromatography. Purified DNA was concentrated by isopropanol precipitation and was then dissolved in Tris–EDTA buffer. Chromatin was assembled using the gradient salt dialysis at 4 °C.
Chromatin was reconstituted by initially titrating across a range of octamer ratios (from 1:12 to 1:24 DNA:octamer molar ratio) in a 20-µl mixture of 0.1–0.3 µM DNA, 20 mM Tris pH 7.5, 2 M
KCl, 1 mM EDTA and 10 mM DTT. For each of these samples, gradient salt dialysis was used at 4 °C in a dialysis device (Thermo Fisher, cat. no. 69572), starting from refolding buffer (20 mM
Tris pH 7.5, 2 M KCl, 1 mM EDTA and 1 mM DTT) to a medium salt buffer containing 20 mM Tris pH 7.5, 250 mM KCl, 1 mM EDTA and 1 mM DTT over 18 h, and the final-step dialysis was carried out
using a low salt buffer containing 20 mM Tris pH 7.5, 2.5 mM KCl, 1 mM EDTA, 1 mM DTT. Quality of chromatinized genes was assessed by 0.8% agarose TBE gel electrophoresis, and the most
appropriate molar ratio of DNA:octamer was selected for large-scale reconstitution. Large-scale reconstitution was conducted as above, except in a volume of 0.2–2 ml using dialysis tubing
(Spectrum, cat. no. 888-11527). To concentrate the assembled chromatin, MgCl2 was added to a final concentration of 20 mM. Then, the mixture was incubated for 15 min at room temperature,
followed by 15 min on ice. The mixture was then centrifuged at 4 °C for 20 min at 20,000 relative centrifugal force, and precipitate was resuspended in the low salt buffer. The concentration
of the nucleosome core particles in the arrays was measured using BCA assay (Thermo Fisher, cat. no. 23252). Chromatinized genes were stored at 4 °C and the quality was assessed by 0.8 %
agarose TBE gel electrophoresis. MICROCOCCAL NUCLEASE DIGESTION Micrococcal nuclease (MNase) digestion was performed as previously described40 with some changes. Specifically, DNA was
diluted to 75 ng µl−1 and chromatinized _ATOH1_ was diluted to 150 ng µl−1 (DNA concentration) in a buffer containing 25 mM HEPES–KOH pH 7.5, 10% glycerol, 100 mM KCl, 3 mM MgCl2, 1 mM EDTA
and 1 mM DTT. Three MNase (NEB, cat. no. M0247S) dilutions were made in a buffer containing 10 mM HEPES pH 7.5, 10 mM KCl, 1.5 mM MgCl2 and 10% glycerol to final concentrations of 45 U µl−1,
15 U µl−1 and 5 U µl−1. Then, 5 µl of the respective MNase dilution was added to 35 µl of the samples and the reaction was started by adding 5 µl of CaCl2 from a 10 mM stock to give a final
concentration of 2 mM. The reaction proceeded at room temperature for 7 min. The reaction was stopped by adding 5 µl EDTA from a 500 mM stock, followed by 100 µl of glycogen stop buffer (20
mM EDTA, 200 mM NaCl, 1% SDS, 0.25 mg ml−1 glycogen). Samples were digested by adding 1 µl of Proteinase K (NEB, cat. no. P8107S) from a 20 mg ml−1 stock followed by incubation for 30 min
at 37 °C. The DNA fragments were purified using a MinElute PCR cleanup kit (Qiagen, cat. no. 28004). After purification, samples were incubated at 55 °C with opened lids for 5 min to
evaporate residual ethanol. DNA fragments were separated on a 1.2% agarose gel in TAE buffer and visualized with GelRed. ANALYTICAL ULTRACENTRIFUGATION All sedimentation velocity experiments
were performed using an Optima analytical ultracentrifuge (Beckman Coulter) at a temperature of 20 °C. Sedimentation velocity runs were recorded by measuring absorbance at 280 or 275 nm at
a rotor speed of 12,000 rpm. Chromatinized _ATOH1_ and _ATOH1_ DNA were diluted in 25 mM Tris pH 7.5, 10 mM KCl, 1 mM EDTA and 1 mM TCEP to final concentrations of 0.025 µM and 41.6 ng µl−1,
respectively. Experiments were performed in a conventional double-sector quartz cell. Solvent density (0.9996 g ml−1 at 20 °C) and viscosity (1.0100 cP at 20 °C) as well as octamer partial
specific volume (0.7443 ml g−1) were computed using the program SEDNTERP41. The _ATOH1_ DNA partial specific volume (0.5 ml g−1) was adopted from the reported values for DNA42. The specific
volumes of octamers and _ATOH_ gene were used to compute the partial specific volume for _ATOH_ arrays (0.65 ml g−1), as previously described43,44. Finally, the sedimentation velocity data
were fitted to a continuous size (c(s)) using the program SEDFIT45. IN VITRO HMTASE ACTIVITY ASSAYS USING RADIOLABELED SAM HMTase activity assays were performed as previously described17,
with some modifications. In brief, for the HMTase reactions with histone H3 as substrates, each 10-µl reaction contained 0.6 µM PRC2, 12 µM H3.1 and five concentrations of
_S_-[methyl-14C]-adenosyl-l-methionine (PerkinElmer, cat. no. NEC363050UC; twofold serial dilutions, starting from 24 µM) were incubated in the reaction buffer A (50 mM Tris–HCl pH 8.0 at 30
°C, 100 mM KCl, 2.5 mM MgCl2, 0.1 mM ZnCl2, 2 mM 2-mercaptoethanol and 0.1 mg ml−1 BSA, 5% v/v glycerol) for 1 h at 30 °C. For the 10-µl reactions with mononucleosomes or nucleosomal arrays
as substrates, 0.6 µM PRC2, 0.6 µM mononucleosomes or 70 ng µl−1 nucleosomal arrays, and three concentrations (6, 3 and 1.5 µM) of 14C-labeled SAM were incubated in reaction buffer B (50 mM
Tris–HCl pH 8.5 at 30 °C, 5 mM MgCl2 and 4 mM DTT) for 2 h at 30 °C. For the assays performed at 0 mM, 50 mM and 100 mM KCl, 10-µl reactions contained 0.6 µM PRC2, 6 µM
_S_-[methyl-14C]adenosyl-l-methionine, either 0.645 µM nucleosomal arrays (where the molar concentration is defined as NCP equivalent) or 1.29 µM H3.1 and reaction buffer B with either 0 mM
KCl, 50 mM KCl or 100 mM KCl final concentration. For the HMTase reactions comparing mononucleosomes that were produced using different reconstitution methods (that is, gradient dialysis and
step dilution), each 10-µl reaction contained 0.6 µM PRC2, 1.2 µM H3 or 0.6 µM mononucleosomes (NCP147 or NCP182 that were reconstituted either by step dilutions or gradient dialysis) and
three concentrations (24, 12 and 6 µM) of _S_-[methyl-14C]adenosyl-l-methionine. All reactions were assayed in a reaction buffer of 50 mM Tris pH 8.5 at 30 °C, 5 mM MgCl2 and 4 mM DTT, and
incubated at 30 °C for 2 h. All the reactions were stopped by adding 4× LDS loading dye (Thermo Fisher Scientific, cat. no. NP0007) to a final concentration of 1× LDS with 1%
2-mercaptoethanol (Sigma-Aldrich, cat. no. M3148) and heating at 95 °C for 5 min. The reactions were then loaded onto 16.5% SDS–PAGE gels and run on ice for 120 min at 160 V. Gels were
stained with InstantBlue Coomassie protein stain (Expedeon, cat. no. ISB1L) before vacuum drying for 1 h at 80 °C. Dried gels were then exposed to a storage phosphor screen for several days
before acquiring radiograms using a Typhoon 5 Imager (GE Healthcare). All experiments were performed in three independent replicates that were carried out on three different days.
Densitometry was carried out using ImageQuant software (GE Healthcare). Relative activity of nucleosomal substrates was obtained by dividing all densitometry values of the same replicate by
the densitometry value of H3 methylation of NCP182 (gradient dialysis) by PRC2 wild-type protein at the highest SAM concentration of a given replicate. Relative activity in the presence of
the H3 substrate was obtained by dividing all densitometry values within a given replicate by the densitometry value for the same substrate as quantified in the presence of the PRC2
wild-type protein at the highest SAM concentration. This was performed for all three replicates for all substrates. The resulting values were then plotted with error bars using GraphPad
Prism 9 software. TIME-COURSE IN VITRO METHYLTRANSFERASE-GLO ASSAY Before the MTase-Glo assay, 5 µM PRC2 was incubated with 10 µM PALI1-K1241me3 peptide and 20 µM SAM for 30 min at 30 °C in
the HMTase buffer (50 mM Tris pH 8.0 at 30 °C, 0.5 mM MgCl2, 0.1% Tween-20, 5 mM DTT and 35 mM KCl). Then, various concentrations of 50, 100 or 200 nM PRC2 and 1,200 nM _ATOH1_ nucleosomal
arrays (concentration defined as NCP molar equivalent) were assayed with 25 µM SAM in the HMTase buffer in a 384-well plate (Sigma, cat. no. M3561). In experiments with an
allosteric-effector peptide, H3K27me3 peptide was added to a final concentration of 50 µM. For each enzyme, either wild type or mutant, a separate reaction without nucleosomal substrate was
set for background subtraction. Reactions were incubated at 30 °C and quenched using 1 µl of 2.45% v/v TFA at 10 time points between 0 to 180 min. Standard curves were created for each
replicate using twofold serial dilutions of SAH from 1 µM to 15.6 nM in the same buffer as the samples. The luminescence signal was developed using the MTase-Glo methyltransferase assay kit
(Promega, cat. no. V7602) and captured using a BMG FLUOstar OPTIMA plate reader (BMG Labtech). Three independent measurements were performed on three different days. RNA-BINDING ASSAYS USING
FLUORESCENCE ANISOTROPY Fluorescence anisotropy (FA) was performed as previously described20. Briefly, 3′ fluorescein labeled G4 24 RNA (UUAGGG)4 was incubated for 2 min at 95 °C in 10 mM
Tris–HCl pH 7.5 and was then snap-cooled on ice for 2 min. RNA was then incubated for 30 min at 37 °C in binding buffer (50 mM Tris–HCl pH 7.5 at 25 °C, 200 mM KCl, 2.5 mM MgCl2, 0.1 mM
ZnCl2, 2 mM 2-mercaptoethanol, 0.1 mg ml−1 BSA (NEB, cat. no. B9000S), 0.05% Nonidet P40 (Roche, cat. no. 11754599001) and 0.1 mg ml−1 fragmented yeast tRNA (Sigma, cat. no. R5636)). After
that, RNA was combined with serial dilutions of the protein for a final reaction volume of 20 µl containing 5 nM fluorescently labeled RNA at the desired final protein concentration. The
mixture was equilibrated at 30 °C for 30 min before measurement. Fluorescence anisotropy data were collected using a PHERAstar plate reader (BMG Labtech) at 30 °C (_λ_ex = 485 nm, _λ_em =
520 nm). The background was subtracted from protein-free samples. _K_d, Hill and standard error values were calculated with GraphPad Prism 9 software using nonlinear regression for specific
binding with Hill slope function. For assaying the RNA-binding activities using different RNAs, the following RNA probes were synthesized as 3′ 6-FAM-labeled RNA by IDT: HOTAIR46, MEG3 (ref.
47) the G4 RNA probe described above and a size-matched G4 mt RNA without G tracts20. FA was performed as described above, except that the binding buffer contained 100 mM KCl. The probe
sequences were as follows: HOTAIR RNA: GGGAGCCCAGAGUUACAGACGGCGGCGAGAGGAAGGAGGGGCGU MEG3 RNA: UGCCCAUCUACACCUCACGAGGGCA G4 RNA: (UUAGGG)4 G4 mt RNA: (UGAGUG)4 ELECTROPHORETIC MOBILITY SHIFT
ASSAY EMSA was carried out as previously described29. Briefly, PRC2 dilutions and Cy5-H2A labeled mononucleosomes (final concentration 5 nM) were incubated at 4 °C for 30 min in binding
buffer (50 mM Tris–HCl, pH 7.5 at 25 °C, 100 mM KCl, 2 mM 2-mercaptoethanol, 0.05% v/v NP-40, 0.1 mg ml−1 BSA, 5% glycerol). The reaction mixtures were then subjected to nondenaturing gel
electrophoresis at 6.6 V cm−1 over a 0.7% agarose gel buffered with 1× TBE at 4 °C for 30 min. The Cy5 dye signals were captured using a Typhoon 5 Imager. Three independent replicates were
carried out on three different days. CELL CULTURE K562 cells were cultured in RPMI 1640 (Merck, cat. no. R8758) growth medium supplemented with 10% FBS (Cellsera, cat. no. AU-FBS/SF) and 1%
(v/v) penicillin–streptomycin (Thermo Scientific, cat. no. 15140122) and were incubated at 37 °C with 5% CO2. Mouse ES cells were cultured on 0.1% gelatin-coated dishes in DMEM medium
supplemented with 20% FBS, 1% (v/v) penicillin–streptomycin, 50 µM β-mercaptoethanol, 1:100 GlutaMAX (Thermo, cat. no. 35050061), 1:100 MEM nonessential amino acids (Thermo, cat. no.
11140076), 1:100 sodium pyruvate (Thermo, cat. no. 11360070), 1:1,000 leukemia inhibitory factor (LIF; produced in-house), 3 µM CHIR99021 (STEMCELL, cat. no. 72054) and 1 µM PD0325901
(STEMCELL, cat. no. 72184). HEK293T cells (for lentiviral production) were cultured in DMEM medium supplemented with 10% FBS, 1% (v/v) penicillin–streptomycin. K562 cells were acquired from
ATCC and all cells were tested periodically for mycoplasma contamination. CRISPR–CAS9 KNOCKOUT OF EZH2 AND RESCUE IN K562 CELLS _EZH2_ gRNA sequence (AATAATCAGGCATACCATCT) and a negative
control gRNA targeting _AAVS1_ locus (CGGGCCCCTATGTCCACTTC) were subcloned into BsmBI linearized pXPR_003 vector (puromycin selection). Flag-EZH2 WT and mutants were subcloned into SmaI
(NEB, cat. no. R0141) linearized pHIV-EGFP (Addgene, cat. no. 21373, EGFP selection using a polycistronic expression system) vector using Gibson Assembly and NEB Stable Competent _E. coli_
(NEB, cat. no. C3040). The plasmids were fully sequenced. Lentiviruses were generated and stored as previously described29. For the generation of Cas9-expressing K562 cells, plasmids for the
polycistronic expression of Cas9 and mCherry were packed into lentiviruses and were transduced into K562 cells. The cells were selected using flow cytometry. For EZH2 depletion and rescue
experiments, 3 × 104 K562 cells were transduced in RPMI and 8 µg ml−1 polybrene using 200 µl lentivirus stock of either _EZH2_ gRNA or _AAVS1_ gRNA, and 200 µl lentivirus stock of either
rescue or empty vector to a final volume of 700 µl in a treated 24-well plate (day 0). On day 2, half of the cell suspension was moved to a 10-cm dish with puromycin added to a final
concentration of 10 µg ml−1. On day 7, cells were sorted by flow cytometry on the basis of EGFP and mCherry signals and seeded into a 10-cm dish. On day 14, cells were trypsinized, washed
twice with PBS by centrifugation at 500 relative centrifugal force for 5 min and were then resuspended in PBS to 1 million per ml before aliquoting them for the downstream applications, as
described below. This entire process, starting from day 0, was carried out independently for each replicate while starting on a different day. For experiments done using the derived cell
lines, see Supplementary Methods. GENERATION OF EZH1/2 DKO AND EZH2 RESCUE MOUSE ES CELL LINES For subcloning of human EZH2 WT or mutants into a pLenti expression vector, full-length open
reading frames (ORFs) of EZH2 WT or mutants were PCR amplified and subcloned into the pCR™8/GW/TOPO Gateway cloning entry vector according to the instructions of the manufacturer (Thermo,
cat. no. K250020). The EZH2 ORFs were subsequently subcloned into Gateway destination vector pLENTI-EFIA-FLAG/HA (a gift from A. Bracken, Trinity College Dublin) using Clonase Gateway LR
Clonase II Plus enzyme (Thermo, cat. no. 12538120) according to the instructions of the manufacturer. The plasmids were fully sequenced. Lentiviruses were generated using HEK293T cells as
described above. The _Ezh1/2_ dKO cell line was generated by treating the _Ezh1_−/−; _Ezh2_f/f;_Rosa26CreERT2_ mouse ES cell line (generated by the laboratory of K. Helin36 with 0.5 µM 4-OHT
(Sigma, cat. no. H7904) for 96 h. For generating the human EZH2 pLenti rescues in the _Ezh1/2_ dKO cell line, 150,000 mouse ES cell were seeded per well on a six-well plate, one day before
the viral transduction. The mouse ES cells were then treated with virus for 48 h before puromycin selection at 1 µg ml−1. For generating the human EZH2 pHIV-EGFP rescues in the _Ezh1/2_ dKO
cell line, the same lentiviral transduction procedure was applied and cells were sorted using flow cytometry on the basis of the EGFP signal after 48 h treatment with virus. For experiments
done using the derived cell lines, see Supplementary Methods. REPORTING SUMMARY Further information on research design is available in the Nature Portfolio Reporting Summary linked to this
article. DATA AVAILABILITY CUT&Tag, RNA-seq and ChIP-Rx data and processed files have been deposited in the NCBI GEO database under accession number GSE239447. Source data are provided
with this paper. CODE AVAILABILITY Data analyses were performed with standard published software, as described in Methods, using scripts that are available via Zenodo at
https://doi.org/10.5281/zenodo.10866993 (ref. 48). REFERENCES * Bhat, P., Honson, D. & Guttman, M. Nuclear compartmentalization as a mechanism of quantitative control of gene expression.
_Nat. Rev. Mol. Cell Biol._ 22, 653–670 (2021). CAS PubMed Google Scholar * Cech, T. R. & Steitz, J. A. The noncoding RNA revolution–trashing old rules to forge new ones. _Cell_ 157,
77–94 (2014). CAS PubMed Google Scholar * Davidovich, C. & Cech, T. R. The recruitment of chromatin modifiers by long noncoding RNAs: lessons from PRC2. _RNA_ 21, 2007–2022 (2015).
CAS PubMed PubMed Central Google Scholar * Blackledge, N. P. & Klose, R. J. The molecular principles of gene regulation by Polycomb repressive complexes. _Nat. Rev. Mol. Cell Biol._
22, 815–833 (2021). CAS PubMed PubMed Central Google Scholar * Pengelly, A. R., Copur, Ö., Jäckle, H., Herzig, A. & Müller, J. A histone mutant reproduces the phenotype caused by
loss of histone-modifying factor Polycomb. _Science_ 339, 698–699 (2013). CAS PubMed Google Scholar * Zhao, J. et al. Genome-wide identification of polycomb-associated RNAs by RIP-seq.
_Mol. Cell_ 40, 939–953 (2010). CAS PubMed PubMed Central Google Scholar * Davidovich, C., Zheng, L., Goodrich, K. J. & Cech, T. R. Promiscuous RNA binding by Polycomb repressive
complex 2. _Nat. Struct. Mol. Biol._ 20, 1250–1257 (2013). CAS PubMed PubMed Central Google Scholar * Wang, X. et al. Targeting of Polycomb repressive complex 2 to RNA by short repeats
of consecutive guanines. _Mol. Cell_ 65, 1056–1067.e5 (2017). CAS PubMed Google Scholar * Almeida, M., Bowness, J. S. & Brockdorff, N. The many faces of Polycomb regulation by RNA.
_Curr. Opin. Genet. Dev._ 61, 53–61 (2020). CAS PubMed PubMed Central Google Scholar * Cifuentes-Rojas, C., Hernandez, A. J., Sarma, K. & Lee, J. T. Regulatory interactions between
RNA and polycomb repressive complex 2. _Mol. Cell_ 55, 171–185 (2014). CAS PubMed PubMed Central Google Scholar * Herzog, V. A. et al. A strand-specific switch in noncoding transcription
switches the function of a Polycomb/Trithorax response element. _Nat. Genet._ 46, 973–981 (2014). CAS PubMed PubMed Central Google Scholar * Kaneko, S., Son, J., Bonasio, R., Shen, S.
S. & Reinberg, D. Nascent RNA interaction keeps PRC2 activity poised and in check. _Genes Dev._ 28, 1983–1988 (2014). CAS PubMed PubMed Central Google Scholar * Beltran, M. et al.
The interaction of PRC2 with RNA or chromatin is mutually antagonistic. _Genome Res._ 26, 896–907 (2016). CAS PubMed PubMed Central Google Scholar * Wang, X. et al. Molecular analysis of
PRC2 recruitment to DNA in chromatin and its inhibition by RNA. _Nat. Struct. Mol. Biol._ 24, 1028–1038 (2017). CAS PubMed PubMed Central Google Scholar * Beltran, M. et al. G-tract RNA
removes Polycomb repressive complex 2 from genes. _Nat. Struct. Mol. Biol._ 26, 899–909 (2019). CAS PubMed PubMed Central Google Scholar * Rinn, J. L. et al. Functional demarcation of
active and silent chromatin domains in human HOX loci by noncoding RNAs. _Cell_ 129, 1311–1323 (2007). CAS PubMed PubMed Central Google Scholar * Long, Y. et al. RNA is essential for
PRC2 chromatin occupancy and function in human pluripotent stem cells. _Nat. Genet._ 52, 931–938 (2020). CAS PubMed PubMed Central Google Scholar * Kraft, K. et al. Polycomb-mediated
genome architecture enables long-range spreading of H3K27 methylation. _Proc. Natl Acad. Sci. USA_ 119, e2201883119 (2022). CAS PubMed PubMed Central Google Scholar * Long, Y. et al.
Conserved RNA-binding specificity of polycomb repressive complex 2 is achieved by dispersed amino acid patches in EZH2. _eLife_ 6, e31558 (2017). PubMed PubMed Central Google Scholar *
Zhang, Q. et al. RNA exploits an exposed regulatory site to inhibit the enzymatic activity of PRC2. _Nat. Struct. Mol. Biol._ 26, 237–247 (2019). CAS PubMed PubMed Central Google Scholar
* Poepsel, S., Kasinath, V. & Nogales, E. Cryo-EM structures of PRC2 simultaneously engaged with two functionally distinct nucleosomes. _Nat. Struct. Mol. Biol._ 25, 154–162 (2018).
CAS PubMed PubMed Central Google Scholar * Kasinath, V. et al. Structures of human PRC2 with its cofactors AEBP2 and JARID2. _Science_ 359, 940–944 (2018). CAS PubMed PubMed Central
Google Scholar * ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. _Nature_ 489, 57–74 (2012). Google Scholar * Finogenova, K. et al. Structural
basis for PRC2 decoding of active histone methylation marks H3K36me2/3. _eLife_ 9, e61964 (2020). CAS PubMed PubMed Central Google Scholar * Kasinath, V. et al. JARID2 and AEBP2 regulate
PRC2 in the presence of H2AK119ub1 and other histone modifications. _Science_ 371, eabc3393 (2021). CAS PubMed PubMed Central Google Scholar * Wang, X. et al. Regulation of histone
methylation by automethylation of PRC2. _Genes Dev._ 33, 1416–1427 (2019). CAS PubMed PubMed Central Google Scholar * Lee, C.-H. et al. Automethylation of PRC2 promotes H3K27 methylation
and is impaired in H3K27M pediatric glioma. _Genes Dev._ 33, 1428–1440 (2019). CAS PubMed PubMed Central Google Scholar * Miller, S. A., Damle, M., Kim, J. & Kingston, R. E. Full
methylation of H3K27 by PRC2 is dispensable for initial embryoid body formation but required to maintain differentiated cell identity. _Development_ 148, dev196329 (2021). CAS PubMed
PubMed Central Google Scholar * Zhang, Q. et al. PALI1 facilitates DNA and nucleosome binding by PRC2 and triggers an allosteric activation of catalysis. _Nat. Commun._ 12, 4592 (2021).
CAS PubMed PubMed Central Google Scholar * Pasini, D., Bracken, A. P., Jensen, M. R., Lazzerini Denchi, E. & Helin, K. Suz12 is essential for mouse development and for EZH2 histone
methyltransferase activity. _EMBO J._ 23, 4061–4071 (2004). CAS PubMed PubMed Central Google Scholar * Kaya-Okur, H. S., Janssens, D. H., Henikoff, J. G., Ahmad, K. & Henikoff, S.
Efficient low-cost chromatin profiling with CUT&Tag. _Nat. Protoc._ 15, 3264–3283 (2020). CAS PubMed PubMed Central Google Scholar * Yu, J.-R., Lee, C.-H., Oksuz, O., Stafford, J. M.
& Reinberg, D. PRC2 is high maintenance. _Genes Dev._ 33, 903–935 (2019). CAS PubMed PubMed Central Google Scholar * Xie, H. et al. Chronic myelogenous leukemia-initiating cells
require Polycomb group protein EZH2. _Cancer Discov._ 6, 1237–1247 (2016). CAS PubMed PubMed Central Google Scholar * Lavarone, E., Barbieri, C. M. & Pasini, D. Dissecting the role
of H3K27 acetylation and methylation in PRC2 mediated control of cellular identity. _Nat. Commun._ 10, 1679 (2019). PubMed PubMed Central Google Scholar * Glancy, E. et al. PRC2.1- and
PRC2.2-specific accessory proteins drive recruitment of different forms of canonical PRC1. _Mol. Cell_ 83, 1393–1411.e7 (2023). CAS PubMed PubMed Central Google Scholar * Højfeldt, J. W.
et al. Accurate H3K27 methylation can be established de novo by SUZ12-directed PRC2. _Nat. Struct. Mol. Biol._ 25, 225–232 (2018). PubMed PubMed Central Google Scholar * Davidovich, C.,
Goodrich, K. J., Gooding, A. R. & Cech, T. R. A dimeric state for PRC2. _Nucleic Acids Res._ 42, 9236–9248 (2014). CAS PubMed PubMed Central Google Scholar * Luger, K., Rechsteiner,
T. J. & Richmond, T. J. Preparation of nucleosome core particle from recombinant histones. _Methods Enzymol._ 304, 3–19 (1999). CAS PubMed Google Scholar * Sabantsev, A., Levendosky,
R. F., Zhuang, X., Bowman, G. D. & Deindl, S. Direct observation of coordinated DNA movements on the nucleosome during chromatin remodelling. _Nat. Commun._ 10, 1720 (2019). PubMed
PubMed Central Google Scholar * Fyodorov, D. V. & Kadonaga, J. T. Chromatin assembly in vitro with purified recombinant ACF and NAP-1. _Methods Enzymol._ 371, 499–515 (2003). CAS
PubMed Google Scholar * Harding, S. E., Rowe, A. J. & Horton, J. C. _Analytical Ultracentrifugation in Biochemistry and Polymer Science_ (The Royal Society of Chemistry, 1992). *
Durchschlag, H. in _Thermodynamic Data for Biochemistry and Biotechnology_ (ed. Hinz, H.-J.) 45–128 (Springer, 1986). * Perkins, S. J. Protein volumes and hydration effects. _Eur. J.
Biochem._ 157, 169–180 (1986). CAS PubMed Google Scholar * Schmidt, C. L. A. Proteins, amino acids and peptides as ions and dipolar ions (Cohn, Edwin J.; Edsall, John T.). _J. Chem.
Educ._ 20, 415 (1943). Google Scholar * Schuck, P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. _Biophys. J._ 78,
1606–1619 (2000). CAS PubMed PubMed Central Google Scholar * Wu, L., Murat, P., Matak-Vinkovic, D., Murrell, A. & Balasubramanian, S. Binding interactions between long noncoding RNA
HOTAIR and PRC2 proteins. _Biochemistry_ 52, 9519–9527 (2013). CAS PubMed Google Scholar * Mondal, T. et al. MEG3 long noncoding RNA regulates the TGF-β pathway genes through formation of
RNA-DNA triplex structures. _Nat. Commun._ 6, 7743 (2015). CAS PubMed Google Scholar * Gail, E., Healy, E., Zhang, Q. & Davidovich, C. Inseparable RNA binding and chromatin
modification activities of a nucleosome-interacting surface in EZH2. _Zenodo_ https://doi.org/10.5281/zenodo.10866993 (2024). Download references ACKNOWLEDGEMENTS We thank the support of the
Monash FlowCore and the MASSIVE HPC facility. Q.Z. is supported by Investigator Grant EL1 (APP1196365) from National Health and Medical Research Council (NHMRC). C.D. is an EMBL-Australia
Group Leader and a Sylvia and Charles Viertel Senior Medical Research Fellow and acknowledges support from the ARC (DP190103407) and the NHMRC (APP1162921, APP1184637, APP2011767 and
APP2020900). This research was funded partially by the Victoria State Government through mRNA Victoria to Q.Z. and C.D. AUTHOR INFORMATION Author notes * These authors contributed equally:
Emma H. Gail, Evan Healy. AUTHORS AND AFFILIATIONS * Department of Biochemistry and Molecular Biology, Biomedicine Discovery Institute, Faculty of Medicine, Nursing and Health Sciences,
Monash University, Clayton, Victoria, Australia Emma H. Gail, Evan Healy, Sarena F. Flanigan, Natasha Jones, Xiao Han Ng, Michael Uckelmann, Vitalina Levina, Qi Zhang & Chen Davidovich *
South Australian immunoGENomics Cancer Institute (SAiGENCI), Faculty of Health and Medical Sciences, University of Adelaide, Adelaide, South Australia, Australia Qi Zhang * EMBL-Australia
at SAiGENCI, Adelaide, South Australia, Australia Qi Zhang * EMBL-Australia, Clayton, Victoria, Australia Chen Davidovich Authors * Emma H. Gail View author publications You can also search
for this author inPubMed Google Scholar * Evan Healy View author publications You can also search for this author inPubMed Google Scholar * Sarena F. Flanigan View author publications You
can also search for this author inPubMed Google Scholar * Natasha Jones View author publications You can also search for this author inPubMed Google Scholar * Xiao Han Ng View author
publications You can also search for this author inPubMed Google Scholar * Michael Uckelmann View author publications You can also search for this author inPubMed Google Scholar * Vitalina
Levina View author publications You can also search for this author inPubMed Google Scholar * Qi Zhang View author publications You can also search for this author inPubMed Google Scholar *
Chen Davidovich View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS Q.Z., S.F.F., E.H., N.J., M.U., V.L. and X.H.N. carried out experiments.
Q.Z., E.H.G., S.F.F., E.H. and X.H.N. analyzed data. Q.Z., E.H.G., E.H. and C.D. wrote the paper. Q.Z., E.H.G., E.H. and C.D. designed the project. Q.Z. and C.D. supervised the project.
CORRESPONDING AUTHORS Correspondence to Qi Zhang or Chen Davidovich. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION
_Nature Genetics_ thanks Luciano Di Croce, Andrea Piunti and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION
SUPPLEMENTARY INFORMATION Supplementary Methods and Figs. 1–7. REPORTING SUMMARY PEER REVIEW FILE SOURCE DATA SOURCE DATA FIG. 1 Unprocessed western blots. RIGHTS AND PERMISSIONS OPEN ACCESS
This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as
long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third
party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the
article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright
holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Gail, E.H., Healy, E., Flanigan,
S.F. _et al._ Inseparable RNA binding and chromatin modification activities of a nucleosome-interacting surface in EZH2. _Nat Genet_ 56, 1193–1202 (2024).
https://doi.org/10.1038/s41588-024-01740-8 Download citation * Received: 12 June 2022 * Accepted: 02 April 2024 * Published: 14 May 2024 * Issue Date: June 2024 * DOI:
https://doi.org/10.1038/s41588-024-01740-8 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative