Play all audios:
ABSTRACT Influenza A virus’s (IAV’s) frequent genetic changes challenge vaccine strategies and engender resistance to current drugs. We sought to identify conserved and essential RNA
secondary structures within IAV’s genome that are predicted to have greater constraints on mutation in response to therapeutic targeting. We identified and genetically validated an RNA
structure (packaging stem–loop 2 (PSL2)) that mediates in vitro packaging and in vivo disease and is conserved across all known IAV isolates. A PSL2-targeting locked nucleic acid (LNA),
administered 3 d after, or 14 d before, a lethal IAV inoculum provided 100% survival in mice, led to the development of strong immunity to rechallenge with a tenfold lethal inoculum, evaded
attempts to select for resistance and retained full potency against neuraminidase inhibitor-resistant virus. Use of an analogous approach to target SARS-CoV-2, prophylactic administration of
LNAs specific for highly conserved RNA structures in the viral genome, protected hamsters from efficient transmission of the SARS-CoV-2 USA_WA1/2020 variant. These findings highlight the
potential applicability of this approach to any virus of interest via a process we term ‘programmable antivirals’, with implications for antiviral prophylaxis and post-exposure therapy.
SIMILAR CONTENT BEING VIEWED BY OTHERS FROM A GENOME-WIDE SCREEN OF RNAI MOLECULES AGAINST SARS-COV-2 TO A VALIDATED BROAD-SPECTRUM AND POTENT PROPHYLAXIS Article Open access 16 March 2023
PYRIMIDINE INHIBITORS SYNERGIZE WITH NUCLEOSIDE ANALOGUES TO BLOCK SARS-COV-2 Article 07 February 2022 ALTERED TMPRSS2 USAGE BY SARS-COV-2 OMICRON IMPACTS INFECTIVITY AND FUSOGENICITY
Article Open access 01 February 2022 MAIN IAV is a segmented RNA virus that causes major morbidity and mortality worldwide. Current antiviral therapies target viral proteins that frequently
mutate, rendering many such therapies inadequate1,2,3. Despite a breadth of knowledge about the viral lifecycle, knowledge of the RNA secondary structure of the genome is limited. Research
on other RNA viruses has revealed genomic RNA to be capable of playing many important roles in viral lifecycles beyond merely encoding amino acid sequences, suggesting that viral RNA
structural elements could be promising therapeutic targets4,5. To the extent that these RNA structural elements are both essential and highly conserved, these features could reduce the
degree of freedom for mutations that are compatible with virus function. This, in turn, could translate into a high barrier for resistance to therapeutics designed to disrupt these RNA
structures. In IAV, genome packaging is one such critical juncture in which RNA structure might serve a central function. The IAV genome consists of eight single-stranded, negative-sense
viral RNA (vRNA) segments that encode a minimum of 14 known viral proteins6. The vRNA, together with nucleoprotein (NP) and the heterotrimeric polymerase complex, comprising PB2, PB1 and PA
proteins, forms the complete viral ribonucleoprotein (vRNP)7. To be fully infectious, IAV virions must incorporate at least one of each segment’s vRNP8. The current paradigm supports a
selective packaging method whereby the eight vRNPs are selected in a hierarchal manner mediated by unique, segment-specific packaging signals present in the terminal and central coding
regions of each vRNA that allow for discrimination between the latter9,10,11. Each vRNP interacts with at least one other partner vRNP to form a supramolecular complex12 probably maintained
by intersegment RNA–RNA and/or protein–RNA interactions hypothesized to guide the packaging process8,13. The mechanism mediating this selection and arrangement is, however, poorly
understood. Curiously, packaging signals exist in regions of high nucleotide conservancy that strongly suppress synonymous codon usage14,15,16. Conservation of the primary sequence beyond
what is required for protein coding suggests the potential for maintenance of RNA structures possessing biological functionalities. Certain synonymous mutations within the polymerase gene,
_PB2_, affect not only its own packaging, but also the incorporation of other segments11,14,17. We hypothesized that _PB2_’s dominant role in the packaging process might be facilitated by
nonprotein elements encoded by the _PB2_ vRNA, including structured RNA elements. To test this hypothesis, we first mapped the RNA secondary structure within _PB2_ that mediates packaging,
and then genetically validated this structure’s role in the viral lifecycle in vitro and IAV pathogenesis in vivo. With this model, we demonstrate proof of concept for a new class of
antiviral therapeutics that can efficiently disrupt packaging and prevent and treat otherwise lethal IAV disease in vivo, as well as enable the development of strong functional immunity in
mice, with a high barrier to resistance. Moreover, we hypothesized that an analogous approach can be applied to any RNA virus of interest, and the recent coronavirus (COVID-19) pandemic
provided an opportunity for us to successfully test this hypothesis by targeting severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in vitro and in vivo. RESULTS SHAPE IDENTIFIES
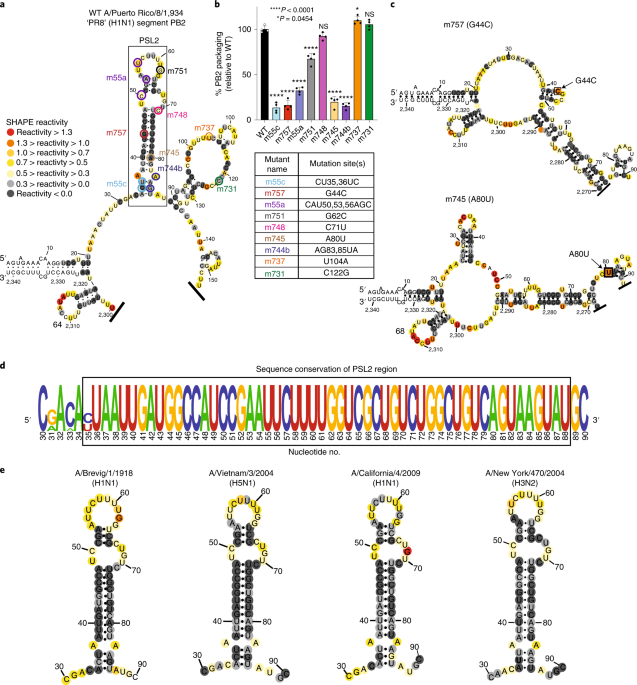
CONSERVED CANDIDATE PB2 PACKAGING SIGNAL To search for structured RNA domains in IAV segments, we first applied selective 2′-hydroxylacylation analyzed by primer extension (SHAPE)18 and
computational modeling to IAV segment _PB2_ genomic vRNA. In vitro transcribed, full-length (−)-sense _PB2_ vRNA from strain A/Puerto Rico/8/1934 (H1N1) PR8 was folded in solution19 and
interrogated using an electrophilic SHAPE reagent that preferentially reacts with nucleotides existing in flexible, single-stranded states18 (Fig. 1). This analysis revealed that much of the
2,341-nucleotide (nt) vRNA is largely unstructured (Supplementary Fig. 1a), as described in previous genome analyses15,20. However, these previous studies did not analyze the last 80
nucleotides of _PB2_’s terminal coding regions. SHAPE-guided modeling suggested several areas in this terminal region that contain stable RNA secondary structures, most notably a stem–loop
motif, named herein as PSL2 (Fig. 1a and Supplementary Fig. 1b; nucleotides 34–87). This region includes a set of nucleotides that were previously implicated in segment _PB2_ packaging
through mutational analysis via an unidentified mechanism (Fig. 1a,b, see circled nucleotides, and Supplementary Table 1)11,14,16. Supporting the hypothesis that these prior mutations act
through disruption of PSL2 structure, SHAPE analysis of the mutants yielded different conformations that all abrogated the wild-type (WT) PSL2 structure (Fig. 1c and Supplementary Fig. 2).
The 60-nt region encompassing PSL2 displays almost 100% sequence conservation at the single-nucleotide level between representative seasonal and pandemic IAV strains of different subtypes
and species origins (Fig. 1d and Supplementary Fig. 1c). Further analysis revealed that this high degree of conservation extends to all known IAV isolates available in public databases
(Supplementary Fig. 1d), suggesting the existence of a strict biological requirement to maintain an intact PSL2 structure. Indeed, the PSL2 stem–loop structure was recovered in SHAPE-guided
modeling of full-length _PB2_ RNA across diverse species and subtypes, including the highly pathogenic avian H5N1 and pandemic 1918 H1N1 strains (Fig. 1e). M2 VALIDATES PSL2 AND PREDICTS NEW
PACKAGING MUTANTS To further test the SHAPE analysis of the PSL2 RNA structure and to uncover additional informative mutations needed for in vivo tests, we applied multidimensional
chemical-mapping methods to the PSL2 segment. Mutate-and-map (M2) analysis couples systematic mutagenesis with high-throughput chemical mapping to produce accurate basepair inferences and
interactions of RNA domains21. By sequentially mutating RNA one nucleotide at a time with its Watson–Crick complement and measuring the impact that this mutagenesis has on chemical
reactivity, pair-wise correlations between close and distant residues can be established. First, M2 measurements confirmed disruption of the chemical reactivity pattern on systematic
mutation of each PSL2 stem residue, including changes at nucleotides previously identified to be relevant for _PB2_ packaging (Fig. 2; see noted fields)11,14. Automated computational
analysis based on these M2 data recovered the SHAPE-guided PSL2 structure with high confidence (Fig. 2 and Supplementary Fig. 3), further validating our structural model. Second, as
predictive tests, we designed compensatory mutations to restore basepairings—albeit not the native sequence—in the WT structure that were disrupted by the initial packaging-defective
mutations (Supplementary Fig. 4). These mutation–rescue variants restored the PSL2 SHAPE pattern, providing in vitro validation of the modeled structure at basepair resolution and suggesting
sequence variants to test the role of PSL2 structure in vivo. To test whether the PSL2 stem–loop structure observed in solution was relevant to virus packaging in the cellular milieu, the
same nine synonymous mutations reported by Gog et al. and Marsh et al. (Fig. 1a,b and Supplementary Table 1), as well as four new synonymous mutations characterized by M2 analysis (Fig.
2a,b), were cloned into plasmids containing the PR8 _PB2_ gene11,14,16. The packaging efficiencies of the nine previously known mutants, now in the PR8 background, were comparable to those
originally described in the WSN33 virus11 (Fig. 1b and Supplementary Table 1). Of these, mutants m55c, m757, m745 and m744b were predicted to show the most notable impairment based on their
location within PSL2’s stem regions (Fig. 1a–c and Supplementary Fig. 2). In contrast, published mutations that have no effect on _PB2_ packaging (for example, m731) mapped to the
unstructured apical loop or fell outside of PSL2 and did not alter its structural integrity (Supplementary Fig. 5a), whereas mutations with minor effects on virus packaging showed only minor
alterations to the structure (Supplementary Fig. 5b)11. The three new synonymous mutants (m74-1, m74-2 and m68) identified by M2 analysis as having a substantial effect on in vitro PSL2
structure (Fig. 2a, see green-marked nucleotides) showed significant loss in _PB2_ WT-like packaging efficiency levels (Fig. 2b). The strong correlation of structure disruption with in
cellulo packaging efficiency observed across these mutants supports a role of PSL2 structure in virus packaging. COMPENSATORY MUTATIONS RESTORE PSL2 STRUCTURE AND FUNCTION To investigate the
functional role of PSL2 in IAV genome packaging, compensatory mutations designed to restore the WT stem–loop structure destroyed by the packaging-defective mutations (Fig. 2c and
Supplementary Fig. 4) were cloned into PR8 expression plasmids to generate mutant rescue viruses. The compensatory mutations rescued not only the virus packaging for segment _PB2_ (Fig. 2d),
but also other segments previously reported to be affected by the deleterious mutations, consistent with the proposed hierarchal role of _PB2_ in IAV packaging (Supplementary Fig.
6a)10,11,17. In addition to recovering _PB2_ packaging, the compensatory mutations gave complete or near-complete rescue of the virus titer loss caused by the defective mutations (Fig. 2e
and Supplementary Fig. 6b). Some nonsynonymous compensatory mutations were able to restore _PB2_ packaging better than others (for example, m745-comp compared with m757-comp). This possibly
reflects incomplete restoration of PB2 protein function through exogenous addition (Fig. 2d,e and Supplementary Fig. 6) because, for nonsynonymous mutations, we also expressed WT PB2 protein
to mitigate the possibility of any impairment in PB2 protein function. We performed computational enumeration and multidimensional mutation–rescue22 (M2R) experiments to identify additional
successful PSL2-defective and compensatory mutant pairs (Fig. 3a,b and Supplementary Fig. 7). Successful M2R was defined when each single mutation alone disrupted the SHAPE-mapped WT PSL2
structure, while the double compensatory mutations recovered its structure (Fig. 3a,b, see boxed electropherograms). Although most successful partners required nonsynonymous changes, we
discovered a single M2R pair of synonymous substitutions that obviated WT PB2 protein addition (Fig. 3b,c and Supplementary Fig. 7). Making each mutation alone (m52 and m65) resulted in
severe packaging defects and, in the case of the m52 mutation, virus titer loss exceeding 4log10—an extreme impairment beyond the 1–2log10 that had been previously reported11,14 for
packaging-defective viruses (Fig. 3d,e and Supplementary Figs. 6 and 8). When introduced together into a doubly mutated m52/65-compensatory virus that restored PSL2 structure, albeit with an
altered sequence, the compensatory mutations restored both packaging efficiency and virus titer to WT levels. To ensure that any loss or subsequent rescue of virus production caused by
these mutations was not due to defects in replication or translation, each M2R pair was tested in transfection-based replicon assays. All mutant PB2 proteins and vRNAs were produced at
comparable WT levels (Supplementary Figs. 9 and 10). As an orthogonal means of testing packaging efficiency, we also assessed the effect of select mutations on vRNA packaging by denaturing
RNA gel, which recapitulated our earlier packaging findings (Supplementary Fig. 11c, Figs. 2d and 3d, and Supplementary Fig. 6). To test the relevance of the PSL2 structure in an in vivo
model, BALB/c mice were intranasally infected with either WT or mutant PR8 viruses harboring point mutations predicted to disrupt or restore PSL2 structure. Mice infected with the
PSL2-disrupting mutations—m745 mutant strain (20% packaging efficiency) or the severely defective mutant virus, m52 (<4% packaging efficiency)—showed reduced or no clinical signs of
illness, respectively, either in weight loss or in survival compared with the phosphate-buffered saline (PBS) control (Fig. 3f,g). In contrast, inclusion of compensatory mutations that
restore the PSL2 structure rescued virus pathogenicity: animals infected with m52/65-comp and m745-comp displayed comparable mortality profiles to mice infected with WT PR8 (Fig. 3f,g). To
the best of our knowledge, these are probably among the first data to indicate that packaging-defective viruses are attenuated in vivo and a genomic IAV RNA secondary structure mediates
influenza disease progression. DESIGNING ANTI-IAV LNA THERAPEUTICS TARGETING THE PSL2 STRUCTURE Given the strong evolutionary conservation of the PSL2 structure (Fig. 1d,e and Supplementary
Fig. 1c,d), we postulated that PSL2-targeted therapeutics could possess broad-spectrum activity across IAV subtypes and strains. Nine antisense oligonucleotides (ASOs) with modified LNA
bases23 were designed against PSL2 to disrupt various regions of the overall RNA secondary structure (Fig. 4a). Two LNAs, LNA8 and LNA9, are identical in sequence to LNA6 and LNA7,
respectively, but possess six or seven unmodified (nonlocked) DNA nucleotide ‘gapmers’ optimized for RNase-H activation that can degrade RNA in RNA–DNA hybrids24. First, to assess the impact
that LNA binding has on PSL2 RNA secondary structure, toeprinting and SHAPE chemical mapping were performed on _PB2_ vRNA in the presence of the LNAs. Sequences encoded in LNA6–LNA9,
corresponding to binding sites on the right 3′-side of the stem–loop structure (Fig. 4a), exhibited the greatest ability to bind and disrupt the WT PSL2 structure (Supplementary Fig. 12). To
test LNA-mediated targeting of PSL2 across different viral subtypes, Madin–Darby canine kidney (MDCK) cells were treated with LNA before infection with either PR8 (H1N1) virus or the
tissue-culture-adapted A/Hong Kong/8/1968 (HK68) (H3N2) virus, and virus inhibition was assessed by plaque assay (Fig. 4b). As predicted by the mutational and LNA chemical-mapping
experiments (Fig. 2 and Supplementary Fig. 12), LNAs directed against only the top loop of PSL2 (LNA1, LNA4), and LNAs solely targeting the 3′-base of PSL2 (for example, LNA3 and LNA5) had
minimal effect on viral titer. In contrast, nucleotide coverage of both the top loop and the middle bulge by LNA6 resulted in >2log10 titer deficits for PR8 (Fig. 4a,b). LNA8, the
RNase-H-activated copy of LNA6, produced even greater antiviral activity against both viruses of up to 3log10. Notably, LNA9, the RNase-H-activated copy of LNA7, possessed the strongest
antiviral capacity, inhibiting virus production by over 4log10 and 3log10 against PR8 and HK68, respectively. Although a labeled version of LNA9 could be clearly visualized in cells
harboring vRNPs (Supplementary Fig. 13), no off-target effect of LNA9 on steady-state levels of viral protein, vRNA, complementary RNA or cellular toxicity after 24 h was observed
(Supplementary Fig. 14). Having identified a potent candidate LNA, we next investigated the treatment time-course and concentration parameters of LNA9’s antiviral activity when administered
prophylactically at 2 and 4 h pre-infection, and therapeutically at 2 and 4 h post-infection. Cells pretreated with the LNA had the most potent antiviral response (<4log10) and displayed
strong virus inhibition even at the lowest concentration tested (1 nM) (Fig. 4c). There was a trend toward decreasing antiviral activity as the time of administration post-infection
increased, but, even at the latest tested time point of addition, >2log10 suppression of viral titer was achieved. Similar efficacy was seen in the presence of a high multiplicity of
infection (MOI) (Supplementary Fig. 15). Furthermore, LNA9 treatment resulted in a dramatic loss of _PB2_ packaging compared with controls (Fig. 4d), an effect that resembled the mutational
studies and extended to the 2009 pandemic virus (pH1N1) and 2012 H3N2 virus, indicating the potential for broad-spectrum antiviral activity. SELECTION OF IAV VARIANTS WITH ESCALATING DRUG
PRESSURE To test the hypothesis that the high conservation of PSL2 reflects a biological constraint against mutation of this structure, and that this could translate into a high barrier to
the development of resistance for PSL2-targeting therapeutics, we determined the susceptibility of WT PR8 virus to LNA9 under conditions designed to promote the development of resistance
over serial virus passaging (Fig. 4f). In parallel, we performed analogous experiments using the neuraminidase inhibitor (NAI) oseltamivir carboxylate (OSLT, Tamiflu). After seven virus
passages in the presence of escalating drug concentrations, the half-maximal effective concentration (EC50) of OSLT increased from 4.1 nM to 100 µM—a >20,000-fold increase (Fig. 4g). In
comparison, after ten virus passages in the presence of LNA9, the EC50 value showed no significant change and remained in the range 16–22 pM (Fig. 4f). To date, we still have been unable to
select for viral mutations capable of generating resistance to LNA9. Moreover, LNA9 was equally effective (EC50 = 18 pM) against a virus strain (A/WSN/33 (H1N1)) containing the H275Y
mutation (N1 numbering system) that confers resistance to OSLT, whereas high-level resistance to OSLT was confirmed, with an EC50 of 53 μM (Fig. 4h,i). These results extend the therapeutic
capabilities of LNA9 and provide strong support for potential treatment of NAI-resistant viruses with PSL2-targeting LNAs. PSL2-TARGETED LNAS PROTECT MICE FROM LETHAL IAV INFECTIONS As a
proof of concept to assess the in vivo efficacy of prophylactic LNA treatment against PSL2, BALB/c mice were intranasally administered a single 20-μg dose of LNA9 or scrambled (Scr.) LNA 1
or 3 d before infection with a lethal dose of PR8 virus. The untreated control mice experienced dramatic weight loss and were humanely sacrificed by days 5 and 6. In contrast, a single
intranasal dose of LNA9 was completely protective when administered 1 d, and even 3 d, before viral infection (Fig. 5a) and showed significantly reduced virus titers in the lungs
(>2.5log10 virus reduction) compared with the Scr. LNA control at 72 h post-infection (Supplementary Fig. 16). In addition to the well-known benefits of LNA antisense gapmer technology
that enables recruitment of RNase-H to degrade the targeted RNA23,24, LNA ASOs have also been reported to show dramatic, long-lasting effects (even >1 month) after the last administered
dose in a variety of disease models25,26,27,28. We hypothesized that PSL2-targeted LNAs might similarly possess long-term prophylactic effects due to the nuclease-resistant property
conferred by the phosphorthioate backbone present in LNAs. To test this hypothesis, we administered a single, increased dose of LNA9 (30 μg) 1 week (day −7) before infection with a lethal
dose of IAV. Although 100% of untreated mice succumbed to the infection, 70% of the LNA9 1-week pretreatment group were protected from lethality (Fig. 5b). To determine whether our
therapeutics could be further optimized for improved efficacy, we explored targeting other sequences in PSL2. Although LNA9 targets nucleotides in the lower 3′-stem of PSL2, our mutational
analyses suggested the importance of the 52- to 65-nt pair in the upper stem (Fig. 3). We hypothesized that an LNA designed against these nucleotides (LNA14) might enhance efficacy (Fig.
4a). Indeed, SHAPE analysis indicated that LNA14 was an even more potent disruptor of PSL2 structure than LNA9 (Fig. 5c,d). In biological confirmation of this, mice treated with LNA14 were
fully protected when given a single dose 1 week before infection with a lethal dose of IAV (Fig. 5e). When we increased the LNA dosage to 40 μg and administered a single LNA14 treatment to
mice 2 weeks (day −14) before IAV infection, the mock-treated mice succumbed to infection between days 7 and 8 due to severe disease, whereas the entire LNA14-treated cohort survived, with
significantly lower disease scores, indicative of minor-to-undetectable disease symptoms and reduced weight loss compared with controls (Fig. 5f–h and Supplementary Table 2). We attribute
the LNAs’ antiviral effect to their direct binding of PSL2, as opposed to nonspecific activation of antiviral pathways. Indeed, LNA14, LNA9 or derivative molecules did not elicit activation
of either interferon (IFN)-stimulated genes or the NF-κB (nuclear factor κ-light-chain-enhancer of activated B cells) pathway in lung or myeloid cells (Supplementary Fig. 17). Given the mild
symptoms observed in LNA-treated and -protected mice, we hypothesized that the resulting highly attenuated infection might be sufficient to enable mice receiving prophylactic LNA treatment
to develop an effective immunization against a secondary infection through production of immunity. To test this hypothesis, mice from the 1-week LNA14 pretreatment-surviving cohort (Fig. 5e)
were challenged alongside age-matched, naive controls, 65 d post-primary infection with ten times the mouse lethal dose (10 LD100) of IAV (Fig. 5i). The secondary challenge had no effect on
weight, clinical score or survival of mice from the LNA14 pretreatment group (a total of 72 d since treatment or 65 d since primary infection), whereas the age-matched controls presented
with rapid disease and were humanely sacrificed by day 6 post-challenge infection (Fig. 5j–l). Blood and spleen cells were analyzed for the presence of virus-specific T cells and the
development of antibodies against viral proteins (Supplementary Fig. 18a,b), which demonstrated stimulation of both the cellular and the humoral arms of the immune system, although their
respective contributions to complete absence of symptoms after a tenfold higher lethal inoculum await definition. After demonstrating PSL2-targeted LNA efficacy in prophylactic models, we
next tested its potential as a post-infection therapeutic. Due to the rapid onset of symptoms and illness in IAV infections, US Food and Drug Administration-approved IAV therapeutics are
most challenged when administered after 48 h of disease onset29. In contrast, we hypothesized that anti-PSL2 LNAs, administered either intravenously or intranasally, could treat IAV well
after an infection has been established. To test this hypothesis, mice infected with a lethal dose of PR8 virus were treated with LNA14, LNA9, Scr. LNA or vehicle control by intravenous
injection 3 d post-infection (day +3) when mice typically become noticeably ill. Although animals treated with controls rapidly succumbed to the infection, 65% of mice treated with LNA9
survived lethal infection and all the LNA14-treated mice survived (Fig. 5m). Next, we tested the therapeutic efficacy of LNA14 via intranasal administration against the 2009 modern pandemic
‘swine’ virus (A/California/04/2009 pH1N1; CA09), which has since become a dominant seasonal strain worldwide. Mice were infected with a lethal dose of CA09, then given a single intranasal
dose of LNA14, Scr. LNA or vehicle control 3 d post-infection. OSLT was used as a therapeutic control and administered by oral gavage twice daily for 5 consecutive days starting at 3 d
post-infection. The virus was 100% lethal against the Scr. LNA and vehicle control mice, and only 2/7 OSLT-treated mice were protected from lethal disease (Fig. 5n,o). In comparison, 6/7
mice in the LNA14-treated group were protected from lethality and collectively had much lower clinical scores than the other cohorts (Fig. 5p). Sampling of lungs taken 2 d after LNA14
treatment of CA09-infected mice (that is, 5 d post-infection) showed significantly reduced virus titers (>6log10 virus reduction) compared with the controls (Supplementary Fig. 19). We
also observed good dose proportionality in the lungs up to the highest dose tested to date (80 μg of LNA14) (Supplementary Fig. 20). Lowering the LNA14 dose down to 5 μg still resulted in
full protection from death (Supplementary Fig. 21), whereas Scr. LNA offered no more protection than vehicle control. PROGRAMMABLE ANTIVIRALS AGAINST SARS-COV-2 RNA STRUCTURES We
hypothesized that we could leverage our approach to targeting IAV and similarly design potent LNAs against SARS-CoV-2. Briefly, we first performed sequence alignments of the entire 30-kb
positive-sense RNA genome of SARS-CoV-2 and related beta-coronaviruses to identify conserved sequence regions predicted to contain RNA secondary structures30. We then focused on highly
conserved nucleotide stretches of sufficient length to enable antisense nucleotide attack, and a high probability of structural stability to identify promising candidate antiviral targets.
SHAPE yielded high-resolution RNA secondary structure maps of these regions and guided LNA design. Highly conserved targets included a stem–loop structure near the end of the SARS-CoV-2
genome (nt 28,743–28,792; Fig. 6a) and the structured domain spanning the terminal 5′-UTR into the ORF1-coding sequence (nt 258–276; Fig. 6b). LNAs were vetted for their ability to disrupt
these structures by chemical mapping of the LNA–RNA interaction (Fig. 6c,d). The LNAs were then screened against a SARS-CoV-2-Nluc reporter virus under Biosafety Level 3 (BSL-3) conditions
(Fig. 6e). The nucleoside analog, EIDD-1931 (ref. 31), was included as a positive control, and Scr. LNA and dimethylsulfoxide (DMSO) treated and nontreated (NT) served as negative controls.
Both LNAs against these target structures inhibited virus replication by as much as 3log10, with the 25 nM LNA-12.8 outperforming the 5 µM EIDD control by nearly 1log10 (Fig. 6e). Next, an
LNA dose–response experiment with expanded replicates was performed in Huh-7.5 cells that were modified to overexpress the human angiotensin-converting enzyme 2 (hACE2) and transmembrane
protease, serine 2 (TMPRSS2) receptors to enable more efficient virus replication32 (Fig. 6f). Indeed, the viral replication signal was enhanced by nearly 2.5log10 compared with Huh-7 cells
in this system (Fig. 6e). Even at the higher virus burden, however, all tested LNA concentrations displayed statistically significant virus inhibition in a dose-dependent manner compared
with the Scr. LNA controls and were nontoxic to the cells at concentrations 10× higher than the highest dose assayed for antiviral activity (Supplementary Fig. 22a). We next tested our LNAs
against the fully intact WT SARS-CoV-2 virus in a lung epithelial cell line expressing hACE2-A549 (Fig. 6g), which confirmed the previous Nluc virus results, whereby LNA treatment displayed
>3log10 (virus inhibition) for both LNAs at 100 nM and LNA-12.8 at 25 nM, and were nontoxic to cells tested up to 1 µM (Fig. 6h and Supplementary Fig. 22b). We next pretreated hACE2-A549
cells with LNAs and then infected them with a new SARS-CoV-2 clinical isolate (SARS-CoV-2/human/USA/OH-UC-1/2020 ON642077) derived from a chronically infected cancer patient, whose sequence
had evolved over a 7-month infection period (Fig. 6h). This clinical isolate harbors multiple mutations including Leu452Arg, Asp614Gly, Glu484Ala/Gln and Asn501Tyr in the spike protein
receptor-binding domain shown to be involved in antibody escape, enhancement of virus replication and increased transmission found in variants of concern including alpha, beta, delta, mu and
omicron33 (https://covid.cdc.gov/covid-data-tracker/#published-sars-cov-2-sequences) (Supplementary Table 3). Despite numerous mutations throughout the viral spike protein-coding region and
elsewhere in the genome, the LNAs retained their high levels of potency against this variant: both 100 nM treatments, as well as the 25 nM LNA-12.8 treatment, demonstrated complete virus
inhibition to the experimental limit of detection, equaling a 5log10 drop-in virus titer (Fig. 6h). Given the high transmissibility of SARS-CoV-2, we sought to test the effectiveness of LNA
treatment to prevent or mitigate SARS-CoV-2 transmission in a Syrian hamster model. In this system, hamsters were pretreated intranasally with a single 100 µg dose of our top SARS-CoV-2 LNA
candidate, LNA-12.8, on 2 consecutive days, day −1 and day 0, before exposure to infected sentinel hamsters (Fig. 6i). Pretreated cohorts were repeatedly exposed to the infected sentinels
for 2 h a day for 3 consecutive days. Daily oropharyngeal swabs were collected. Then 4 d post-initial exposure, lungs were collected and virus titers of LNA-12.8-treated versus
vehicle-treated animals were compared (Fig. 6j). LNA-12.8 dramatically reduced viral titers by over 3log10 compared with vehicle in exposed animals, with two hamsters containing no
detectable virus in their lungs (Fig. 6j). As shown in Fig. 6k, viral titers in the oropharyngeal swabs showed parallel reductions in response to LNA treatment to the viral titer reductions
observed in the lungs, with all hamsters showing undetectable oropharyngeal viral titers by day 4. Collectively, our identification, design and targeting of highly conserved RNA secondary
structures in IAV and SARS-CoV-2, are proof-of-principle evidence toward a new model of rapid and effective antiviral development. DISCUSSION We describe in the present study the discovery
and characterization of an RNA stem–loop structure, PSL2, that serves as a packaging signal for genome segment _PB2_. PSL2 is conserved across all known influenza A isolates. Knowledge of
PSL2’s RNA secondary structure helps explain previously discovered packaging-defective mutations and enables rational design of more potently disrupting mutations. Compensatory mutations
that restore PSL2’s structure (but not primary sequence) rescue virus packaging and titer loss in vitro and restore lethality in vivo, thus providing strong genetic validation of PSL2’s
importance in influenza biology and disease. LNAs designed to disrupt PSL2 structure dramatically inhibit IAV in vitro against viruses of different strains and subtypes. In contrast to NAIs,
such as OSLT, and the endonuclease inhibitor, baloxavir marboxil (Xofluza), which are vulnerable to drug-resistant mutations34,35,36, our PSL2-targeting LNAs exhibit a high barrier to the
development of resistance and are equally effective against WT and NAI-resistant viruses. In vivo, intranasal dosing of LNAs results in potent antiviral efficacy and prevents mortality in
mice, even with a single dose administered 2 weeks before infection with a lethal IAV inoculum. Moreover, PSL2-targeting LNAs also enable the surviving mice to develop vigorous immunity. In
therapeutic models, a single LNA dose given 3 d after infection provides complete protection from death. Together, these results have exciting implications for the development of a new class
of pan-genotypic, anti-IAV therapies for prophylaxis of, treatment of established and ‘just-in-time’ universal vaccination against an IAV infection. In addition, because traditional
vaccines take weeks to provide full protection, a co-administered single dose of LNA14 could provide protection during this vulnerability window. We envision several paths to the clinic for
PSL2-targeted therapies like LNA14 against IAV, including aerosol delivery to outpatients or intravenous administration for severely ill hospitalized patients. Importantly, incorporating the
virus’s RNA secondary structure into the LNA design allowed us to achieve far greater inhibition than ASOs designed against the same viral genomic sequence, but which relied only on primary
nucleotide sequence homology for their design37. Although we initially focused on IAV, an analogous approach can be taken for virtually any virus of interest. Indeed, we were able to
rapidly identify highly conserved RNA secondary structures in SARS-CoV-2 (ref. 30) and then design appropriate LNAs leveraging the lessons learned in targeting IAV—a process we now term
‘programable antivirals’. Our lead LNAs demonstrated greater in vitro activity against SARS-CoV-2 than the nucleoside analog-positive controls at 200× the LNA concentration. Moreover, the
LNAs were highly effective against a clinical isolate harboring mutations that have been associated with decreased susceptibility to recent COVID-19 vaccines33, with titer reductions of up
to 5log10 at 25 nM concentrations of LNA. Finally, our top anti-SARS-CoV-2 LNA exhibited profound in vivo activity by successfully decreasing and in some cases completely preventing virus
transmission in hamsters. Limitations of our study include the very preliminary nature of the SARS-CoV-2 data and the need for further experiments, including assessment of therapeutic
application of the LNA (which is the subject of another paper). Taken together, this programmable antiviral strategy now excitingly offers a highly adaptable potential solution to current
and future viruses of concern, especially respiratory pandemic viruses. Their broad-spectrum potential can enable advanced stockpiling and prepositioning, with the ability to fill the void
before a possible vaccine might be developed and deployed. Programmable antivirals can also offer protection against vaccine-resistant virus strains, as well as provide immediate protection
during the months needed for vaccine-induced immunity to ramp up. METHODS CELLS AND VIRUSES HEK293T (CRL-1573), Vero E6 (CRL-1586) and MDCK-NLB-2 (CCL-34) cells were obtained from American
Type Culture Collection (ATCC) and were maintained according to ATCC instructions. A549-Dual (a549d-nfis) and THP1-Dual (thpd-nfis) were obtained from InvivoGen and maintained according to
the manufacturer’s instructions. All cell lines used in the present study were routinely checked for _Mycoplasma_ contamination (MycoAlert Mycoplasma Detection Kit, Lonza) and were
authenticated by the respective vendors. All cell lines used for SARS-CoV-2 experimentation, including Huh-7, ACE2-TMPRSS2-Huh-7.5 and ACE2-A549 cells, were maintained at 37 °C in complete
Dulbecco’s modified Eagle’s medium (DMEM; Gibco) containing 10% fetal bovine serum (FBS; Invitrogen), penicillin and streptomycin (Gibco) and Hepes buffer (Gibco). The ACE2-A549 cells were
specially engineered to overexpress the ACE2 receptor in human alveolar basal epithelial cells (A549), whereas the ACE2-TMPRSS2-Huh-7.5 cells were a kind gift of the Catherine Blish
laboratory and engineered to overexpress both hACE2 and human TMPRSS2 receptors. A549-Dual and THP1-Dual cells were purchased from InvivoGen. WT influenza A/PR/8/34 (PR8) H1N1 virus
(ATCC-VR-95) and the tissue-culture-adapted PR8 virus (ATCC-VR-1469) were purchased from ATCC. PR8 mutant viruses were generated using an eight-plasmid reverse genetic system as previously
described38. Tissue-cultured adapted influenza A/Hong Kong/8/68 (HK68) H3N2 virus (ATCC-VR-1679), A/Virginia/ATCC6/2012 (H3N2) virus (ATCC-VR-1811), A/Virginia/ATCC1/2009TC (H1N1) virus
(ATCC-VR-1736) and A/Wisconsin/33 (H1N1) virus (VR-1520) were purchased from ATCC. A/California/4/2009 (pH1N1) virus was kindly gifted by E. Govorkova from St. Jude Children’s Research
Hospital (Memphis, USA). Viruses were grown and amplified in 10-d-old, specific, pathogen-free, research-grade chicken embryos at 35 °C (Charles River Laboratories; SPAEAS). WT recombinant
SARS-CoV-2 was prepared and handled as described39. The recombinant SARS-CoV-2-Nluc virus is an authentic, fully replicating virus in which ORF7a has been deleted and replaced with Nluc40.
The SARS-CoV-2 clinical variant was isolated from a 54-year-old man with melanoma and lymphoma, who received a confirmed COVID-19 diagnosis by PCR in March 2020. Virus sequencing was
performed on virus isolated from a nasopharyngeal swab sample in virus transport medium taken after the patient had been infected for 7 months. PLASMID CONSTRUCTS AND CLONING Plasmids were
used containing the WT _PB2_ segments from influenza viruses A/Puerto Rico/8/34 (H1N1) (PR8), A/New York/470/2004 (H3N2) (NY470), A/New York/312/2001 (H1N1) (NY312), A/Brevig Mission/1/1918
(H1N1) (1918), A/California/04/2009 (H1N1) (CA09) and A/Vietnam/03/2004 (H5N1) (VN1203). For the generation of PR8 packaging mutant vRNA, we utilized a Stratagene QuickChange XL
site-directed mutagenesis kit for mutagenesis of a pDZ plasmid containing the _PB2_ gene of PR8 (ref. 38). Sequences of each mutated construct were confirmed by automated sequencing. The
eight-plasmid pBD rescue system for A/WSN/33 (H1N1) was kindly donated by A. Mehle. The H275Y NA mutant was generated by QuickChange mutagenesis from the bidirectional pBD plasmids, as
described above. REVERSE GENETICS AND VIRUS TITRATIONS Recombinant A/Puerto Rico/8/34 (PR8) virus and recombinant A/WSN/33 (WSN) virus were generated using eight-plasmid reverse genetic
systems38. Briefly, 1 × 106 cells of a HEK293T/MDCK co-culture were Lipofectamine 3000 (Invitrogen) transfected with 1 μg of one of each of the eight segments contained within plasmids that
utilize a bidirectional dual Pol I/II promoter system for the simultaneous synthesis of genomic vRNA and messenger RNA. For rescue of compensatory _PB2_ mutant viruses where a nonsynonymous
change was required, a WT PB2 protein expression plasmid (Pol II) was co-transfected during virus rescue. Supernatants were collected 24 h post-transfection. PR8 rescue viruses were then
inoculated into the allantoic cavities of 10-d-old chicken embryos. WSN rescue viruses were passaged subsequent times on MDCK cells. Rescue of recombinant viruses was assessed by
hemagglutination (HA) activity. Each newly rescued virus was further plaque titered and mutations were confirmed by sequencing of mutated genes. Plaque assays were carried out on confluent
MDCK cells as described previously41. HA assays were carried out in 96-well round-bottomed plates at room temperature, using 50 μl of virus dilution and 50 μl of a 0.5% suspension of turkey
red blood cells (LAMPIRE Biological Laboratories) in PBS. ISOLATION OF PACKAGED VRNAS To analyze packaged vRNA for PR8 mutated viruses, 10-d-old eggs were inoculated with approximately 1,000
p.f.u. of recombinant virus and incubated for 72 h. Allantoic fluid was harvested and supernatant was dual clarified by low-speed centrifugation. Clarified supernatant was then layered on a
30% sucrose cushion and ultracentrifuged at 30,000 r.p.m. for 2.5 h (Beckman Rotor SW41). Pelleted virus was resuspended in PBS and TRIzol (Invitrogen) extracted. Precipitated vRNA was
resuspended in a final volume of 20 μl of 10 mM Tris-HCl, pH 8.0 and stored at −80 °C. Virus supernatant from LNA-treated cells was harvested 48 h post-infection and subjected to low-speed
centrifugation at 1,000 r.p.m., then 10,000 r.p.m. Isolation continued as indicated above. QUANTITATIVE PCR ANALYSIS OF PACKAGED VRNAS Approximately 200 ng of extracted vRNA was reverse
transcribed using a universal 3′-primer (5′-AGGGCTCTTCGGCCAGCRAAAGCAGG) and Superscript III reverse transcriptase (Invitrogen). The reverse transcription (RT) product was diluted
approximately 10,000-fold and used as a template for quantitative (q)PCR. Separate PCRs were then carried out as previously described42 with segment-specific primers. The 10-μl reaction
mixture contained 1 μl of diluted RT product, a 0.5 μM primer concentration and SYBR Select Master Mix (Applied Biosystems) which included SYBR GreenER dye, 200 μM deoxynucleoside
triphosphates, heat-labile UDG (uracil-_N_-glycosylase), optimized SYBR Green Select Buffer and AmpliTaq DNA polymerase UP enzyme. Relative vRNA concentrations were determined by analysis of
cycle threshold values, total vRNA amount within a sample was normalized to the level of HA vRNA and then percentages of incorporation were calculated relative to the levels of WT vRNA
packaging. Viral packaging results represent the averaged levels of vRNA incorporation ± s.d. derived from two independent virus purifications, with vRNA levels quantified in triplicate.
DENATURING RNA GEL Extracted viral RNA (100–300 ng) was diluted with equal volume of NOVEX TBE–Urea sample buffer and incubated at 70 °C for 10 min before separation on a 6% TBE–Urea gel for
18 h at a constant voltage of 80 V. RNA from each sample was run in several dilutions to enable clear visualization of the genomic RNA without over-saturation of the band’s signal. The RNA
was visualized by staining for 30 min in 0.5× Tris/borate/EDTA (TBE) buffer supplemented with 0.5 mg ml−1 of ethidium bromide followed by visualization in a GelDoc EZ Imager system (BioRad).
Silver staining was performed using the SilverXpress silver stain kit (Invitrogen) according to the manufacturer’s instruction. Silver staining and ethidium bromide staining were compared
and shown to have the same linear range of detection (data not shown); ethidium bromide was selected for lane visualization due to a higher background signal in silver staining. The band
intensity in each lane was determined using Image Lab Software (BioRad) and an analysis of the intensity of each genomic band relative to the total intensity of all genome segments was
determined and normalized to the intensity of the HA band relative intensity. STRAND-SPECIFIC RT–QPCR MDCK cells transfected with 1 M LNA9 or Scr. LNA were infected with PR8 virus at an MOI
of 0.1 24 h post-transfection. Then 8 h post-infection total cellular RNA was extracted in TRIzol reagent (Invitrogen) and the RNA was purified using the Direct-Zol RNA mini-prep (Zymo
Research) according to the manufacturer’s protocol. RT and qPCR were performed according to the literature43. Complementary DNAs of the influenza vRNA and complementary viral RNA (cRNA) were
synthesized with tagged primers to add an 18- to 20-nt tag that was unrelated to the influenza virus at the 5′-end (cRNAtag: 5′-GCT AGC TTC AGC TAG GCA TC-3′; vRNAtag: 5′-GGC CGT CAT GGT
GGC GAA T-3′). Hot-start RT with the tagged primer was performed as described in Kawakami et al.43 using saturated trehalose. A 5.5-μl mixture containing 200 ng of total RNA sample and 10
pmol of tagged primer was heated for 10 min at 65 °C, chilled immediately on ice for 5 min and then heated again at 60 °C. After 5 min, 14.5 μl of preheated reaction mixture (4 μl of First
Strand buffer (5×, Invitrogen), 1 μl of 0.1 M dithiothreitol, 1 μl of dNTP mix (10 mM each), 1 μl of Superscript III reverse transcriptase (200 U μl−1, Invitrogen), 1 μl of RNasin Plus RNase
inhibitor (40 U μl−1, Promega) and 6.5 μl of saturated trehalose) was added and incubated for 1 h. RT–qPCR was performed with PowerUp SYBR Green SuperMix (Applied Biosystems) on a BioRad
CFX96 Real-Time System. Then 7 µl of a tenfold dilution of the cDNA was added to the qPCR reaction mixture (10 μl of SYBR Green SuperMix (2×), 1.5 μl of forward primer (10 μM) and 1.5 μl of
reverse primer (10 μM)). The cycle conditions of qPCR were 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. The qPCR primers were: PR8 segment 1 (_PB2_) cRNA:
forward: 5′-TCC ACC AAA GCA AAG TAG AAT GC-3′; reverse: 5′-GCT AGC TTC AGC TAG GCA TCA GTA GAA ACA AGG TCG TTT TTA AAC-3′; PR8 segment 1 (PB2) vRNA: forward: 5′-GGC CGT CAT GGT GGC GAA TAG
ACG AAC AGT CGA TTG CCG AAG C-3′, reverse: 5′-AGT ACT CAT CTA CAC CCA TTT TGC-3′; PR8 segment 4 (HA) cRNA: forward: 5′-CTG TAT GAG AAA GTA AAA AGC C-3′, reverse: 5′-GCT AGC TTC AGC TAG GCA
TCA GTA GAA ACA AGG GTG TTT TTC-3′; and PR8 segment 4 (HA) vRNA: forward: 5′-GGC CGT CAT GGT GGC GAA TAG GAT GAA CTA TTA CTG GAC CTT GC-3′, reverse: 5′-TCC TGT AAC CAT CCT CAA TTT GGC-3′.
ANIMALS All animal studies were performed in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals and approved by the Stanford University
Administrative Panel on Laboratory Animal Care and by the Utah State University Institutional Animal Care and Use Committee. Animals were housed in disposable cages connected to an Innorack
IVC-ventilated rodent housing system under 12 h light:dark cycle at 24 °C and 25–30% humidity. Healthy age-matched female BALB/c mice aged 6–8 weeks (Jackson Laboratories) were randomly
separated into groups for infection/treatment or used as uninfected/nontreated controls. Treatment groups were not blinded to the investigators. Mice were identified with tag numbers
throughout the experiment. The 10-week-old female golden Syrian hamsters (LVG strain, Charles River Laboratories) were separated into groups for exposure studies with an additional sentinel
group that was inoculated directly with virus. IN VIVO MOUSE INFECTIONS Mice were lightly anesthetized with isoflurane and intranasally infected with 50 μl of virus preparation at 1 LD100—a
concentration of approximately 1,000 p.f.u. for virus-packaging mutant experiments and 900 p.f.u. for LNA treatment experiments. Weights and clinical scores were assessed daily, and animals
were humanely sacrificed when a clinical score of 5 was recorded (see Supplementary Table 2 for clinical score determination). Kaplan–Meier survival curves were generated using GraphPad
Prism. IN VIVO MOUSE IAV ANTIVIRAL ASSAYS ‘In vivo-ready’ LNAs were custom designed and ordered from QIAGEN (formally Exiqon) and later from IDT. For intranasal delivery, in vivo-ready LNA
was mixed in complexes with in vivo_-_JetPEI transfection reagent (Polyplus) according to the manufacturer’s protocol to the indicated final concentration in 50–75 μl of 5% glucose solution.
Mice were then lightly anesthetized with isoflurane and 50–75 μl of the solution was delivered intranasally. For retro-orbital delivery, in vivo-ready LNA was mixed in complexes with in
vivo_-_JetPEI transfection reagent (Polyplus) according to the manufacturer’s protocol to the indicated final concentration in 200 μl of 5% glucose solution. Mice were then anesthetized and
the solution was delivered by retro-orbital injection. OSLT (Sigma-Aldrich, catalog no. SML1606) was prepared in sterile water and administered to mice at a dose of 10 mg kg−1 twice daily by
oral gavage (totaling 20 mg kg−1 d−1), with 8 h between dosing intervals. PREVENTION OF SARS-COV-2 TRANSMISSION IN SYRIAN HAMSTERS Infection of donor sentinel hamsters (nontreated):
6-week-old WT golden Syrian hamsters were intranasally infected with 1 × 104.3 CCID50 (cell culture infectious dose 50%) of SARS-CoV-2 (USA_WA1/2020 strain) in a 100-µl volume. LNA
pretreatment group: 6-week-old golden Syrian hamsters (_n_ = 5) were pretreated by intranasal instillation with a 200-µl volume containing 100 µg of LNA-12.8 on day −1 and day 0 before
exposure to the SARS-CoV-2-infected sentinel hamsters. Vehicle-treated hamsters (_n_ = 4) received PBS by intranasal nebulization. LNA- and vehicle-treated hamsters were co-housed and
exposed to the infected sentinels for 2 h per d for 3 consecutive days. Then, 4 d after the initial exposure, the lungs were harvested and virus titers were determined by CCID50 in
triplicate. Statistical analysis was performed using an unpaired Student’s _t_-test with GraphPad Prism 9 software. LNA DESIGN AND PREPARATION Oligonucleotides containing LNAs were custom
synthesized by Exiqon and later by IDT. Capitalized letters denote LNA. Lower-case letters denote typical (nonlocked) DNA nucleotides. All oligonucleotides contain phosphorothioate
internucleoside linkages. LNA8 and -9 were designed as LNA gapmers to contain a stretch of six or seven DNA nucleotides optimized for RNase-H recruitment. Sequences of all LNAs are shown
below: LNA1: 5′-AccAaaAGaaT-3′ LNA2: 5′-TggCcATcaaT-3′ LNA3: 5′-TagCAtActtA-3′ LNA4: 5′-CCAAAAGA-3′ LNA5: 5′-CATACTTA-3′ LNA6: 5′-CagaCaCGaCCaaAA-3′ LNA7: 5′-TAcTtaCTgaCagCC-3′ LNA8:
5′-AGAcacgaccaaAAG-3′ LNA9: 5′-TACTtactgacaGCC-3′ LNA14: 5′-CGACcaaaagaATTC-3′ SCR. LNA (NEGATIVE CONTROL): 5′-AACACGTCTATACGC-3′ SARS-CoV-2-directed LNAs: LNA-12.8: 5′-AGGAagttgtagCACG-3′
LNA-14.3: 5′-GCTctccatcttaCCT-3′ IN VITRO LNA ANTIVIRAL ASSAYS For all experiments, LNAs were reconstituted in RNase-free water at 100 μM stock solutions, aliquoted and stored at −20 °C
before single use. Lipofectamine 3000 (Life Technologies) was used to transfect LNA into cells at indicated concentrations per the manufacturer’s protocol. For IAV prophylactic antiviral
assays, 1 × 106 MDCK cells were plated in 6-well plates 24 h before being transfected with the indicated LNA. Cells were then infected at the indicated time points with 0.01 MOI of PR8
(H1N1) or HK68 (H3N2) virus. For post-infection therapeutic assessment, MDCK cells were infected with PR8 or HK68 before LNA transfection as described above. Then 48 h post-infection,
supernatant was collected and the viral titer was determined by plaque assay in triplicate. For SARS-CoV-2-Nluc assays: 1 d before transfection, Huh-7 or ACE2-TMPRSS2-Huh-7.5 was plated on
96-well clear-bottomed plates to 60–70% confluency at the time of treatment with the LNA ASOs LNA-12.8, LNA-14.3 or Scr. LNA. Lipofectamine 3000 (Life Technologies) was used to transfect LNA
ASOs into cells at 25 nM or 100 nM final concentration, according to the manufacturer’s protocol. Cells were then infected with SARS-CoV-2 reporter virus expressing nanoluciferase
(SARS-CoV-2-Nluc) at an MOI of 0.3 for 1 h, after which the virus was removed and fresh medium was added. Recombinant SARS-CoV-2-Nluc is a fully replicating virus in which ORF7 has been
deleted and replaced with Nluc. Thus, the measurement of Nluc expression is a surrogate marker of virus replication enabling the screening of antiviral compounds. A nucleoside analog
β-d-_N_4-hydroxycytidine, EIDD-1931, with potent activity against SARS-CoV-2, was included as a positive control. A DMSO control was included as a mock-treated, negative control. Data were
graphed and analyzed in Prism v.8 and v.9 by GraphPad. Statistical analysis of the data from each cell type was computed as an ordinary one-way analysis of variance (ANOVA) using Dunnett’s
multiple comparison test against the DMSO control or Scr. LNA control from each cell type, where indicated. SARS-CoV-2 plaque assay experiments were performed in 24-well plates: 24-well
plates were seeded with 1 × 105 Vero E6 cells per well in DMEM, 10% FBS and 1× antibiotic/antimycotic 24 h before Lipofectamine 3000 transfection of LNA. Scr. LNA was included as a negative
control. Then 12 h post-transfection, LNA-transfection medium was removed, and cells were infected at an MOI of 0.01 with either recombinant WT SARS-CoV-2 (PMID: 32526206) or a SARS-CoV-2
clinical isolate derived from a nasopharyngeal swab taken from a chronically infected cancer patient 7 months after the initial confirmatory COVID-19 PCR test. After adding virus inoculum,
plates were incubated at 37 °C for 1 h, after which input virus was removed, wells were washed with 1 ml of medium and 1 ml of ‘infection medium’ (DMEM, 5% FBS, 1× antibiotic/antimycotic)
was added. After 48 h at 37 °C, 200 µl of culture supernatant was collected and stored at −80 °C until quantitation of infectious virus by plaque assay. Briefly, Vero E6 cells were seeded at
500,000 cells per well per 2 ml in 6-well plates. After 24 h, samples were thawed at room temperature and serially diluted in PBS. Medium was removed from the six-well plates and serial
dilutions were added to the plate, incubated for 1 h at 37°C and then overlaid with DMEM, 5% FBS, 1× antibiotic/antimycotic and 0.9% agarose. After 72 h, neutral red stain was added to each
plate, incubated for 3 h and plaques were counted. The number of plaque-forming units (p.f.u.) per milliliter was generated using the following formula: p.f.u. ml−1 = number of plaques ×
serial dilution factor × 5. LNA TREATMENT AND IAV-PACKAGING EFFICIENCY DETERMINATION Briefly, T75 flasks of 80% confluent MDCK cells were transfected with 100 nM of Scr. LNA, LNA9 or mock
untreated by Lipofectamine 3000 transfection, according to the manufacturer’s protocol. Then 12 h post-transfection, cells were infected with 0.01 MOI of WT TC-adapted PR8 virus. After 1 h,
virus was removed and the cells were washed with PBS; 48-h post-infection supernatants were collected and RNA was isolated as described in isolation of packaged vRNAs and assay methods. IN
VITRO DRUG SELECTION LNA9 selection: 80–90% confluent MDCK cells in 12-well plates were transfected in duplicate with a starting concentration of 0.01 nM (~½EC50) LNA9 for passage 1 by
Lipofectamine transfection (see above). Then 12 h post-transfection, cells were washed with PBS and infected with an MOI of 0.01 of WT PR8 virus. After 1-h incubation at 37 °C, cells were
washed and virus growth medium was added. Cells were incubated until 50% cytopathic effect (CPE) was evident (48–72 h). Virus supernatant was harvested, low-speed centrifuge clarified,
aliquoted, plaque titered and stored at −80 °C. The virus supernatant was then continuously serially passaged in the presence of escalating concentrations of LNA9 (0.01 nM to 100 nM). If no
CPE were evident, the drug concentration was lowered and the added virus concentration increased until 50% CPE occurred. OSLT selection: confluent MDCK cells in 12-well plates were infected
with an MOI of 0.01 of PR8 virus. After adsorption for 1 h, cells were washed with PBS and OSLT (Sigma-Aldrich, catalog no. Y0001340) was added to the virus growth medium at a starting
concentration of 1 nM (~½EC50). Drug selection proceeded as described above, with escalating concentrations of OSLT (0.01 nM to 250 μM) at each subsequent passage. EC50 DETERMINATION For
LNA9, the EC50 was defined as the concentration of drug effective in reducing the percentage of virus titer to 50% of that for the no-drug control. In brief, the EC50 was determined by
seeding 5 × 105 MDCK cells in each well of a 12-well plate and incubating overnight at 37 °C under 5% CO2. Cells were then transfected with LNA9 as described above at concentrations from
0.01 nM to 10 μM. Plates were incubated at 37 °C for 12 h before infection with 0.01 MOI of WT PR8, serially passaged LNA-treated virus, WSN33 WT or WSN33 H275Y NAI-resistant virus. Then 48
h post-infection, supernatants were collected, centrifuge clarified, aliquoted and stored at −80 °C. The viral titer for each drug dilution was performed by plaque assay in duplicate. The
EC50 was the concentration of LNA9 yielding a percentage titer of 50% of that without drug. For OSLT, the EC50 was defined as the concentration of drug reducing the total percentage of
plaques to 50% of that for the no-drug control, determined by plaque reduction assay1. Briefly, confluent MDCK cells in 12-well plates were infected with approximately 100 p.f.u. of WT PR8,
serially passaged OSLT-treated virus, WSN33 WT or WSN33 H275Y NAI-resistant virus and incubated for 1 h at 37 °C. Cells were then washed with PBS and a 50:50 mix of 1% agarose: 2× virus
growth DMEM containing varying concentrations of drug (0.1 nM to 1 mM) was added to the cells. Plates were harvested 72 h later, stained with Crystal Violet and plaques were counted. The
EC50 was the concentration of OSLT reducing the total percentage of plaques to 50% of that without drug. All results were plotted in GraphPad Prism to generate EC50 curves. IN VITRO
TRANSCRIPTION OF FULL-LENGTH IAV VRNA For each WT isolate (PR8, 1918, VN1203, NY470, NY312 and CA09) and PR8 packaging mutant clones, _PB2_ cDNA was amplified from plasmid-using,
segment-specific primers under a T7 promoter. Amplified cDNA was gel purified using an Invitrogen DNA gel kit. The vRNAs were then produced by in vitro transcription, using T7-MEGAscript
kit. The vRNAs for SHAPE were purified by MEGAclear (Thermo Fisher Scientific, catalog no. AM1908) with purity and length verified by capillary electrophoresis. THE SINGLE FLUOROPHORE-SHAPE
1D ANALYSIS OF FULL-LENGTH IAV VRNA In vitro transcribed _PB2_ vRNA was folded (100 mM NaCl, 2.5 mM MgCl2, 65 °C for 1 min, 5-min cooling at room temperature, 37 °C for 20–30 min) in 100 mM
Hepes, pH 8. The 2′-acylation with _N_-methylisatoic anhydride18 and RT primer extension were performed at 45 °C for 1 min, 52 °C for 25 min and 65 °C for 5 min, as previously described44.
6-Carboxyfluorescein (6FAM) was used for all labeled primers (primer sequences available on request). Exceptions to these protocols were as follows: (1) RNA purification after acylation was
performed using RNA C&C columns (Zymo Research), rather than ethanol precipitation; (2) before and after SHAPE primer buffer was added, the mixture was placed at room temperature for 2–5
min, which enhanced RT yields significantly; (3) DNA purification was performed using Sephadex G-50 size exclusion resin in 96-well format, then concentrated by vacuum centrifugation,
resulting in a more significant removal of primer; and (4) 2 pmol of RNA was used in ddGTP (2′,3′-dideoxyguanosine-5′-triphosphate) RNA-sequencing reactions. The ABI 3100 Genetic Analyzer
(50-cm capillaries filled with POP-6 matrix) was set with the following parameters: voltage 15 kV, _T_ = 60 °C, injection time = 15 s. The GeneScan program was used to acquire the data for
each sample, which consisted of purified DNA resuspended in 9.75 μl of Hi-Di-Formamide, to which 0.25 μl of ROX500 internal size standard (ABI catalog no. 602912) was added. PeakScanner
parameters were set to the following parameters: smoothing=none; window size=25; size calling=local southern; baseline window=51; peak threshold=15. Fragments 250 and 340 were
computationally excluded from the ROX500 standard45. The data from PeakScanner were then processed into SHAPE data by using FAST (fast analysis of SHAPE traces), a customized algorithm
developed in our lab19. FAST automatically corrects for signal differences due to handling errors, adjusts for signal decay and converts fragment length to nucleotide position, using a ddGTP
ladder as an external sizing standard and the local Southern blotting method5,19. This algorithm embedded in the RNAstructure program is freely available at
http://med.stanford.edu/glennlab/download.html. RNAstructure parameters: slope and intercept parameters of 2.6 and −0.8 kcal mol−1 were initially tried, as suggested46; however, we found
that smaller intercepts closer to 0.0 kcal mol−1 (for example, ~−0.3) produced fewer less optimal structures (within a maximum energy difference of 10%). We speculate that this minor
parameter difference may be due to the precise fitting achieved between experimental and control datasets by the automated FAST algorithm. FAST was written in ANSI C/C++ and is integrated
into RNAstructure with FAST, which requires MFC (Microsoft Foundation Classes). RNA structures were drawn and colored using RNAViz 2 (ref. 47) and finalized in Adobe Illustrator. IAV PSL2
CONSTRUCT DESIGN, RNA SYNTHESIS AND CHEMICAL MODIFICATION FOR M2 EXPERIMENTS Double-stranded DNA templates were prepared by PCR assembly of DNA oligomers designed by an automated MATLAB
script as previously described (available at https://primerize.stanford.edu)48. Constructs for M2 include all single mutants to the Watson–Crick counterpart. Compensatory mutants for M2R
were designed based on basepairing in the proposed secondary structure22. In vitro transcription reactions, RNA purification and quantification steps were as described previously48.
One-dimensional (1D) chemical mapping, M2 and M2R were carried out in the 96-well format as described previously48,49,50. Briefly, RNA was heated up and cooled to remove secondary structure
heterogeneity, then folded properly and incubated with SHAPE reagent (5 mg ml−1 of 1-methyl-7-nitroisatoic anhydride (1M7))51; modification reaction was quenched and RNA was recovered by
poly(dT) magnetic beads (Ambion) and FAM-labeled Tail2-A20 primer; RNA was washed by 70% ethanol twice and resuspended in double-distilled water (ddH2O), followed by RT to cDNA and heated
NaOH treatment to remove RNA. The final cDNA library was recovered by magnetic bead separation, rinsed, eluted in Hi-Di-Formamide (Applied Biosystems) with ROX350 ladder and loaded to a
capillary electrophoresis sequencer (ABI 3100). Data processing, structural modeling and data deposition: the HiTRACE software package v.2.0 was used to analyze CE data (both MATLAB toolbox
and web server available52,53). Trace alignment, baseline subtraction, sequence assignment, profile fitting, attenuation correction and normalization were accomplished as previously
described54,55. Sequence assignment was accomplished manually with verification from sequencing ladders. Data-driven secondary structure models were obtained using the Fold program of the
RNAstructure package v.5.4 (ref. 56) with pseudo-energy slope and intercept parameters of 2.6 kcal mol−1 and –0.8 kcal mol−1. Two-dimensional _z_-score matrices for M2 datasets and
helix-wise bootstrapping confidence values were calculated as described previously22,48. The _z_-score matrices were used as basepair-wise, pseudo-free energies with a slope and intercept of
1.0 kcal mol−1 and 0 kcal mol−1. Secondary structure images were generated by VARNA57. These chemical-mapping datasets, including 1D mapping, M2 and M2R have been deposited at the RNA
Mapping DataBase (RMDB: http://rmdb.stanford.edu)58, accession nos.: PSL2IAV_1M7_0001, PSL2IAV_RSQ_0001. SHAPE ANALYSIS OF LNA-TARGETED IAV VRNA A truncated DNA template of PR8 virus segment
_PB2_ containing nucleotides 1–88 was prepared by PCR assembly of DNA oligomers, and in vitro transcription reactions, RNA purification and quantification steps were as described
previously48. The 1D SHAPE chemical mapping was performed in a 96-well plate format as described above, with the following exception: once RNA was denatured and refolded as described, 100 nM
of each prepared LNA was added to the folded RNA and incubated with 5 mg ml−1 of SHAPE reagent 1M7. Modification quenching, RNA recovery, re-suspension, RT, cDNA-sequencing and data
processing were performed as described50. SHAPE 1D ANALYSIS OF NONTREATED AND LNA-TREATED SARS-COV-2 RNA RNA was folded (0.5 M Na Hepes, pH 8; 90 °C for 3 min, 12-min cooling at room
temperature, 50 °C for 20 min, 12-min cooling at room temperate) in 100 mM MgCl2 with or without LNA and with or without SHAPE reagent 1M7 modification. RNA was purified and quenched with
magnetic beads (0.5 M 2-(_N_-morpholino)ethanesulfonic acid sodium salt, pH 6, FAM-A20 tail 2 primer, 5 M NaCl and Ampure beads) and reverse transcribed at 48 °C for 40 min, followed by a
0.4 M NaOH acid quench to improve signal intensity. The resulting cDNA was resuspended in ROX Hi-Di-Formamide and diluted for capillary electrophoresis analysis. As an internal control, the
RNA was created with GAGUA hairpins in 5′- and 3′-termini designed to be reactive in the presence of 1M7. The results were analyzed using the HiTRACE method52 and standardized by the
Kladwang et al. method55. PBMCS AND SPLENOCYTE ISOLATION Whole blood was collected from mice into heparinized tubes. The whole blood was overlaid on top Ficoll-Paque medium and centrifuged
at 400_g_ for 40 min at room temperature. The top layer containing plasma and platelets was removed and the peripheral blood mononuclear cells (PBMCs) at the interphase of the Ficoll layer
were collected. The mononuclear cells were diluted in PBS and pelleted by centrifugation at 500_g_ for 15 min after an additional wash with PBS. The pellet was suspended in PBS. Splenocytes
were isolated by manually grinding the spleen over a 40-μm cell strainer. The cells were transferred several times through the strainer and processed further as described above. TruStain fcX
(anti-mouse CD16/32) antibody specific for FcyR III/II (1 μg per 1 × 106 cells) was used to block nonspecific staining, followed by staining with Zombie Aqua viability kit (BioLegend).
Antibody staining was performed by mixing the cells with an antibody mix containing BD Horizon PE-CF594 Rat Anti-Mouse CD45 (BD Biosciences), PE/Cy7 anti-mouse CD3 (BioLegend), APC/Cy7
anti-mouse CD8a (BioLegend) and H-2Kd Influenza HA Tetramer-IYSTVASSL-PE (MBL International). All antibodies were diluted at a ratio of 2 ml of each antibody per 1 × 106 cells and HA
tetramer was the diluter at a ratio of 5 ml per 1 × 106 cells. Splenocytes were isolated from a mouse spleen by immersing the spleen in Hanks’ balanced salt solution (HBSS) with 10% FBS and
washing with HBSS to remove blood. The spleen was placed in a 40-mm cell strainer and the tissue was mashed with a syringe plunger to break down the tissue and dislodge the cells. After
complete disruption of the organ, the cells were run through the strainer into a conical tube. The cell suspension was pelleted by centrifugation at 350_g_ for 10 min at 4 °C. The pellet was
resuspended in water for 20 s to disrupt red blood cells followed by addition of 2× PBS solution. The cells were pelleted and subjected to staining as described for PBMCs above. FLOW
CYTOMETRY After staining, the cells were fixed and subjected to flow cytometry using a BD LSR II flow cytometer (BD Biosciences) equipped with 488-nm, 405-nm, 640-nm and 532-nm lasers. Data
were collected using BD FACSDiva software (BD Biosciences) and analyzed using FlowJo software (TreeStar). The gating strategy for positive tetramer cells was as follows: forward scatter area
(FSC)-H/FSC-A gate used to collect cells and a gate for live cells was then generated. The live cells were gated for CD3+/CD45+ cells and the positive cells were gated to determine the
CD8+/HA-tetramer+ cells. Acquired data was analyzed using a FlowJo software (TreeStar). LNA EFFECT ON IFN AND NF-KB PATHWAYS THP1-Dual or A549-Dual cells (InvivoGen) were transfected in
triplicate with LNA9, LNA14 or derivatives thereof, or poly(I:C) (R&D Systems) using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s protocol. Treatment
with recombinant human tumor necrosis factor α (hTNF-α; R&D Systems), recombinant human IFN (hIFN-γ; R&D Systems), recombinant hIFN-α (R&D Systems) or lipopolysaccharide (LPS)
from O111:B4 _Escherichia coli_ (Sigma-Aldrich) was used as a positive control for activation of the two pathways. Supernatant from the cultures were collected 24–32 h
post-transfection/treatment to measure luciferase signal or secreted alkaline phosphatase activity as indicators of IFN pathway stimulation or NF-κB pathway stimulation, respectively.
Results were normalized to reflect fold induction relative to NT cells. IL-6 AND TNF-Α ELISA Supernatants from THP1-Dual cells (InvivoGen) were transfected in triplicate with LNA9, LNA14 or
poly(I:C) (R&D Systems). Treatment with recombinant hIFN-γ (R&D Systems), recombinant hIFN-α (R&D Systems) or LPS from O111:B4 _E. coli_ (Sigma-Aldrich) was used as a positive
control to cause induction of interleukin (IL)-6 or TNF-α secretion. Supernatants were collected 24 h post-transfection/treatment and assayed for TNF-α and IL-6 concentration by ELISA
(Thermo Fisher Scientific) according to the ELISA kit protocol. STATISTICAL ANALYSES We expressed the data as the mean ± s.d. or mean ± s.e.m. where indicated. We used Student’s _t_-test (to
compare two samples) or ANOVA (to compare multiple samples) as analyzed by GraphPad Prism (v.8 and v.9) for statistical analysis. We performed Kaplan–Meier log(rank) tests for survival
analyses. We considered all _P_ values >0.05 not to be significant. REPORTING SUMMARY Further information on research design is available in the Nature Research Reporting Summary linked
to this article. DATA AVAILABILITY Data for 1D SHAPE experiment is available in source data of this manuscript. Chemical-mapping datasets for mutate-and-map and mutation/rescue experiments
are available at http://rmdb.stanford.edu, accession nos. PSL2IAV_1M7_0001, PSL2IAV_RSQ_0001. Source data are provided with this paper. All the data that support the findings of the present
study are available from the corresponding author upon request. CODE AVAILABILITY DNA oligomers automated design script is available as MATLAB script at https://primerize.stanford.edu. A
code to analyze map-and-mutate datasets is available as MATLAB script for download at https://ribokit.github.io/HiTRACE. A tutorial is available at http://hitrace.org. One-dimensional SHAPE
with FAST algorithm embedded in the RNAstructure program is freely available at http://med.stanford.edu/glennlab/download.html. REFERENCES * Memoli, M. J., Hrabal, R. J., Hassantoufighi, A.,
Eichelberger, M. C. & Taubenberger, J. K. Rapid selection of oseltamivir- and peramivir-resistant pandemic H1N1 virus during therapy in 2 immunocompromised hosts. _Clin. Infect. Dis._
50, 1252–1255 (2010). Article CAS PubMed Google Scholar * Hai, R. et al. Influenza A (H7N9) virus gains neuraminidase inhibitor resistance without loss of in vivo virulence or
transmissibility. _Nat. Commun._ 4, 2854 (2013). Article PubMed Google Scholar * Hayden, F. G. & de Jong, M. D. Emerging influenza antiviral resistance threats. _J. Infect. Dis._ 203,
6–10 (2011). Article PubMed PubMed Central Google Scholar * Romero-Lopez, C. & Berzal-Herranz, A. Unmasking the information encoded as structural motifs of viral RNA genomes: a
potential antiviral target. _Rev. Med. Virol._ 23, 340–354 (2013). Article CAS PubMed PubMed Central Google Scholar * Pang, P. S. et al. Structural map of a microRNA-122: hepatitis C
virus complex. _J. Virol._ 86, 1250–1254 (2012). Article CAS PubMed PubMed Central Google Scholar * Palese, P. & Shaw, M. L. Orthomyxoviridae. In _Fields Virology_ 5th edn (eds
Knipe, D. M. & Howley, P. M.) 1647–1689 (Lippincott Williams & Wilkins, 2007). * Compans, R. W., Content, J. & Duesberg, P. H. Structure of the ribonucleoprotein of influenza
virus. _J. Virol._ 10, 795–800 (1972). Article CAS PubMed PubMed Central Google Scholar * Noda, T. & Kawaoka, Y. Structure of influenza virus ribonucleoprotein complexes and their
packaging into virions. _Rev. Med. Virol._ 20, 380–391 (2010). Article CAS PubMed PubMed Central Google Scholar * Hutchinson, E. C., von Kirchbach, J. C., Gog, J. R. & Digard, P.
Genome packaging in influenza A virus. _J. Gen. Virol._ 91, 313–328 (2010). Article CAS PubMed Google Scholar * Gao, Q. et al. The influenza A virus PB2, PA, NP, and M segments play a
pivotal role during genome packaging. _J. Virol._ 86, 7043–7051 (2012). Article CAS PubMed PubMed Central Google Scholar * Marsh, G. A., Rabadan, R., Levine, A. J. & Palese, P.
Highly conserved regions of influenza a virus polymerase gene segments are critical for efficient viral RNA packaging. _J. Virol._ 82, 2295–2304 (2008). Article CAS PubMed Google Scholar
* Fournier, E. et al. A supramolecular assembly formed by influenza A virus genomic RNA segments. _Nucleic Acids Res._ 40, 2197–2209 (2012). Article CAS PubMed Google Scholar *
Gavazzi, C. et al. An in vitro network of intermolecular interactions between viral RNA segments of an avian H5N2 influenza A virus: comparison with a human H3N2 virus. _Nucleic Acids Res._
41, 1241–1254 (2013). Article CAS PubMed Google Scholar * Gog, J. R. et al. Codon conservation in the influenza A virus genome defines RNA packaging signals. _Nucleic Acids Res._ 35,
1897–1907 (2007). Article CAS PubMed PubMed Central Google Scholar * Moss, W. N., Priore, S. F. & Turner, D. H. Identification of potential conserved RNA secondary structure
throughout influenza A coding regions. _Rna_ 17, 991–1011 (2011). Article CAS PubMed PubMed Central Google Scholar * Liang, Y., Huang, T., Ly, H. & Parslow, T. G. Mutational
analyses of packaging signals in influenza virus PA, PB1, and PB2 genomic RNA segments. _J. Virol._ 82, 229–236 (2008). Article CAS PubMed Google Scholar * Muramoto, Y. et al. Hierarchy
among viral RNA (vRNA) segments in their role in vRNA incorporation into influenza A virions. _J. Virol._ 80, 2318–2325 (2006). Article CAS PubMed PubMed Central Google Scholar *
Wilkinson, K. A., Merino, E. J. & Weeks, K. M. Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE): quantitative RNA structure analysis at single nucleotide resolution.
_Nat. Protoc._ 1, 1610–1616 (2006). Article CAS PubMed Google Scholar * Pang, P. S., Elazar, M., Pham, E. A. & Glenn, J. S. Simplified RNA secondary structure mapping by automation
of SHAPE data analysis. _Nucleic Acids Res._ 39, e151 (2011). Article PubMed PubMed Central Google Scholar * Priore, S. F., Moss, W. N. & Turner, D. H. Influenza A virus coding
regions exhibit host-specific global ordered RNA structure. _PLoS ONE_ 7, e35989 (2012). Article CAS PubMed PubMed Central Google Scholar * Kladwang, W. & Das, R. A mutate-and-map
strategy for inferring base pairs in structured nucleic acids: proof of concept on a DNA/RNA helix. _Biochemistry_ 49, 7414–7416 (2010). Article CAS PubMed Google Scholar * Tian, S.,
Cordero, P., Kladwang, W. & Das, R. High-throughput mutate-map-rescue evaluates SHAPE-directed RNA structure and uncovers excited states. _RNA_ 20, 1815–1826 (2014). Article CAS PubMed
PubMed Central Google Scholar * Vester, B. & Wengel, J. LNA (locked nucleic acid): high-affinity targeting of complementary RNA and DNA. _Biochemistry_ 43, 13233–13241 (2004).
Article CAS PubMed Google Scholar * Kurreck, J., Wyszko, E., Gillen, C. & Erdmann, V. A. Design of antisense oligonucleotides stabilized by locked nucleic acids. _Nucleic Acids Res._
30, 1911–1918 (2002). Article CAS PubMed PubMed Central Google Scholar * Straarup, E. M. et al. Short locked nucleic acid antisense oligonucleotides potently reduce apolipoprotein B
mRNA and serum cholesterol in mice and non-human primates. _Nucleic Acids Res._ 38, 7100–7111 (2010). Article CAS PubMed PubMed Central Google Scholar * Staedel, C. et al. Inhibition of
gastric tumor cell growth using seed-targeting LNA as specific, long-lasting microRNA inhibitors. _Mol. Ther. Nucleic Acids_ 4, e246 (2015). Article CAS PubMed PubMed Central Google
Scholar * Javanbakht, H. et al. Liver-targeted anti-HBV single-stranded oligonucleotides with locked nucleic acid potently reduce HBV gene expression in vivo. _Mol. Ther. Nucleic Acids_ 11,
441–454 (2018). Article CAS PubMed PubMed Central Google Scholar * Hillebrand, F. et al. Gymnotic delivery of LNA mixmers targeting viral SREs induces HIV-1 mRNA degradation. _Int. J.
Mol. Sci._ 20, 1088 (2019). Article CAS PubMed Central Google Scholar * _Influenza Treatment_ (Centers for Disease Control and Prevention, National Center for Immunization and
Respiratory Diseases, 2019). * Rangan, R. et al. RNA genome conservation and secondary structure in SARS-CoV-2 and SARS-related viruses: a first look. _RNA_ 26, 937–959 (2020). Article CAS
PubMed PubMed Central Google Scholar * Sheahan, T.P., et al. An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 in human airway epithelial cell cultures and multiple
coronaviruses in mice. _Sci. Transl. Med._ 12, abb5883 (2020). * Wang, R. et al. Genetic screens identify host factors for SARS-CoV-2 and common cold coronaviruses. _Cell_ 184, 106–119.e114
(2021). Article CAS PubMed Google Scholar * _SARS-CoV-2 Variant Classifications and Definitions_ (Centers for Disease Control & Prevention, National Center for Immunization and
Respiratory Diseases, Division of Viral Diseases, 2022); https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-classifications.html * Bloom, J. D., Gong, L. I. & Baltimore, D.
Permissive secondary mutations enable the evolution of influenza oseltamivir resistance. _Science_ 328, 1272–1275 (2010). Article CAS PubMed PubMed Central Google Scholar * Takashita,
E. et al. Detection of influenza A (H3N2) viruses exhibiting reduced susceptibility to the novel cap-dependent endonuclease inhibitor baloxavir in Japan. _Eur. Surveill._ 24, 1800698 (2019).
Article Google Scholar * Hurt, A. C. et al. Characteristics of a widespread community cluster of H275Y oseltamivir-resistant A(H1N1)pdm09 influenza in Australia. _J. Infect. Dis._ 206,
148–157 (2012). Article CAS PubMed PubMed Central Google Scholar * Giannecchini, S., Clausi, V., Nosi, D. & Azzi, A. Oligonucleotides derived from the packaging signal at the 5′ end
of the viral PB2 segment specifically inhibit influenza virus in vitro. _Arch. Virol._ 154, 821–832 (2009). Article CAS PubMed Google Scholar * Hoffmann, E., Neumann, G., Kawaoka, Y.,
Hobom, G. & Webster, R. G. A DNA transfection system for generation of influenza A virus from eight plasmids. _Proc. Natl Acad. Sci. USA_ 97, 6108–6113 (2000). Article CAS PubMed
PubMed Central Google Scholar * Hou, Y. J. et al. SARS-CoV-2 reverse genetics reveals a variable infection gradient in the respiratory tract. _Cell_ 182, 429–446.e414 (2020). Article CAS
PubMed PubMed Central Google Scholar * Dinnon, K. H. 3rd et al. A mouse-adapted model of SARS-CoV-2 to test COVID-19 countermeasures. _Nature_ 590, E22 (2021). Article CAS PubMed
Google Scholar * Szretter, K. J., Balish, A. L. & Katz, J. M. Influenza: propagation, quantification, and storage. _Curr. Protoc. Microbiol._ CHAPTER 15, Unit 15G, 11 (2006). Google
Scholar * Marsh, G. A., Hatami, R. & Palese, P. Specific residues of the influenza A virus hemagglutinin viral RNA are important for efficient packaging into budding virions. _J.
Virol._ 81, 9727–9736 (2007). Article CAS PubMed PubMed Central Google Scholar * Kawakami, E. et al. Strand-specific real-time RT-PCR for distinguishing influenza vRNA, cRNA, and mRNA.
_J. Virol. Methods_ 173, 1–6 (2011). Article CAS PubMed Google Scholar * Mortimer, S. A. & Weeks, K. M. Time-resolved RNA SHAPE chemistry: quantitative RNA structure analysis in
one-second snapshots and at single-nucleotide resolution. _Nat. Protoc._ 4, 1413–1421 (2009). Article CAS PubMed PubMed Central Google Scholar * Akbari, A. et al. Improved DNA fragment
length estimation in capillary electrophoresis. _Electrophoresis_ 29, 1273–1285 (2008). Article CAS PubMed Google Scholar * Deigan, K. E., Li, T. W., Mathews, D. H. & Weeks, K. M.
Accurate SHAPE-directed RNA structure determination. _Proc. Natl Acad. Sci. USA_ 106, 97–102 (2009). Article CAS PubMed Google Scholar * De Rijk, P., Wuyts, J. & De Wachter, R.
RnaViz 2: an improved representation of RNA secondary structure. _Bioinformatics_ 19, 299–300 (2003). Article PubMed Google Scholar * Kladwang, W., VanLang, C. C., Cordero, P. & Das,
R. A two-dimensional mutate-and-map strategy for non-coding RNA structure. _Nat. Chem._ 3, 954–962 (2011). Article CAS PubMed PubMed Central Google Scholar * Kladwang, W., Cordero, P.
& Das, R. A mutate-and-map strategy accurately infers the base pairs of a 35-nucleotide model RNA. _RNA_ 17, 522–534 (2011). Article CAS PubMed PubMed Central Google Scholar *
Cordero, P., Kladwang, W., VanLang, C. C. & Das, R. The mutate-and-map protocol for inferring base pairs in structured RNA. _Methods Mol. Biol._ 1086, 53–77 (2014). Article CAS PubMed
PubMed Central Google Scholar * Mortimer, S. A. & Weeks, K. M. A fast-acting reagent for accurate analysis of RNA secondary and tertiary structure by SHAPE chemistry. _J. Am. Chem.
Soc._ 129, 4144–4145 (2007). Article CAS PubMed Google Scholar * Yoon, S. et al. HiTRACE: high-throughput robust analysis for capillary electrophoresis. _Bioinformatics_ 27, 1798–1805
(2011). Article CAS PubMed Google Scholar * Kim, H., Cordero, P., Das, R. & Yoon, S. HiTRACE-Web: an online tool for robust analysis of high-throughput capillary electrophoresis.
_Nucleic Acids Res._ 41, W492–W498 (2013). Article PubMed PubMed Central Google Scholar * Kim, J. et al. A robust peak detection method for RNA structure inference by high-throughput
contact mapping. _Bioinformatics_ 25, 1137–1144 (2009). Article CAS PubMed Google Scholar * Kladwang, W. et al. Standardization of RNA chemical mapping experiments. _Biochemistry_ 53,
3063–3065 (2014). Article CAS PubMed Google Scholar * Mathews, D. H. et al. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA
secondary structure. _Proc. Natl Acad. Sci. USA_ 101, 7287–7292 (2004). Article CAS PubMed PubMed Central Google Scholar * Darty, K., Denise, A. & Ponty, Y. VARNA: interactive
drawing and editing of the RNA secondary structure. _Bioinformatics_ 25, 1974–1975 (2009). Article CAS PubMed PubMed Central Google Scholar * Cordero, P., Lucks, J. B. & Das, R. An
RNA Mapping DataBase for curating RNA structure mapping experiments. _Bioinformatics_ 28, 3006–3008 (2012). Article CAS PubMed PubMed Central Google Scholar Download references
ACKNOWLEDGEMENTS We thank H. Jin and her team at MedImmune for the kind donation of chicken eggs and technical guidance at the start of the project. We thank the Stanford in vitro BSL-3
Service Center and its director J. Garhyan for assistance with the SARS-CoV-2 research. We thank additional members of R.D.’s lab, I. N. Zheludev, R. Rangan and H. Wayment-Steele, for
meaningful discussions of the SARS-CoV-2 structures and design of original SARS-CoV-2 RNA constructs for SHAPE experiments. The work was supported in part by a Mona M. Burgess Stanford BIO-X
Interdisciplinary Graduate Fellowship (to R.J.H.), the National Institutes of Health (NIH) Graduate Training Grant (no. 5T32AI007328-24 to R.J.H.), NIH Training Grant (no. 5T32DK007056 to
E.A.P.) and the following grants to J.S.G.: NIH research grants (nos. R56A1111460, U19A1109662, RO1AI132191 and U19AI171421), an influenza Harrington Scholar Innovator grant and grant no.
W81XWH1810647 from the US Army Medical Research Acquisition Activity, Department of Defense, Open Philanthropy and Good Ventures Fund, Fastgrants, a COVID-19 Harrington Scholar Innovator
Grant and the Dr. Tri Cao Nguyen Fund for Pandemic Preparedness. The work was also supported in part by the Intramural Research Program of the National Institute of Allergy and Infectious
Diseases, NIH. The data presented in this manuscript are tabulated in the main paper and the supplementary materials. Chemical-mapping datasets for M2 and M2R experiments have been deposited
at the RMDB (http://rmdb.stanford.edu). AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * ViRx@Stanford, Stanford Medicine, Stanford, CA, USA Rachel J. Hagey, Menashe Elazar, Edward A. Pham
& Jeffrey S. Glenn * Department of Microbiology & Immunology, Stanford University School of Medicine, Stanford, CA, USA Rachel J. Hagey & Jeffrey S. Glenn * Department of
Medicine, Division of Gastroenterology and Hepatology, Stanford University School of Medicine, Stanford, CA, USA Rachel J. Hagey, Menashe Elazar, Edward A. Pham, Lily Ben-Avi, Claire
Bernardin-Souibgui, Matthew F. Yee, Meirav Vilan Rabinovitch, Benjamin Fram, Grace Lam, Josephine Devera, Khanh Nguyen, Anming Xiong, Talia Avisar, Ping Liu, Phillip S. Pang & Jeffrey S.
Glenn * Department of Biochemistry, Stanford University, Stanford, CA, USA Siqi Tian, Wipapat Kladwang & Rhiju Das * Department of Epidemiology, University of North Carolina at Chapel
Hill, Chapel Hill, NC, USA Fernando R. Moreira, Rita M. Meganck & Timothy P. Sheahan * Division of Infectious Diseases and Geographic Medicine, Department of Medicine, Stanford
University, Stanford, CA, USA Aimee Beck, Arjun Rustagi & Catherine A. Blish * Institute for Antiviral Research, Department of Animal, Dairy, and Veterinary Sciences, Utah State
University, Logan, UT, USA Scott A. Gibson & Brett L. Hurst * Department of Medicine, Biomedical Informatics Research, Stanford University School of Medicine, Stanford, CA, USA Steven
Schaffert & Purvesh Khatri * Department of Internal Medicine, Division of Infectious Diseases, University of Cincinnati College of Medicine, Cincinnati, OH, USA Carl J. Fichtenbaum *
Department of Microbiology and Immunology, University of Texas Medical Branch, Galveston, TX, USA Chien-Te Tseng * Center for Biodefense and Emerging Diseases, Galveston National Laboratory,
University of Texas Medical Branch, Galveston, TX, USA Chien-Te Tseng * Viral Pathogenesis and Evolution Section, Laboratory of Infectious Diseases, National Institute of Allergy and
Infectious Diseases, National Institutes of Health, Bethesda, MD, USA Jeffery K. Taubenberger * Chan Zuckerberg Biohub, San Francisco, CA, USA Catherine A. Blish * Department of Physics,
Stanford University, Stanford, CA, USA Rhiju Das * Veterans Administration Medical Center, Palo Alto, CA, USA Jeffrey S. Glenn Authors * Rachel J. Hagey View author publications You can also
search for this author inPubMed Google Scholar * Menashe Elazar View author publications You can also search for this author inPubMed Google Scholar * Edward A. Pham View author
publications You can also search for this author inPubMed Google Scholar * Siqi Tian View author publications You can also search for this author inPubMed Google Scholar * Lily Ben-Avi View
author publications You can also search for this author inPubMed Google Scholar * Claire Bernardin-Souibgui View author publications You can also search for this author inPubMed Google
Scholar * Matthew F. Yee View author publications You can also search for this author inPubMed Google Scholar * Fernando R. Moreira View author publications You can also search for this
author inPubMed Google Scholar * Meirav Vilan Rabinovitch View author publications You can also search for this author inPubMed Google Scholar * Rita M. Meganck View author publications You
can also search for this author inPubMed Google Scholar * Benjamin Fram View author publications You can also search for this author inPubMed Google Scholar * Aimee Beck View author
publications You can also search for this author inPubMed Google Scholar * Scott A. Gibson View author publications You can also search for this author inPubMed Google Scholar * Grace Lam
View author publications You can also search for this author inPubMed Google Scholar * Josephine Devera View author publications You can also search for this author inPubMed Google Scholar *
Wipapat Kladwang View author publications You can also search for this author inPubMed Google Scholar * Khanh Nguyen View author publications You can also search for this author inPubMed
Google Scholar * Anming Xiong View author publications You can also search for this author inPubMed Google Scholar * Steven Schaffert View author publications You can also search for this
author inPubMed Google Scholar * Talia Avisar View author publications You can also search for this author inPubMed Google Scholar * Ping Liu View author publications You can also search for
this author inPubMed Google Scholar * Arjun Rustagi View author publications You can also search for this author inPubMed Google Scholar * Carl J. Fichtenbaum View author publications You
can also search for this author inPubMed Google Scholar * Phillip S. Pang View author publications You can also search for this author inPubMed Google Scholar * Purvesh Khatri View author
publications You can also search for this author inPubMed Google Scholar * Chien-Te Tseng View author publications You can also search for this author inPubMed Google Scholar * Jeffery K.
Taubenberger View author publications You can also search for this author inPubMed Google Scholar * Catherine A. Blish View author publications You can also search for this author inPubMed
Google Scholar * Brett L. Hurst View author publications You can also search for this author inPubMed Google Scholar * Timothy P. Sheahan View author publications You can also search for
this author inPubMed Google Scholar * Rhiju Das View author publications You can also search for this author inPubMed Google Scholar * Jeffrey S. Glenn View author publications You can also
search for this author inPubMed Google Scholar CONTRIBUTIONS R.J.H. and J.S.G. conceived and designed the study. R.J.H., M.E., L.B., M.R., C.B.S, T.P.S., F.R.M, R.M.M., A.B., K.N. and S.T.
performed the experiments. R.J.H., M.E., L.B., S.T., T.P.S. and C.B.S analyzed the data. M.E., L.B., M.Y. and T.A. assisted with mouse experiments. E.A.P. and R.J.H. conceived the idea of
targeting PSL2 and SARS-CoV-2-conserved RNA secondary structure using LNAs and designed antisense oligonucleotides. R.J.H, E.A.P. and M.E. designed LNA experiments and coordinated in vitro
and in vivo SARS-CoV-2 experiments. B.F. performed sequencing and assisted in experimental prep. M.Y. and E.A.P. designed the in vitro assessment of LNAs in myeloid cells and M.Y. performed
the experiments. J.D. and G.L. assisted in pharmacokinetics determination. P.L. assisted in cloning of IAV control constructs. W.K. provided important technical assistance on SHAPE
experiments. A.X. assisted with experimental prep. P.S.P. contributed to the initial conception of the study. T.P.S., F.R.M. and R.M.M. assisted with SARS-CoV-2 experiments. A.R. and C.A.B.
established the BSL-3 screening. A.R. performed the screening. C.J.F. isolated and characterized the SARS-CoV-2 clinical variant. B.L.H. managed the SARS-CoV-2 hamster study. S.A.G.
performed the virus titrations. S.S. and P.K. provided bioinformatics and statistical analyses. C.T. was instrumental in setting up in vivo SARS-CoV-2 assays and conditions. J.K.T. offered
valuable expertise and coordinates before publication, discussed results and provided critical reagents. R.D. and J.S.G. provided experimental feedback and assisted in the analysis of the
results. R.J.H., J.S.G., M.E., E.A.P, S.T. and R.D. wrote the paper. CORRESPONDING AUTHOR Correspondence to Jeffrey S. Glenn. ETHICS DECLARATIONS COMPETING INTERESTS J.S.G., R.J.H., E.P. and
M.E. are inventors on patents pertaining to the materials presented in this article that has been filed with the US Patent and Trademark Office by Stanford University. PEER REVIEW PEER
REVIEW INFORMATION _Nature Medicine_ thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: Alison Farrell, in collaboration with the
_Nature Medicine_ team. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION Supplementary Figs. 1–22 and Tables 1–3. REPORTING SUMMARY SUPPLEMENTARY DATA 1 Supplementary Fig. 10 Source data, full blot.
SUPPLEMENTARY DATA 2 Supplementary Fig. 11a Source data, full blot. SUPPLEMENTARY DATA 3 Supplementary Fig. 14a Source data, full blot. SUPPLEMENTARY DATA 4 Supplementary Fig. 18 Source
data, gating strategy. SOURCE DATA SOURCE DATA FIG. 1 IAV SHAPE reactivities for Fig. 1 and supplementary figures. SOURCE DATA FIG. 6A,B SARS-CoV-2 SHAPE reactivity. RIGHTS AND PERMISSIONS
Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Hagey, R.J., Elazar, M., Pham, E.A. _et al._ Programmable antivirals targeting critical conserved viral RNA secondary structures
from influenza A virus and SARS-CoV-2. _Nat Med_ 28, 1944–1955 (2022). https://doi.org/10.1038/s41591-022-01908-x Download citation * Received: 21 March 2017 * Accepted: 20 June 2022 *
Published: 18 August 2022 * Issue Date: September 2022 * DOI: https://doi.org/10.1038/s41591-022-01908-x SHARE THIS ARTICLE Anyone you share the following link with will be able to read this
content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative