Play all audios:
ABSTRACT Pathogen genome sequencing during epidemics enhances our ability to identify and understand suspected clusters and investigate their relationships. Here, we combine genomic and
epidemiological data of the 2022 mpox outbreak to better understand early viral spread, diversification and transmission dynamics. By sequencing 52% of the confirmed cases in Portugal, we
identified the mpox virus sublineages with the highest impact on case numbers and fitted them into a global context, finding evidence that several international sublineages probably emerged
or spread early in Portugal. We estimated a 62% infection reporting rate and that 1.3% of the population of men who have sex with men in Portugal were infected. We infer the critical role
played by sexual networks and superspreader gatherings, such as sauna attendance, in the dissemination of mpox virus. Overall, our findings highlight genomic epidemiology as a tool for the
real-time monitoring and control of mpox epidemics, and can guide future vaccine policy in a highly susceptible population. SIMILAR CONTENT BEING VIEWED BY OTHERS GENOMIC EPIDEMIOLOGY
REVEALS TRANSMISSION PATTERNS AND DYNAMICS OF SARS-COV-2 IN AOTEAROA NEW ZEALAND Article Open access 11 December 2020 EPIDEMIOLOGICAL AND GENOMIC EVOLUTION OF THE ONGOING OUTBREAK OF CLADE
IB MPOX VIRUS IN THE EASTERN DEMOCRATIC REPUBLIC OF THE CONGO Article Open access 11 February 2025 RAPID INCIDENCE ESTIMATION FROM SARS-COV-2 GENOMES REVEALS DECREASED CASE DETECTION IN
EUROPE DURING SUMMER 2020 Article Open access 14 October 2021 MAIN Mpox is a viral zoonosis caused by mpox virus (MPXV), a member of the genus Orthopoxvirus that also includes variola virus
(which causes smallpox), vaccinia virus, camelpox virus and cowpox virus, all of which are pathogenic to humans1,2,3,4,5. MPXV was discovered in 1958, when a non-lethal rash disease broke
out in captive cynomolgus monkeys in Copenhagen, Denmark6. The first case in humans was reported in 1970 in the Democratic Republic of Congo, where a 9-month-old infant presented with what
appeared to be an early-stage smallpox rash7. MPXV infection is often caused by spill-over events from animals (such as small rodents and non-human primates) to humans1,2,3,4,5. However,
mpox can also be transmitted from person to person through direct contact with lesions, body fluids and respiratory secretions, or contact with contaminated material1,4,8. Mpox is endemic in
West and Central Africa, where several outbreaks have occurred in recent decades1,2,3,4,9,10,11,12. Nevertheless, until 2022, only small clusters or sporadic cases of mpox had been recorded
outside of endemic regions. These cases were generally travel-related and linked to countries with endemic mpox1,2,3,4,12,13,14,15,16,17 or related to imported small mammals18, but had
limited subsequent human-to-human sustained transmission. Presently, a large multi-country mpox outbreak is ongoing worldwide. The first cases were reported in May 2022, with 88,600
laboratory-confirmed cases and 152 deaths being reported in 113 member states across all six World Health Organization (WHO) regions by 2 August 2023 (ref. 19). On 23 July 2022, the WHO
Director-General declared this outbreak a public health emergency of international concern20. The outbreak has disproportionately affected men who have sex with men (MSM), and MSM with mpox
frequently had skin lesions in the anogenital area, suggesting the amplification of transmission through sexual networks21,22,23. MPXV genomes from the 2022 outbreak belong to clade IIb24,25
according to the recently proposed nomenclature25. This clade, within the formerly designated ‘West African’ clade, is characterized by less severe symptoms and a lower lethality rate than
clade I (formerly ‘Congo Basin’ clade)25,26. Additionally, as clade IIb viruses have shown sustained human-to-human transmission, a new subclade was designated ‘hMPXV1’ (ref. 25,27). Within
this subclade, a hierarchy of lineages (starting with ‘A’) similar to SARS-CoV-2 lineage labels was proposed. The MPXV causing the current outbreak was classified as lineage B.1, which has
been further subdivided into sublineages B.1.1, B.1.2, and so on (https://github.com/mpxv-lineages)25. The first comparative genomic analyses of these genomes revealed that the MPXV causing
the 2022 multi-country outbreak shares a recent common ancestor with MPXV sequences connected to Nigeria (A.1 lineage)24, where a large outbreak occurred in 2017 and 2018 (ref. 12,16,28).
More recently, other hMPXV1 lineages (for example, A.2.1, A.2.2, A.2.3 or A.3) have been sporadically detected in countries without endemic mpox from a wide latitude (for example, the United
States, Vietnam, Egypt and the United Kingdom), supporting the aforementioned link to the epidemic in Nigeria and that other human-to-human transmission chains are probably ongoing besides
the recognized large B.1 outbreak27,29. However, the number of sequences reported throughout the years (until 2022) is still very limited, hindering the establishment of precise origins,
evolutionary routes and clone dissemination. The sequences collected during the 2022 outbreak diverge by around 50 single-nucleotide polymorphisms (SNPs) from pre-outbreak sequences24. This
indicates an unexpectedly high substitution rate for the slow-evolving double-stranded DNA Orthopoxvirus. In-depth mutational analysis suggested the action of host APOBEC3 (apolipoprotein B
mRNA-editing catalytic polypeptide-like 3) enzymes in viral evolution (owing to the mutating bias of these SNPs: GA>AA and TC>TT base substitutions) and enabled us to estimate that the
introduction of hMPXV1 into the human population occurred around 2016, probably followed by ‘silent’ human-to-human transmission27. Signs of potential MPXV–human adaptation in the
microevolution of the current outbreak were also investigated early on24,30,31, but the adaptive value of the APOBEC3 hypermutation in the long term is still uncertain27. Since the beginning
of the 2022 outbreak, and mirroring the open science practices observed in the case of SARS-CoV-2, researchers from several countries immediately started to share MPXV genomes in public
databases (for example, GISAID, GenBank), and existing platforms (such as Nextstrain (https://nextstrain.org/monkeypox/hmpxv1) and GenSpectrum (https://mpox.genspectrum.org/)) were rapidly
adapted. This collaborative environment facilitated the execution of phylogenetics-based spatiotemporal studies and the surveillance of the genetic variability of MPXV by tracking its spread
and assessing evolutionary traits potentially linked to its adaptation to the human host or immune evasion24,27,30,31,32. Since the first confirmed cases in Portugal on 17 May 2022 (ref.
24,33), the country has embarked on a major effort of to sequence the MPXV and share data, and has been able to obtain the genome sequences for more than 50% of all reported mpox cases in
the country, as of January 2023. In addition, in Portugal, MPXV infection was included in the National Epidemiological Surveillance System (SINAVE), which gathers demographic, clinical,
epidemiological and laboratory data of all mandatory notifiable diseases. Nevertheless, even with comprehensive epidemiological surveillance systems and wide availability of diagnostics
tests, how transmission occurs remains uncertain. Mathematical models are particularly useful to understand partly observed infectious disease phenomena34. In particular, epidemic models
like the SEIR (Susceptible-Exposed-Infectious-Recovered) model divide the population into different groups based on infection status and can be used to estimate time-dependent parameters
such as the time-varying reproduction number (_R__t_) and the expected number of susceptible, infected and recovered cases throughout the time series35. To date, the early transmission
dynamics of the international MPXV outbreak have not been well characterized. Here, we combined epidemiological and genomic data to better understand the transmission dynamics of MPXV,
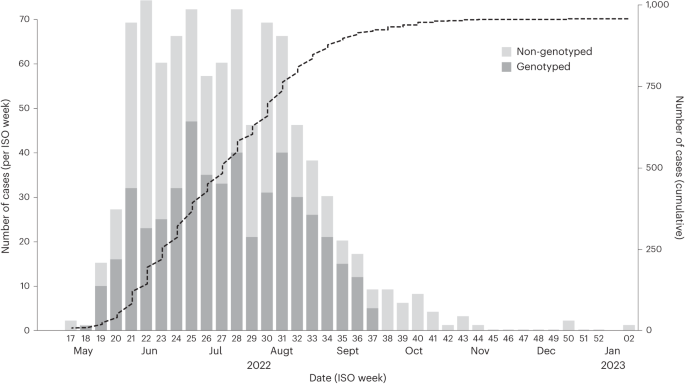
providing evidence that can help to tailor public health messages and guide vaccine policy. RESULTS CASES AND SEQUENCE SAMPLING The first mpox cases in Portugal were laboratory-confirmed on
17 May 2022, and the epidemic peaked around 17 July 2022, followed by a decreasing trend in the number of cases. A total of 951 cases were confirmed in Portugal, as of 10 January 2023 (Fig.
1). To understand the genomic epidemiology of MPXV in Portugal during the 2022 multi-country outbreak, viral genome sequences were obtained from 495 individuals with a PCR-positive test. The
dates of sample collection of the 495 genotyped cases spanned from 4 May (ISO week 18) to 16 September (ISO week 37) 2022, representing a large sequence sampling of 54.2% (495 out of 914)
of all confirmed mpox cases in Portugal during this period (corresponding to 52.1% of total confirmed cases, as of 10 January 2023) (Fig. 1). Demographic variables of sex and age were
available for 447 out of the 495 studied mpox cases. Most of these individuals (99.1%; 443 out of 447) identified as male. A majority were in the age bracket 30–39 years (44.1%; 197 out of
447), followed by the age brackets 20–29 years (28.4%; 127 out of 447) and 40–49 years (20.4%; 91 out of 447). Human immunodeficiency virus (HIV) infection status was reported as positive in
42.9% of individuals with mpox who provided their status (168 out of 392). Among males who reported their sexual orientation (_n_ = 344), 96.5% (332 out of 344) self-identified as MSM. To
better understand the introduction and transmission dynamics of MPXV in Portugal, epidemiological data were collected with particular focus on travel history, exposure settings and
transmission routes that could be complemented by the available genotyping data (Supplementary Table 1). TRANSMISSION DYNAMICS To understand the transmission intensity and estimate key
epidemiological parameters of this outbreak in Portugal, we used case incidence data to fit a time-discrete SEIR model (Fig. 2). We observed an estimated _R__t_ of 2.25 (95% credible
interval (CrI), 1.59–2.95) at the beginning of the time series (earliest reported onset of symptoms, 22 April 2022), increasing to a maximum of 2.70 (95% CrI, 2.11–3.30) on 10 May 2022 (Fig.
2a). We estimated a subsequent decline in _R__t_, falling below 1 at the end of June 2022. The model accounted for travel history and was able to reconstruct well the true number of cases
(Fig. 2b), with an estimated reporting rate of 0.62 (95% CrI, 0.43–0.83). We also estimated the proportions of the MSM population in Portugal that were susceptible (Fig. 2c), infected (Fig.
2d) and recovered (Fig. 2e). The model calculated that 1.3% (_n_ = 1,370; 95% CrI, 818–2,228) of MSM were infected during this period, with 98.7% (95% CrI, 98.1–99.0) of the MSM population
still susceptible to infection at the end of this time series. The model estimated the epidemic peak, defined as the highest daily incidence of confirmed cases, to occur on 8 June 2022 with
15 infections (95% CrI, 10–24), before faltering slightly and subsequently declining rapidly from the end of July 2022. In parallel, we conducted a sensitivity analysis with no travel
history, across different infectious periods of 14, 21 and 28 days (\(\sigma\)) ∈ {0.071, 0.048, 0.036} and with different infection seeds and found largely consistent model estimates
(Supplementary Figs. 1, 2 and 3). IDENTIFICATION OF SUBCLUSTERS Of the 495 MPXV genome sequences from Portugal analyzed in the present study, 494 belonged to the B.1 lineage (and
sublineages) associated with the large multi-country outbreak. One sequence (PT0428; date of collection, 1 August 2022) belonged to sublineage A.2.3 (not linked to the large outbreak), thus
representing an independent introduction of the virus into the country. The individual to whom these data relate self-identifies as MSM, and reported travel history during the incubation
period. Although the individual is from a country endemic for mpox in the West Africa region, local health authorities were unable to determine the specific countries visited by the
individual during the relevant period. As of 2 November 2022, the publicly available Nextstrain hMPXV1 phylogenetic tree
(https://nextstrain.org/groups/neherlab/PT-MPXV-transmission/2022-11-02) included 2,714 outbreak sequences (18.2% from Portugal) from 28 countries across Europe, Asia, Africa, Oceania, South
America and North America (Supplementary Table 1). Of the 2,714 outbreak sequences, 377 were phylogenetically placed at the outbreak basal level (that is, identical consensus sequences with
no extra mutations). This considerably high number of ‘root’ sequences was not unexpected because viral dissemination at the global level might have occurred very rapidly, probably
triggered by multiple superspreader events, and led to a rapid clonal expansion with less opportunity for mutation diversification. Most of the sequences, 2,126 (78.3%), belonged to genetic
subclusters, defined as subbranches with at least two sequences diverging from the 2022 outbreak basal level by at least one SNP. This subcluster definition (that is, one SNP above the
outbreak ‘root’) is aligned with criteria being applied to designate international MPXV sublineages according to the nomenclature proposed in https://github.com/mpxv-lineages. A total of 182
genetic subclusters were identified in the global tree, with 52 of them (78.3%) including at least two Portuguese sequences (hereafter called ‘Portuguese subclusters’) (Supplementary Table
2). The Portuguese subclusters comprised 66.6% (329 out of 494) of sequences from Portugal, meaning that around two-thirds of the mpox cases could be, at least phylogenetically, linked to
another case(s) detected in Portugal. In total, 96 out of the 494 Portuguese sequences (19.4%) were phylogenetically placed at the outbreak basal level. The remaining 69 sequences (14.0%)
formed ‘singleton’ branches (_n_ = 49) or were integrated within international clusters with a single Portuguese sequence (_n_ = 20) (Supplementary Tables 1 and 2). SIZE, TIMESPAN AND
INTERNATIONAL ‘LINKAGE’ OF PORTUGUESE SUBCLUSTERS The Portuguese subclusters showed considerable diversity in terms of size, timespan and inclusion of sequences from other countries (Fig.
3a). Seven Portuguese subclusters contained at least ten sequences from Portugal, with cluster 172 (corresponding to the internationally designated sublineage B.1.9) including the highest
number of Portuguese sequences (41 Portuguese and 3 international sequences). In particular, 25 out of the 52 Portuguese subclusters (48.1%) included international sequences (Fig. 3a and
Supplementary Table 2). The observation that 27 Portuguese subclusters (51.9%) exclusively included sequences from Portugal should be interpreted with caution because of the high
discrepancies in sequence sampling between countries. It also suggests that, as expected, some ‘sublineages’ (that is, genetic subclusters) had a more restricted circulation in Portugal,
while others showed considerable international spread. Among the latter, we highlight the considerable circulation in Portugal of the internationally designated sublineages B.1.1 (_n_ = 15),
B.1.5 (_n_ = 16) and B.1.7 (_n_ = 25), which are among the five largest Portuguese subclusters (Fig. 3a and Supplementary Table 2). Nonetheless, in contrast to sublineage B.1.9, with 93% of
sequences from Portugal, all of these international lineages included less than 30% Portuguese sequences, as of 2 November 2022 (Fig. 3a and Supplementary Tables 1 and 2). Other
internationally designated sublineages detected in Portugal were B.1.2, B.1.3, B.1.8, B.1.11 and B.1.14, and these included even less Portuguese sequences (each with less than ten Portuguese
sequences and/or with Portuguese sequences representing less than 10% of the cluster size). For instance, the largest cluster at the global level, corresponding to sublineage B.1.2,
included 214 sequences, with only four sequences (1.9%) being from Portugal (Fig. 3a and Supplementary Tables 1 and 2). In addition, we assessed the timespan of the subclusters, that is, the
time (in days) between the earliest and latest detection of a given sublineage at national and international levels (Fig. 3a,b and Supplementary Table 2). At the international level, as of
2 November 2022, the largest time interval (172 days) was observed for sublineage B.1.3, which has been detected in 13 countries since 28 April 2022 (Fig. 3b). It is noteworthy that the
earliest occurrence of several Portuguese subclusters that involved international sequences was observed in a Portuguese case, suggesting that the emergence of some outbreak sublineages (or,
at least, the early dissemination of some sublineages at a global level) occurred in Portugal (Fig. 3a). At the national level, 24 out of the 52 Portuguese subclusters included mpox cases
detected in a time interval equal to or above 30 days (Fig. 3a and Supplementary Table 2). The largest time interval between two Portuguese cases of the same cluster was observed for cluster
174 (corresponding to the international sublineage B.1.5), with a total of 109 days (between ISO weeks 21 and 36, 2022) (Fig. 3b). Given the estimated substitution rate of the
outbreak-causing MPXV27 and its spread through sexual networks1,22, it is worth noting that a genetic subcluster does not necessarily represent a sustained transmission chain. To address
this challenge of detecting multiple introductions, we assessed whether individuals within the same subcluster reported travel history (within the incubation period of MPXV) to different
countries and then assessed whether those countries were represented by sequences in the same subcluster. Travel history was reported for 85 out of 494 cases (17.2%), with 43 of them
belonging to subclusters with international sequences (Supplementary Tables 1 and 2). The names of travel-related countries (that is, countries from the travel history inquiry, known for 84
out of 85 cases) were reported by at least one case for 25 Portuguese subclusters (17 having international sequences) and 6 international subclusters with a single Portuguese sequence. Not
unexpectedly, for 14 Portuguese subclusters, more than one country of travel was reported by different individuals whose sequences were in the same subcluster, thus providing strong evidence
for more than one introduction of those 14 sublineages into the country (Fig. 3c and Supplementary Table 2). For instance, this scenario was identified for the four internationally
designated sublineages showing the highest circulation in Portugal (B.1.1, B.1.5, B.1.7 and B.1.9). Most notably, for 11 subclusters, we found at least one match between a travel-related
country and a country with sequences in those subclusters, with that country being the most or second most represented in terms of the number of sequences for seven subclusters (Fig. 3c and
Supplementary Table 2). In addition to the inclusion of travel history, epidemiological data on sexual contact with tourists, travelers or visitors was reported in 101 out of 494 cases (20%)
(Supplementary Table 1) and could indirectly inform about potential introductions or reintroductions of the virus into Portugal. Most of these cases, 50 out of 101 (49.5%), belonged to
subclusters with international sequences, namely 20 Portuguese subclusters and 6 international subclusters with a single Portuguese sequence. The name of the tourism-related country (that
is, countries linked to sexual contacts with tourists, travelers or visitors) was specified by 50 individuals. In total, sexual contact with tourists from a known country was confirmed by at
least one Portuguese individual for 21 subclusters (13 with international sequences). Similarly to the scenario observed after the inclusion of travel history data, we found subclusters
with more than one tourism-related country (reported by different individuals) in the same cluster (potentially reflecting additional independent introductions), as well as subclusters in
which a given tourism-related country matched a country with sequences in that cluster (Fig. 3c and Supplementary Table 2). TRANSMISSION ROUTES AND EXPOSURE CONTEXT OF PORTUGUESE SUBCLUSTERS
Detailed epidemiological data on the most probable transmission routes and on possible exposure in the 21 days before symptom onset were available for most of the studied cases (399 and 318
out of the 494 cases, respectively) (Supplementary Table 1). Sexual contact was reported as a possible route of transmission in 95.2% (380 out of 399) of cases, followed by non-sexual
person-to-person transmission (3.3%; 13 out of 399), healthcare-associated transmission (0.5%; 2 out of 399) and ‘other’ (1.0%; 4 out of 399). Regarding the exposure context, most cases
reported exposure at events involving sexual contacts, either small (such as private party or club, or sauna) (49.7%; 158 out of 318) or large (such as festivals) (2.8%; 9 out of 138).
Household exposure was reported by 84 out of 318 cases (26.4%), 79 of which reported sexual transmission. Additional exposure contexts included small (3.5%) and large (1.6%) events without
sexual contacts, workplace (0.9%), healthcare services (0.6%) and ‘other’ (14.1%) (Supplementary Table 1). Within each Portuguese subcluster, small events involving sexual contacts were
reported as the most frequent exposure context in 34 subclusters, followed by household exposure (most frequently reported in ten small subclusters with two to seven cases per subcluster)
(Fig. 3c and Supplementary Table 2). The in-depth epidemiological investigation also recorded sauna attendance in the 21 days before symptom onset for 76 out of the 494 studied individuals.
Notably, 71 out of the 76 cases reporting ‘sauna’ (93%) were phylogenetically placed at the outbreak basal level (‘root’) (_n_ = 22) or were part of subclusters (_n_ = 49), with 33 belonging
to subclusters with international sequences (Supplementary Table 1). In total, 23 out of the 52 Portuguese subclusters included at least one case reporting ‘sauna’ (Fig. 3c and
Supplementary Table 2). In particular, sauna attendance was reported in at least one case for the top six subclusters in terms of the number of Portuguese sequences. We could trace back the
attendance of 28 of the 76 individuals who reported sauna attendance to one particular sauna (‘sauna1’) located in the Lisbon and Tagus Valley geographic region. These cases were found
across 11 Portuguese subclusters, eight of which included international sequences (Fig. 3c). Sexual contacts with multiple or anonymous individuals were also reported by 283 out of 494 cases
(57.3%), with nine subclusters exclusively involving cases that reported this variable (Fig. 3c and Supplementary Table 2). DISCUSSION The estimated substitution rate of the MPXV that
caused the 2022 outbreak (around six substitutions per genome per year)27 and its transmission characteristics, associated with sexual networks and superspreader events providing the
opportunity for rapid outbreak dissemination21,22,23, challenge the identification and tracking of singular transmission chains, as well as the distinction between continuous within-country
transmission from multiple introductions. Still, our transmission model and genetic clustering analysis with extensive epidemiological data integration provide evidence regarding the
transmission dynamics of MPXV. Despite the complexity of identifying the epidemiological parameters of MPXV in this outbreak, we fitted an SEIR model accounting for travel history and
obtained a reasonable fit. Owing to the long infectious period of MPXV2, we expected that _R__t_ would fall below 1 before cases start declining, as observed in this outbreak. The fast
decline of the _R__t_ in Portugal can be explained by three main factors: contact tracing procedures, behavioral changes and vaccination. Contact tracing procedures were implemented as early
as May 2022, when the first cases were reported. Intimate contacts and sexual partners of cases were identified, when possible, and were advised to stay in isolation for the maximum
duration of the incubation period. During the pre-smallpox-vaccination period, in June 2022, the public health strategy focused on mobilizing the MSM population for prevention and control
measures and on identifying groups with high risk of MPXV transmission, particularly those who would have a high number of sexual partners. This particular group of the MSM population might
have played a key role in viral transmission in this outbreak36,37, as supported by the present study. Targeted public health messages to the MSM population and community stakeholders
working in the field of sexually transmitted diseases (STDs) probably led to behavior changes, as seen by the rapid decrease of the _R__t_. Following the approval of the smallpox vaccine for
emergency use in the mpox outbreak, ring vaccination started in Portugal in July 2022, consolidating the decrease in transmission risk. The difference between the estimated and the observed
epidemic peaks probably translates to a lag of reporting. One of the unique features of this outbreak is that most cases self-identify as MSM. We estimate that only 1.3% of the MSM
population in Portugal38 was infected with the virus, leaving an almost entirely susceptible MSM population with the potential for future epidemics. This could be corroborated by a targeted
seroprevalence study in Portugal, as well as in other high incidence-reporting countries. Recent studies have suggested the role of both cryptic and asymptomatic transmission, which could
explain, partly, the proportion of cases not observed and not reported33,39,40,41. This model emphasizes the value of transmission models, which enables us to estimate the true underlying
size of the epidemic, accounting for reporting probabilities as well as susceptible population proportions. Our transmission model has potential limitations in the assumptions adopted, which
are important to consider when interpreting the results. Firstly, we assumed a homogeneous mixing of the entire MSM population living in Portugal, and we did not include network data among
the MSM population because of the incompleteness of these data in the inquiries. Secondly, we relied on laboratory-confirmed case data as an indicator of epidemic size and used travel
history as an approximation for the imported cases. Lastly, the model does not account for the possibility that some exposed individuals are perhaps less susceptible to the MPXV (for
example, smallpox-vaccinated individuals), and the low number of vaccinated individuals during the period of this time series (less than 500) hampered the inclusion of the effect of
vaccination on these dynamics. Regarding the genomic epidemiological analysis, we were able to identify the B.1 sublineages (that is, genetic subclusters) with the highest impact on the mpox
epidemic in Portugal and assess whether their circulation seemed to be higher in Portugal (owing to the lack of international sequences) or revealed multi-country dissemination (Fig. 3).
Moreover, the inclusion of travel data, as well as data on sexual contacts with tourists, travelers or visitors, complemented the phylogenetic data to support the existence of multiple
introductions of the same sublineage, even though different scenarios were observed. For example, sublineage B.1.9, which had intense circulation in Portugal since the beginning of the
epidemic (Fig. 3), was found to have been reintroduced at a later stage after the epidemic peak. By contrast, other sublineages seemed to be introduced multiple times throughout the
epidemic, although each introduction probably seeded limited transmission chains (for example, B.1.5 and B.1.7). Supporting the reliability of the disclosed international linkage, we found a
link between travel-related or tourism-related countries and the countries with (a high proportion of) sequences in those subclusters. For instance, we found one travel-related introduction
of sub-lineage B.1.2 from the United States, and this country represented around half (105 out of 214) of all B.1.2 sequences reported worldwide during the study period. In another example,
Germany was the most frequently represented country (114 out of 161 sequences) within cluster 181 (corresponding to sublineage B.1.1), which was consistent with the travel history reported
by one Portuguese case linked to that cluster. Still, it is noteworthy that travel history data also allowed the identification of other potential independent B.1.1 introductions from
Brazil, Canada and Italy, which corroborates the complexity of such inferences, even with the vast epidemiological data and high sequence sampling rates (Supplementary Tables 1 and 2). Also,
in a few instances, the lack of matches could have been underestimated because of the low number of sequences available in GenBank from some countries. For example, several cases across
different subclusters reported travel history to Spain and Brazil, but only a few matches were found. Attendance at small venues involving sexual contacts, namely saunas (in particular
‘sauna1’), was found to be among the most reported exposure contexts within the studied population, which is consistent with previous reports1,21,22,32,42,43. We cannot directly point to
saunas as the main triggers for the extensive dissemination of MPXV, as it is not possible to ascertain whether these locations were the actual settings of transmission for all of these
cases. However, our study clearly supports the important role of such potential superspreader events in outbreak dissemination21,22,38, as these cases were frequently integrated into large
subclusters (for example, they were reported for the six largest subclusters) and were commonly found across subclusters with international sequences (for example, almost half of the cases
reporting sauna attendance were linked to these subclusters). From another perspective, the potential epidemiological linkage of a particular setting (for example, ‘sauna1’) to several
subclusters clearly shows the difficulties of untying the complex transmission networks associated with this multi-country outbreak. These challenges are highlighted not only by the
considerably high number of cases reporting sexual contact with multiple or anonymous individuals, but also because almost all subclusters (49 out of 52) involved cases attending different
hospitals or STD clinics. For instance, each of the five largest subclusters involved cases from at least 11 different hospitals or STD clinics, with potential negative impact on
epidemiological data collection, despite the existence of standardized case investigation forms. Furthermore, these challenges cannot be disconnected from the intrinsic subjectivity
associated with the interviews, which are dependent on both recall bias and the participants’ willingness to provide the requested information, considering the exposure context (sexual
networks). Future efforts to develop methods to improve the collection of this information while protecting individuals’ privacy are warranted to increase the added value of genomic
epidemiology. This is well reflected by the fact that the epidemiological inquiries were able to collect information about person-to-person contact for only 13 pairs of individuals among the
494 studied cases. This information is pivotal to complement the genomic data to track transmission. Indeed, the phylogenetic data clearly discard a direct or indirect epidemiological
connection between the cases only for two out of the 13 pairs (Supplementary Table 1). This concordance (that is, most known contacts were confirmed in the same genetic subclusters) suggests
that our clustering analysis might provide a linkage between most cases within the same cluster. We are aware that because of the large size of the dataset and MPXV genome, occasional
sequence or phylogenetic artifacts (for example, reversions, spurious mutations or false homoplasies) might insert inconsistencies into the clustering. Still, our conservative clustering
approach and inspection of the Portuguese data suggests that this effect may be minimal, reinforcing the presented global clustering scenario, which would be difficult to uncover with only
epidemiological data. An unexpected finding was the detection of a non-outbreak-related sequence (PT0428) from sublineage A.2.3. This case, also self-identified as MSM, could be linked to
the West Africa region and parallels other recent detections of non-B.1 lineages across the world27. By unveiling concurrent human-to-human transmission and broad dissemination of multiple
hMPXV-1 lineages (outside the B.1 outbreak), these recent findings corroborate the idea that the historical paradigm of MPXV ecology, evolution and epidemiology has changed, posing new
challenges for the prevention and control of mpox24,27,29,30. By taking advantage of a large sequence sampling and the collection of vast epidemiological data, we shed light on the
introductions and transmission dynamics of MPXV in Portugal. As one of the countries that reported the first cases and, similarly to the United Kingdom42 and Spain38, one of the most
affected countries during the early stages of the 2022 outbreak24,33, it is very likely that Portugal’s epidemics played an important role in the early and widespread dissemination of MPXV
worldwide. This is supported by our study, as the emergence of some outbreak sublineages, or their early global dissemination, most probably occurred in Portugal. Our study also estimates
that only 62% of the true case incidence was observed and that 1.3% of the MSM population was infected during this period, leaving the possibility for similar epidemics to occur. We also
consolidated the key role of events or venues with superspreader potential and sexual networks in MPXV transmission and identified the sublineages with the highest impact on outbreak spread
in Portugal, while emphasizing their spatiotemporal landscape and international linkage. Our study is an integrative genomic epidemiology analysis in the context of the 2022 worldwide MPXV
outbreak. Despite lacking individual network contact data to associate the corresponding genetic subclusters and transmission chains, our combined approach may prove to be crucial to
untangle and respond to novel and emerging viral threats, even in contexts as complex as the one faced during the 2022 multi-country mpox outbreak. This study leverages evidence about mpox
transmission that can support and guide future public health interventions, including vaccine strategies. METHODS This research complies with all relevant ethical regulations. The planning,
conduct and reporting of this study was in accordance with the Declaration of Helsinki, as revised in 2013. Ethical approval for the use of surveillance data was not required due to the
National Health Authority (Directorate-General of Health) permit to access and use surveillance data for communicable disease outbreak investigations in the public interest. At laboratory
level, the Portuguese National Institute of Health (INSA) is the national reference laboratory, being the Portuguese laboratory authorized by the Directorate-General of Health (through the
Technical orientation no. 004/2022 of 31 May 2022) to process the samples for identification and genetic characterization of MPXV. All samples subjected to viral genetic characterization
were processed in an anonymized fashion. This study was approved by the INSA’s ethical committee ‘Comissão de Ética para Saúde’. EPIDEMIOLOGICAL DATA COLLECTION Demographic and
epidemiological data were collected by the Directorate-General of Health, through SINAVE and the main STD clinics. Data were collected by the attending physician by conducting both
face-to-face and phone interviews using the surveillance-standardized case investigation form. Demographic variables that were included were sex and age. Risk practice variables included
travel history, contact with a confirmed case, self-identifying as MSM, number of sexual partners, having anonymous and/or multiple sexual partners, engaging in sexual activities with
tourists and attending sex venues (sauna, or public or private parties). Countries reported in the travel history inquiry or linked to sexual contacts with tourists, travelers or visitors
were referred to as ‘travel-related’ or ‘tourism-related’ countries throughout the manuscript, respectively. Details are presented in Supplementary Table 1. STATISTICAL ANALYSES TRANSMISSION
DYNAMICS We fitted a discrete-time SEIR model to the reported case data using a daily time step. We assumed an incubation period of 5.6 days (ref. 44) and an infectious period of 21 days
(ref. 2,3). For the number of susceptible cases, we used the estimates for the MSM Portuguese population45. The population was assumed to be completely susceptible, and the model was seeded
with ten infections at the beginning of the case time series. We estimated a time-varying reproduction number \(R(t)\) as a random walk function with a fixed standard deviation of 0.1. The
rates of change of each compartment are described in equations (1–4). Here, S, E, I and R represent the susceptible, exposed, infectious and recovered proportions of the population.
Additionally, \(\alpha\) is the rate of onset of infectiousness, \(\sigma\) is the rate of recovery from infection and \(\beta \left(t\right)\) is the time-varying transmission rate,
calculated as \(\sigma R(t)\). $$\frac{{dS}}{{dt}}=-\beta (t)I\left(t\right)S(t)$$ (1) $$\frac{{dE}}{{dt}}=\beta I\left(t\right)S\left(t\right)-\,\alpha E(t)$$ (2)
$$\frac{{dI}}{{dt}}=\,\alpha E\left(t\right)-\,\sigma I(t)$$ (3) $$\frac{{dR}}{{dt}}=\,\sigma I(t)$$ (4) The expected number of reported cases at day _t_, \(C(t)\) is calculated as shown in
equation (5), where \(\rho\) is the estimated time-constant reporting rate and \(N\) is the size of the population. The model was fit using a negative binomial likelihood with estimated
over-dispersion parameter, \(\varphi\). $$C\left(t\right)=N\rho \frac{{dI}}{{dt}}$$ (5) The model was fitted in a Bayesian framework using Hamiltonian Monte Carlo No-U-Turn sampling in the
CmdStanR package. The model was run for 2,000 iterations with two chains and a warm-up period of 1,000 iterations. Sensitivity results across different infectious periods of 14, 21 and 28
days (\(\sigma\)) ∈ {0.071, 0.048, 0.036}, with different infection seeds and without accounting for the importations are shown in Supplementary Figs. 1, 2 and 3. We assessed convergence
using the R-hat statistic for each parameter. DNA EXTRACTION, SEQUENCING AND GENOME CONSENSUS GENERATION All biological samples were received by the Emergency Response and Biopreparedness
Unit at INSA and were screened for MPXV using real-time PCR targeting the _rpo18_ gene46, on a CFX Opus Real-Time PCR System (Biorad), with viral genome sequencing being attempted for
available samples with real-time PCR threshold cycle (Ct) ≤ 30. The first ten mpox genome sequences (PT0001 to PT0010) were generated as described in a previous publication24. For samples
PT0011 to PT0048, with the exception of the DNA extraction (which was conducted using the MagMAX Viral/Pathogen Nucleic Acid Isolation kit in a KingFisher Extractor), the same procedure
(Nextera XT library preparation and shotgun metagenomics by 2 × 150 bp paired-end sequencing on an Illumina NextSeq 2000 apparatus) was applied. After the development of an amplicon-based
sequencing (PrimalSeq) approach47, all subsequent samples (PT0049–PT0595) were processed using an adapted version of this protocol and primer scheme
(https://doi.org/10.17504/protocols.io.5qpvob1nbl4o/v2). Specifically, DNA amplification was performed using 12.5 µl of NEBNext Q5 Hot Start HiFi PCR Master Mix (New England Biolabs), 3.7 µl
of each primer pool (1.5 µM per pool in the final reaction) (in separate reactions), 3.8 µl of nuclease-free water and 5 µl of template DNA in a final reaction volume of 25 µl. PCR
amplification conditions were 3 min at 98 °C, followed by 35 cycles of 15 s at 98 °C and 5 min at 63 °C. PCR products of each sample were then pooled and subjected to clean-up with Agencourt
AMPure XP (Beckman Coulter, catalog no. A63880) using a 1:1 volume ratio. Dual-indexed libraries were constructed according to the Nextera XT library preparation guide (Illumina) with minor
modifications. Normalization of libraries was performed using a bead-based procedure or a standard procedure according to the concentration values of each library following parallel
capillary electrophoresis in the Fragment Analyzer instrument (Agilent). Library pools were denatured and diluted before loading according to the manufacturer’s protocols. Paired-end
sequencing (2 × 150 bp) was performed on Illumina NextSeq 550 or NextSeq 2000 instruments. Reference-based genome consensus sequences were obtained using the INSaFLU pipeline v.1.5.2
(https://insaflu.insa.pt/; https://github.com/INSaFLU; https://insaflu.readthedocs.io)48, following the same procedure as previously described24. Sample details are presented in
Supplementary Table 1. PHYLOGENETICS AND IDENTIFICATION OF OUTBREAK SUBCLUSTERS To identify and characterize the subclusters of outbreak-related Portuguese sequences within the framework of
the international MPXV genetic diversity, a global IQ-TREE phylogenetic tree was built using the Nextstrain49 hMPXV-1 build (https://github.com/nextstrain/monkeypox; commit
https://github.com/nextstrain/monkeypox/tree/06ae223e34aff74402e9e4caf5d4322c7c99aad3) over curated genome sequences retrieved from the National Center for Biotechnology Information (NCBI)
GenBank Virus collection (https://www.ncbi.nlm.nih.gov/labs/virus/vssi/#). Sequence and metadata curation involved Nextclade, Augur and custom scripts
(https://github.com/nextstrain/monkeypox/tree/06ae223e34aff74402e9e4caf5d4322c7c99aad3/ingest). MPXV sublineage classification followed the international nomenclature proposed in
https://github.com/mpxv-lineages. As of 2 November 2022, the ‘big’ MPXV Nextstrain public dataset (available for navigation at
https://nextstrain.org/groups/neherlab/PT-MPXV-transmission/2022-11-02) included 2,774 sequences, with the B.1 subbranch including 2,721 outbreak-related sequences collected in 28 countries
across Europe, Asia, Africa, Oceania, South America and North America (Supplementary Table 1). This 2022 outbreak subtree included 502 Portuguese MPXV genome sequences, of which 494
(representing 18.2% (494 out of 2,714) of the global B.1 sequence dataset, as of 2 November 2022) were used in this study (Supplementary Table 1). Eight Portuguese sequences were excluded
from the clustering analysis because of same-patient redundancy and/or uncertainty about patient identification and/or sample collection date). The global outbreak B.1 subtree and respective
metadata (Supplementary Dataset 1), including enriched epidemiological data collected for the Portuguese samples (Supplementary Table 1), as described above, were used to identify and
characterize all genetic subclusters (defined as any subbranch with at least two sequences diverging from the outbreak basal level by at least one SNP) using ReporTree v.1.0.1
(https://github.com/insapathogenomics/ReporTree)50. Specifically, ReporTree was requested to cut the tree at one single threshold level from the outbreak basal level (‘root’) using the
‘root-dist’ method available through the ‘TreeCluster’ analysis mode51, and setting a distance unit (‘-d’ argument) corresponding to less than one SNP. ReporTree was also asked to report
statistics for all derived subclusters and key metadata variables. A simplified command line string is as follows: _reportree.py -m metadata.tsv -t outbreak_subtree.nwk –analysis treecluster
--method-threshold root_dist-1 -d 0.000002 --columns_summary_report metadata_1,metadata_n --metadata2report metadata_1,metadata_n –out reportree_. Advanced and integrative visualization and
assessment of the phylogenetic tree, together with genomic, epidemiological and spatiotemporal data was performed with Auspice
(https://nextstrain.org/groups/neherlab/PT-MPXV-transmission/2022-11-02). In addition, https://auspice.us/ was used to interactively explore and interpret the identified subclusters (the
JSON file, the ‘divergence’ outbreak B.1 tree and metadata with subclusters are available for navigation in Supplementary Dataset 1). In particular, genetic subclusters including at least
two Portuguese sequences (‘Portuguese subclusters’) were thoroughly inspected, with the cluster-defining SNP being identified and checked for main homoplasies. The ‘_mutation_profile_’
python script (https://github.com/insapathogenomics/mutation_profile) was applied to screen whether the cluster-defining SNPs follow signatures potentially compatible with APOBEC3-mediated
viral genome editing (namely, GA>AA and TC>TT replacements, which were observed in 86% of those SNPs) (Supplementary Table 2). No clustering inferences were taken for the sequences
categorized in the ‘outbreak basal level’ (‘root’), because of the outbreak characteristics (see Results) and because one could not exclude the possibility that such ‘under-divergence’ may
be due to the lack of genome sequence completeness of some Portuguese and non-Portuguese sequences and/or bioinformatics artefacts (for example, reversions) across the multiple pipelines
used worldwide. The identified sublineages were also characterized in terms of timespan; that is, the time (in days) between the earliest and latest detection at both national and
international levels. For 21 Portuguese patients, the genotyped sample did not correspond to the patient’s earliest MPXV-positive sample (see Supplementary Table 1). Therefore, to avoid bias
in the evaluation of the timespan between the first and latest detection of a given sublineage in Portugal, we used the date of collection of the first PCR-positive sample instead of the
date of collection of the first genotyped sample for the timespan analysis (reflected in Fig. 3). REPORTING SUMMARY Further information on research design is available in the Nature
Portfolio Reporting Summary linked to this article. DATA AVAILABILITY MPXV reads mapping to the reference sequence MPXV-UK_P2 (GenBank accession no. MT903344.1) were deposited in the
European Nucleotide Archive (ENA) (BioProject accession no. PRJEB53055). Assembled consensus sequences were deposited in the NCBI under the accession numbers detailed in Supplementary Table
1. CODE AVAILABILITY Software code used in bioinformatics analysis is available at INSaFLU v.1.5.2 (https://insaflu.insa.pt/; code: https://github.com/INSaFLU/INSaFLU), ReporTree v.1.0.1
(https://github.com/insapathogenomics/ReporTree), get_mutation_profile (https://github.com/insapathogenomics/mutation_profile), Nextstrain (https://github.com/nextstrain/mpox;
https://github.com/nextstrain/monkeypox/tree/06ae223e34aff74402e9e4caf5d4322c7c99aad3; https://github.com/nextstrain/monkeypox/tree/06ae223e34aff74402e9e4caf5d4322c7c99aad3/ingest), and MPXV
lineages (https://github.com/mpxv-lineages). Software code used in the transmission modelling is available at https://github.com/marianaperezduque/mpox_2022. REFERENCES * European Centre
for Disease Prevention and Control. Monkeypox multi-country outbreak – second update, 18 October.
https://www.ecdc.europa.eu/en/publications-data/monkeypox-multi-country-outbreak-second-update (2022) * World Health Organization. Mpox (monkeypox).
https://www.who.int/news-room/fact-sheets/detail/monkeypox (2019) * Centers for Disease Control and Prevention Update: multistate outbreak of monkeypox – Illinois, Indiana, Kansas, Missouri,
Ohio, and Wisconsin, 2003. _MMWR Morb. Mortal. Wkly Rep._ 52, 561–564 (2003). Google Scholar * Bunge, E. et al. The changing epidemiology of human mpox-A potential threat? A systematic
review. _PLoS Negl. Trop. Dis._ 16, e0010141 (2022). Article PubMed PubMed Central Google Scholar * World Health Organization. WHO recommends new name for monkeypox disease. (WHO,
accessed 2 Aug 2023); https://www.who.int/news/item/28-11-2022-who-recommends-new-name-for-monkeypox-disease (2022) * Magnus, P. et al. A pox-like disease in cynomolgus monkeys. _Acta
Pathol. Microbiol. Scand._ 46, 156–176 (1959). Article Google Scholar * Ladnyj, I. D. et al. A human infection caused by monkeypox virus in Basankusu Territory, Democratic Republic of the
Congo. _Bull. World Health Organ._ 46, 593–597 (1972). CAS PubMed PubMed Central Google Scholar * Shchelkunov, S. N. An increasing danger of zoonotic orthopoxvirus infections. _PLoS
Pathog._ 9, e1003756 (2013). Article PubMed PubMed Central Google Scholar * Hutin, Y. J. et al. Outbreak of human monkeypox, Democratic Republic of Congo, 1996 to 1997. _Emerg. Infect.
Dis._ 7, 434–438 (2001). Article CAS PubMed PubMed Central Google Scholar * Meyer, H. et al. Outbreaks of disease suspected of being due to human monkeypox virus infection in the
Democratic Republic of Congo in 2001. _J. Clin. Microbiol._ 40, 2919–2921 (2002). Article PubMed PubMed Central Google Scholar * Durski, K. N. et al. Emergence of Monkeypox - West and
Central Africa, 1970-2017. _MMWR Morb. Mortal. Wkly Rep._ 67, 306–310 (2018). Article PubMed PubMed Central Google Scholar * Yinka-Ogunleye, A. et al. Outbreak of human monkeypox in
Nigeria in 2017-18: a clinical and epidemiological report. _Lancet Infect. Dis._ 19, 872–879 (2019). Article PubMed PubMed Central Google Scholar * Vaughan, A. et al. Two cases of
monkeypox imported to the United Kingdom, September 2018. _Euro Surveill._ 23, 1800509 (2018). Article PubMed PubMed Central Google Scholar * Yong, S. E. F. et al. Imported Monkeypox,
Singapore. _Emerg. Infect. Dis._ 26, 1826–1830 (2020). Article PubMed PubMed Central Google Scholar * Erez, N. et al. Diagnosis of imported Monkeypox, Israel, 2018. _Emerg. Infect. Dis._
25, 980–983 (2019). Article PubMed PubMed Central Google Scholar * Mauldin, M. R. et al. Exportation of monkeypox virus from the African continent. _J. Infect. Dis._ 225, 1367–1376
(2022). Article CAS PubMed Google Scholar * Kraemer, M. U. G. et al. Tracking the 2022 monkeypox outbreak with epidemiological data in real-time. _Lancet Infect. Dis._ 22, 941–942
(2022). * Reed, K. D. et al. The detection of Monkeypox in humans in the Western Hemisphere. _N. Engl. J. Med._ 350, 342–350 (2004). Article CAS PubMed Google Scholar * 2022-23 Monkeypox
Outbreak: Global Trends (WHO, accessed 2 Aug 2023); https://worldhealthorg.shinyapps.io/mpx_global * Second meeting of the International Health Regulations (2005) (IHR) Emergency Committee
regarding the multi-country outbreak of monkeypox (WHO, accessed 2 Aug 2023);
https://www.who.int/news/item/23-07-2022-second-meeting-of-the-international-health-regulations-(2005)-(ihr)-emergency-committee-regarding-the-multi-country-outbreak-of-monkeypox *
Thornhill, J. P. et al. Monkeypox virus infection in humans across 16 countries — April–June 2022. _N. Engl. J. Med._ 387, 679–691 (2022). Article CAS PubMed Google Scholar * Vaughan, A.
M. et al. A large multi-country outbreak of monkeypox across 41 countries in the WHO European Region, 7 March to 23 August 2022. _Euro Surveill._ 27, 2200620 (2022). * Kozlov, M. Monkeypox
goes global: why scientists are on alert. _Nature_ 606, 15–16 (2022). Article CAS PubMed Google Scholar * Isidro, J. et al. Phylogenomic characterization and signs of microevolution in
the 2022 multi-country outbreak of monkeypox virus. _Nat. Med._ 28, 2220–2221 (2022). Article CAS PubMed PubMed Central Google Scholar * Happi, C. et al. Urgent need for a
non-discriminatory and non-stigmatizing nomenclature for monkeypox virus. _PLoS Biol._ 20, e3001769 (2022). Article CAS PubMed PubMed Central Google Scholar * McCollum, A. M. et al.
Human monkeypox. _Clin. Infect. Dis._ 58, 260–267 (2014). Article PubMed Google Scholar * O’Toole, A. N. et al. Putative APOBEC3 deaminase editing in MPXV as evidence for sustained human
transmission since at least 2016. Preprint at _bioRxiv_ https://doi.org/10.1101/2023.01.23.525187 (2022) * Alakunle, E. et al. Monkeypox virus in Nigeria: infection biology, epidemiology,
and evolution. _Viruses_ 12, 1257 (2020). Article CAS PubMed PubMed Central Google Scholar * Luna, N. et al. Monkeypox virus (MPXV) genomics: a mutational and phylogenomic analyses of
B.1 lineages. _Travel Med. Infect. Dis._ 52, 102551 (2023). Article CAS PubMed PubMed Central Google Scholar * Gigante, C. M. et al. Multiple lineages of monkeypox virus detected in the
United States, 2021-2022. _Science_ 378, 560–565 (2022). Article CAS PubMed PubMed Central Google Scholar * Crystal, M. et al. Genomic deletions and rearrangements in monkeypox virus
from the 2022 outbreak, USA. Preprint at _bioRxiv_ https://doi.org/10.1101/2022.09.16.508251 (2022) * Pfaff, F. et al. Monkeypox genomic surveillance will challenge lessons learned from
SARS-CoV-2. _Lancet_ 400, 22–23 (2022). Article CAS PubMed PubMed Central Google Scholar * Perez Duque, M. et al. Ongoing monkeypox virus outbreak, Portugal, 29 April to 23 May 2022.
_Euro Surveill._ 27, 2200424 (2022). Article PubMed PubMed Central Google Scholar * Held, LHens, N et al. In _Handbook of Infectious Disease Data Analysis_ 1st edn (Chapman and Hall/CRC,
2019). Book Google Scholar * Vynnycky, E & White, R. _An Introduction to Infectious Disease Modelling_ (Oxford Univ. Press, 2010). * Endo, A. et al. Heavy-tailed sexual contact
networks and monkeypox epidemiology in the global outbreak, 2022. _Science_ 378, 90–394 (2022). Article CAS PubMed Google Scholar * Murayama, H. et al. Accumulation of immunity in
heavy-tailed sexual contact networks shapes mpox outbreak sizes. _J. Infect. Dis._ 4, jiad254 (2023). Article Google Scholar * Iñigo Martínez, J. et al. Monkeypox outbreak predominantly
affecting men who have sex with men, Madrid, Spain, 26 April to 16 June 2022. _Euro Surveill._ 27, 2200471 (2022). Article PubMed PubMed Central Google Scholar * De Baetselier et al.
Retrospective detection of asymptomatic monkeypox virus infections among male sexual health clinic attendees in Belgium. _Nat. Med._ 28, 2288–2292 (2022). Article PubMed PubMed Central
Google Scholar * Ferré, V. et al. Detection of monkeypox virus in anorectal swabs from asymptomatic men who have sex with men in a sexually transmitted infection screening program in Paris,
France. _Ann. Intern. Med._ 175, 1491–1492 (2022). Article PubMed Google Scholar * Maruotti, A. et al. Estimating the undetected infections in the monkeypox outbreak. _J. Med. Virol._
95, e28099 (2023). Article CAS PubMed Google Scholar * Vivancos, R. et al. Community transmission of monkeypox in the United Kingdom, April to May 2022. _Euro Surveill._ 27, 2200422
(2022). Article CAS PubMed PubMed Central Google Scholar * Antinori, A. et al. Epidemiological, clinical and virological characteristics of four cases of monkeypox support transmission
through sexual contact, Italy, May 2022. _Euro Surveill._ 27, 2200421 (2022). Article CAS PubMed PubMed Central Google Scholar * Madewell, Z. et al. Serial interval and incubation
period estimates of monkeypox virus infection in 12 jurisdictions, United States, May–August 2022. _Emerg. Infect. Dis._ 29, 818–821 (2023). Article PubMed PubMed Central Google Scholar
* Marcus, U. et al. Estimating the size of the MSM populations for 38 European countries by calculating the survey-surveillance discrepancies (SSD) between self-reported new HIV diagnoses
from the European MSM internet survey (EMIS) and surveillance-reported HIV diagnoses among MSM in 2009. _BMC Public Health_ 13, 919 (2023). Article Google Scholar * Nitsche, A. et al.
Detection of orthopoxvirus DNA by real-time PCR and identification of variola virus DNA by melting analysis. _J. Clin. Microbiol._ 42, 1207–1213 (2004). Article CAS PubMed PubMed Central
Google Scholar * Chen, N.F.G. et al. Development of an amplicon-based sequencing approach in response to the global emergence of mpox. _PLoS Biol._ 21, e3002151 (2023). * Borges, V. et
al. INSaFLU: an automated open web-based bioinformatics suite ‘from-reads’ for influenza whole-genome-sequencing-based surveillance. _Genome Med._ 10, 46 (2018). Article PubMed PubMed
Central Google Scholar * Hadfield, J. et al. Nextstrain: real-time tracking of pathogen evolution. _Bioinformatics_ 34, 4121–4123 (2018). Article CAS PubMed PubMed Central Google
Scholar * Mixão, V. et al. ReporTree: a surveillance-oriented tool to strengthen the linkage between pathogen genetic clusters and epidemiological data. _Genome Med._ 15, 43 (2023). Article
PubMed PubMed Central Google Scholar * Balaban, M. et al. TreeCluster: Clustering biological sequences using phylogenetic trees. _PLoS ONE_ 14, e0221068 (2019). Article CAS PubMed
PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS We gratefully acknowledge the engagement and willingness of all participants to share information critical to the
investigation. We are grateful to the authors and laboratories that originated and submitted the genetic sequences released in GenBank. The acquisition of equipment associated with
whole-genome sequencing used in this study (including the Illumina NextSeq 2000) was funded by the HERA (Human and Environmental Risk Assessment) project (Grant/2021/PHF/23776), supported by
the European Commission through the European Centre for Disease Prevention and Control (ECDC), and partially funded by the GenomePT project (POCI-01-0145-FEDER-022184), supported by COMPETE
2020 (Operational Programme for Competitiveness and Internationalisation (POCI)), Lisboa Portugal Regional Operational Programme (Lisboa2020), Algarve Portugal Regional Operational
Programme (CRESC Algarve2020) under the PORTUGAL 2020 Partnership Agreement through the European Regional Development Fund (ERDF), and by the Portuguese Science and Technology Foundation
(FCT). This study was also supported by the ERINHA-Advance project (funded by the European Union’s Horizon 2020 Research & Innovation program, grant agreement no. 824061) and benefited
from co-funding from the European Union’s Horizon 2020 Research and Innovation programme under grant agreement no. 773830 (One Health European Joint Programme), in particular by the
co-funding of the post-doctoral fellowships of J.S.D. and V.M. and the development of INSaFLU. We also thank M. Pinheiro (iBiMED at the Universidade de Aveiro) for his continuous support in
updating the INSaFLU platform and the Infraestrutura Nacional de Computação Distribuída (INCD) for providing computational resources for testing it. INCD was funded by the FCT and FEDER
under the project 22153-01/SAICT/2016. M.P.D. is funded by the Gates Cambridge Scholarship (no. OPP1144). The funders had no role in study design, data collection and analysis, decision to
publish or preparation of the manuscript. The GAT-Intendente team also thanks A. Vasques, L. Fortuna, J. Moreira, I. Correia and Á. Baginha. AUTHOR INFORMATION Author notes * These authors
contributed equally: Vítor Borges, Mariana Perez Duque. AUTHORS AND AFFILIATIONS * Genomics and Bioinformatics Unit, Department of Infectious Diseases, National Institute of Health Doutor
Ricardo Jorge (INSA), Lisbon, Portugal Vítor Borges, Rita Ferreira, Daniel Sobral, Luís Coelho, Alexandra Nunes, Joana Isidro, Miguel Pinto, João Dourado Santos, Verónica Mixão & João
Paulo Gomes * Epidemiology and Statistics Division, Directorate-General of Health, Lisbon, Portugal Mariana Perez Duque, João Vieira Martins, Vítor Cabral Veríssimo & Berta Grau *
Pathogen Dynamics Group, Department of Genetics, University of Cambridge, Cambridge, United Kingdom Mariana Perez Duque & Megan O’Driscoll * Public Health Emergency Centre,
Directorate-General of Health, Lisbon, Portugal Paula Vasconcelos & Berta Grau * Emergency Response and Biopreparedness Unit, Department of Infectious Diseases, National Institute of
Health Doutor Ricardo Jorge (INSA), Lisbon, Portugal Ana Pelerito, Isabel Lopes de Carvalho, Maria Sofia Núncio & Rita Cordeiro * National Reference Laboratory for Sexually Transmitted
Infections, Department of Infectious Diseases, National Institute of Health Doutor Ricardo Jorge (INSA), Lisbon, Portugal Maria José Borrego, Raquel Rocha, Sílvia Lopo, Raquel Neves &
Paula Palminha * Biozentrum, University of Basel, Basel, Switzerland Cornelius Roemer & Richard A. Neher * Swiss Institute of Bioinformatics, Basel, Switzerland Cornelius Roemer &
Richard A. Neher * Veterinary and Animal Research Centre (CECAV), Faculty of Veterinary Medicine, Lusófona University, Lisbon, Portugal Alexandra Nunes & João Paulo Gomes * Technology
and Innovation Unit, Department of Human Genetics, National Institute of Health Doutor Ricardo Jorge (INSA), Lisbon, Portugal Daniela Santos, Silvia Duarte & Luís Vieira * Technical
Board, National Institute of Health Doutor Ricardo Jorge (INSA), Lisbon, Portugal Fátima Martins * Department Coordination, Department of Infectious Diseases, National Institute of Health
Doutor Ricardo Jorge (INSA), Lisbon, Portugal Jorge Machado * Public Health Unit, ACES Cascais, ARSLVT, Cascais, Portugal Vítor Cabral Veríssimo * ECDC Fellowship Programme, Field
Epidemiology path (EPIET), European Centre for Disease Prevention and Control (ECDC), Solna, Sweden Berta Grau * Directorate of Information and Analysis, Directorate-General of Health,
Lisbon, Portugal Berta Grau & André Peralta-Santos * Comprehensive Health Research Centre (CHRC), Escola Nacional de Saúde Pública, Universidade NOVA de Lisboa, Lisbon, Portugal André
Peralta-Santos * Serviço de Dermatovenereologia, Consulta de DST, Centro Hospitalar Universitário de Lisboa Central, Lisbon, Portugal José Neves, Margarida Caldeira, Mafalda Pestana &
Cândida Fernandes * Serviço de Doenças Infeciosas, Hospital de Curry Cabral, Centro Hospitalar Universitário de Lisboa Central, Lisbon, Portugal João Caria, Raquel Pinto, Diana Póvoas &
Fernando Maltez * Instituto Gulbenkian de Ciência, Oeiras, Portugal Diana Póvoas * Unidade de Doenças Sexualmente Transmissíveis da Lapa, Lisbon, Portugal Ana Isabel Sá & Mafalda Brito
Salvador * GAT - Grupo de Ativistas em Tratamentos, Av. Paris, Lisbon, Portugal Eugénio Teófilo, Miguel Rocha, Virginia Moneti & Luis Miguel Duque * GAT - Grupo de Ativistas em
Tratamentos, Intendente, Lisbon, Portugal Francisco Ferreira e Silva & Teresa Baptista * Serviço de Doenças Infeciosas e Medicina Tropical, Hospital de Egas Moniz, Centro Hospitalar de
Lisboa Ocidental, Lisbon, Portugal Joana Vasconcelos, Sara Casanova, Kamal Mansinho & João Vaz Alves * Serviço de Dermatovenereologia, Hospital Garcia de Orta, Almada, Portugal João
Alves & António Silva * Dermatology Department, Centro Hospitalar Universitário Lisboa Norte EPE, Lisbon, Portugal Miguel Alpalhão, Cláudia Brazão, Diogo Sousa & Paulo Filipe *
Dermatology Research Unit (PFilipe Lab), Instituto de Medicina Molecular João Lobo Antunes, University of Lisbon, Lisbon, Portugal Miguel Alpalhão & Paulo Filipe * Dermatology University
Clinic, Faculty of Medicine, University of Lisbon, Lisbon, Portugal Miguel Alpalhão & Paulo Filipe * Serviço de Infeciologia, Hospital Professor Doutor Fernando Fonseca, Amadora,
Portugal Patrícia Pacheco, Francesca Peruzzu & Rita Patrocínio de Jesus * Serviço Infeciologia do CHUP, Largo Professor Abel Salazar, Porto, Portugal Luís Ferreira & Josefina Mendez
* Serviço de Doenças Infeciosas, Hospital Pedro Hispano – ULS Matosinhos, Matosinhos, Portugal Sofia Jordão, Frederico Duarte, Maria João Gonçalves & Eduarda Pena * Serviço de Doenças
Infeciosas, Centro Hospitalar Universitário de São João, Porto, Portugal Claúdio Nunes Silva, André Rodrigues Guimarães & Margarida Tavares * National Health Authority,
Directorate-General of Health, Lisbon, Portugal Graça Freitas Authors * Vítor Borges View author publications You can also search for this author inPubMed Google Scholar * Mariana Perez
Duque View author publications You can also search for this author inPubMed Google Scholar * João Vieira Martins View author publications You can also search for this author inPubMed Google
Scholar * Paula Vasconcelos View author publications You can also search for this author inPubMed Google Scholar * Rita Ferreira View author publications You can also search for this author
inPubMed Google Scholar * Daniel Sobral View author publications You can also search for this author inPubMed Google Scholar * Ana Pelerito View author publications You can also search for
this author inPubMed Google Scholar * Isabel Lopes de Carvalho View author publications You can also search for this author inPubMed Google Scholar * Maria Sofia Núncio View author
publications You can also search for this author inPubMed Google Scholar * Maria José Borrego View author publications You can also search for this author inPubMed Google Scholar * Cornelius
Roemer View author publications You can also search for this author inPubMed Google Scholar * Richard A. Neher View author publications You can also search for this author inPubMed Google
Scholar * Megan O’Driscoll View author publications You can also search for this author inPubMed Google Scholar * Raquel Rocha View author publications You can also search for this author
inPubMed Google Scholar * Sílvia Lopo View author publications You can also search for this author inPubMed Google Scholar * Raquel Neves View author publications You can also search for
this author inPubMed Google Scholar * Paula Palminha View author publications You can also search for this author inPubMed Google Scholar * Luís Coelho View author publications You can also
search for this author inPubMed Google Scholar * Alexandra Nunes View author publications You can also search for this author inPubMed Google Scholar * Joana Isidro View author publications
You can also search for this author inPubMed Google Scholar * Miguel Pinto View author publications You can also search for this author inPubMed Google Scholar * João Dourado Santos View
author publications You can also search for this author inPubMed Google Scholar * Verónica Mixão View author publications You can also search for this author inPubMed Google Scholar *
Daniela Santos View author publications You can also search for this author inPubMed Google Scholar * Silvia Duarte View author publications You can also search for this author inPubMed
Google Scholar * Luís Vieira View author publications You can also search for this author inPubMed Google Scholar * Fátima Martins View author publications You can also search for this
author inPubMed Google Scholar * Jorge Machado View author publications You can also search for this author inPubMed Google Scholar * Vítor Cabral Veríssimo View author publications You can
also search for this author inPubMed Google Scholar * Berta Grau View author publications You can also search for this author inPubMed Google Scholar * André Peralta-Santos View author
publications You can also search for this author inPubMed Google Scholar * José Neves View author publications You can also search for this author inPubMed Google Scholar * Margarida
Caldeira View author publications You can also search for this author inPubMed Google Scholar * Mafalda Pestana View author publications You can also search for this author inPubMed Google
Scholar * Cândida Fernandes View author publications You can also search for this author inPubMed Google Scholar * João Caria View author publications You can also search for this author
inPubMed Google Scholar * Raquel Pinto View author publications You can also search for this author inPubMed Google Scholar * Diana Póvoas View author publications You can also search for
this author inPubMed Google Scholar * Fernando Maltez View author publications You can also search for this author inPubMed Google Scholar * Ana Isabel Sá View author publications You can
also search for this author inPubMed Google Scholar * Mafalda Brito Salvador View author publications You can also search for this author inPubMed Google Scholar * Eugénio Teófilo View
author publications You can also search for this author inPubMed Google Scholar * Miguel Rocha View author publications You can also search for this author inPubMed Google Scholar * Virginia
Moneti View author publications You can also search for this author inPubMed Google Scholar * Luis Miguel Duque View author publications You can also search for this author inPubMed Google
Scholar * Francisco Ferreira e Silva View author publications You can also search for this author inPubMed Google Scholar * Teresa Baptista View author publications You can also search for
this author inPubMed Google Scholar * Joana Vasconcelos View author publications You can also search for this author inPubMed Google Scholar * Sara Casanova View author publications You can
also search for this author inPubMed Google Scholar * Kamal Mansinho View author publications You can also search for this author inPubMed Google Scholar * João Vaz Alves View author
publications You can also search for this author inPubMed Google Scholar * João Alves View author publications You can also search for this author inPubMed Google Scholar * António Silva
View author publications You can also search for this author inPubMed Google Scholar * Miguel Alpalhão View author publications You can also search for this author inPubMed Google Scholar *
Cláudia Brazão View author publications You can also search for this author inPubMed Google Scholar * Diogo Sousa View author publications You can also search for this author inPubMed Google
Scholar * Paulo Filipe View author publications You can also search for this author inPubMed Google Scholar * Patrícia Pacheco View author publications You can also search for this author
inPubMed Google Scholar * Francesca Peruzzu View author publications You can also search for this author inPubMed Google Scholar * Rita Patrocínio de Jesus View author publications You can
also search for this author inPubMed Google Scholar * Luís Ferreira View author publications You can also search for this author inPubMed Google Scholar * Josefina Mendez View author
publications You can also search for this author inPubMed Google Scholar * Sofia Jordão View author publications You can also search for this author inPubMed Google Scholar * Frederico
Duarte View author publications You can also search for this author inPubMed Google Scholar * Maria João Gonçalves View author publications You can also search for this author inPubMed
Google Scholar * Eduarda Pena View author publications You can also search for this author inPubMed Google Scholar * Claúdio Nunes Silva View author publications You can also search for this
author inPubMed Google Scholar * André Rodrigues Guimarães View author publications You can also search for this author inPubMed Google Scholar * Margarida Tavares View author publications
You can also search for this author inPubMed Google Scholar * Graça Freitas View author publications You can also search for this author inPubMed Google Scholar * Rita Cordeiro View author
publications You can also search for this author inPubMed Google Scholar * João Paulo Gomes View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS
V.B., M.P.D. and J.P.G. contributed to study design and development of concept. R.C. coordinated the laboratory diagnosis. J.V.M, P.V., R.F., V.C.V., B.G., A.P.S., J.N., M.C., M. Pestana,
C.F., J.C., R.P., D.P., F. Maltez, A.I.S., M.B.S., E.T., M.R., V. Moneti, L.M.D., F.F.S., T.B., J.V., S.C., K.M., J.V.A., J.A., A.S., M.A., C.B., D. Sousa, P.F., P. Pacheco, F.P., R.P.J.,
L.F., J. Mendez, S.J., F.D., M.J.G., E.P., C.N.S., A.R.G., M.T. and G.F. contributed to epidemiological data collection and preparation. R.F., A.P., I.L.C, M.S.N., M.J.B., R.R., S.L., R.N.,
P. Palminha, L.C., A.N., F. Martins, J. Machado and R.C. contributed to the collection and processing of clinical specimens. R.F., L.C., J.I., D. Santos, S.D. and L.V. performed wet lab
sequencing procedures. V.B., M.P.D., D. Sobral., C.R., R.A.N., M.O’D., J.I., M. Pinto, J.D.S. and V. Mixão contributed to genomic and epidemiological data analysis. V.B., M.P.D. and J.P.G.
coordinated the study and were the main contributors for manuscript writing. All authors critically reviewed the manuscript for intellectual content, approved the final version of the
manuscript for submission and agreed to be accountable for all aspects of the work. CORRESPONDING AUTHOR Correspondence to João Paulo Gomes. ETHICS DECLARATIONS COMPETING INTERESTS B.G. is a
fellow of the ECDC Fellowship Programme, supported financially by the ECDC. The views and opinions expressed herein do not state or reflect those of the ECDC. The ECDC is not responsible
for the data and information collation and analysis and cannot be held liable for conclusions or opinions drawn. All the other authors have no competing interests. PEER REVIEW PEER REVIEW
INFORMATION _Nature Medicine_ thanks Juan David Ramirez and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling editor: Alison Farrell,
in collaboration with the _Nature Medicine_ team. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and
institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION Supplementary Figs. 1–3 REPORTING SUMMARY SUPPLEMENTARY TABLES 1 AND 2 Supplementary Table 1. Public MPXV
genome sequences (hMPXV-1, Clade IIb, Lineage B.1 and sublineages) used in this study and available metadata for the 494 outbreak PT cases. Supplementary Table 2. Characterization of the
Portuguese subclusters according to the size, timespan, international linkage and transmission dynamics. SUPPLEMENTARY DATA 1 Nextstrain JSON file, “divergence” outbreak B.1 tree and
metadata with subclusters for navigation in https://auspice.us/. RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License,
which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link
to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless
indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or
exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints
and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Borges, V., Duque, M.P., Martins, J.V. _et al._ Viral genetic clustering and transmission dynamics of the 2022 mpox outbreak in Portugal.
_Nat Med_ 29, 2509–2517 (2023). https://doi.org/10.1038/s41591-023-02542-x Download citation * Received: 05 February 2023 * Accepted: 08 August 2023 * Published: 11 September 2023 * Issue
Date: October 2023 * DOI: https://doi.org/10.1038/s41591-023-02542-x SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry,
a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative