Play all audios:
ABSTRACT Cohesin structures the genome through the formation of chromatin loops and by holding together the sister chromatids. The acetylation of cohesin’s SMC3 subunit is a dynamic process
that involves the acetyltransferase ESCO1 and deacetylase HDAC8. Here we show that this cohesin acetylation cycle controls the three-dimensional genome in human cells. ESCO1 restricts the
length of chromatin loops, and of architectural stripes emanating from CTCF sites. HDAC8 conversely promotes the extension of such loops and stripes. This role in controlling loop length
turns out to be distinct from the canonical role of cohesin acetylation that protects against WAPL-mediated DNA release. We reveal that acetylation controls the interaction of cohesin with
PDS5A to restrict chromatin loop length. Our data support a model in which this PDS5A-bound state acts as a brake that enables the pausing and restart of loop enlargement. The cohesin
acetylation cycle hereby provides punctuation in the process of genome folding. SIMILAR CONTENT BEING VIEWED BY OTHERS SMC3 ACETYLATION, PDS5 AND SCC2 CONTROL THE TRANSLOCASE ACTIVITY THAT
ESTABLISHES COHESIN-DEPENDENT CHROMATIN LOOPS Article 16 June 2022 CONFORMATIONAL DYNAMICS OF COHESIN/SCC2 LOADING COMPLEX ARE REGULATED BY SMC3 ACETYLATION AND ATP BINDING Article Open
access 22 September 2023 H3K56 ACETYLATION REGULATES CHROMATIN MATURATION FOLLOWING DNA REPLICATION Article Open access 02 January 2025 MAIN Cohesin plays an important role in
three-dimensional (3D) genome organization through the formation and enlargement of chromatin loops1,2,3,4,5,6,7. This process requires the activity of cohesin’s ATPase and its regulator
SCC2NIPBL (refs. 1,2,3,7). Loop length is restricted by the cohesin release factor WAPL3,4,5. Together these proteins keep the looping process dynamic due to continuous cycles of
cohesin-dependent formation of loops, their enlargement, and DNA release. In mammals, the position of cohesin-dependent loops is determined by the architectural protein CTCF, which restricts
chromatin loops to distinct chromosome domains, also known as topologically associated domains (TADs)8,9,10. Recent work shows that CTCF acts as an anchor point to stabilize cohesin on
chromatin and promote the formation and/or maintenance of CTCF-anchored loops11,12. A CTCF mutant deficient in anchoring still displays TAD boundaries11, suggesting that anchoring may not
fully explain the mechanism by which CTCF controls chromatin looping. CTCF appears also to act as a boundary to prevent passage beyond CTCF sites11,12,13,14, but whether this boundary
function is mediated via cohesin-CTCF anchoring or a different molecular mechanism remains poorly understood. The cohesin acetyltransferase ESCO1 acetylates the cohesin SMC3 subunit and
localizes to CTCF sites12,15,16. ESCO1 was recently shown to stabilize cohesin on chromatin and promote the formation of CTCF-anchored loops. ESCO1 was proposed to do so by protecting
cohesin against WAPL-mediated release12, similar to the mechanism by which cohesin is protected to maintain sister chromatid cohesion9,17. A study in budding yeast, however, showed that
cohesin acetylation restricts chromatin loop length independently of WAPL18. The mechanism by which cohesin acetylation regulates chromatin looping therefore remains unclear. While
acetylation is important for locking cohesin on DNA, the role of deacetylation is less well understood. Cohesin deacetylation by the deacetylase HDAC8 (Hos1 in budding yeast) is required for
recycling of cohesin complexes for the next round of sister chromatid cohesion19,20,21. HDAC8 is present throughout the cell cycle21, suggesting that it might play a role beyond recycling
of cohesive cohesin complexes. If cohesin acetylation indeed stabilizes cohesin at CTCF sites, HDAC8-mediated deacetylation could provide a mechanism to enable further loop enlargement
beyond CTCF. In this study we explore the role of the cohesin acetylation cycle in controlling the 3D genome. We show that cohesin acetylation regulates chromosome folding by restricting the
length of architectural stripes and chromatin loops. Cohesin acetylation appears to control genome organization independently of its canonical role in protecting against WAPL. We find that
cohesin acetylation rather converts cohesin into a PDS5A-bound state to restrict the length of chromatin loops. RESULTS COHESIN ACETYLATION LIMITS THE LENGTH OF STRIPES AND LOOPS To assess
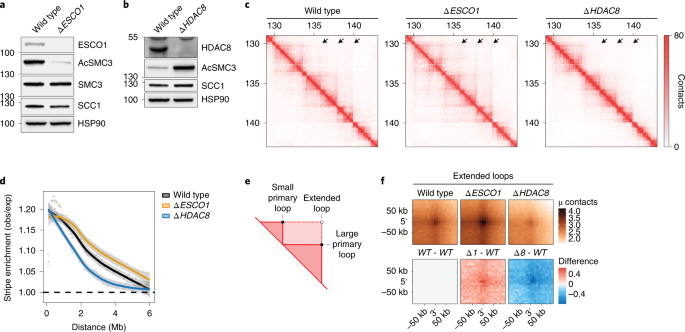
whether the cohesin acetylation cycle regulates the 3D genome, we generated knockout cells for either ESCO1 or HDAC8 in the human HAP1 cell line using CRISPR–Cas9 technology (Fig. 1a,b). We
found that ESCO1 in these cells is responsible for the vast majority of SMC3 acetylation (Fig. 1a) while _∆HDAC8_ cells, as expected, exhibited increased levels of acetylated SMC3 (Fig. 1b).
To specifically study the role of the cohesin acetylation cycle in chromatin looping, in contrast to its role in sister chromatid cohesion, we performed Hi-C analysis in G1-sorted cells.
This also enabled us to assess looping in cells that lack cohesin acetylation, because cohesin’s other acetyltransferase, ESCO2, is absent in G1 (ref. 22). Hi-C matrices of wild-type cells
displayed expected features, including TADs, loops connecting CTCF sites, and architectural stripes. Such stripes are thought to be formed by monodirectional loop extrusion by cohesin that
is anchored to CTCF. Interestingly, we observed notable differences in these stripes in cells with altered cohesin acetylation levels. While cells lacking ESCO1 showed an increase in the
length of stripes, _∆HDAC8_ cells displayed shorter stripes (Fig. 1c). To assess these stripes genome wide, we performed an aggregate stripe analysis that quantifies the distance of the
contacts emanating from CTCF sites. These analyses showed that, relative to the wild type, the architectural stripes of _∆ESCO1_ cells are enriched for longer-range contacts, while these are
depleted in _∆HDAC8_ cells (Fig. 1d and Extended Data Fig. 1b). We found that this looping defect in _∆HDAC8_ cells is dependent on ESCO1 (Extended Data Fig. 1d,e). Cohesin acetylation thus
seems to limit the length of architectural stripes. We then investigated whether _∆ESCO1_ cells harbor CTCF-anchored loops that extend even beyond those found in wild-type cells. We
therefore scored the formation of ‘extended loops’, which are predicted to be formed when loops are enlarged beyond those computationally detected in wild-type cells (Fig. 1e)3. Extended
loops indeed were more abundant in _∆ESCO1_ cells (Fig. 1f), whereas such extended loops were decreased in _∆HDAC8_ cells (Fig. 1f and Extended Data Figs. 1c and 5a,d). We note that the
effects on architectural stripes were clearer than those on CTCF-anchored loops. The combined findings that _∆ESCO1_ cells displayed longer architectural stripes and more pronounced extended
loops, while _∆HDAC8_ cells displayed shorter architectural stripes and loops, indicate that cohesin acetylation limits the degree to which loops can be enlarged. ACETYLATION CONTROLS LOOP
LENGTH INDEPENDENTLY OF WAPL Previous studies revealed that the cohesin-release factor WAPL restricts the extension of chromatin loops3,4. We show that ESCO1, to some degree, is also
important in restricting the size of chromatin loops. If cohesin acetylation simply protects cohesin against WAPL-mediated release, one would not expect to find an increase in long-range
interactions in _∆ESCO1_ cells. To directly test whether ESCO1 and WAPL might regulate looping independently, we generated double-knockout cells for _ESCO1_ and _WAPL_, and performed Hi-C
analyses in G1-sorted cells (Fig. 2a,b). These analyses revealed that deletion of _ESCO1_ exacerbates the _∆WAPL_ phenotype (Fig. 2a). In comparison with _∆WAPL_ cells, _∆ESCO1/∆WAPL_ cells
displayed longer architectural stripes and harbored more pronounced extended loops (Fig. 2c,d and Extended Data Fig. 2a,b). We thus find that cohesin acetylation restricts the size of
chromatin loops in a manner that is at least partially independent of WAPL. ACETYLATION CONVERTS COHESIN INTO A PDS5A-BOUND STATE To identify which factors are key to the mechanism by which
cohesin acetylation controls loop length, we performed a haploid genetic screen23. To find genetic interactors of HDAC8, we compared control HAP1 cells with _∆HDAC8_ HAP1 cells (Fig. 3a).
This screen revealed that _∆HDAC8_ cells specifically benefit from losing PDS5A (Fig. 3b, Extended Data Fig. 3a,b and Supplementary Table 3). PDS5A is a regulatory cohesin subunit that
inhibits cohesin’s ATPase activity24,25. To test whether cohesin acetylation affects the binding of cohesin to PDS5A, we performed coimmunoprecipitation experiments. Pulldown of the core
cohesin component SMC1 revealed that PDS5A is more frequently bound to cohesin in _∆HDAC8_ cells in comparison with wild-type cells (Fig. 3c and Extended Data Fig. 3d). Cohesin acetylation
apparently converts cohesin into a PDS5A-bound state. It remains unclear whether we identified PDS5A as a hit in our screen due to a role for this factor in DNA looping, or rather due to a
role in for example sister chromatid cohesion. COHESIN ACETYLATION CONTROLS LOOP LENGTH THROUGH PDS5A To assess whether PDS5A is key to the mechanism by which cohesin acetylation restricts
loop length, we deleted _PDS5A_ in _∆HDAC8_ cells (Fig. 4a). Hi-C analyses on G1-sorted cells revealed that _∆HDAC8_/_∆PDS5A_ cells display an increase in both extended loops and the length
of architectural stripes, which is comparable to what is observed in _∆PDS5A_ cells (Fig. 4b–e and Extended Data Fig. 2c,d). Notably, _∆HDAC8_/_∆PDS5A_ cells retained high levels of cohesin
acetylation (Fig. 4a). Together, these findings suggest that cohesin acetylation by itself does not prevent loop enlargement, and that the restrictive role of cohesin acetylation in DNA
looping requires PDS5A. Interestingly, single knockouts for _PDS5A_ already displayed a distinct chromatin looping phenotype (Fig. 4b). _∆PDS5A_ cells showed an increase in extended loops,
which appeared to be at the expense of primary loops (Fig. 4c,d). The difference plot, however, revealed that this increase in long-range interactions is not specific to CTCF sites, as we
did not see a clear focal enrichment at the loop anchor (Fig. 4d). Likewise, _∆PDS5A_ cells showed an increase in the length of stripes, while this signal was less enriched close to CTCF
sites (Fig. 4e). These findings suggest that PDS5A not only promotes the formation of CTCF-anchored loops, but also restricts the enlargement of chromatin loops genome wide. NO PROMINENT
ROLE FOR PDS5B IN 3D GENOME ORGANIZATION Cells deficient for the PDS5A paralog PDS5B displayed no evident changes in primary loops, extended loops, or the length of architectural stripes
(Extended Data Fig. 4a–d and Extended Data Fig. 2e,f). Our observation that only _∆PDS5A_ cells, and not _∆PDS5B_ cells, display such phenotypes could be explained by differences in
abundance, because we found that PDS5A is considerably more abundant than PDS5B (Extended Data Fig. 4e,f). Chromatin looping in HAP1 cells thus appears to be largely controlled by PDS5A.
DISCUSSION Taken together, we find that the cohesin acetylation cycle regulates genome folding, and does so by modulating the length of loops and architectural stripes. While ESCO1 prevents
the extension of such loops and stripes, HDAC8 promotes this extension (Fig. 5a). This role in controlling loop length turns out to be distinct from the canonical role of cohesin acetylation
that protects against WAPL-mediated DNA release. We show that the cohesin acetylation cycle instead controls the binding of PDS5A to regulate loop enlargement. Our findings indicate that
cohesin acetylation regulates the looping process at CTCF sites through PDS5A. These findings fit with earlier work in HeLa cells showing that combined depletion of PDS5A and PDS5B decreased
the amount of CTCF loops4. Importantly, we find that cohesin acetylation and PDS5A do not control the presence of architectural stripes, but rather their length. This suggests that the
acetylation cycle does not control CTCF anchoring by itself. It is more likely that a PDS5A-dependent brake mechanism allows for the pausing of loop enlargement at CTCF sites. This mechanism
in turn could enable CTCF to act as a boundary, albeit transiently. Such a PDS5A brake mechanism appears to not be limited to CTCF sites, because Pds5 likewise inhibits the loop enlargement
process in yeast, which have no CTCF18,26. We suggest that cohesin acetylation and PDS5 binding thus represent an ancient regulatory mechanism, which is taken advantage of by CTCF to
control loop length. But how, mechanistically speaking, could the cohesin acetylation cycle and PDS5A binding then control looping? Earlier work showed that SCC2NIPBL stimulates cohesin’s
ATPase, whereas PDS5 inhibits cohesin’s ATPase activity24,25. This fits well with the finding that cohesin can initiate and enlarge DNA loops only in the presence of SCC2NIPBL1,2 but not in
the presence of PDS5 (ref. 1). Because PDS5 and SCC2NIPBL compete for the same binding interface on SCC1 (refs. 25,27), regulation of this exchange could provide a mechanism to control the
loop enlargement process. Recent structural work reveals that SCC2NIPBL binds cohesin at multiple interfaces, including SMC3’s ATPase head28,29,30,31,32. Binding to this latter interface is
observed in cohesin’s unacetylated state28,29,30. A key part of this interface contains the two lysines that are acetylated by cohesin’s acetyltransferases. Acetylation of these lysines
neutralizes their charge and has been proposed to disfavor interaction with SCC2NIPBL (ref. 30). However, mutant cohesin complexes in which these lysines are replaced by ‘acetyl-mimicking’
glutamines are barely impaired in their ATPase activity29. We indeed found that SCC2NIPBL can still bind to acetylated cohesin complexes (Extended Data Fig. 3e). Together this would indicate
that cohesin acetylation does not intrinsically compromise SCC2NIPBL binding, and that cohesin acetylation can be compatible with ATPase activity. Correspondingly, we found that cohesin
acetylation by itself does not restrict chromatin looping, but only does so in the presence of PDS5A. Cryo-EM structures of yeast cohesin show that Pds5 binds to the Smc3 ATPase head32. Our
data suggest that acetylation of SMC3 actually enhances the binding of cohesin to PDS5A. Further support for this model comes from observations in yeast that nonacetylatable Smc3 mutants
display reduced Pds5 binding (see the paper by Bastié et al.33), and that acetylation promotes the stability of Pds5 on chromatin34,35. The interconnection between SCC2NIPBL and PDS5, and
how these proteins regulate the activities of cohesin, remains incompletely understood. Future studies will be needed to assess whether PDS5 binding to acetylated cohesin indeed prevents
SCC2NIPBL binding to the SMC3 ATPase head, and whether SCC2NIPBL may then remain connected to the complex through another interface. It would also be relevant to test whether
acetylation-dependent PDS5 binding indeed inhibits SCC2NIPBL-stimulated ATPase activity. Such experiments could include in vitro ATPase assays on acetylated cohesin complexes, in which the
amount of PDS5 is titrated until SCC2NIPBL-stimulated ATPase activity is inhibited, similar to previous experiments using unacetylated cohesin complexes32. While questions remain regarding
SCC2NIPBL, our data support the model where the binding of PDS5 to cohesin stabilizes the complex in a conformation that prevents ATP hydrolysis and further loop enlargement. By promoting
the binding of PDS5, cohesin acetylation could maintain this conformation and thereby convert enlarging loops into static loops. HDAC8-mediated deacetylation in turn could alleviate this
paused state to restart the looping reaction (Fig. 5b). This key regulatory mechanism turns out to be conserved from yeast to humans (see the paper by Bastié et al.33). The modulation of
cohesin’s looping activity by cohesin acetylation could therefore be a universal mechanism that controls genome topology across the eukaryotic tree of life. METHODS CELL CULTURE AND GENE
EDITING HAP1 cells36 were cultured in Iscove’s modified Dulbecco’s medium (Invitrogen) supplemented with 10% fetal calf serum (FCS; Clontech), 1% UltraGlutamin (Lonza) and 1%
penicillin-streptomycin (Invitrogen). Knockout cell lines were generated either by insertion of a resistance cassette in the first coding exon after the start ATG or by generation of small
out-of-frame indels using CRISPR–Cas9 technology. CRISPRs targeting _ESCO1_ (5’-CATGAGTACAAGGTCATCAA-3’ and 5’-AGCTTAACCGGAGATCACAA-3’), _HDAC8_ (5’-CAGTGGGCAGTCGCTGGTCC-3’ and
5’-CGGGACTATAGATATAAACC-3’), _PDS5A_ (5’-GTGGCGTCGTGAGTGCCGACGGG-3’ and 5’-GGAAGATCGCTTACCCTCCG-3’) and _PDS5B_ (5’-TCTGATATTTCCTTGACCCC-3’) were cloned into px330 (Addgene plasmid no.
42230). _∆WAPL_ cells were generated as previously described3, and used as a parental cell line to generate double-knockout cells for _WAPL_ and _ESCO1_. Either blasticidin or puromycin
resistance cassettes were used, as described previously23. Knockout cell lines were confirmed by PCR genotyping and immunoblotting analysis. The following oligos were used for ESCO1:
5’-CCAGGACACAAAAATCCTCTTC-3’ and 5’-CTTCATCTCATTCTTTTTCGGG-3’; for HDAC8: 5’-TAGGGCAACAAGGATGGTTAGT-3’ and 5’-TTTCTTGGGATTACAGGCAGAT-3’; for PDS5A: 5’-ACTGTGAACCAAAAGTTGTCCC-3’ and
5’-ATCAAAATCCGTCCAGACACTT-3’; and for PDS5B: 5’-GTTACAAATTTTGGTTGGTGGG-3’ and 5’-CCTCTGCCCTACACAGATGTAA-3’. A list of cell lines used in this manuscript is given in Supplementary Table 1. To
endogenously tag the C terminus of SCC1RAD21 with a HALO-tag, we used an approach previously described32 with slight adaptations. Because _∆PDS5A_ and _∆PDS5B_ cells were already resistant
to puromycin, we cotransfected pRS-BLAST at a 1:10 ratio to the px459 plasmid. Transfected clones were selected using 10 μg ml–1 blasticidin for 2 days. Colonies were picked when clearly
visible. Integration of the HALO-tag at the correct location was then confirmed by PCR genotyping and immunoblotting analysis. Wild-type cells with SCC1-HALO tagged were generated as
previously described32. IMMUNOBLOTTING Cells were pelleted at 500_g_ for 3 min and resuspended in RIPA buffer consisting of 10 mM Tris-Cl (pH 8.0), 1 mM EDTA, 0.5 mM EGTA, 1% Triton X-100,
0.1% sodium deoxycholate, 0.1% SDS and 140 mM NaCl, supplemented with protease inhibitors (Roche). Lysates were vortexed for 30 s and incubated on ice for 30 min. Lysates were spun at
20,000_g_ for 10 min at 4 °C, and supernatants were quantified by Bradford analysis (Bio-Rad). Denatured proteins (10–20 μg) were loaded and run on polyacrylamide gels and transferred to
nitrocellulose membranes. Membranes were blocked with 5% w/v milk in TBS-Tween (TBS-T, 0.1%). Primary and secondary antibody incubations were performed in 5% milk in TBS-Tween (0.1%) and
membranes were washed with TBS-T, except for SCC2NIPBL, which was incubated in 5% bovine serum albumin (Sigma) in TBS-T (0.1%). The signal was developed with Clarity Western ECL Substrate
(Bio-Rad) or Immobilon western Chemiluminescent HRP substrate (Millipore) on the ChemiDoc Imaging System (Bio-Rad). COIMMUNOPRECIPITATION Cells were pelleted at 500_g_ for 5 min and
resuspended in lysis buffer consisting of 50 mM Tris (pH 7.5), 5 mM EDTA, 150 mM NaCl and 0.1% NP-40, supplemented with protease inhibitors (Roche) and phosphatase inhibitors (Sigma, 1:100).
Cells were incubated for 30 min on ice. Lysates were supplemented with Ambion DNase I (Invitrogen, 1:100) and Benzonase Nuclease (Millipore, 600 U ml–1) and incubated for 4 h on a rotator
at 4 °C. Lysates were spun at 20,000_g_ for 10 min at 4 °C and supernatants were mixed with two volumes of TNENG buffer, consisting of 50 mM Tris (pH 7.5), 5 mM EDTA, 150 mM NaCl, 0.1% NP-40
and 10% glycerol, supplemented with protease inhibitors (Roche). Supernatants were quantified by Bradford analysis (Bio-Rad). Protein lysate (300 μg for SMC1 IP and 750 μg for SCC2 IP) was
used, along with 3 μg of antibody. Lysates were mixed with 30 μl of Protein G Dynabeads (Invitrogen) and incubated overnight at 4 °C while tumbling. Beads were washed three times with wash
buffer consisting of 50 mM Tris (pH 7.5), 5 mM EDTA, 150 mM NaCl and 0.1% NP-40, and proteins were denatured using Laemmli buffer at 95 °C for 10 min. Coimmunoprecipitation was checked by
immunoblotting analysis, as described above. ANTIBODIES Coimmunoprecipitation experiments were performed with the following antibodies: SMC1 (Bethyl, no. A300-055A) and SCC2 (Bethyl, no.
A301-779A). Immunoblots were performed using the following antibodies and dilutions: HSP90 (Santa Cruz, no. sc13119 F8, 1:10,000), ESCO1 (a kind gift from S. Rankin37, 1:1,500), HDAC8
(Sigma-Aldrich, no. WH0055869M1, 1:1,000), AcSMC3 (a kind gift from K. Shirahige38, 1:1,500), WAPL (Santa Cruz, no. sc365189, 1:1,000), SMC1 (Bethyl, no. A300-055A, 1:2,000), SMC3 (Bethyl,
no. A300-060A-5, 1:2,000), SCC1 (Millipore, no. 05-908, 1:1,000), PDS5A (Bethyl, no. A300-089A, 1:1,000), PDS5B (Bethyl, no. A300-538A, 1:500), SCC2 (Santa Cruz, no. sc374625, 1:1,000), SCC4
(Abcam, no. ab46906, 1:1,000), Actin (Abcam, no. ab6276, 1:5,000) and Tubulin (Abcam, no. ab18251, 1:10.000). Secondary antibodies Goat-anti-Mouse-PO (DAKO, no. P0447) and
Goat-anti-Rabbit-PO (DAKO, no. P0448) were used at 1:2,000 dilution. FLUORESCENCE RECOVERY AFTER PHOTOBLEACHING Cells with endogenously tagged SCC1 were grown on LabTekII-chambered cover
glass (Thermo Scientific Nunc). To be able to specifically perform fluorescence recovery after photobleaching (FRAP) on G1 cells, cells were transfected with DNA helicase B fragment fused
with near-infrared fluorescent protein (DHB-iRFP) using FuGENE Transfection Reagent 2–3 days before imaging. On the day of imaging, cells were incubated for 30 min with 300 nM HALO-ligand
JF549 (Promega). Cells were washed three times with normal medium and incubated for 30 min to allow removal of excess ligand. The medium was replaced with Leibovitz L-15 imaging medium
(Invitrogen), then FRAP analysis was performed on a Leica SP5 confocal microscope with a ×63/1.4 numerical aperture oil objective using the LAS-AF FRAP-Wizard. G1 cells were selected based
on nuclear localization of DHB-iRFP, as described in ref. 11. Five images were taken before bleaching, then half of the nucleus was photobleached using five pulses of 100% transmission of
the 561-nm laser. After bleaching, ten frames were taken every 2 s and subsequently 120 frames were taken every 10 s. Fluorescence intensity was measured in bleached and unbleached areas by
user-defined regions in ImageJ v.2.1.0/1.53k. Recovery was quantified by calculating the difference in intensity between bleached and unbleached regions. To ensure that we quantified cells
with homogeneous SCC1 distribution, we excluded those in which the difference in intensity between bleached and unbleached areas was already >10% in prebleaching frames. MS ANALYSIS For
protein digestion, frozen cell pellets were lysed in boiling guanidine (GuHCl) lysis buffer as previously described39. Protein concentration was determined with a Pierce Coomassie (Bradford)
Protein Assay Kit (Thermo Scientific), according to the manufacturer’s instructions. After dilution to 2 M GuHCl, aliquots corresponding to 200 μg of protein were digested twice (4 h and
overnight) with trypsin (Sigma-Aldrich) at 37 °C, enzyme/substrate ratio 1:75. Digestion was quenched by the addition of trifluoroacetic acid (final concentration 1%), after which peptides
were desalted on a Sep-Pak C18 cartridge (Waters). Samples were dried in a vacuum centrifuge and reconstituted in 2% formic acid for mass spectrometry (MS) analysis. Peptide mixtures were
loaded directly on the analytical column (ReproSil-Pur 120 C18-AQ, 1.9 μm, 75 μm × 500 mm, packed in-house) and analyzed by nano liquid chromatography–tandem MS (LC–MS/MS) on an Orbitrap
Fusion Tribrid mass spectrometer equipped with a Proxeon nLC1000 system (Thermo Scientific). Solvent A was 0.1% formic acid/water and solvent B was 0.1% formic acid/80% acetonitrile.
Peptides were eluted from the analytical column at a constant flow of 250 nl min–1 in a 270-min gradient, containing a 250-min stepped increase from 3 to 35% solvent B followed by a 20-min
wash in 80% solvent B. Raw data were analyzed by Proteome Discoverer (v.2.5.0.400, Thermo Scientific) using standard settings. MS/MS data were searched against the Human Swissprot database
(20,395 entries, release 2021_04) using Sequest HT. The maximum permitted precursor mass tolerance was 50 ppm and 0.6 Da for fragment ion masses. Trypsin was chosen as cleavage specificity,
allowing two missed cleavages. Carbamidomethylation (C) was set as a fixed modification, while oxidation (M) was used as variable modifications. False discovery rates for peptide and protein
identification were set to 1% and, as an additional filter, Sequest HT XCorr>1 was set. For wild-type cells, protein peptide spectrum match (PSM) values of PDS5A and PDS5B of three
biological replicates were averaged and displayed in the panel. HI-C ANALYSIS Hi-C libraries were prepared as previously described40, with the protocol adapted slightly for G1 analyses. An
asynchronous pool of cells was first crosslinked using 2% formaldehyde for 10 min at room temperature and quenched with 2 M glycine. The 10% smallest cells were then sorted based on forward
and side scatter using a BD FACSAria II. Five million cells were collected for Hi-C analysis and then processed according to a protocol following crosslinking. To assess sorting efficiency,
0.5 million sorted cells and 0.5 million asynchronous cells were permeabilized for 10 min using 0.1% triton in PBS. Cells were stained with DAPI (Sigma-Aldrich) and assayed on a BD LSR
Fortessa Machine. Plots were generated with FlowJo (v.10). The gating strategy is depicted in Supplementary Fig. 1a. Raw sequence data were mapped and processed using HiC-Pro v.2.9 and v.3.0
(ref. 41), with hg19 as reference; juicebox-ready files were generated using Juicebox-pre (juicer tools v.1.9.8)42 (see Supplementary Table 2 for the number of valid pairs per sample and
the percentage of _cis_ contacts). For visualization and downstream analyses, contact matrices were ICE normalized43 and normalized to 100 million contacts per sample. For Hi-C analysis on
asynchronous cells, we first subsampled the data to obtain amounts of reads equal to the sample with the lowest amount of reads. To visualize the genome-wide effects of our knockout cells,
we performed APA40 using the loops previously identified in wild-type HAP1 cells3. APA was performed as implemented in GENOVA v.1.0 (ref. 44). In brief, for the set of loop coordinates a
square submatrix was selected centred at these locations, including a 100-kb flanking region upstream and downstream. These submatrices were then averaged to obtain a mean contact map for
these locations. The difference plots were obtained as the difference of mean contact maps in comparison with the indicated control cell line. We performed a similar analysis for extended
loops, as described previously3. Extended loops are defined as those formed when the 5’ loop anchor is combined with every 3’ loop anchor in a 3-Mb region that is not the primary loop
itself. APA scores for primary and extended loops were measured using the quantify function with size 3 in GENOVA v.1.0 to obtain the mean signal intensity of each loop. For analysis of
differences in stripes, we used an aggregate analysis similar to those described above, as has been done previously45. We selected the top 10,000 CTCF chromatin immunoprecipitation
sequencing peaks in wild-type HAP1 cells11 as the center location for a submatrix of 6 Mb. To account for differences in distance from the diagonal, the signal was normalized to the expected
signal per sample at that distance before averaging them to obtain mean contact maps. In this way we specifically assessed enrichment at stripes, rather than looking at the ‘general’
differences in loop length between genotypes. To quantify differences in stripes across samples, the signal from 3’ and 5’ average stripes was fit to a polynomial surface using local
fitting, with stats::loess(), to include the 95% confidence interval in gray. GENOME-WIDE SYNTHETIC VIABILITY SCREEN _∆HDAC8_ HAP1 cells were mutagenized using a gene-trap retrovirus
produced in HEK293T cells (obtained from ATCC) and concentrated either by ultracentrifugation as described previously23 or employing centrifugal ultrafiltration devices. Here,
retrovirus-containing medium was harvested on two consecutive days, filtered (0.45 μm) and concentrated using Amicon Ultra-15 Centrifugal Filter Units with 100 K MWCO (Merck-Millipore). The
virus concentrates from both harvests were combined, supplemented with 8 μg ml–1 protamine sulfate (Sigma) and used to infect ~40 million HAP1 cells. To map fitness genes in _ΔHDAC8_ HAP1
cells, mutagenized cells were passaged for an additional 14 days after gene-trap virus infection. Cells were then harvested and fixed with BD fix buffer I (BD Biosciences) for 10 min at 37
°C. After washing with FACS buffer (10% FCS in PBS), cells were stained with DAPI (1 μg ml–1) for 1 h at room temperature to visualize G1 cells. Twenty-four million G1 haploid cells were
sorted using a BD FACSAria Fusion, followed by genomic DNA extraction and library preparation as described in ref. 23. The gating strategy is depicted in Supplementary Fig. 1b. Insertion
mapping and data analysis were performed as described previously23 with certain modifications. Sequence reads (50 base pairs) were aligned to the human genome (v.hg38) using Bowtie,
resulting in a unique alignment to the human genome with zero or one mismatch. Insertions were assigned to protein-coding genes. For every gene, the transcript containing the longest open
reading frame was used and unique alignments in intronic regions between the transcription initiation site and stop codon were counted. Genes enriched for gene-trap insertions in either the
sense or antisense orientation were identified using a false-discovery-rate-corrected binomial test (step 1, _P_ value cutoff 0.05), and genes that deviated in _∆HDAC8_ cells from wild-type
control cells were identified by a bidirectional Fisher’s exact test with all independent control datasets (step 2, _P_ value cutoff 0.05). An odds ratio cutoff of 0.7 was applied using the
aggregated wild-type control datasets with a greater Fisher’s test. To find genes whose inactivation rescued the growth defect of _∆HDAC8_ cells, we focused on fitness enhancers. These genes
are defined by an increase in sense orientation integrations observed in _∆HDAC8_ cells but not in wild-type controls (step 1) and show a bias in the sense/antisense ratio compared with all
control datasets (step 2). This yielded PDS5A as the strongest and most important fitness enhancer. REPORTING SUMMARY Further information on research design is available in the Nature
Research Reporting Summary linked to this article. DATA AVAILABILITY The Hi-C and genetic screen data generated have been deposited in GEO, accession no. GSE174628. The proteomics data have
been deposited in the PRIDE database, accession no. PXD032185. Source data are provided with this paper. Any other relevant data are available from the corresponding author upon reasonable
request. CODE AVAILABILITY Hi-C data were analyzed with GENOVA, which can be downloaded here: https://github.com/deWitLab/GENOVA. REFERENCES * Davidson, I. F. et al. DNA loop extrusion by
human cohesin. _Science_ 366, 1338–1345 (2019). Article CAS PubMed Google Scholar * Kim, Y., Shi, Z., Zhang, H., Finkelstein, I. J. & Yu, H. Human cohesin compacts DNA by loop
extrusion. _Science_ 366, 1345–1349 (2019). Article CAS PubMed PubMed Central Google Scholar * Haarhuis, J. H. I. et al. The cohesin release factor WAPL restricts chromatin loop
extension. _Cell_ 169, 693–707 (2017). Article CAS PubMed PubMed Central Google Scholar * Wutz, G. et al. Topologically associating domains and chromatin loops depend on cohesin and are
regulated by CTCF, WAPL, and PDS5 proteins. _EMBO J._ 36, 3573–3599 (2017). Article CAS PubMed PubMed Central Google Scholar * Gassler, J. et al. A mechanism of cohesin-dependent loop
extrusion organizes zygotic genome architecture. _EMBO J._ 36, 3600–3618 (2017). Article CAS PubMed PubMed Central Google Scholar * Rao, S. S. P. et al. Cohesin loss eliminates all loop
domains. _Cell_ 171, 305–309 (2017). Article CAS PubMed PubMed Central Google Scholar * Schwarzer, W. et al. Two independent modes of chromatin organization revealed by cohesin
removal. _Nature_ 551, 51–56 (2017). Article PubMed PubMed Central Google Scholar * Merkenschlager, M. & Nora, E. P. CTCF and cohesin in genome folding and transcriptional gene
regulation. _Annu. Rev. Genomics Hum. Genet._ 17, 17–43 (2016). Article CAS PubMed Google Scholar * Yatskevich, S., Rhodes, J. & Nasmyth, K. Organization of chromosomal DNA by SMC
complexes. _Annu. Rev. Genet._ 53, 445–482 (2019). Article CAS PubMed Google Scholar * van Ruiten, M. S. & Rowland, B. D. On the choreography of genome folding: a grand pas de deux
of cohesin and CTCF. _Curr. Opin. Cell Biol._ 70, 84–90 (2021). Article PubMed CAS Google Scholar * Li, Y. et al. The structural basis for cohesin-CTCF-anchored loops. _Nature_ 578,
472–476 (2020). Article CAS PubMed PubMed Central Google Scholar * Wutz, G. et al. ESCO1 and CTCF enable formation of long chromatin loops by protecting cohesin STAG1 from WAPL. _eLife_
9, 9906–9933 (2020). Article Google Scholar * Nora, E. P. et al. Molecular basis of CTCF binding polarity in genome folding. _Nat. Commun_. 11, 5612 (2020). * Davidson, I. F. et al. Rapid
movement and transcriptional re-localization of human cohesin on DNA. _EMBO J._ 35, 2671–2685 (2016). Article CAS PubMed PubMed Central Google Scholar * Minamino, M. et al. Esco1
acetylates cohesin via a mechanism different from that of Esco2. _Curr. Biol._ 25, 1694–1706 (2015). Article CAS PubMed Google Scholar * Rahman, S., Jones, M. J. K. & Jallepalli, P.
V. Cohesin recruits the Esco1 acetyltransferase genome wide to repress transcription and promote cohesion in somatic cells. _Proc. Natl Acad. Sci. USA_ 112, 11270–11275 (2015). Article CAS
PubMed PubMed Central Google Scholar * Uhlmann, F. SMC complexes: from DNA to chromosomes. _Nat. Rev. Mol. Cell Biol._ 17, 399–412 (2016). Article CAS PubMed Google Scholar *
Dauban, L. et al. Regulation of cohesin-mediated chromosome folding by eco1 and other partners. _Mol. Cell_ 77, 1279–1293 (2020). Article CAS PubMed Google Scholar * Beckouët, F. et al.
An Smc3 acetylation cycle is essential for establishment of sister chromatid cohesion. _Mol. Cell_ 39, 689–699 (2010). Article PubMed PubMed Central CAS Google Scholar * Borges, V. et
al. Hos1 deacetylates Smc3 to close the cohesin acetylation cycle. _Mol. Cell_ 39, 677–688 (2010). Article CAS PubMed Google Scholar * Deardorff, M. A. et al. HDAC8 mutations in Cornelia
de Lange syndrome affect the cohesin acetylation cycle. _Nature_ 489, 313–317 (2012). Article CAS PubMed PubMed Central Google Scholar * Lafont, A. L., Song, J. & Rankin, S.
Sororin cooperates with the acetyltransferase Eco2 to ensure DNA replication-dependent sister chromatid cohesion. _Proc. Natl Acad. Sci. USA_ 107, 20364–20369 (2010). Article CAS PubMed
PubMed Central Google Scholar * Blomen, V. A. et al. Gene essentiality and synthetic lethality in haploid human cells. _Science_ 350, 1092–1096 (2015). Article CAS PubMed Google Scholar
* Murayama, Y. & Uhlmann, F. Biochemical reconstitution of topological DNA binding by the cohesin ring. _Nature_ 505, 367–371 (2014). Article CAS PubMed Google Scholar * Petela, N.
J. et al. Scc2 is a potent activator of cohesin’s ATPase that promotes loading by binding scc1 without Pds5. _Mol. Cell_ 70, 1134–1148 (2018). Article CAS PubMed PubMed Central Google
Scholar * Costantino, L., Hsieh, T.-H. S., Lamothe, R., Darzacq, X. & Koshland, D. Cohesin residency determines chromatin loop patterns. _eLife_ 9, e59889 (2020). Article CAS PubMed
PubMed Central Google Scholar * Kikuchi, S., Borek, D. M., Otwinowski, Z., Tomchick, D. R. & Yu, H. Crystal structure of the cohesin loader Scc2 and insight into cohesinopathy. _Proc.
Natl Acad. Sci. USA_ 113, 12444–12449 (2016). Article CAS PubMed PubMed Central Google Scholar * Higashi, T. L. et al. A structure-based mechanism for DNA entry into the cohesin ring.
_Mol. Cell_ 79, 917–933.e9 (2020). Article CAS PubMed PubMed Central Google Scholar * Collier, J. E. et al. Transport of DNA within cohesin involves clamping on top of engaged heads by
Scc2 and entrapment within the ring by Scc3. _eLife_ 9, 531–536 (2020). Article Google Scholar * Shi, Z., Gao, H., Bai, X.-C. & Yu, H. Cryo-EM structure of the human cohesin-NIPBL-DNA
complex. _Science_ 368, 1454–1459 (2020). Article CAS PubMed Google Scholar * Bauer, B. W. et al. Cohesin mediates DNA loop extrusion by a ‘swing and clamp’ mechanism. _Cell_ 184,
5448–5464 (2021). Article CAS PubMed PubMed Central Google Scholar * Petela, N. J. et al. Folding of cohesin’s coiled coil is important for Scc2/4-induced association with chromosomes.
_eLife_ 10, e67268 (2021). * Bastié, N. et al. Smc3 acetylation, Pds5 and Scc2 control the translocase activity that establishes cohesin-dependent chromatin loops. _Nat. Struct. Mol. Biol._
https://doi.org/10.1038/s41594-022-00780-0 * Chan, K. L. et al. Cohesin’s DNA exit gate is distinct from its entrance gate and is regulated by acetylation. _Cell_ 150, 961–974 (2012).
Article CAS PubMed PubMed Central Google Scholar * Chapard, C., Jones, R., van Oepen, T., Scheinost, J. C. & Nasmyth, K. Sister DNA entrapment between juxtaposed Smc heads and
kleisin of the cohesin complex. _Mol. Cell_ 75, 224–237 (2019). Article CAS PubMed PubMed Central Google Scholar * Carette, J. E. et al. Ebola virus entry requires the cholesterol
transporter Niemann-Pick C1. _Nature_ 477, 340–343 (2011). Article CAS PubMed PubMed Central Google Scholar * Alomer, R. M. et al. Esco1 and Esco2 regulate distinct cohesin functions
during cell cycle progression. _Proc. Natl Acad. Sci. USA_ 114, 9906–9911 (2017). Article CAS PubMed PubMed Central Google Scholar * Nishiyama, T. et al. Sororin mediates sister
chromatid cohesion by antagonizing Wapl. _Cell_ 143, 737–749 (2010). Article CAS PubMed Google Scholar * Jersie-Christensen, R. R., Sultan, A. & Olsen, J. V. Simple and reproducible
sample preparation for single-shot phosphoproteomics with high sensitivity. _Methods Mol. Biol._ 1355, 251–260 (2016). Article CAS PubMed Google Scholar * Rao, S. S. P. et al. A 3D map
of the human genome at kilobase resolution reveals principles of chromatin looping. _Cell_ 159, 1665–1680 (2014). Article CAS PubMed PubMed Central Google Scholar * Servant, N. et al.
HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. _Genome Biol._ 16, 259 (2015). Article PubMed PubMed Central CAS Google Scholar * Durand, N. C. et al. Juicer
provides a one-click system for analyzing loop-resolution Hi-C experiments. _Cell Syst._ 3, 95–98 (2016). Article CAS PubMed PubMed Central Google Scholar * Imakaev, M. et al. Iterative
correction of Hi-C data reveals hallmarks of chromosome organization. _Nat. Methods_ 9, 999–1003 (2012). Article CAS PubMed PubMed Central Google Scholar * van der Weide, R. H. et al.
Hi-C analyses with GENOVA: a case study with cohesin variants. _NAR Genom. Bioinform_. 3, lqab040 (2021). * Hansen, A. S. et al. Distinct classes of chromatin loops revealed by deletion of
an RNA-binding region in CTCF. _Mol. Cell_ 76, 395–411 (2019). Article CAS PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS We thank our colleagues for helpful
discussions. We thank H. Teunissen, T. van den Brand and M. Panarotto-Périclès for technical assistance, K. Shirahige (The University of Tokyo) for the acetylated SMC3 antibody, S. Rankin
(Oklahoma Medical Research Foundation) for the ESCO1 antibody, T. Perrakis for advice on protein structure, M. Vermeulen and I. Santos-Barriopedro for experiments not included in the
manuscript and the NKI Genomics Core facility and Flow Cytometry facility for assistance. M.S.v.R. was supported by Boehringer Ingelheim Fonds; M.S.v.R., D.G., Á.S.C. and B.D.R. by the
European Research Council (no. ERC 772471—‘CohesinLooping’); J.H.I.H. and L.W. by the Dutch Cancer Society (no. KWF 11665); L.H. and M.A. by the Dutch NWO X-omics Initiative (no.
184.034.019); E.d.W. by the European Research Council (nos. ERC 637587—‘HAP-PHEN’ and ERC 865459—‘FuncDis3D’); and T.R.B. by the Dutch Research Council (no. 016.Vici.170.033). AUTHOR
INFORMATION Author notes * These authors contributed equally: Démi van Gent, Ángela Sedeño Cacciatore. AUTHORS AND AFFILIATIONS * Division of Cell Biology, The Netherlands Cancer Institute,
Amsterdam, the Netherlands Marjon S. van Ruiten, Démi van Gent, Ángela Sedeño Cacciatore, Laureen Willems, Judith H. I. Haarhuis & Benjamin D. Rowland * Division of Biochemistry, Oncode
Institute, The Netherlands Cancer Institute, Amsterdam, the Netherlands Astrid Fauster, Maarten L. Hekkelman & Thijn R. Brummelkamp * Proteomics Facility, The Netherlands Cancer
Institute, Amsterdam, the Netherlands Liesbeth Hoekman & Maarten Altelaar * Biomolecular Mass Spectrometry and Proteomics, Bijvoet Center for Biomolecular Research, Utrecht Institute for
Pharmaceutical Sciences, Utrecht University and Netherlands Proteomics Centre, Utrecht, the Netherlands Maarten Altelaar * Division of Gene Regulation, Oncode Institute, The Netherlands
Cancer Institute, Amsterdam, the Netherlands Elzo de Wit Authors * Marjon S. van Ruiten View author publications You can also search for this author inPubMed Google Scholar * Démi van Gent
View author publications You can also search for this author inPubMed Google Scholar * Ángela Sedeño Cacciatore View author publications You can also search for this author inPubMed Google
Scholar * Astrid Fauster View author publications You can also search for this author inPubMed Google Scholar * Laureen Willems View author publications You can also search for this author
inPubMed Google Scholar * Maarten L. Hekkelman View author publications You can also search for this author inPubMed Google Scholar * Liesbeth Hoekman View author publications You can also
search for this author inPubMed Google Scholar * Maarten Altelaar View author publications You can also search for this author inPubMed Google Scholar * Judith H. I. Haarhuis View author
publications You can also search for this author inPubMed Google Scholar * Thijn R. Brummelkamp View author publications You can also search for this author inPubMed Google Scholar * Elzo de
Wit View author publications You can also search for this author inPubMed Google Scholar * Benjamin D. Rowland View author publications You can also search for this author inPubMed Google
Scholar CONTRIBUTIONS M.S.v.R., D.G. and J.H.I.H. performed wet-laboratory cell biology experiments. M.S.v.R. and A.F. performed the genetic screen. A.F. and M.L.H. analyzed screen data.
M.S.v.R. and L.W. prepared Hi-C samples. Á.S.C. and D.G. analyzed Hi-C data. L.H. and M.A. performed mass spectrometry analysis. T.R.B., E.d.W. and B.D.R. provided supervision. M.S.v.R. and
B.D.R. wrote the manuscript with input from all authors. CORRESPONDING AUTHOR Correspondence to Benjamin D. Rowland. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing
interests. PEER REVIEW PEER REVIEW INFORMATION _Nature Structural & Molecular Biology_ thanks the anonymous reviewers for their contribution to the peer review of this work. Sara Osman
was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team. ADDITIONAL INFORMATION PUBLISHER’S NOTE
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. EXTENDED DATA EXTENDED DATA FIG. 1 THE LOOPING DEFECT OF _∆HDAC8_ CELLS
IS ESCO1-DEPENDENT. (A) The 10% smallest cells were sorted to obtain G1 cells. FACS plots showing the DNA content of unsorted (asynchronous) and sorted (G1) cells. Cells were fixed and
stained with DAPI. (B) Aggregate stripe analysis to quantify the signal enrichment emanating from CTCF sites at 100-kb resolution. The architectural stripe phenotype is observed in
independent clones. (C) APA analysis reveals that the extended loop phenotype is also observed in independent clones. Differential APA plots for extended loops compared to wild type (WT).
_∆ESCO1_ (_∆1_) cells show an increase in extended loops. _∆HDAC8_ (_∆8_) cells show a decrease in extended loops. (D) Western blot analysis of the indicated genotypes. This experiment was
performed twice with similar results. (E) Aggregate stripe analysis to quantify the signal enrichment emanating from CTCF sites at 100-kb resolution. The short stripes in _∆HDAC8_ cells are
rescued upon ESCO1 deletion. This phenotype is also observed in a replicate Hi-C experiment in an independent _∆ESCO1/∆HDAC8_ clone (dashed line). (F) Hi-C contact matrices for asynchronous
cells of the indicated genotypes. A locus at chromosome 2 is shown at 10-kb resolution. Matrices were normalized to 100 million contacts per sample. (G) APA for extended loops. Differential
APA plots for extended loops compared to wild type (WT). Asynchronous _∆HDAC8_ (∆8) cells show a decrease in extended loops. (H) Aggregate stripe analysis to quantify the signal enrichment
emanating from CTCF sites at 100-kb resolution. Asynchronous _∆HDAC8_ cells display shorter stripes in comparison to asynchronous wild type cells. Source data EXTENDED DATA FIG. 2 HI-C
REPLICATES IN G1 CELLS. (A) Aggregate peak analysis (APA) reveals that the extended loop phenotype is also observed in a replicate Hi-C experiment. _∆ESCO1/∆WAPL_ cells show an increase in
extended loops in comparison to _∆WAPL_ cells. (B) The extended stripe phenotypes for _∆WAPL_ and _∆ESCO1/∆WAPL_ cells are also observed in a replicate Hi-C experiment. The aggregate stripe
analysis quantifies the signal enrichment emanating from CTCF sites at 100-kb resolution. (C) APA plots show that the extended loop phenotypes are also observed in a replicate Hi-C
experiment. For _∆PDS5A_ (_∆5_ _A_) cells we used an independent clone, for _∆HDAC8/∆PDS5A_ (_∆8/∆5A_) cells we used the same clone. (D) The extended stripe phenotypes for _∆PDS5A_ and
_∆HDAC8/∆PDS5A_ cells are also observed in a replicate Hi-C experiment in the clones described in (c). The aggregate stripe analysis quantifies the signal enrichment emanating from CTCF
sites at 100-kb resolution. (E) APA in an independent _∆PDS5B_ clone confirms that PDS5B does not regulate the formation of extended loops. (F) Hi-C analysis in an independent _∆PDS5B_ clone
confirms that PDS5B does not regulate the length of architectural stripes. EXTENDED DATA FIG. 3 HAPLOID GENETIC SCREEN IN ∆_HDAC8_ CELLS. (A) Plot depicting the screen results for wild type
cells and _∆HDAC8_ cells. Several cohesin regulators are highlighted. (B) Gene-trap integration patterns for PDS5A in anti-sense (blue) or sense (red) orientation in wild type and _∆HDAC8_
cells. _∆HDAC8_ cells harbour an increase in sense insertions along the entire gene. (C) Gene-trap integration patterns for SCC2NIPBL in anti-sense (blue) or sense (red) orientation in wild
type and _∆HDAC8_ cells. The sense insertions in _∆HDAC8_ cells appear to be tolerated until exon 10, while exons 11 - 47 appear to remain essential. This pattern much resembles the pattern
found in _∆WAPL_ cells3. (D) Pulldown experiment on the core cohesin subunit SMC1 in cells lacking _ESCO1_ (_∆1_) or _HDAC8_ (_∆8_). We find that cohesin in _∆HDAC8_ cells is enriched for
binding to PDS5A, PDS5B, and WAPL. Cohesin’s binding to these factors appears to be less evidently affected in _∆ESCO1_ cells. We note that in wild type cells only a small fraction of
cohesin complexes is acetylated. These low acetylation levels could explain why it is relatively difficult to assess differences in binding of the mentioned proteins in _∆ESCO1_ cells. This
experiment was performed 3 times with similar results. (E) Pulldown experiment on the cohesin regulator SCC2NIPBL in cells lacking _ESCO1_ (_∆1_) or _HDAC8_ (_∆8_). The upper three rows
belong to one experiment and the lower two rows to another experiment. Both a short exposure (se) and long exposure (le) are shown for the core cohesin subunit SMC1. We find that the amount
of cohesin acetylation does not affect SCC2NIPBL binding to cohesin. We also find that SCC2NIPBL pulls along acetylated cohesin complexes, suggesting that cohesin acetylation and cohesin’s
binding to SCC2NIPBL are not mutually exclusive. This experiment was performed 3 times with similar results. Source data EXTENDED DATA FIG. 4 PDS5B DOES NOT CONTROL CHROMATIN LOOPING IN HAP1
CELLS. (A) Hi-C contact matrices for G1 cells of the indicated genotypes. A locus at chromosome 5 is shown at 10-kb resolution. Matrices were normalized to 100 million contacts per sample.
_∆PDS5B_ cells do not display chromatin looping defects. (B) Aggregate stripe analysis to quantify the signal enrichment emanating from CTCF sites at 100-kb resolution. PDS5B does not
control the length of stripes. (C) Aggregate peak analysis (APA) for primary loops. PDS5B does not regulate primary loops. (D) APA for extended loops. _∆PDS5B_ cells do not show an increase
in extended loops. (E) The RNA read counts of PDS5A and PDS5B in wild type HAP1 cells, from11. Mean and standard deviation are shown of three biological replicates (grey circles depict
replicates). (F) The PSM counts of PDS5A and PDS5B in whole cell proteomics in wild type HAP1 cells. Mean and standard deviation are shown of three biological replicates (grey circles depict
replicates). (G) Western blot analysis of wild type and _∆PDS5A_ cells with either untagged or tagged SCC1-HALO. (H) Quantification of the FRAP experiment in SCC1-HALO tagged G1 cells. Mean
and standard deviation for 17 wild type cells and 17 _∆PDS5A_ cells, measured over 5 independent experiments. (I) Example images of cells used in (h) at the indicated time points after
photobleaching. White scale bar is 5 µm. Note that the ∆_PDS5A_ cells display a ‘vermicelli’-like SCC1 localization. (J) Western blot analysis of wild type and _∆PDS5B_ cells with either
untagged or tagged SCC1-HALO. (K) Quantification of the FRAP experiment in SCC1-HALO tagged G1 cells. Mean and standard deviation for 12 wild type cells and 12 _∆PDS5B_ cells, measured over
4 independent experiments. (L) Example images of cells used in (k) at the indicated time points after photobleaching. White scale bar is 5 µm. Source data EXTENDED DATA FIG. 5 OVERVIEW HI-C
ANALYSES OF DIFFERENT GENOTYPES. (A) Hi-C contact matrices for G1 cells of the indicated genotypes. A locus at chromosome 4 is shown at 10-kb resolution. Matrices were normalized to 100
million contacts per sample. (B) Hi-C contact matrices for G1 cells of the indicated genotypes. A locus at chromosome 4 is shown at 10-kb resolution. Matrices were normalized to 100 million
contacts per sample. These Hi-C libraries were less deeply sequenced than the Hi-C libraries presented in (a). (C) Aggregate peak analysis (APA) for primary loops using the same scale for
all genotypes. The bottom right value depicts the APA score. (D) APA for extended loops using the same scale for all genotypes. The bottom right value depicts the APA score. SUPPLEMENTARY
INFORMATION SUPPLEMENTARY INFORMATION Supplementary Fig. 1 and Tables 1 and 2. REPORTING SUMMARY SUPPLEMENTARY TABLE Gene-trap integration genetic screen. Numbers of gene-trap integrations
per genotype in sense and antisense orientation. SOURCE DATA SOURCE DATA FIG. 1 Unprocessed immunoblots. SOURCE DATA FIG. 2 Unprocessed immunoblots. SOURCE DATA FIG. 3 Unprocessed
immunoblots. SOURCE DATA FIG. 4 Unprocessed immunoblots. SOURCE DATA EXTENDED DATA FIG. 1 Unprocessed immunoblots. SOURCE DATA EXTENDED DATA FIG. 3 Unprocessed immunoblots. SOURCE DATA
EXTENDED DATA FIG. 4 Unprocessed immunoblots. RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use,
sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative
Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated
otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds
the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and
permissions ABOUT THIS ARTICLE CITE THIS ARTICLE van Ruiten, M.S., van Gent, D., Sedeño Cacciatore, Á. _et al._ The cohesin acetylation cycle controls chromatin loop length through a PDS5A
brake mechanism. _Nat Struct Mol Biol_ 29, 586–591 (2022). https://doi.org/10.1038/s41594-022-00773-z Download citation * Received: 15 December 2021 * Accepted: 05 April 2022 * Published: 16
June 2022 * Issue Date: June 2022 * DOI: https://doi.org/10.1038/s41594-022-00773-z SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get
shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative