Play all audios:
ABSTRACT Substrate polyubiquitination drives a myriad of cellular processes, including the cell cycle, apoptosis and immune responses. Polyubiquitination is highly dynamic, and obtaining
mechanistic insight has thus far required artificially trapped structures to stabilize specific steps along the enzymatic process. So far, how any ubiquitin ligase builds a proteasomal
degradation signal, which is canonically regarded as four or more ubiquitins, remains unclear. Here we present time-resolved cryogenic electron microscopy studies of the 1.2 MDa E3 ubiquitin
ligase, known as the anaphase-promoting complex/cyclosome (APC/C), and its E2 co-enzymes (UBE2C/UBCH10 and UBE2S) during substrate polyubiquitination. Using cryoDRGN (Deep Reconstructing
Generative Networks), a neural network-based approach, we reconstruct the conformational changes undergone by the human APC/C during polyubiquitination, directly visualize an active E3–E2
pair modifying its substrate, and identify unexpected interactions between multiple ubiquitins with parts of the APC/C machinery, including its coactivator CDH1. Together, we demonstrate how
modification of substrates with nascent ubiquitin chains helps to potentiate processive substrate polyubiquitination, allowing us to model how a ubiquitin ligase builds a proteasomal
degradation signal. SIMILAR CONTENT BEING VIEWED BY OTHERS PROFILING UBIQUITIN SIGNALLING WITH UBIMAX REVEALS DNA DAMAGE- AND SCFΒ-TRCP1-DEPENDENT UBIQUITYLATION OF THE ACTIN-ORGANIZING
PROTEIN DBN1 Article Open access 14 December 2023 MECHANISM OF MILLISECOND LYS48-LINKED POLY-UBIQUITIN CHAIN FORMATION BY CULLIN-RING LIGASES Article Open access 07 February 2024
DEUBIQUITINATING ENZYMES AND THE PROTEASOME REGULATE PREFERENTIAL SETS OF UBIQUITIN SUBSTRATES Article Open access 18 May 2022 MAIN The processivity of molecular machines is paramount to the
countless biological processes that enable life. Molecular machines, such as polymerases, ribosomes or proteasomes, commonly use nucleotide-directed motors to potentiate key steps in
processive reactions, for example, translocation of substrates1,2. Recent advances in cryogenic electron microscopy (cryo-EM) have enabled the description of the physical properties of these
machines3,4. Thus far, conformations seem to be Boltzmann distributed, without large thermodynamic barriers, and molecules can sample the full space, implying microscopic reversibility at
every conformational step5. Directionality comes from kinetic asymmetries in processes that stabilize specific conformational states, for example, nucleotide hydrolysis6. The fundamentally
important ubiquitin (Ub) ligases do not directly bind or hydrolyze nucleotides as a part of their mechanism but still maintain forward-driven, processive activity7,8,9,10,11,12. The complete
conformational landscape for any of the >600 Ub ligases during polyubiquitination is unknown13. Our current understanding of how these enzymes maintain processivity and ensure that
ubiquitination proceeds in a directional manner is therefore incomplete. Substrate ubiquitination requires Ub to be passed along a cascade of E1, E2 and E3 enzymes14. E3 enzymes serve as a
hub for substrate recognition and directly mediate the transfer of Ub from the E2 to the substrate15. A key Ub ligase known as the anaphase-promoting complex/cyclosome (APC/C) drives the
cell cycle by recognizing important substrates, for example, cyclin B and securin, and ubiquitinating them to mark them for proteasomal degradation16,17. Degradation of such substrates
controls cell cycle timing, including mitosis and G1 maintenance18,19. A key determining factor in the timing of APC/C substrate degradation is the efficiency of Ub signal formation on the
substrate and whether it occurs in a processive or distributive manner8,9,11,20. This Ub signal can vary through modification of numerous lysines on the substrate or through different Ub
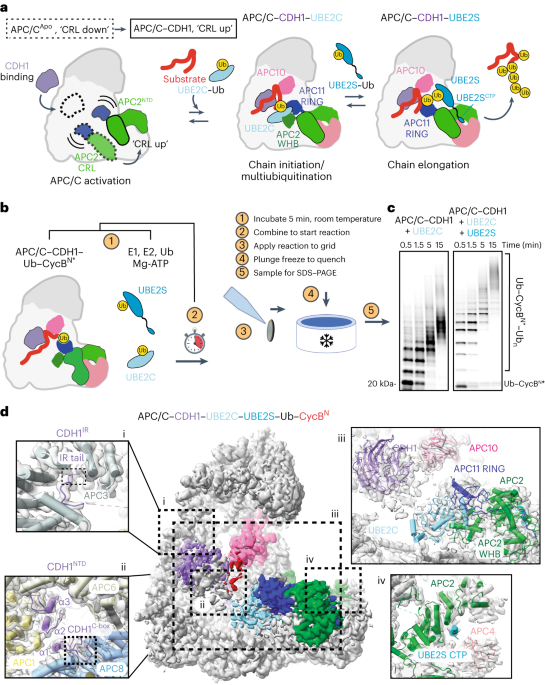
linkages (for example, K11, K48, K63 or K11/K48 branched chains)21,22,23. During a processive ubiquitination cycle, the substrate binds to APC/C coactivators (CDC20 in mitosis and CDH1 in
interphase) and APC10 (refs. 8,24,25). The binding of the coactivator elicits a conformational change in the cullin–RING (CRL) subunits APC2–APC11, which promotes the binding of the first
E2, UBE2C/UBCH10, that adds Ub directly to the substrate (Fig. 1a)26,27,28. UBE2C can either continue to modify substrate lysines or make short chains21,29,30,31. A second E2, UBE2S, extends
the K11-linked Ub chains on the substrate32,33,34. These reactions can all occur during a single substrate-binding event while the E2s require multiple rounds of transient binding and
catalysis to modify the substrate. A proteasome degradation signal is typically regarded as a substrate that is modified with four or more Ubs35. A structure of an E3 building such signal
remains elusive. So far, the structures of APC/C with either UBE2C or UBE2S, and other E3–E2 pairs, have mostly relied on chemical crosslinking approaches to lock the dynamic enzymes in
place23,27,36,37,38,39,40. These studies allow for high-resolution structural determination of active catalytic architectures but limit our understanding of the conformational landscape and
driving forces of E3-mediated polyubiquitination. A single molecule study of APC/C-dependent polyubiquitination revealed that substrate-linked Ub(s) enhance the processivity of the
ubiquitination reaction by improving the substrate binding affinity or processive affinity amplification7. However, the precise coordination of Ub-binding sites, of E2 binding and of Ub
transfer by the APC/C or any E3 to build a proteasome degradation signal remains largely undescribed. In this Article, we performed time-resolved cryo-EM (TR-EM) experiments to watch the
APC/C in action and understand its structural and biophysical properties during substrate polyubiquitination. By combining TR-EM with in vitro ubiquitination assays and total internal
reflection fluorescence (TIRF) microscopy, our work provides the framework for a sculpted energy landscape, where particularly low-energy (highly populated) intermediates are not observed
along the reaction path of APC/C-dependent polyubiquitination. In general, macromolecular machines such as the APC/C coordinate dynamic equilibrium within flexible domains with stochastic
binding of E2s to carry out their activity. These types of molecular systems therefore exhibit highly multimodal heterogeneity. We show that the APC/C couples the thermal noise power of its
flexible domains with transient stabilizing interactions and with the emerging Ub-modified substrate to drive processivity. Allosteric changes upon binding of coactivators and the chain
elongating E2 UBE2S potentiate this processivity. Together, these interactions allow us to create a model for how APC/C builds a proteasomal degradation signal. RESULTS STRUCTURE OF THE
APC/C ACTIVELY UBIQUITINATING A SUBSTRATE The APC/C and its E2s can perform a vast number of different ubiquitination reactions21,22,23,29,30,31,32,33,34,41. Trapping each of these
individual structural states has required detailed biochemical knowledge about the system to stabilize the reaction intermediates23,27,36,37,38,42. Given the multiple substrate and Ub lysine
target sites, the number of potential ubiquitination states that would require artificial trapping is huge. Furthermore, generating structures of later stages of substrate ubiquitination to
help define the determinants of processivity would require complex protein engineering to form different Ub chain linkage types. We therefore sought to solve structures of the APC/C and its
E2s performing polyubiquitination over a time course to examine its conformational heterogeneity, identify unknown states of polyubiquitination and uncover interactions between the
substrate-linked Ub and the APC/C machinery. To generate TR-EM samples of the APC/C during substrate ubiquitination, we preincubated two mixtures, one containing the APC/C, CDH1 and
substrate, and a second containing the E1, UBE2C, Mg-ATP and Ub (Fig. 1b). For a substrate, the fluorescently labeled CycB N-terminus (Ub-CycBN*) containing a fused Ub was used to ensure
that additional observed interactions resulted from the formation of polyubiquitination rather than the initial substrate priming step (Extended Data Fig. 1a). To examine the impact of UBE2S
on the structural landscape of APC/CCDH1, a second reaction was performed that included UBE2C and UBE2S. The two mixtures were combined to start the reaction, and samples were taken from
the reaction at specific timepoints (0.5, 1.5, 5 and 15 min), applied to a grid and plunge frozen. Visualization of the reaction on sodium dodecyl sulfate–polyacrylamide gel electrophoresis
(SDS–PAGE) showed an expected difference between the two time courses, with the presence of both E2s resulting in longer Ub chains than UBE2C alone (Fig. 1c). Both reactions showed
differences in levels of Ub modification between the initial and later stages of the reaction. Single-particle electron microscopy (EM) data were then collected from each grid at each
timepoint (Extended Data Fig. 1b). The data collected on a Titan Krios resulted in 25,380 and 25,900 movies for the reaction mixtures containing APC/CCDH1–Ub–CycBN–UBE2C and
APC/CCDH1–Ub–CycBN–UBE2C–UBE2S, respectively (Extended Data Fig. 1b,c). After multiple rounds of classification to filter each dataset down to ~600,000–700,000 particles, we were able to
solve high-resolution consensus structures (3–4 Å) for each dataset (Extended Data Fig. 1d–f and Table 1). Because of prior work, we were able to build the structure of APC/C interacting
with its coactivator CDH1 and UBE2C for both datasets (Fig. 1d and Extended Data Fig. 1f). The coactivator N- and C-terminal tails are also resolved in their respective binding sites, with
the CDH1 IR tail localized on APC3 and the CDH1 C-box bound to APC8 (Fig. 1d, i and ii) (ref. 27). The D-box of CycB is seen bound to the CDH1–APC10 groove and UBE2C is clasped by the APC11
RING and APC2 WHB (Fig. 1d, iii) (refs. 23,27,38). However, a key difference between these two structures (Fig. 1d and Extended Data Fig. 1f) and previous work is that UBE2C is not
artificially trapped through chemical crosslinking or through genetic fusion to either APC11 or the UBE2S C-terminus23,27,38. Density for the UBE2S C-terminal peptide (CTP) is visible in the
groove formed by APC2–APC4 in the dataset containing both E2s (Fig. 1d, iv) (refs. 23,27). Therefore, our structures validate prior work that relied on chemical crosslinking with UBE2C and
UBE2S and reveal structures of an E3–E2–E2 complex actively ubiquitinating a substrate, indicating that both E2s can bind the APC/C simultaneously23,27,38. When comparing the structure of
APC/C–CDH1–UBE2C from this study to those of the same complex from two previous studies (EMD-2925 and EMD-2929), the structures were similar, with map-to-map correlations of either 0.76 or
0.81 (Extended Data Fig. 1g)23,27,38. When we compared the structures of APC/C–CDH1–UBE2C and APC/C–CDH1–UBE2C–UBE2S generated from the two datasets in this study, we observed a map-to-map
correlation of 0.93 (Extended Data Fig. 1h). In other words, the reaction that contained UBE2S did not reveal extra density for the UBE2S catalytic core. The observed density for the
UBE2SCTP in our APC/C–CDH1–UBE2C–UBE2S structure did agree with structures from previous studies where it was resolved (Extended Data Fig. 1i). Ultimately, this is consistent with the
previous kinetic work suggesting that the UBE2SCTP provides the affinity for UBE2S to bind to the APC/C, suggesting that the intrinsic kinetic differences (on and off rates) between E2 and
E3 interactions are a potential limiting factor to solving structures of E3–E2 pairs actively ubiquitinating a substrate23,27,42,43. These differences could be due to either binding
interactions or the type of ubiquitination occurring, that is, ubiquitinating the target directly versus building a polyubiquitin chain. TOTAL HETEROGENEITY REVEALS A SCULPTED ENERGY
LANDSCAPE To provide further insight into the conformational landscape of the APC/C and its E2s and understand processive polyubiquitination, we examined the structural heterogeneity of the
APC/C during catalysis in the two datasets using cryoDRGN (Deep Reconstructing Generative Networks)3. The cryoDRGN method learns a mapping of particle images into a low-dimensional
continuous vector space, termed the latent space, and an associated neural network volume representation that can reconstruct a 3D density volume from any point in this latent space,
allowing for the visualization of a unique 3D map for each particle (Extended Data Fig. 2a). The full dataset’s latent space representation can be visualized in 2D using dimensionality
reduction techniques such as principal component analysis (PCA) and uniform manifold approximation and projection (UMAP)44. The distribution of structures can be further analyzed by
_k_-means clustering on the latent space representation, with particles classified into various states based on their cluster assignments. For each of the two time courses, particles across
all timepoints were combined and used to train the neural network. UMAP visualization of the latent space for both time courses revealed a continuous, featureless surface, suggesting a
convoluted landscape with high degrees of overlap between the distribution of structures across time points (Extended Data Fig. 2b,c). A comprehensive sampling of 500 structures from the
latent space at the _k_-means cluster centers (_k_ = 500) showed largely similar compositional and conformational variability between the datasets and recapitulated several known APC/C
conformations (Extended Data Fig. 2b,c). Trajectories generated along the principal components of the latent space captured the broad conformational changes of the APC/C. For example, the
trajectory along the first component (PC1) shows the continuous motion through which the APC2 CRL domain (the C-terminus of APC2) transitions from the ‘CRL down’ to the ‘CRL up’ conformation
(Fig. 2a). Along the second component (PC2), the APC2 CRL can be visualized transitioning from an intermediate, partially raised ‘CRL up’ conformation to the fully raised ‘CRL up’
conformation with clear density for UBE2C present (Extended Data Fig. 2d and Supplementary Video 1). By applying PCA on the 500 volumes generated at the _k_-means cluster centers, we were
able to uncover interesting aspects of conformational variability of the APC/C (Extended Data Fig. 3). Along the first volume PC trajectory, we can trace the binding of the coactivator
together with the simultaneous displacement of the APC2 CRL domain from the ‘down’ to the ‘up’ conformation, resulting in a ~15 Å displacement. The second volume PC trajectory follows a
CDH1-bound state where the CRL is partially in the ‘up’ conformation and undergoes an ~11 Å displacement to the ‘CRL up’ conformation. The third and fourth PCs map heterogeneity at the
coactivator and the catalytic core, with CDH1 displacements of ~10 and ~11 Å visualized, respectively. Changes in density present near the coactivator and contacts made and broken between
the components involved in the catalytic core (CDH1, UBE2C, APC11, APC2CRL and APC10) are also visualized, demonstrating the power of cryoDRGN for visualizing complex heterogeneity. Extra
density, particularly near the coactivator and UBE2C visualized in PC3 and PC4, suggests heterogeneity at the catalytic core due to the presence of substrate and Ub. A systematic analysis of
the 500 cryoDRGN volumes revealed four major conformational states: APC/C with the APC2CRL domain in a ‘CRL down’ conformation, an APC/CCDH1-‘CRL up’ conformation, and two categories of
states where either APC7, located at the top of the APC, or the APC2 CRL–APC11 RING domains showed high degrees of variability (labeled ‘Partial APC7 and ‘CRL unresolved’, respectively)
(Fig. 2b and Extended Data Fig. 4). While the biological relevance of the ‘Partial APC7’ and ‘CRL unresolved’ states is currently unclear, some differences within these states can be noted.
For example, the classes where APC2–APC11 are unresolved all contained CDH1. Additionally, extra density was observed around the coactivator and the APC2 N-terminal domain remained clearly
resolved, suggesting the potential for dynamics within the APC2–APC11 domains that are not captured by our methods. The mapping of the four major states onto the UMAP visualization of the
particle distribution in the latent space revealed a convoluted landscape with some degree of overlap between the states (Fig. 2c,d). Interestingly, the mapping of the distribution of the
states relative to one another was preserved in the landscapes of both datasets. Between the two datasets, the one containing only UBE2C showed higher fractions of particles in the ‘Partial
APC7’ and ‘CRL unresolved’ states, suggesting a potential role for UBE2S in stabilizing these domains or the APC/C overall (Fig. 2e,f). Both datasets showed similar levels of ‘CRL down’
particles. Overall, the distribution of particles within the states changed relatively little across the timepoints, suggesting an energy landscape void of new highly populated APC/C
intermediates (∆∆_G_ < 2 kBT) in the mixture in any of the possible conformational states at a given time and regardless of the extent of overall substrate ubiquitination. This
observation suggests that polyubiquitination is not a process with large energy barriers, and instead the reactions are stochastic, with thermal noise and allostery due to the simultaneous
binding of multiple components driving the conformational states. Thus, directionality in the processivity of the reaction needs to be introduced by different means than energy barriers.
UBE2C ASSOCIATION WITH APC/C IS INCREASED BY CDH1 AND UBE2S Further analysis of the volumes from _k_-means clustering major states revealed additional characteristics about APC/C
conformational dynamics. The ‘CRL down’ major state clusters could be subdivided into two substates: an apo state that showed no density near APC10 that could be attributed to CDH1 and a
small minority of clusters that did show density consistent with CDH1 bound (Fig. 3a and Extended Data Fig. 5a). The much higher level of ‘CRL down’ in the apo ‘CDH1 unbound’ state is
consistent with previous data showing that coactivator binding mediates the transition from the ‘CRL down’ to the ‘CRL up’ states26,28. For the ‘CRL up’ state, where we expect that allostery
through binding is a directing force, we examined if the composition of these states is altered by the presence of UBE2S, as suggested previously45. When UBE2S is present, the UBE2C-bound
fraction is slightly increased at each timepoint (Fig. 3b and Extended Data Fig. 5b). In the reaction without UBE2S, UBE2C is present in ~40% of the particles. When UBE2S is added, UBE2C is
present in ~50–60% of the particles over the time course. This finding suggests that UBE2S improves the association of UBE2C with the APC/C and potentiates multiubiquitination. To
investigate cooperative interactions between the APC/CCDH1-associated E2s on a single-molecule level, we developed a TIRF microscopy-based system. Briefly, a fluorescently labeled,
biotinylated substrate (biotin–CycBN*) was immobilized within flow chambers functionalized with neutravidin (Fig. 3c). UBE2C labeled with JaneliaFluor549 (UBE2C-JF549) was mixed with
ubiquitination reaction components, including unlabeled APC/C, then added to the flow cells immediately before data acquisition. These tagged and labeled reagents retained their
functionality in substrate ubiquitination reactions (Extended Data Fig. 5c). Within the flow chamber, APC/CCDH1 will bind the immobilized substrate and recruit UBE2C–JF549 from solution to
the slide surface for ubiquitination. Therefore, fluorescence signal events at the slide surface provide a readout of UBE2C–JF549 recruitment by APC/CCDH1. We observed minimal background
fluorescence from the functionalization process, robust signal from immobilized biotin–CycBN*, and minimal signal from the fluorescent substrate in the UBE2C channel, indicating that our
TIRF system allows for a clear delineation between substrate and E2 signals at the slide surface (Extended Data Fig. 5d–h). Next, we sought to assess how the UBE2SCTP impacts UBE2C
recruitment by titrating a UBE2SCTP 18mer peptide into the reaction. Addition of the UBE2SCTP substantially increased the total events of UBE2C localizing to the slide surface, indicating
that the UBE2SCTP enhances UBE2C recruitment to APC/CCDH1-bound substrate (Fig. 3d, Extended Data Fig. 5f and Supplementary Video 2). This result supports prior work that UBE2S is capable of
stimulating UBE2C activity through its unique binding arrangement on the APC/C45. Altogether, we demonstrate coordination between UBE2C and the UBE2SCTP at APC/CCDH1 and present a flexible
system to study substrate-dependent interactions with single-molecule resolution. Furthermore, this system corroborates our structural data that UBE2S assists in stabilizing the ‘CRL up’
conformation, permitting UBE2C to bind the APC/C. APC/CCDH1–UBE2C STRUCTURES MEDIATING POLYUBIQUITINATION So far, most structures of RING E3s bound to an E2 involve an unmodified substrate
mimicking substrate priming or a monoubiquitinated substrate to mimic monoubiquitination, multiubiquitination (targeting multiple lysines on a substrate), or specific chain elongation
(Extended Data Fig. 6a)23,27,36,37,38,39,40. Since our datasets involve the entire polyubiquitination process, we rationalized that localized classification approaches would help us identify
APC/C structures further along polyubiquitination than ever before. This processing resulted in unexpected insights into RING E3–E2-dependent polyubiquitination. Local classification using
a mask around the active site resulted in a set of unprecedented structures for APC/C–UBE2C-mediated polyubiquitination. In addition to known interactions, multiple additional Ub
interactions were identified. First, a Ub is bound to the RING domain on the opposite side of the UBE2C-binding site (Fig. 4a, left). This finding is particularly interesting as prior work
relied on a tight-binding ubiquitin variant (UbV) to observe this interaction23. Next, we observed unexpected density consistent with the size and shape of Ub at the coactivator, suggesting
a potential Ub-binding site (Fig. 4a, center left). Additional density is observed between the coactivator and the C-terminal region of UBE2C known as the Helix-Turn-Helix (HTH) (Fig. 4a,
center right). Intriguingly, a subset of particles contained density where seemingly a di-Ub contacts both the coactivator and the E2 simultaneously (Fig. 4a, right). As Ub binding can
modulate the activity of E2s and E3s, these potential Ub interactions with either APC11 RING, UBE2C or CDH1 could have unappreciated effects on APC/C-dependent ubiquitination. Since we
previously identified that Ub can interact with the APC11 RING domain and the HTH from other E2s (refs. 23,46), we sought to examine the function of the unexpected Ub-binding site on CDH1.
As tight-binding UbVs have been tremendously helpful in disentangling the regulation of the several enzymes of the Ub system23,47,48,49,50, including the APC11 RING domain, we selected a
library of UbVs against both APC/C coactivators, CDH1 and CDC20 (Fig. 4b). This process revealed a UbV (henceforth known as UbVCDH1) that was substantially enriched during the selection
(Fig. 4c). To validate that UbVCDH1 interacts with the coactivators in a manner similar to wild-type Ub, we performed a series of tests. First, a co-pulldown assay found that a GST–UbVCDH1
fusion bound both CDC20 and CDH1 better than the GST–Ub fusion (Fig. 4d). Because the binding site of UbVCDH1 is potentially in proximity to the KEN-box degron binding site, we expected
UbVCDH1 to inhibit APC/C-mediated ubiquitination. Consistent with this hypothesis, UbVCDH1 inhibited APC/C substrate degradation in mitotic extracts of HeLaS3 cells (Fig. 4e). Conversely,
substrate degradation rates were not influenced by the presence or absence of excess Ub upon the addition of UBE2C (Extended Data Fig. 6b). UbVCDH1 was then used as surrogate to determine
the structure of APC/CCDH1 bound to Ub. To optimize UbVCDH1 for structural studies, we tested if UbVCDH1 could be fused to either the KEN-box or D-box of Hsl1, an APC/C substrate, to further
strengthen its interaction with the APC/C. As expected, the UbVCDH1–Hsl1 D-box fusion markedly inhibited APC/C-mediated substrate ubiquitination compared to either the UbVCDH1 alone or the
UbVCDH1-Hsl1 KEN-box fusion (Fig. 4f and Extended Data Fig. 6c). Therefore, APC/CCDH1 bound to UbVCDH1–Hsl1 D-box was subjected to cryo-EM. As expected, the Hsl1 D-box was bound between
APC10 and CDH1 and the UbVCDH1 was bound to CDH1 near the KEN-box binding site (Fig. 4g and Extended Data Fig. 6d). These data are consistent with the ubiquitination assays showing that the
UbVCDH1–Hsl1 KEN-box fusion weakly inhibited the APC/C-mediated substrate ubiquitination compared to UbVCDH1 alone (Fig. 4f and Extended Data Fig. 6c). MULTIPLE UB-BINDING SITES PROMOTE
PROCESSIVE UBIQUITINATION With the identification of multiple Ub-binding sites, we wanted to explore their contributions to APC/C-dependent ubiquitination. In addition to UbVCDH1, another
UbV (henceforth known as UbVRING) was previously developed to bind to the APC11 RING exosite and was used to trap structures of the APC/C with UBE2C and UBE2S (ref. 23). To specifically
examine how these Ub-binding sites synergize to promote ubiquitination, we used these two UbVs to specifically disrupt these Ub-dependent interactions (Fig. 5a). First, we performed
single-encounter experiments to specifically interrogate the different effects of the two UbVs in either substrate priming (the addition of the first Ub) or processive polyubiquitination.
APC/C, CDH1 and a fluorescently labeled substrate are preincubated separately from a mixture of E1, E2, MgATP, Ub and a large excess of nonfluorescent substrate (Fig. 5b). Therefore, only
the substrate that is preincubated with APC/C is modified during a single binding event. As expected, the UbVRING reduced the number of Ubs added to the substrate during processive
ubiquitination but did not disrupt substrate priming (Fig. 5c,d and Extended Data Fig. 7a). Surprisingly, the UbVCDH1 inhibited both reactions even though the fluorescent substrate is
preincubated with APC/CCDH1. We then performed substrate-independent experiments to determine if the observed inhibition is specific to substrate ubiquitination or another step in the Ub
cascade. Both the APC/C-dependent hydrolysis of an oxyester version of UBE2C~Ub and di-Ub synthesis by APC/C–UBE2S were unimpeded by the UbVCDH1 (Extended Data Fig. 7b,c). Therefore, we
tested the ubiquitination of several substrates in multiple turnover experiments and found that all substrates were inhibited by UbVCDH1 (Extended Data Fig. 7d). To specifically test the
degron dependence of these effects, we purified degron variants of Ub–Securin. Interestingly, the addition of both UbVs inhibits Ub–Securin ubiquitination when both degrons were mutated,
indicating that the UbVs are preventing substrate recruitment through the added Ub (Fig. 5e and Extended Data Fig. 7e). However, UbVCDH1 inhibited Ub–Securin KENMut/DMut ubiquitination more
than the UbVRING when UBE2C was used as the E2, suggesting that the UbVCDH1 has additional inhibitory mechanisms. Our experiments suggested that UbVCDH1 could also serve as an acceptor,
receiving Ub from UBE2C instead of UBE2S. To support our hypothesis, a fluorescently labeled version of UbVCDH1 (*UbVCDH1) was subjected to ubiquitination by APC/C–UBE2C. Indeed, *UbVCDH1
could be modified in both a CDH1- and APC2 WHB-dependent manner (Fig. 5f). These results reveal that the CDH1 and RING Ub-binding sites promote substrate binding and improve the processivity
of the APC/C. Additionally, the coactivator-bound Ub can also serve as an acceptor for Ub chain formation by UBE2C, but not UBE2S (Extended Data Fig. 7f). Given that these interactions with
Ub are probably very weak on their own, we turned to an innovative technique called mechanically transduced immunosorbent assay (METRIS) to examine the role of Ub-binding sites in providing
additional affinity for the substrate51. METRIS is a single-particle method that can provide relative, but quantitative, assessments of binding with high statistical power. Biotinylated
APC/CCDH1 was used to coat magnetic beads, then added to a functionalized surface of either CycBN or Ub–CycBN, and subjected to a rotating magnetic field. The distance the bead rolls is
indicative of the strength of the protein–protein interaction and is noted as the rolling parameter (RP) (Fig. 5g). Ub–CycBN displayed a significant increase in its RP (0.21 ± 0.007)
compared to CycBN (0.15 ± 0.004), supporting the idea that Ub enhances substrate binding (Fig. 5h). Additionally, when the UbVs were added to competitively remove this noted increase, only
UbVCDH1 substantially reduced the RP to 0.19 ± 0.007, which is still higher than the RP of APC/CCDH1 to CycBN (Fig. 5h). To focus on the role of CDH1 in promoting the binding between APC/C
and Ub, we performed experiments in the presence or absence of CDH1 and only used Ub on the functionalized surface. As expected, the addition of CDH1 to the APC/C enhanced the RP between the
APC/C and Ub from 0.14 ± 0.004 to 0.17 ± 0.006 (Fig. 5i). Furthermore, the addition of both UbVs completely eliminated this improvement in the RPs to background levels (0.13 ± 0.003),
suggesting that both the APC11 RING and CDH1 contribute to Ub binding. Combined, these findings suggest that multimodal interactions between substrate-linked Ubs and the coactivators, as
well as the APC/C itself, work together to stabilize the emerging Ub chain and facilitate the processivity and efficiency with which the APC/C modifies its substrates (Fig. 5j). DISCUSSION
To interpret molecular mechanisms, data from structural biology approaches, for example, X-ray crystallography or cryo-EM, are combined with information from biochemical or biophysical
assays. Structural studies deliver static snapshots of chemically or thermodynamically stabilized machines. Biochemical assays can only monitor a restricted set of atomic arrangements within
the machine at the same time. E3 Ub ligases are under intense regulation through transiently binding cofactors and perform many tasks, including recruiting different substrates, interacting
with multiple E2s and forming different types of Ub chains. Therefore, trying to connect biochemical assays with structural studies to understand processive reactions remains challenging.
Furthermore, understanding the driving forces and resolving key intermediate states of an evolving, Ub-modified substrate remains largely elusive. To combat this problem, we performed TR-EM
experiments of the APC/C actively polyubiquitinating a substrate with its E2s. Instead of chemically trapping the APC/C in a distinct polyubiquitination architecture, we examined APC/CCDH1
modifying Ub-CycBN over a time course through manual plunge freezing. Using advanced deep-learning approaches for analyzing structural heterogeneity, we were able to generate a functional
conformational landscape, revealing a sculpted energy landscape without high-energy populated intermediates, suggesting the absence of new thermodynamic barriers for APC/C-mediated
polyubiquitination with either UBE2C or both E2s. These landscapes are stochastic with few minima or barriers. Instead, we hypothesize that thermal noise is the primary driving force and
changes in the substrate, Ub and E2 interactions result in subtle allosteric changes to the APC/C scaffold that are difficult to distinctly classify. In short, there is one mode of action of
the APC/C that can mediate several tasks, and subtle changes in the immediate environment, such as the ubiquitination state of the substrate or E2 bindings, modulate its function. This type
of landscape can also be readily tunable by additional regulators. Since the APC/C is tightly regulated by multiple kinases, phosphatases and inhibitors, each component may only slightly
alter the conformational landscape or result in different conformational pathways during different stages of the cell cycle18,52. While there are methodological limitations inherent to any
structural method, for example, the air–water interface in cryo-EM, we reproduced several known states on the basis of structures that have been prepared in different ways from different
groups, providing support for our workflow. Future work performing this type of analysis on analogous E3s can reveal how different binding partners and post-translational modifications, such
as cullin neddylation, stabilize their active states40,53. It was previously shown by single-molecule studies of APC/C-dependent ubiquitination that substrate-linked Ubs either help drive
further catalysis through processive affinity amplification or track the Ub chain7,54. Our analysis revealed one previously discovered Ub-binding site on APC11 and two potential Ub-binding
sites on the coactivator and UBE2C HTH23. We speculate that these potential Ub-binding sites could be used to enhance the processivity of polyubiquitination by increasing the substrate
binding affinity for the APC/C, as suggested by our METRIS data. However, these Ub-binding sites could have additional roles during polyubiquitination. For example, Ub binding to the
UBE2CHTH might also allosterically modulate UBE2C activity. The Ub–coactivator interaction could similarly have multiple implications. First, because UbVCDH1 can serve as an acceptor, the
coactivator could be presenting the Ub in a particular orientation for short chain elongation by UBE2C. Alternatively, the Ub-binding sites might be positioned in a manner to allow the
substrate lysine to be modified by UBE2C instead of the Ub. This hypothesis is potentially interesting as the Ub-binding site on the coactivator is near the substrate KEN-box binding site.
Since the APC/C coactivators are a key point of regulation by the cell and facilitate substrate recruitment to the APC/C, these additional Ub-binding interactions are probably subject to a
series of small changes in the reaction environment and may facilitate the variety of polyubiquitination outcomes of APC/C-dependent reactions, for example, cell cycle progression. Taken
together, Ub-binding sites on the substrate receptor may be a recurring theme across E3 ligases as it may help control the extent of polyubiquitination. Several known interactions were not
present in our current processing, suggesting there are probably many additional insights present in this dataset. For example, Ub binding to the APC2 WHB was not observed in our
refinements47. Additionally, while we observed UBE2C bound to the APC/C in both datasets, the UBE2S core was never observed even though the UBE2SCTP was clearly visible. The UBE2S core binds
very weakly to APC2, but the UBE2SCTP binds tightly to the APC2–APC4 groove23,27,42. Nonetheless, both E2s were bound in the APC/CCDH1–UBE2C–UBE2S polyubiquitination dataset. As substrate
priming is typically regarded as the slow step, while chain elongation is relatively quicker, our work suggests that intrinsic differences in the reaction and/or binding kinetics of the E2s
is a limiting factor for this type of approach for different E2–E3 pairs10,12,55. In conclusion, we examined APC/C-mediated substrate polyubiquitination through an innovative approach,
revealing the conformational landscape for substrate polyubiquitination by the APC/C. The APC/C is regulated throughout the cell cycle, and a sculpted landscape is perhaps necessary to
finely tune its activity in different contexts. Furthermore, the processivity of polyubiquitination does not come directly through nucleotide binding and/or hydrolysis but rather the Ub
binding to the E3. It is well established that E3 ligases are modulated by post-translational modifications and protein–protein interactions. Therefore, as we revealed several fundamental
insights into APC/C-mediated polyubiquitination in relatively few experiments, we expect this method will be useful for the future studies of other E3s. METHODS PROTEIN PURIFICATION
Recombinant APC/C was expressed in High Five insect cells (Thermo Scientific) as previously described56. APC/C with a Twin-Strep(II)-tag on the APC4 C-terminus was purified via affinity
chromatography, ion-exchange chromatography and size exclusion chromatography (SEC). CDH1, UBE2C, UBE2S, UBA1, Ub, methylated Ub, Ub–CycB, Ub–Securin, Securin and CycB were purified as
described42. For enzyme assays, CDH1 and CDC20 were expressed in baculovirus-infected insect cells with an N-terminal Myc(3x)-Hexahistidine tag and Flag(3x) tag, respectively. CDH1 and CDC20
were then subjected to Ni-NTA or anti-Flag M2 resin, respectively, and the N-terminal tags were removed by HRV 3C protease. The proteins were further purified by ion-exchange chromatography
and SEC. For phage display, both CDH1 and CDC20 were purified using an uncleavable Myc(3x)-Hexahistidine tag. GST–UbVCDH1 and GST–Ub for co-pulldown experiments were purified using
GST-affinity chromatography and SEC. Fluorescent UbVCDH1 was conjugated to a fluorescent LPETGG peptide via a sortase-dependent reaction and purified by buffer exchange, Ni-NTA resin and gel
filtration to remove sortase and unconjugated peptide. UbVCDH1–Hsl1–degron fusions were designed to contain either the Hsl1 D-box (HHHHHHENLYFQSGGGMQIFVKTRKGTSITLEVEPSDTIENVK
AKIQDKEGIPPDQQRLRFAGKQLEDGRTLSDYNIHNASSLYLRSPLRALAGGSGSGSSSYLEEQKPKRAALSDITNSFNKMN) or the Hsl1 KEN-box
(HHHHHHENLYFQSGGGRLRISGVSTNKENEGPEYPTKIGSGSGSGSMQIFVKTRKGTSITLEVEPSDTIENVKAKIQDKEGIPPDQQRLRFAGKQLEDGRTLSDYNIHNASSLYLRSPLRALAG). Both UbVCDH1–Hsl1 D-box and UbVCDH1–Hsl1 KEN-box were purified
using nickel-affinity chromatography and SEC. GST–TEV–AviTag–Cyclin B (1–88)–Cys–6xHis or GST–TEV–AviTag–Ub–Cyclin B (1–88)–Cys-–6xHis constructs were generated as the substrate to
immobilize for TIRF microscopy or METRIS. Once purified, the substrate was biotinylated and fluorescently labeled at the C-terminal cysteine using Alexa488–maleimide. A UBE2C–LPETGG–6xHis
construct was expressed in BL21 (DE3) RIL _Escherichia_ _coli_ and purified. UBE2C was conjugated to a polyethylene glycol (PEG) modified peptide (GGGG–PEG–k(N3)NH2) through a
sortase-dependent reaction and repurified. The UBE2C–LPETGG–PEG–k(N3)NH2 was fluorescently labeled with JF549 through click chemistry by reacting 50 μM of UBE2C with 250 μM CuSO4, 1.25 mM
THPTA and 2.5-fold excess functionalized JF549 dye for 1.5 h on ice. The reaction was quenched with 5 mM ethylenediaminetetraacetic acid and purified using SEC. SYNTHESIS OF REACTIVE JANELIA
DYE JF549 JF549 CO2H (Tocris, 5 mg, 0.0088 mmol) was reacted with Alkyne–PEG–2NH2 (2.52 mg, 0.0176 mmol) in the presence of diisopropylcarbodiimide (1.633 µl, 0.0106 mmol),
_N_-hydroxysuccinimide (1.52 mg, 0.0132 mmol) and diisopropylethylamine (4.59 µl, 0.026 mmol) in 500 µl of dimethylformamide (Supplementary Fig. 1). The reaction was allowed to stir at room
temperature overnight. JF549-Alkyne was purified by high-performance liquid chromatography using a linear gradient of 0–100% A:B (A: 95% H2O:5% CH3CN + 0.1% trifluoroacetic acid; B: 95%
CH3CN:5% H2O + 0.1% trifluoroacetic acid). The solvent was removed by rotary evaporation and further dried under high vacuum. Yield: 4.02 mg, 0.0069 mmol, 78.8%. Nuclear magnetic resonance
(NMR) spectra were recorded on a 400 MHz spectrometer. NMR (400 MHz, CDCl3): _δ_ 2.40 (t, 4_J_ = 2.0 Hz, 1H, alkynyl CH), 2.54 (qt, 3_J_ = 7.6 Hz, 4H, azetidinyl b-CH2), 3.60–3.66 (m, 8H,
-OCH2CH2OCH2CH2NH-), 4.12 (d, 4_J_ = 2.4 Hz, 2H, propargylic CH2), 4.22 (t, 3_J_ = 7.6 Hz, 8H, azetidinyl a-CH2), 6.28 (d, 4_J_ = 2.0 Hz, 2H), 6.34 (dd, 3_J__1_ = 9.2 Hz, _4__J__2_ = 2.0 Hz,
2H), 6.95 (d, 3_J_ = 9.2 Hz, 2H), 7.64 (br t, 3_J_ = 4.8 Hz, 1H, CONH-), 7.72 (s, 1H), 8.00 (d, 3_J_ = 8.4 Hz, 1H), 8.24 (d, 3_J_ = 8.4 Hz, 1H). Exact mass 580.24421 _m_/_z_. Observed mass
580.24484 _m_/_z_. CRYO-EM SAMPLE PREPARATION Time-resolved EM reaction components were combined in two mixes at the following concentrations: 0.7 µM APC/C, 0.7 µM CDH1, 1.25 µM Ub–CycBN*
(mix 1) and 2.5 µM UBE2C, ± 2.5 µM UBE2S, 0.2 µM E1, 16 µM Ub and 5 mM Mg-ATP (mix 2). Mixes 1 and 2 were incubated for 5 min at room temperature, then combined to start the reaction.
Samples were taken at 0.5 s, 1.5 min, 5 min and 15 min, applied to a quantifoil (1.2/1.3) 400 Cu mesh grid, blotted using a Leica plunge freezer and plunge frozen in liquid ethane. Samples
were taken for SDS–PAGE at the time of plunge freezing and visualized using a Typhoon fluorescent scanner. APC/C–CDH1–UbVCDH1–Hsl1 D-box complex was prepared by incubating 0.7 µM APC/C with
1.5 µM CDH1, 90 µM UbVCDH1–Hsl1 D-box and 0.005% fluorinated octyl maltoside. The mixture was applied to a quantifoil (1.2/1.3) 200 Cu mesh grid, blotted using a Leica plunge freezer for 2 s
and plunge frozen in liquid ethane. CRYO-EM DATA ACQUISITION The grids were imaged using a Titan Krios (Max-Planck Institute for Multidisciplinary Science) on a K2 detector. Images were
collected at a nominal magnification of 180,000× and a pixel size of 0.82 Å per pixel. Images contained 39 frames with a total dose of 42 e− Å−2. A total of 25,380 micrographs were collected
for the APC/C–CDH1–UBE2C dataset and 25,900 micrographs for the APC/C–CDH1–UBE2C–UBE2S dataset (Extended Data Fig. 1b). EM samples containing APC/C–CDH1–UbVCDH1–Hsl1 D-box were imaged using
a Titan Krios G4 (Vienna) with a cold field emission gun and a post-column Selectris energy filter (ThermoFisher) with a 10 V slit, width and a Falcon 4i direct electron detector
(ThermoFisher). Images were collected at a magnification of 180,000× and a pixel size of 0.951 Å per pixel, with a cumulative total dose of 40 e− Å−2. IMAGE PROCESSING Images for both
datasets were motion-corrected using MotionCorr2 in Relion57,58. Contrast transfer function (CTF) correction was done in CryoSPARC using CTFFIND4 (refs. 59,60,61). 2D classification was
carried out in CryoSPARC in three iterative rounds to filter out contamination and bad particles. The particles in the best classes were used to generate an ab initio model, with 661,289
particles in the APC/C–CDH1–UBE2C and 774,933 particles in the APC/C–CDH1–UBE2C–UBE2S dataset. This initial model was then used as a reference for 3D refinement in CryoSPARC, generating a
4.0 Å model for the APC/C–CDH1–UBE2C dataset and 3.5 Å model for the APC/CCDH1–UBE2C–UBE2S dataset (Fig. 1d and Extended Data Fig. 1f). A local resolution map for both final models was
generated using the local resolution estimation tool in CryoSPARC (Extended Data Fig. 1d). Images for the APC/C–CDH1–UbVCDH1–Hsl1 D-box dataset were preprocessed (patch motion correction and
CTF estimation) using CryoSPARC Live. 2D classification was carried out in CryoSPARC in 30 iterative rounds to filter out contamination and bad particles. 3D classification was then used to
select for particles containing both CDH1 and UbVCDH1–Hsl1 D-box density. Particles in the best class were 3D refined in CryoSPARC, generating a model at 3.88 Å (Fig. 4g). MODEL BUILDING
Cryo-EM density maps generated using 3D homogeneous refinement were sharpened using deepEMhancer and fit with existing models of APC/C using flexible fitting tools62. Initial atomic
coordinates for both the APC/C–CDH1–UBE2C and APC/C–CDH1–UBE2C–UBE2S–Ub–CycBN models were generated using the cryo-EM structure of APC/C–CDH1 (Protein Data Bank (PDB) 7qe7). Models were
fitted using the fit-in-map rigid-body fitting tool in UCSF ChimeraX and refined using Isolde63,64. Atomic coordinates for UbVCDH1–Hsl1 D-box were generated using ColabFold structure
prediction65. The cryo-EM density map for APC/C–CDH1–UbVCDH1–Hsl1 D-box was fitted using rigid body fitting using PDB 5a31 to fit the backbone of the APC/C and CDH1 and the
ColabFold-generated structure of UbVCDH1–Hsl1 D-box. CRYODRGN HETEROGENEITY ANALYSIS For cryoDRGN analysis, particles from each dataset were downsampled in CryoSPARC to a 200-pixel box size
(2.35 Å per pixel) and used together with CTF parameters and poses from the 3D refinement to train a cryoDRGN eight-dimensional latent variable model for 60 epochs. The encoder and decoder
architectures consisted of three hidden layers with 1,024 nodes. The model was analyzed using the cryoDRGN analysis tools described in Zhong et al.3, to visualize the latent space and
generate representative volumes and trajectories. _k_-Means clustering with _k_ = 500 was used to analyze the latent embeddings of the particles, with volumes generated at the cluster
centers using the decoder network to sample the heterogeneity. The latent embedding of the particle distribution was visualized using data dimensionality reduction techniques such as PCA and
UMAP (Extended Data Fig. 2b). The cryoDRGN ‘analyze_landscape’ tool was used to carry out PCA on the volumes generated using _k_-means (_k_ = 500) clustering (Extended Data Fig. 3a). The
500 volumes generated by the _k_-means clustering analysis were assigned manually to one of four major states (‘CRL down’, ‘CRL up’, ‘CRL unresolved’ and ‘Partial APC7). Particles from each
of these four major states were extracted and 3D refinements were generated using homogeneous refinement in CryoSPARC, resulting in maps at resolutions of 4.8 Å, 4.1 Å, 4.2 Å and 4.3 Å,
respectively (Fig. 2b and Extended Data Fig. 4a). The minor states ‘CRL down, CDH1 bound’, ‘CRL down, CDH1 unbound’, ‘CRL up, partial’, ‘CRL up, no UBE2C’ and ‘CRL up, UBE2C bound’ were
assigned by visual inspection of the major states. COVERSLIP PREPARATION Glass coverslips (Corning) were cleaned, functionalized through silanization and PEGylation, and assembled into flow
chambers for TIRF microscopy experiments. All buffers were 0.22 μm sterile filtered. Coverslips were sonicated for 30 min in 1 M KOH, 30 min in 200-proof EtOH, then 20 min in deionized (DI)
water. Coverslips were silanized by sonicating for 5 min in MeOH, rocked for 12 min in a solution of 93.5% MeOH, 5% acetic acid and 1.5% (3-aminopropyl) triethoxysilane, sonicated for 1 min,
rocked in solution for another 12 min, then sonicated for 60 min in DI water. Pairs of coverslips were sandwiched together with a solution of 100 mM NaHCO3, 500 mM K2SO4, 18% succinimidyl
valerate–PEG and 0.4% biotin–PEG, then incubated in the dark overnight in humid conditions. Glass slides (Fisher Scientific) were modified with diamond drill bits to have three pairs of
holes on opposite sides of the slide. Thin strips of double-sided tape were laid across the width of the slide on the outside of the holes and between each pair. Functionalized coverslips
were washed with DI water, dried and placed on top of the taped slide, then sealed with coverslip sealant to create flow chambers. Once assembled, flow chambers were incubated with 100 mg
ml−1 bovine serum albumin (BSA) in assay buffer (20 mM HEPES pH 8 and 200 mM NaCl, 0.22 μm filtered) for 30 min, then a 0.5 mg ml−1 neutravidin and 100 mg ml−1 BSA mixture for 5 min, then a
50 nM Cyclin B–Alexa488 and 20 mg ml−1 BSA mixture for 2 min. The chambers were washed after each incubation step. TIRF MICROSCOPY TIRF microscopy was performed on an IX81 Olympus microscope
equipped with a 100× TIRF objective (UPLAPO100XOHR, numerical aperture 1.5, Olympus), four solid-state OBIS lasers (Coherent OBIS: 405 nm, 488 nm, 561 nm and 647 nm; controlled via
CoherentConnection software), and two scientific complementary metal–oxide–semiconductor cameras for dual-color imaging. The microscope was controlled using MicroManager software66. An
initial image of the immobilized substrate within flow chambers was taken with the 488 nm laser at 4 mW power to verify substrate attachment and determine the region of interest for
subsequent analysis. Experiment movies were collected using the 561 nm laser at 2 mW power with 200 ms exposure over a 10 min period after adding reaction components to the flow chamber.
Experimental reactions were made by premixing a phosphomimetic version of APC/C36,67 and CDH1 (mix 1), and premixing BSA, UBE2C–JF549 and Ub (mix 2). Final reaction mixtures contained 30 nM
pE APC, 100 nM CDH1, 20 mg ml−1 BSA, 200 nM UBE2C–JF549, 5 μM Ub, 100 nM UBA1, 0.8% d-glucose, 5.6 mg ml−1 glucose oxidase, 0.34 mg ml−1 catalase, 2 mM TROLOX and a titration of UBE2S CTP.
These mixtures were combined with 5 mM ATP-MgCl2 and added to flow chambers before data acquisition. To validate that our system, ubiquitination of 50 nM biotin–CycBN–A488 was monitored over
a short time course using SDS–PAGE (Extended Data Fig. 5c). TIRF MICROSCOPY DATA ANALYSIS For each condition, a set of _N_ = 3 experiments were carried out. ImageJ/FIJI was used to extract
a standardized region of interest from experimental movies. In MATLAB 2023a, images were background-subtracted, and single molecules were isolated using an algorithm based on á trous wavelet
decomposition68. Long-lasting events were linked across frames using a well-established algorithm69. Our analysis utilized the MATLAB Curve Fitting Toolbox, Image Processing Toolbox,
Microscopy Image Browser, and the Statistics and Machine Learning Toolbox. For each condition, outliers were removed using the ROUT method in GraphPad Prism (Q = 1%) (ref. 70). Detected
binding events for each experiment were normalized relative to the condition without APC/C. LOCAL HETEROGENEITY ANALYSIS Particles assigned the ‘CRL up’ major state were extracted and
imported into CryoSPARC for further analysis61. Particles were classified into separate 3D classes using the ‘3D Classification Beta’ tool for local classification using a mask including
only the catalytic core of the APC/C (CDH1, APC10, UBE2C, APC11 and the APC2CRL domains). Particles from each class were further refined using 3D homogeneous refinement and fitted using
rigid body fitting in UCSF ChimeraX. UBIQUITINATION ASSAYS In general, fluorescent substrate ubiquitination assays were performed at room temperature, quenched by SDS-containing sample
buffer, separated by SDS–PAGE, and visualized using a Typhoon Fluorescence scanner. Ubiquitination assays testing inhibition of substrate polyubiquitination were carried out by mixing 70 nM
APC/C, 0.5 µM CDH1, 5 mM ATP/MgCl2, 1 µM E1, 1 µM UBE2C, 0.5 µM Ub–CycBN*, ±30 µM UbVCDH1, ±Hsl1 D-box, ±Hsl1 KEN-box, ±UbVCDH1–Hsl1 D-box or ±UbVCDH1–Hsl1 KEN-box. A total of 125 µM Ub was
added to start the reaction and quenched after 15 min. Single-encounter assay conditions were carried out by first incubating two separate mixes for 5 min: mix 1 containing APC/C–CDH1,
either CycBN* or a version of CycBN* containing a single lysine and ±30 µM UbVRING or UbVCDH1, and mix 2 containing 0.1 µM E1, 5 µM UBE2C, 5 mM ATP/MgCl2, excess unlabeled Hsl1 D-box at 48
µM, and either Ub or methyl-Ub at 125 µM. The two mixtures were combined and then quenched after 3 min. Bands containing either total modified substrate or substrate modified with >4 Ubs
were quantified and normalized relative to the wild-type levels of ubiquitination. Values were plotted using GraphPad Prism. Substrate ubiquitination reactions testing Ub–Securin degron
mutants were carried out by combining 70 nM APC/C, 0.5 µM CDH1, 5 mM ATP/MgCl2, 1 µM E1, 1 µM UBE2C, 0.5 µM Ub–Securin*, ±30 µM UbVCDH1, ±30 µM UbVRING, and 125 µM Ub added to start the
reaction, then quenched after 15 min. Ubiquitination assays testing different E2 and substrate combinations were carried out in a similar manner with 70 nM APC/C, 0.5 µM CDC20, 5 mM
ATP/MgCl2, 1 µM E1, 1 µM E2 (UBE2C, UBCH5B and/or UBE2S), 0.5 µM substrate (CycBN* or Ub-CycBN*, Securin* or Ub-Securin*) and ±30 µM UbVCDH1. *UbVCDH1 ubiquitination reactions were carried
out using 50 nM APC/C or APC/C∆WHB, ±0.5 µM CDH1, 7 µM *UbVCDH1, 2 µM UBE2C and/or UBE2S, 1 µM E1, 5 mM ATP/MgCl2 and 125 µm Ub, and carried out at 0, 30 and 60 min timepoints. Intrinsic
ability of APC/C to catalyze the hydrolysis of Ub from UBE2C was tested using an oxyester-linked UBE2C~Ub (where ~ denotes oxyester linkage) in the presence and absence of coactivator or
UbVCDH1. Reaction mixtures contained 1 µM APC/C, 5 µM UBE2C~Ub and ±10 µM UbVCDH1 and were quenched after 0, 3 or 5 h at room temperature and visualized using SDS–PAGE and Coomassie
staining. Di-Ub synthesis assays were carried out as described previously, with the addition of ±10 µM UbVCDH1(ref. 23). PHAGE DISPLAY The UbV library was based upon the same design as
library 2 generated by Ernst et al.50. Phage display selection was carried out according to established protocols48. Briefly, 1 μM purified CDH1 and CDC20 were coated on 96-well MaxiSorp
plates (Thermo Scientific) by incubating overnight at 4 °C. Five rounds of selection using the phage-displayed UbV library were performed against immobilized proteins. The coated wells were
blocked with PBS-B (phosphate-buffered saline (PBS) supplemented with 1% BSA) and incubated at 4 °C for 1 h. The original phage display library (first round) or the enriched input from the
previous round (subsequent rounds) was added to each coated well and incubated for 1 h at room temperature. Non-bound phages were removed by washing the coated wells with PBS-T (PBS
supplemented with 0.05% Tween-20). Washing stringency was increased with each round. To elute the bound phage, 0.1 M HCl was added to each coated well and incubated for 5 min at room
temperature. The pH was neutralized by adding 1 M Tris–HCl (pH 11), and BSA was added to eluted phage to a final concentration of 1%. Amplifying the eluted phage to generate input phage for
subsequent rounds was achieved by superinfecting _E. coli_ OmniMax with both eluted phage and helper phage. After the fifth round of selection, phage generated in rounds 4 and 5 were
analyzed by phage ELISA to obtain crude binding data and their corresponding phagemids were subjected to Sanger sequencing to obtain UbV sequences and assess binder enrichment. The CDC20
selection produced the coactivator-binding UbV used in this study. APC/C SUBSTRATE DEGRADATION ASSAY USING CP HELAS3 EXTRACTS The degradation assay using a checkpoint (CP) cell-free extract
was performed essentially as described previously45,71. HeLaS3 cells were grown at 37 °C with 5% CO2 in Dulbecco’s modified Eagle medium (DMEM, Gibco) supplemented with 10% fetal bovine
serum. Confluent cells were seeded in 15 cm plates at 3 million cells per plate and treated the day after with 2 mM thymidine for 24 h. Thymidine-containing medium was removed, and cells
were washed with warm PBS twice and drug-free DMEM once, and released in DMEM for 4 h. Then, cells were treated with 100 ng ml−1 of nocodazole in DMEM for 11 h to obtain an early mitotic
(prometaphase) population and collected for extract preparation. Mitotic cells (>95%) were collected by ‘shake off’, pelleted at 277_g_, washed with PBS once then resuspended in 1 ml of
ice-cold PBS and pelleted in Eppendorf tubes. Cell pellets from 4 × 15 cm dishes were lysed in 600 µl of SB buffer (20 mM HEPES pH 7.5, 1.5 mM MgCl2, 1 mM dithiothreitol, 5 mM KCl, 2 μg ml−1
pepstatin, 2 μg ml−1 apoprotinin, 10 μg ml−1 leupeptin, 1 mM 4-(2 aminoethyl) benzenesulfonyl fluoride and 1 mM Na3VO4) supplemented with 20 μl of energy mix (375 mM creatine phosphate, 50
mM ATP and 50 mM MgCl2, pH 8.0). Cells were incubated for 30 min on ice and flipped every 5 min before being frozen in liquid nitrogen then quickly thawed at 30 °C, and the procedure was
repeated one more time. DNA was sheared by passing the lysate seven times through a 20 1/2 G and twice through a 25 3/8 G. The lysate was centrifugated at 3,075_g_ at 4 °C for 5 min first,
and its supernatant was then centrifuged at 24,104_g_ at 4 °C for 30 min. The resulting extract was aliquoted and either stored at −80 °C or used immediately. Typically, a master mix made of
50 μl of CP extracts was supplemented with 12 μl of energy mix, 12 μl of either 850 μM Ub or UbVCDH1 and 3 μl of 40 μM UBE2C. The master mix was diluted 1:1 in fresh SB buffer minus
proteases inhibitors, and reactions were incubated at 30 °C. Aliquots were taken out at the indicated times, stopped with sample buffer, boiled and analyzed by SDS–PAGE and western blot.
METRIS The METRIS experiments were performed similarly to previously described methods51. The APC/C harbored a multipurpose tag on the C-terminus of APC4 that included a HaloTag, AviTag and
Twin-Strep(II)-tag. After ion exchange chromatography described above, the APC/C was biotinylated and subjected to SEC. Ub was biotinylated on an N-terminal cysteine using biotin maleimide.
Biotinylated Ub, CycB or Ub-CycB was added to fully saturate and coat streptavidin high-capacity eight-well strips and left to bind for 2 h (Sigma-Aldrich). The wells were then washed with a
blocking buffer and left for 30 min after which they were washed again. The biotinylated APC/C was added in 50-fold excess to the theoretical capacity of streptavidin-coated ferromagnetic
beads (Spherotech). Biotin-APC/C (3 μM) and CDH1 (15 μM) were incubated with ferromagnetic beads for 2 hours and the solution of magnetic beads was diluted 5,000-fold when the solution of
magnetic beads was inserted into the well and magnetized using a permanent neodymium magnet. The well was placed into the Helmholtz-coil like apparatus and the rotating magnetic field was
actuated at a frequency of 1 Hz for all experiments. Data acquisition was achieved using a camera for visualization, video capture and subsequent analysis described previously51. The RP is
the distance the bead travels (∆_x_) divided by the maximum theoretical displacement of a sphere, calculated using the following equation: $${{\mathrm{RP}}}=\frac{\Delta x}{\pi D\tau \omega
}$$ where _D_ is the particle diameter, _ω_ is the frequency of the rotating magnetic field and _τ_is the actuation time. REPORTING SUMMARY Further information on research design is
available in the Nature Portfolio Reporting Summary linked to this article. DATA AVAILABILITY The cryo-EM density maps of APC/C–CDH1–UBE2C–Ub–CycBN and APC/C–CDH1–UBE2C–UBE2S–Ub–CycBN were
deposited in the Electron Microscopy Data Bank under accession numbers EMD-41140 and EMD-41142, respectively. The corresponding atomic coordinates were deposited in the RCSB Protein Data
Bank under accession numbers 8TAR (APC/C–CDH1–UBE2C–Ub–CycBN) and 8TAU (APC/C–CDH1–UBE2C–UBE2S–Ub–CycBN). The raw EM data, trained cryoDRGN models and generated volumes from this study are
available on Electron Microscopy Public Image Archive database (EMPIAR-11660 and EMPIAR-11661). Unprocessed images and numerical raw data are provided as source data. Additional data
available upon request. Source data are provided with this paper. CHANGE HISTORY * _ 02 NOVEMBER 2023 In the version of the article originally published, some links for the Supplementary
information were mislabelled, but have now been corrected in the HTML version of the article. _ REFERENCES * Alberts, B. & Miake-Lye, R. Unscrambling the puzzle of biological machines:
the importance of the details. _Cell_ 68, 415–420 (1992). CAS PubMed Google Scholar * Frank, J. The translation elongation cycle-capturing multiple states by cryo-electron microscopy.
_Philos. Trans. R. Soc. Lond. B_ 372, 20160180 (2017). Google Scholar * Zhong, E. D., Bepler, T., Berger, B. & Davis, J. H. CryoDRGN: reconstruction of heterogeneous cryo-EM structures
using neural networks. _Nat. Methods_ 18, 176–185 (2021). CAS PubMed PubMed Central Google Scholar * Kaledhonkar, S. et al. Late steps in bacterial translation initiation visualized
using time-resolved cryo-EM. _Nature_ 570, 400–404 (2019). CAS PubMed PubMed Central Google Scholar * Fischer, N., Konevega, A. L., Wintermeyer, W., Rodnina, M. V. & Stark, H.
Ribosome dynamics and tRNA movement by time-resolved electron cryomicroscopy. _Nature_ 466, 329–333 (2010). CAS PubMed Google Scholar * Astumian, R. D. Kinetic asymmetry allows
macromolecular catalysts to drive an information ratchet. _Nat. Commun._ 10, 3837 (2019). PubMed PubMed Central Google Scholar * Lu, Y., Wang, W. & Kirschner, M. W. Specificity of the
anaphase-promoting complex: a single-molecule study. _Science_ 348, 1248737 (2015). PubMed PubMed Central Google Scholar * Buschhorn, B. A. et al. Substrate binding on the APC/C occurs
between the coactivator Cdh1 and the processivity factor Doc1. _Nat. Struct. Mol. Biol._ 18, 6–13 (2011). CAS PubMed Google Scholar * Meyer, H. J. & Rape, M. Processive ubiquitin
chain formation by the anaphase-promoting complex. _Semin. Cell Dev. Biol._ 22, 544–550 (2011). CAS PubMed PubMed Central Google Scholar * Kleiger, G., Saha, A., Lewis, S., Kuhlman, B.
& Deshaies, R. J. Rapid E2–E3 assembly and disassembly enable processive ubiquitylation of cullin–RING ubiquitin ligase substrates. _Cell_ 139, 957–968 (2009). CAS PubMed PubMed
Central Google Scholar * Rape, M., Reddy, S. K. & Kirschner, M. W. The processivity of multiubiquitination by the APC determines the order of substrate degradation. _Cell_ 124, 89–103
(2006). CAS PubMed Google Scholar * Pierce, N. W., Kleiger, G., Shan, S. O. & Deshaies, R. J. Detection of sequential polyubiquitylation on a millisecond timescale. _Nature_ 462,
615–619 (2009). CAS PubMed PubMed Central Google Scholar * Deshaies, R. J. & Joazeiro, C. A. RING domain E3 ubiquitin ligases. _Annu. Rev. Biochem._ 78, 399–434 (2009). CAS PubMed
Google Scholar * Haakonsen, D. L. & Rape, M. Branching out: improved signaling by heterotypic ubiquitin chains. _Trends Cell Biol._ 29, 704–716 (2019). CAS PubMed Google Scholar *
Baek, K., Scott, D. C. & Schulman, B. A. NEDD8 and ubiquitin ligation by cullin–RING E3 ligases. _Curr. Opin. Struct. Biol._ 67, 101–109 (2020). PubMed PubMed Central Google Scholar *
Peters, J. M. SCF and APC: the Yin and Yang of cell cycle regulated proteolysis. _Curr. Opin. Cell Biol._ 10, 759–768 (1998). CAS PubMed Google Scholar * King, R. W., Deshaies, R. J.,
Peters, J. M. & Kirschner, M. W. How proteolysis drives the cell cycle. _Science_ 274, 1652–1659 (1996). CAS PubMed Google Scholar * Bodrug, T., Welsh, K. A., Hinkle, M., Emanuele, M.
J. & Brown, N. G. Intricate regulatory mechanisms of the anaphase-promoting complex/cyclosome and its role in chromatin regulation. _Front. Cell Dev. Biol._ 9, 687515 (2021). PubMed
PubMed Central Google Scholar * Primorac, I. & Musacchio, A. Panta rhei: the APC/C at steady state. _J. Cell Biol._ 201, 177–189 (2013). CAS PubMed PubMed Central Google Scholar *
Williamson, A. et al. Regulation of ubiquitin chain initiation to control the timing of substrate degradation. _Mol. Cell_ 42, 744–757 (2011). CAS PubMed PubMed Central Google Scholar *
Kirkpatrick, D. S. et al. Quantitative analysis of in vitro ubiquitinated cyclin B1 reveals complex chain topology. _Nat. Cell Biol._ 8, 700–710 (2006). CAS PubMed Google Scholar * Meyer,
H. J. & Rape, M. Enhanced protein degradation by branched ubiquitin chains. _Cell_ 157, 910–921 (2014). CAS PubMed PubMed Central Google Scholar * Brown, N. G. et al. Dual RING E3
architectures regulate multiubiquitination and ubiquitin chain elongation by APC/C. _Cell_ 165, 1440–1453 (2016). CAS PubMed PubMed Central Google Scholar * da Fonseca, P. C. et al.
Structures of APC/C(Cdh1) with substrates identify Cdh1 and Apc10 as the D-box co-receptor. _Nature_ 470, 274–278 (2011). PubMed Google Scholar * Visintin, R., Prinz, S. & Amon, A.
CDC20 and CDH1: a family of substrate-specific activators of APC-dependent proteolysis. _Science_ 278, 460–463 (1997). CAS PubMed Google Scholar * Li, Q. et al. WD40 domain of Apc1 is
critical for the coactivator-induced allosteric transition that stimulates APC/C catalytic activity. _Proc. Natl Acad. Sci. USA_ 113, 10547–10552 (2016). CAS PubMed PubMed Central Google
Scholar * Chang, L., Zhang, Z., Yang, J., McLaughlin, S. H. & Barford, D. Atomic structure of the APC/C and its mechanism of protein ubiquitination. _Nature_ 522, 450–454 (2015). CAS
PubMed PubMed Central Google Scholar * Chang, L. F., Zhang, Z., Yang, J., McLaughlin, S. H. & Barford, D. Molecular architecture and mechanism of the anaphase-promoting complex.
_Nature_ 513, 388–393 (2014). CAS PubMed PubMed Central Google Scholar * Aristarkhov, A. et al. E2-C, a cyclin-selective ubiquitin carrier protein required for the destruction of mitotic
cyclins. _Proc. Natl Acad. Sci. USA_ 93, 4294–4299 (1996). CAS PubMed PubMed Central Google Scholar * Yu, H., King, R. W., Peters, J. M. & Kirschner, M. W. Identification of a novel
ubiquitin-conjugating enzyme involved in mitotic cyclin degradation. _Curr. Biol._ 6, 455–466 (1996). CAS PubMed Google Scholar * Dimova, N. V. et al. APC/C-mediated multiple
monoubiquitylation provides an alternative degradation signal for cyclin B1. _Nat. Cell Biol._ 14, 168–176 (2012). CAS PubMed PubMed Central Google Scholar * Wu, T. et al. UBE2S drives
elongation of K11-linked ubiquitin chains by the anaphase-promoting complex. _Proc. Natl Acad. Sci. USA_ 107, 1355–1360 (2010). CAS PubMed PubMed Central Google Scholar * Garnett, M. J.
et al. UBE2S elongates ubiquitin chains on APC/C substrates to promote mitotic exit. _Nat. Cell Biol._ 11, 1363–1369 (2009). CAS PubMed PubMed Central Google Scholar * Williamson, A. et
al. Identification of a physiological E2 module for the human anaphase-promoting complex. _Proc. Natl Acad. Sci. USA_ 106, 18213–18218 (2009). CAS PubMed PubMed Central Google Scholar *
Martinez-Fonts, K. et al. The proteasome 19S cap and its ubiquitin receptors provide a versatile recognition platform for substrates. _Nat. Commun._ 11, 477 (2020). CAS PubMed PubMed
Central Google Scholar * Yamaguchi, M. et al. Cryo-EM of mitotic checkpoint complex-bound APC/C reveals reciprocal and conformational regulation of ubiquitin ligation. _Mol. Cell_ 63,
593–607 (2016). CAS PubMed PubMed Central Google Scholar * Alfieri, C. et al. Molecular basis of APC/C regulation by the spindle assembly checkpoint. _Nature_ 536, 431–436 (2016). CAS
PubMed PubMed Central Google Scholar * Brown, N. G. et al. RING E3 mechanism for ubiquitin ligation to a disordered substrate visualized for human anaphase-promoting complex. _Proc. Natl
Acad. Sci. USA_ 112, 5272–5279 (2015). CAS PubMed PubMed Central Google Scholar * Streich, F. C. Jr. & Lima, C. D. Structural and functional insights to ubiquitin-like protein
conjugation. _Annu. Rev. Biophys._ 43, 357–379 (2014). CAS PubMed Central Google Scholar * Baek, K. et al. NEDD8 nucleates a multivalent cullin–RING–UBE2D ubiquitin ligation assembly.
_Nature_ 578, 461–466 (2020). CAS PubMed PubMed Central Google Scholar * Yau, R. G. et al. Assembly and function of heterotypic ubiquitin chains in cell-cycle and protein quality
control. _Cell_ 171, 918–933 e920 (2017). CAS PubMed PubMed Central Google Scholar * Brown, N. G. et al. Mechanism of polyubiquitination by human anaphase-promoting complex: RING
repurposing for ubiquitin chain assembly. _Mol. Cell_ 56, 246–260 (2014). CAS PubMed PubMed Central Google Scholar * Frye, J. J. et al. Electron microscopy structure of human
APC/C(CDH1)–EMI1 reveals multimodal mechanism of E3 ligase shutdown. _Nat. Struct. Mol. Biol._ 20, 827–835 (2013). CAS PubMed PubMed Central Google Scholar * McInnes, L., Healy, J. &
Melville, J. UMAP: uniform manifold approximation and projection for dimension reduction. Preprint at https://arxiv.org/abs/1802.03426 (2018). * Martinez-Chacin, R. C. et al. Ubiquitin
chain-elongating enzyme UBE2S activates the RING E3 ligase APC/C for substrate priming. _Nat. Struct. Mol. Biol._ 27, 550–560 (2020). CAS PubMed PubMed Central Google Scholar * Welsh, K.
A. et al. Functional conservation and divergence of the helix-turn-helix motif of E2 ubiquitin-conjugating enzymes. _EMBO J._ 41, e108823 (2022). CAS PubMed Google Scholar * Watson, E.
R. et al. Protein engineering of a ubiquitin-variant inhibitor of APC/C identifies a cryptic K48 ubiquitin chain binding site. _Proc. Natl Acad. Sci. USA_ 116, 17280–17289 (2019). CAS
PubMed PubMed Central Google Scholar * Zhang, W. & Sidhu, S. S. Generating intracellular modulators of E3 ligases and deubiquitinases from phage-displayed ubiquitin variant libraries.
_Methods Mol. Biol._ 1844, 101–119 (2018). CAS PubMed Google Scholar * Zhang, W. et al. System-wide modulation of HECT E3 ligases with selective ubiquitin variant probes. _Mol. Cell_ 62,
121–136 (2016). CAS PubMed PubMed Central Google Scholar * Ernst, A. et al. A strategy for modulation of enzymes in the ubiquitin system. _Science_ 339, 590–595 (2013). CAS PubMed
Google Scholar * Petell, C. J. et al. Mechanically transduced immunosorbent assay to measure protein–protein interactions. _eLife_ 10, e67525 (2021). CAS PubMed PubMed Central Google
Scholar * Kataria, M. & Yamano, H. Interplay between phosphatases and the anaphase-promoting complex/cyclosome in mitosis. _Cells_ 8, 814 (2019). CAS PubMed PubMed Central Google
Scholar * Duda, D. M. et al. Structural insights into NEDD8 activation of cullin–RING ligases: conformational control of conjugation. _Cell_ 134, 995–1006 (2008). CAS PubMed PubMed
Central Google Scholar * Kelly, A., Wickliffe, K. E., Song, L., Fedrigo, I. & Rape, M. Ubiquitin chain elongation requires E3-dependent tracking of the emerging conjugate. _Mol. Cell_
56, 232–245 (2014). CAS PubMed Google Scholar * Scott, D. C. et al. Two distinct types of E3 ligases work in unison to regulate substrate ubiquitylation. _Cell_ 166, 1198–1214 e1124
(2016). CAS PubMed PubMed Central Google Scholar * Jarvis, M. A. et al. Measuring APC/C-dependent ubiquitylation in vitro. _Methods Mol. Biol._ 1342, 287–303 (2016). PubMed PubMed
Central Google Scholar * Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. _eLife_ 7, e42166 (2018). PubMed PubMed Central Google
Scholar * Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. _Nat. Methods_ 14, 331–332 (2017). CAS PubMed PubMed Central
Google Scholar * Bepler, T., Kelley, K., Noble, A. J. & Berger, B. Topaz-Denoise: general deep denoising models for cryoEM and cryoET. _Nat. Commun._ 11, 5208 (2020). CAS PubMed
PubMed Central Google Scholar * Rohou, A. & Grigorieff, N. CTFFIND4: fast and accurate defocus estimation from electron micrographs. _J. Struct. Biol._ 192, 216–221 (2015). PubMed
PubMed Central Google Scholar * Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. _Nat.
Methods_ 14, 290–296 (2017). CAS PubMed Google Scholar * Sanchez-Garcia, R. et al. DeepEMhancer: a deep learning solution for cryo-EM volume post-processing. _Commun. Biol._ 4, 874
(2021). PubMed PubMed Central Google Scholar * Croll, T. I. ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. _Acta Crystallogr. D_
74, 519–530 (2018). CAS Google Scholar * Pettersen, E. F. et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. _Protein Sci._ 30, 70–82 (2021). CAS
PubMed Google Scholar * Mirdita, M. et al. ColabFold: making protein folding accessible to all. _Nat. Methods_ 19, 679–682 (2022). CAS PubMed PubMed Central Google Scholar * Edelstein,
A. D. et al. Advanced methods of microscope control using muManager software. _J. Biol. Methods_ 1, e10 (2014). PubMed Google Scholar * Qiao, R. et al. Mechanism of APC/CCDC20 activation
by mitotic phosphorylation. _Proc. Natl Acad. Sci. USA_ 113, E2570–E2578 (2016). CAS PubMed PubMed Central Google Scholar * Olivo-Marin, J. Extraction of spots in biological images using
multiscale products. _Pattern Recognit._ 35, 1989–1996 (2002). Google Scholar * Dinsmore, A. D., Weeks, E. R., Prasad, V., Levitt, A. C. & Weitz, D. A. Three-dimensional confocal
microscopy of colloids. _Appl. Opt._ 40, 4152–4159 (2001). CAS PubMed Google Scholar * Motulsky, H. J. & Brown, R. E. Detecting outliers when fitting data with nonlinear regression—a
new method based on robust nonlinear regression and the false discovery rate. _BMC Bioinformatics_ 7, 123 (2006). PubMed PubMed Central Google Scholar * Williamson, A., Jin, L. &
Rape, M. Preparation of synchronized human cell extracts to study ubiquitination and degradation. _Methods Mol. Biol._ 545, 301–312 (2009). CAS PubMed Google Scholar Download references
ACKNOWLEDGEMENTS We thank H. Stark for providing access to his Titan Krios and E. Schliep for assistance with it at the Max-Planck Institute for Multidisciplinary Science. We also thank H.
Kotisch for cryo-EM data recording on the Titan Krios at the IMP and the Electron Microscopy Facility at Vienna BioCenter Core Facilities for access to their instruments and training.
Additionally, we thank the UTSW-UNC Center for Cell Signaling Analysis (RM1GM145399) for dissemination and resources. The UbV library was generously provided by S. Sidhu (University of
Toronto), and UbVCDH1 was selected in the lab of W. Zhang at University of Guelph, supported by a Canadian Institutes for Health Research (CIHR) Project Grant (PJT-162249). Our work is
supported by NIH T32GM008570 (D.L.B. and T.B.) and NSF DGE-1650116 (T.B.); NIH T32GM135095 and the American Heart Association 23PRE1025746 (K.A.W.); NIH P30CA016086 (UNC High-throughput
peptide Synthesis Facility and Array Facility); NIH R35GM122596 (K.M.H.); NIH R01GM120309 and the American Cancer Society RSG-18-220-01-TBG (M.J.E.); WWTF-LS19-029 and Boehringer Ingelheim
(S.J.A., I.G. and D.H.); NIH R35GM128855 and UCRF (N.G.B.). Other than providing resources, the funders had no role in study design, data collection and analysis, or preparation of the
manuscript. For the purpose of Open Access, the author has applied a CC BY public copyright licence to any Author Accepted Manuscript (AAM) version arising from this submission. AUTHOR
INFORMATION Author notes * Bei Liu Present address: College of Future Technology, National Biomedical Imaging Center, Peking University, Beijing, China AUTHORS AND AFFILIATIONS * Department
of Biochemistry and Biophysics and Lineberger Comprehensive Cancer Center, University of North Carolina, Chapel Hill, NC, USA Tatyana Bodrug & Derek L. Bolhuis * Department of
Pharmacology and Lineberger Comprehensive Cancer Center, University of North Carolina, Chapel Hill, NC, USA Tatyana Bodrug, Kaeli A. Welsh, Derek L. Bolhuis, Ethan Paulаkonis, Raquel C.
Martinez-Chacin, Bei Liu, Nicholas Pinkin, Thomas Bonacci, Liying Cui, Pengning Xu, Michael J. Emanuele, Klaus M. Hahn & Nicholas G. Brown * Department of Molecular and Cellular Biology,
College of Biological Science, University of Guelph, Guelph, Ontario, Canada Olivia Roscow & Wei Zhang * Research Institute of Molecular Pathology, Vienna BioCenter, Vienna, Austria
Sascha Josef Amann, Irina Grishkovskaya & David Haselbach * Department of Chemistry, University of the Pacific, Stockton, CA, USA Joseph S. Harrison * School of Engineering, California
Polytechnic State University Humboldt, Arcata, CA, USA Joshua P. Steimel * Department of Computer Science, Princeton University, Princeton, NJ, USA Ellen D. Zhong Authors * Tatyana Bodrug
View author publications You can also search for this author inPubMed Google Scholar * Kaeli A. Welsh View author publications You can also search for this author inPubMed Google Scholar *
Derek L. Bolhuis View author publications You can also search for this author inPubMed Google Scholar * Ethan Paulаkonis View author publications You can also search for this author inPubMed
Google Scholar * Raquel C. Martinez-Chacin View author publications You can also search for this author inPubMed Google Scholar * Bei Liu View author publications You can also search for
this author inPubMed Google Scholar * Nicholas Pinkin View author publications You can also search for this author inPubMed Google Scholar * Thomas Bonacci View author publications You can
also search for this author inPubMed Google Scholar * Liying Cui View author publications You can also search for this author inPubMed Google Scholar * Pengning Xu View author publications
You can also search for this author inPubMed Google Scholar * Olivia Roscow View author publications You can also search for this author inPubMed Google Scholar * Sascha Josef Amann View
author publications You can also search for this author inPubMed Google Scholar * Irina Grishkovskaya View author publications You can also search for this author inPubMed Google Scholar *
Michael J. Emanuele View author publications You can also search for this author inPubMed Google Scholar * Joseph S. Harrison View author publications You can also search for this author
inPubMed Google Scholar * Joshua P. Steimel View author publications You can also search for this author inPubMed Google Scholar * Klaus M. Hahn View author publications You can also search
for this author inPubMed Google Scholar * Wei Zhang View author publications You can also search for this author inPubMed Google Scholar * Ellen D. Zhong View author publications You can
also search for this author inPubMed Google Scholar * David Haselbach View author publications You can also search for this author inPubMed Google Scholar * Nicholas G. Brown View author
publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS T.B., D.L.B., K.A.W., and L.C. prepared the reagents for APC/C-dependent polyubiquitination assays and
METRIS experiments. T.B., K.A.W., D.L.B., E.P., and R.C.M.-C. performed the ubiquitination assays with advice from N.G.B. K.A.W. executed the TIRF microscopy experiments with assistance from
D.L.B., B.L., N.P., P.X., and K.M.H. O.R. and W.Z. engineered the UbVCDH1 using phage display. T.B. conducted the cell extract experiments with advice from M.J.E. and N.G.B. I.G. and S.J.A.
assisted with the preparation, collection, and analysis of the APC/CCDH1 sample with the UbVCDH1-Hsl1 D-box. J.S.H. and J.P.S. performed the METRIS experiments and analyzed the data. T.B.,
K.A.W., D.L.B., E.P., R.C.M.-C., B.L., N.P., K.M.H., E.D.Z., D.H. and N.G.B. assisted with experimental design and data analysis. T.B., D.H., and N.G.B. conceived the project. T.B., D.H.,
and N.G.B. wrote the manuscript with assistance from all authors. CORRESPONDING AUTHORS Correspondence to David Haselbach or Nicholas G. Brown. ETHICS DECLARATIONS COMPETING INTERESTS The
authors declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Nature Structural & Molecular Biology_ thanks Hongtao Yu and the other, anonymous, reviewer(s) for their
contribution to the peer review of this work. Dimitris Typas was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the
editorial team. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. EXTENDED DATA
EXTENDED DATA FIG. 1 PROCESSING AND FITTING OF THE APC/C FROM THE TR-EM DATASETS. A – Diagram depicting domain map of Ub-CycBN*. B, C – Cryo-EM data processing workflow showing
representative micrographs from the APC/C-CDH1-UBE2C and APC/C-CDH1-UBE2C-UBE2S datasets (B). Particle detection was optimized using Topaz59 and filtered using 3 rounds of 2D classification.
Representative 2D projections from the final 2D classification step show the APC/C in multiple orientations (C). D – Local resolution map of the consensus refinements from the datasets. E –
FSC curves showing resolution of the 3D density maps from the consensus refinement from the APC/C-CDH1-UBE2C and APC/C-CDH1-UBE2C-UBE2S datasets. Lines show the 0.143 gold-standard and 0.5
cut-off with values denoting resolution at 0.143 cut-off. F – Similar to Fig. 1d, fitted model of APC/C-CDH1-UBE2C. G – Overlay of APC/C-CDH1-UBE2C electron density map from this study with
published maps from previous studies. Values shown indicate map-to-map correlation of maps from previous studies (EMD-2925 and EMD-2929) with APC/C-CDH1-UBE2C from this data after gaussian
filtering27,38. H – Overlay of electron density maps from the APC/C-CDH1-UBE2C and APC/C-CDH1-UBE2C-UBE2S consensus refinements, value shown indicates map-to-map correlation. I – Models
showing UBE2S-CTP bound to the APC2/APC4 groove in previously described datasets (EMD-2925; EMD-3433)23,27 and here in the APC/C-CDH1-UBE2C-UBE2S dataset. EXTENDED DATA FIG. 2 HETEROGENEITY
ANALYSIS AND CLASSIFICATION OF PARTICLES FROM THE TR-EM DATASETS USING CRYODRGN AND PCA. A – Diagram depicting the cryoDRGN data processing workflow. Particle images are mapped into a
continuous low-dimensional vector space, producing a latent embedding, z. A volume decoder can then reconstruct a unique 3D volume given any value of z, for example a particle’s mapped
location in latent space. Principle Component Analysis (PCA) can be applied to either the set of particle latent embeddings for a dataset or a set of reconstructed 3D volumes to map the
heterogeneity of the particles or volumes in a continuous manner. B, C – UMAP (left) and PCA (right) depiction of the latent space with representative volumes shown at indicated locations,
for APC/C-CDH1-UBE2C (B) and APC/C-CDH1-UBE2C-UBE2S (C), explained variance (EV) noted in parentheses. D – PC trajectory along the second component showing the transition between a ‘CRL
Partial’ to ‘CRL Up, E2 Bound’ state. Values in parentheses denote explained variance (EV). Densities CDH1, APC2, APC11, and UBE2C are colored as purple, green, blue, and cyan, respectively.
EXTENDED DATA FIG. 3 MOVEMENT OF THE APC/C DURING CATALYSIS, SHOWN IN CRYODRGN VOLUME PCA TRAJECTORIES. A – Volume PCA trajectories showing conformational changes along first four
components to visualize displacement of CDH1 and the APC2 CRL along continuous conformational trajectories. Density changes localized near CDH1 and at the APC/C actives site shown in black
stripes. B – Expected variance plotted for the first four components shown in A. C – Histograms showing distributions of volumes along the PCs shown in A. EXTENDED DATA FIG. 4 FSC CURVES AND
UMAP PARTICLE DISTRIBUTIONS OVER TR-EM DATASETS. A – FSC curves depicting the resolution of the consensus 3D refinements shown in Fig. 2b. Lines show the 0.143 gold-standard and 0.5 cut-off
with values denoting resolution at 0.143 cut-off. B – UMAP visualization of the particle distributions in the latent space plotted separately by timepoint. EXTENDED DATA FIG. 5 DEVELOPMENT
OF TIRF-BASED APC/C-DEPENDENT SUBSTRATE UBIQUITINATION ASSAY FOLLOWING FLUORESCENTLY-LABELED UBE2C. A, B – UMAP representation showing the particle distribution of the sub-states in the ‘CRL
Down’ (A) and ‘CRL Up’ (B) major states. C – TIRF microscopy movies capture substrate-E2 interactions prior to complete substrate depletion. Experimental mixtures for the given conditions
were split evenly between microscopy flow cells for data capture (Fig. 3d) and a ubiquitination reaction with 50 nM Biotin-CycBN*-A488 spiked in, shown above, that was ran on an SDS-PAGE gel
and fluorescently scanned. The banding of the ubiquitinated substrate is distorted due to the high BSA concentration needed for microscopy. The experiments were performed independently
three times. D – Duration of detected binding events in seconds across multiple experiments and with increasing concentration of UBE2S CTP. E – There is minimal background signal from slide
functionalization (left) and robust signal from immobilized substrate (center left) in the Substrate channel, as well as minimal background fluorescence from the substrate in the UBE2C
channel (center right). The addition of UBE2C to immobilized substrate without APC/C-CDH1 shows minimal non-specific UBE2C binding (right). F – Example images showing UBE2C binding events
upon addition of APC/C-CDH1 and increasing levels of UBE2S CTP. G – Montage showing representative time series of a UBE2C binding event in the UBE2C channel at 200 ms intervals. H – Montage
(top) depicting duration of individual binding events in experiments described in (C) indicated by red or blue asterisk. Trace of raw intensity values (bottom) showing individual UBE2C
binding events; red or blue asterisks denote corresponding binding events shown in montage (top). Source data EXTENDED DATA FIG. 6 PREVIOUS ARTIFICIALLY-TRAPPED APC/C STRUCTURES MIMICKING
SUBSTRATE UBIQUITINATION. A – Fitted cryoEM models from previous studies of the APC/C showing monoubiquitination (left), multi-ubiquitination (center), and polyubiquitination (right)23,38.
An Hsl1 truncation harboring its KEN- and D-box was used as a substrate where a lysine was replaced with cysteine and crosslinked to the active site of UBE2C to mimic substrate priming
(left). A similar Hsl1 truncation was genetically fused to the UbVRING, purified, and crosslinked to UBE2C and Ub to mimic multi-monoubiquitination (center). For chain elongation, a
UbVRING-Hsl1 D-box chimera harboring a K11C substitution was crosslinked to the active site of UBE2S (right). B – Similar to Fig. 4e, immunoblotting of APC/C-dependent substrate degradation
in mitotic HeLa cell extracts reveals that this process is not inhibited by the addition of excess Ub, in contrast to the UbVCDH1. The experiments were performed independently three times. C
– Similar to Fig. 4f, Securin ubiquitination is inhibited more strongly by the UbVCDH1-Hsl1 D-box fusion, as monitored by fluorescent scanning of APC/C-CDH1-UBE2C-dependent ubiquitination
reactions. The experiments were performed independently three times. D –Cryo-EM map of APC/C-CDH1-UbVCDH1-Hsl1 D-box rigid body fitted with a surface representation model of UBE2C bound to
APC2 (green)/APC11(blue). Left, UbVCDH1 (orange) is in position to receive Ub from UBE2C (cyan). Right, cryo-EM map fitted with atomic models of CDH1 (purple) and UbVCDH1 (orange)-Hsl1 D-box
(red). Yellow rectangle indicates the KEN-box binding site of CDH1, similar to where UbVCDH1 binds. Uncropped gels representative of n = 3 independent experiments for C available in source
data. Source data EXTENDED DATA FIG. 7 UB BINDING TO COACTIVATOR SERVES MULTIPLE PURPOSES DURING APC/C-DEPENDENT POLYUBIQUITINATION. A – Quantitation of CycBN* modified with >4 Ubs from
single encounter assays in Fig. 5c. Error bars: SEM. The experiments were performed independently three times. B – Substrate-independent, APC/C-dependent hydrolysis of an oxyester-linked Ub
from UBE2C C114S is not impeded by the addition of UbVCDH1, monitored by Coomassie-stained SDS-PAGE gel. The experiments were performed independently three times. C – Synthesis of di-Ub by
APC/C-UBE2S is not inhibited by UbVCDH1 as revealed by scanning of an SDS-PAGE gel where the acceptor Ub* is fluorescently labeled. The experiments were performed independently three times.
D – UbVCDH1 inhibits APC/C-CDH1-dependent ubiquitination activity of all substrate and E2 combinations tested, monitored by fluorescent scanning. The experiments were performed independently
three times. E – Ubiquitination of Ub-Securin and Ub-Securin DMut as shown in Fig. 5c. The experiments were performed independently three times. F – Similar to Fig. 5f, ubiquitination of
*UbVCDH1 requires UBE2C and does not occur with UBE2S alone, as monitored by fluorescent scanning. Uncropped gels representative of n≥3 independent experiments for B-F available in source
data. Source data SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION Supplementary Fig. 1. REPORTING SUMMARY SUPPLEMENTARY VIDEO 1 Trajectories from the PCA of the particle distribution in
the latent space show volumes generated along PC1 and PC2 of the latent space and map out the CRL transitioning from the ‘CRL down’ to the ‘CRL up’ conformation as well as heterogeneity near
the coactivator and active site. SUPPLEMENTARY VIDEO 2 Representative movies of UBE2C–JF549 signal across a titration of UBE2SCTP. All movies are taken using the UBE2C channel monitoring
UBE2C–JF549 binding events in flow cells with immobilized Biotin–CycBN*–A488. Exposure: 200 ms. The first 300 frames of 3,000-frame movies are shown. SOURCE DATA SOURCE DATA FIG. 1
Unprocessed western blots and/or gels. SOURCE DATA FIG. 3 Statistical source data. SOURCE DATA FIG. 4 Unprocessed western blots and/or gels. SOURCE DATA FIG. 5 Unprocessed western blots
and/or gels. SOURCE DATA FIG. 5 Statistical source data. SOURCE DATA EXTENDED DATA FIG. 5 Statistical source data. SOURCE DATA EXTENDED DATA FIG. 5 Unprocessed western blots and/or gels.
SOURCE DATA EXTENDED DATA FIG. 6 Unprocessed western blots and/or gels. SOURCE DATA EXTENDED DATA FIG. 7 Unprocessed western blots and/or gels. SOURCE DATA EXTENDED DATA FIG. 7 Statistical
source data. RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution
and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if
changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the
material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to
obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS
ARTICLE Bodrug, T., Welsh, K.A., Bolhuis, D.L. _et al._ Time-resolved cryo-EM (TR-EM) analysis of substrate polyubiquitination by the RING E3 anaphase-promoting complex/cyclosome (APC/C).
_Nat Struct Mol Biol_ 30, 1663–1674 (2023). https://doi.org/10.1038/s41594-023-01105-5 Download citation * Received: 20 December 2022 * Accepted: 21 August 2023 * Published: 21 September
2023 * Issue Date: November 2023 * DOI: https://doi.org/10.1038/s41594-023-01105-5 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get
shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative