Play all audios:
ABSTRACT Translesion DNA synthesis (TLS) is a cellular process that enables the bypass of DNA lesions encountered during DNA replication and is emerging as a primary target of chemotherapy.
Among vertebrate DNA polymerases, polymerase κ (Polκ) has the distinctive ability to bypass minor groove DNA adducts in vitro. However, Polκ is also required for cells to overcome major
groove DNA adducts but the basis of this requirement is unclear. Here, we combine CRISPR base-editor screening technology in human cells with TLS analysis of defined DNA lesions in _Xenopus_
egg extracts to unravel the functions and regulations of Polκ during lesion bypass. Strikingly, we show that Polκ has two main functions during TLS, which are differentially regulated by
Rev1 binding. On the one hand, Polκ is essential to replicate across a minor groove DNA lesion in a process that depends on PCNA ubiquitylation but is independent of Rev1. On the other hand,
through its cooperative interaction with Rev1 and ubiquitylated PCNA, Polκ appears to stabilize the Rev1–Polζ extension complex on DNA to allow extension past major groove DNA lesions and
abasic sites, in a process that is independent of Polκ’s catalytic activity. Together, our work identifies catalytic and noncatalytic functions of Polκ in TLS and reveals important
regulatory mechanisms underlying the unique domain architecture present at the C-terminal end of Y-family TLS polymerases. SIMILAR CONTENT BEING VIEWED BY OTHERS STRUCTURE AND MECHANISM OF
B-FAMILY DNA POLYMERASE Ζ SPECIALIZED FOR TRANSLESION DNA SYNTHESIS Article 17 August 2020 CRYO-EM STRUCTURE OF THE REV1–POLΖ HOLOCOMPLEX REVEALS THE MECHANISM OF THEIR COOPERATIVITY IN
TRANSLESION DNA SYNTHESIS Article 08 May 2024 STRUCTURAL BASIS OF ERROR-PRONE DNA SYNTHESIS BY DNA POLYMERASE _Θ_ Article Open access 28 February 2025 MAIN Lesions generated in DNA can
impede DNA synthesis. High-fidelity replicative polymerases are unable to accommodate DNA lesions in their catalytic site because of the resultant distortion in DNA geometry1,2. This often
leads to replication fork uncoupling, activation of the replication checkpoint and replication stress3. DNA lesions encountered during replication can be bypassed by DNA damage tolerance
(DDT) mechanisms4. Two distinct DDT mechanisms have been described: template switching (TS), which relies on a recombination-based process to copy genetic information from the undamaged
sister chromatid, and translesion DNA synthesis (TLS), which involves specialized DNA polymerases to synthesize across damaged bases5. Unlike replicative polymerases, TLS polymerases exhibit
poor processivity and low fidelity. This is attributed to the lack of proofreading activity and the presence of a flexible catalytic site that can accommodate damaged DNA bases1,2.
Ubiquitylation of proliferating cell nuclear antigen (PCNA) on K164 stimulates both TLS and TS processes6,7,8. This is because most enzymes participating in DDT contain PCNA-binding and
ubiquitin-binding motifs that bind to ubiquitylated PCNA and are thereby targeted to lesion sites9. In vertebrates, the TLS polymerases operating during replication are polymerase η (Polη),
Polι, Polκ and Rev1, which are classified under the Y-family, and Polζ, which is a member of the B-family. Although a single polymerase may catalyze both steps of DNA lesion bypass
(incorporation of a nucleotide opposite the DNA lesion and the subsequent extension past the lesion), TLS often involves the actions of an ‘inserter’ and an ‘extender’ (ref. 9). An example
of an insertion polymerase is Polη, which effectively bypasses ultraviolet (UV)-induced thymine–thymine cyclobutane pyrimidine dimers (CPDs)10,11,12. In contrast, Polζ is a multisubunit TLS
polymerase known for its ability to extend mismatched DNA termini, which can originate from nucleotides inserted by Y-family polymerases13,14. This process is observed during the bypass of
UV-induced 6–4 photoproducts, cisplatin-induced intrastrand crosslinks, and M.HpaII DNA–protein crosslinks (DPCs). In each of these scenarios, Polζ extends the mispaired nucleotide(s)
inserted by Polη15,16,17,18. Despite its deoxycytidyl transferase activity, Rev1 is best known for its scaffolding role on Polζ and other Y-family polymerases19,20. In fact, Rev1 is an
integral component of Polζ in yeast and in _Xenopus_ egg extracts21,22. Rev1 also stimulates the recruitment of Y-family polymerases to lesion sites. This process can occur independently of
PCNA ubiquitylation and offers TLS polymerases an alternative recruitment platform19,23. Consistent with this, Y-family polymerases contain C-terminal regions composed of multiple
protein–protein interaction domains that mediate binding to PCNA, ubiquitin and Rev1. However, the interplay between Rev1 and ubiquitylated PCNA in targeting TLS polymerases to lesion sites
remains unclear. In contrast to Polη, Rev1 and Polζ, the role of Polκ in TLS remains poorly defined. Polκ shares homology with bacterial DinB24,25. In vitro, both DinB and Polκ can bypass
adducts located in the minor groove of DNA, which do not fit in the active site of other TLS polymerases26,27. This is because Polκ contains a unique N-clasp and polymerase-associated domain
(PAD) that allow its catalytic core to open toward the minor groove of DNA28,29. Minor groove adducts include _N_2-adducted guanosines generated by alkylating agents such as benzo[a]pyrene
and acylfuvenes26,30,31. Similarly, illudin S and mitomycin C (MMC) generate minor groove DNA lesions, albeit with different chemistries32,33. Consistent with this, Polκ-deficient cells are
exquisitely sensitive to both illudin S and MMC34,35,36. However, formal proof that Polκ enables the bypass of minor groove DNA lesions during replication is still lacking. Intriguingly,
Polκ-deficient cells are also sensitive to major groove DNA-damaging agents, such as UV radiation and cisplatin35,36,37,38, despite Polκ being unable to synthesize across UV lesions in
vitro39. This suggests that Polκ may function in TLS independently of its catalytic activity. This was proposed on the basis of the sensitivity to different DNA-damaging agents of cells
expressing a catalytic-deficient Polκ40 but the mechanism underlying this function is unknown. In addition, Polκ has also been involved in nucleotide excision repair (NER) in both mammalian
cells and _Xenopus_ egg extracts36,37,41. Polκ was also suggested to participate in the restart of stalled replication forks and the activation of the replication checkpoint42,43,44. How
Polκ is recruited to DNA lesions remains unclear. Like all Y-family polymerases, Polκ contains a long and flexible C-terminal end that mediates interactions with ubiquitylated PCNA45. In
fact, Polκ contains two PCNA-interacting protein (PIP) motifs and two ubiquitin-binding zinc finger (UBZ) domains that may differently contribute to Polκ targeting to lesion sites46.
Moreover, if Polκ can be targeted by PCNA ubiquitylation, why does it also contain a conserved Rev1-interacting region (RIR)? In this regard, the Polκ–Rev1 interaction was shown to mediate
the formation of a stable Polκ–Rev1–Polζ complex in vitro47 but the relevance of this complex is also unknown. In summary, while Polκ catalytic function has been studied in vitro, the roles
and regulations of Polκ during DNA replication remain elusive, particularly with regard to its function across major groove DNA lesions. Here, we elucidate the roles and regulations of Polκ
during DNA replication across defined DNA lesions. By combining clustered regularly interspaced short palindromic repeats (CRISPR) base-editor screening technology in human cells with TLS
analysis of defined DNA lesions in _Xenopus_ egg extracts, we demonstrate that TLS across a minor groove DNA lesion requires Polκ’s catalytic activity. While this function depends on Polκ’s
interaction with ubiquitin and PCNA, it is independent of Rev1. In contrast, we find that a Polκ–Rev1 interaction is essential to stimulate the bypass of major groove DNA lesions and abasic
(AP) sites that require Polζ-mediated extension. Using its flexible C-terminal end, which can cooperatively bind to Rev1 and ubiquitylated PCNA, Polκ appears to stabilize the Rev1–Polζ
complex on damaged chromatin, allowing extension past DNA lesions in a process that is independent of Polκ’s catalytic activity. Collectively, our results unravel the multifaceted functions
and regulations of Polκ during TLS. RESULTS POLΚ IS REQUIRED TO BYPASS MINOR GROOVE ADDUCTS Polκ-deficient cells are exquisitely sensitive to agents that damage the minor groove of DNA such
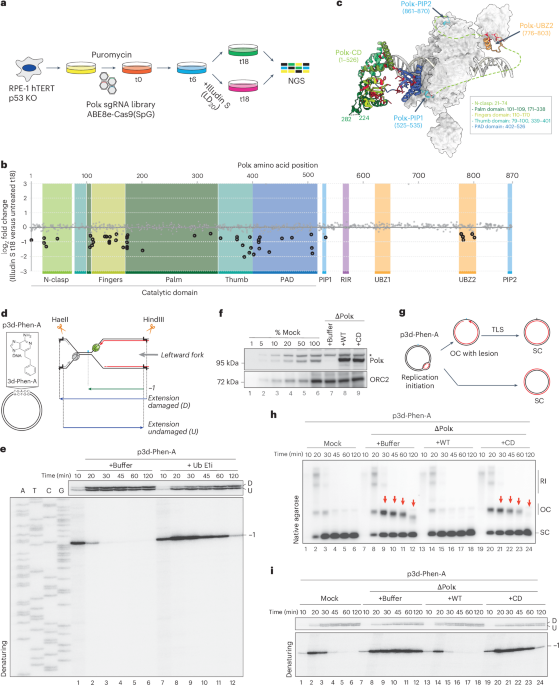
as illudin S35. To understand the role of Polκ’s functional domains in counteracting illudin S-mediated damage, we first performed a CRISPR–Cas9 base-editor tiling screen in the presence and
absence of illudin S. We designed a single guide RNA (sgRNA) lentiviral library aimed at introducing missense mutations along the Polκ coding sequence. The sgRNA library was cloned into
ABE8e-SpG48, which introduces A→G conversions in defined proximity to the sgRNA-binding site. Using this library, we targeted >50% of Polκ amino acids with point substitutions
(Supplementary Table 1). Briefly, RPE1-hTERT _p53_−/− cells were transduced with the lentiviral sgRNA library. After selection, cells were cultured in the presence or absence of a low dose
of illudin S for 12 additional days. Subsequently, sgRNAs were quantified by next-generation sequencing to identify edits that conferred sensitivity to illudin S (Fig. 1a and Supplementary
Table 1). Consistent with Polκ’s unique ability to bypass minor groove DNA adducts in vitro, numerous sgRNAs predicted to cause point substitutions in the catalytic domain of Polκ severely
sensitized cells to illudin S without affecting the untreated condition (Fig. 1b and Extended Data Fig. 1a). These included sgRNAs targeting the N-clasp, PAD and palm and finger domains,
which are essential for Polκ’s TLS properties (Fig. 1b,c and Extended Data Fig. 1b,c)28,29,49. Outside of the catalytic domain, sgRNAs targeting Polκ’s PIP1 (known to bind PCNA) and UBZ2
(known to bind ubiquitin) were the only sgRNAs that also sensitized cells to illudin S (Fig. 1b,c and Extended Data Fig. 1b,c). In contrast, Polκ’s RIR, UBZ1 and PIP2 did not score, despite
the screen containing several sgRNAs targeting these regions. This suggests that these domains may be dispensable to confer illudin S resistance, although we cannot exclude a low editing
efficiency resulting in a lack of effect. Taken together, these results suggest that Polκ is targeted to minor groove DNA lesions by binding to ubiquitylated PCNA (through its PIP1 and UBZ2)
in a process that may be independent of Rev1. To validate our screen and monitor replication across minor groove DNA lesions, we used the _Xenopus_ egg extract system, which recapitulates
DNA replication and TLS15,50. To this end, we generated a plasmid containing a site-specific 3-deaza-3-phenethyl-adenosine acylfulvene minor groove DNA adduct (p3d-Phen-A) (Fig. 1d, left)31
and replicated it in egg extracts supplemented with [α-32P]-labeled deoxyadenosine triphosphate ([α-32P]dATP). We then tested whether the bypass of this lesion requires TLS by comparing
p3d-Phen-A replication with a control plasmid (pCTRL) in the presence or absence of a ubiquitin E1 inhibitor15. While the E1 inhibitor did not affect pCTRL replication (Extended Data Fig.
1d, lanes 1–12), it stabilized open circular (OC) molecules during p3d-Phen-A replication (lanes 13–24). This stabilization suggests a TLS defect in the absence of de novo ubiquitylation,
leading to the accumulation of OC molecules originating from the adducted parental strand (Extended Data Fig. 1e). To understand how the minor groove adduct is bypassed, we analyzed
replication intermediates on a denaturing gel following digest with HaeII and HindIII (Fig. 1d, right). During the replication of p3d-Phen-A, we observed a specific −1 product (Fig. 1e,
lanes 1 and 2). This product disappeared after 20–30 min, correlating with the appearance of extended products (Fig. 1e, lanes 2–6). This is consistent with nascent-strand synthesis first
stalling at the adduct and then bypassing the lesion through TLS. In contrast, in the presence of the ubiquitin E1 inhibitor, nascent-strand synthesis also stalled at −1 but persisted up to
2 h, suggesting severe TLS inhibition (Fig. 1e, lanes 7–12). Next, we investigated whether Polκ is required for bypass of the minor groove adduct. To this end, we depleted Polκ from extracts
and replicated p3d-Phen-A (Fig. 1f and Extended Data Fig. 1f). In the absence of Polκ, conversion of OC to supercoiled (SC) molecules was impaired (Figs. 1g,h, lanes 7–12, red arrows) and
nascent strands persisted at −1 (Fig. 1i, lanes 7–12), indicating the absence of TLS. Polκ-depleted extracts were also supplemented with recombinant Polκ wild type (WT) or a catalytically
inactive mutant harboring D199A and E200A substitutions (CD) (Fig. 1f and Extended Data Fig. 1g)24, which is deficient in DNA synthesis (Extended Data Fig. 1h). TLS was restored by the
addition of Polκ WT but not Polκ CD (Fig. 1h,i, lanes 13–24), confirming that the catalytic activity of Polκ is required to bypass the minor groove DNA lesion. Thus, _Xenopus_ egg extracts
are an ideal system to study the functions and regulations of Polκ during TLS. POLΚ BYPASS OF A MINOR GROOVE LESION IS INDEPENDENT OF REV1 We next examined the functional domains of Polκ
that are required for bypassing the minor groove adduct. To this end, we generated different point mutants of Polκ deficient in PCNA, Rev1 or ubiquitin binding (Fig. 2a)45,51,52,53, which we
added back to Polκ-depleted extracts. Consistent with our tiling screen, the Polκ depletion effect was rescued by the addition of Polκ RIR* (Fig. 2b, lanes 13–16, and Extended Data Fig.
2a,b). Moreover, depletion of Rev1 had no impact on the replication of p3d-Phen-A, which was exclusively dependent on Polκ (Fig. 2c). Thus, Polκ’s catalytic activity across the minor groove
DNA lesion is independent of Rev1. Analysis of Polκ’s PCNA-interacting motifs revealed that both PIP1 and PIP2 contribute to Polκ-mediated bypass. As seen in Fig. 2d (lanes 13–16 and 17–20),
addition of Polκ PIP1 or PIP2 mutant partially restored TLS but not as efficiently as the WT protein (Extended Data Fig. 2c,d). Moreover, addition of a double PIP mutant further impaired
Polκ’s function, leading to nascent strands persisting at −1 for 120 min (Fig. 2d, lanes 21–24, and Extended Data Fig. 2d). We conclude that both PIPs can contribute to Polκ’s TLS activity
across the minor groove adduct. However, in line with previous work showing that PIP1 but not PIP2 is required for Polκ-mediated synthesis54,55 and our base-editor screen where PIP1 but not
PIP2 targeting sensitized cells to illudin S, we envision that Polκ’s PIP1 is the primary PCNA interactor during catalysis. We hypothesize that PIP2 may act as a recruitment or structural
stabilizer by binding to a second PCNA molecule (Discussion). Lastly, analysis of Polκ’s UBZs further agreed with our screen and revealed that UBZ2 but not UBZ1 is important for Polκ’s
function (Fig. 2e and Extended Data Fig. 2e,f). We conclude that Polκ’s catalytic activity across a minor groove lesion is independent of Rev1 and may solely depend on PCNA ubiquitylation
(Discussion). POLΚ’S RIR IS REQUIRED TO BYPASS A MAJOR GROVE DPC Rev1 is a scaffolding protein that facilitates the recruitment of Y-family TLS polymerases to damaged DNA through their RIR
domain9. Surprisingly, we found that Polκ’s RIR is dispensable for bypassing a minor groove adduct, prompting the question of when the Polκ–Rev1 interaction becomes necessary. We previously
showed that both Polκ and Rev1 are synchronously enriched on DNA during replication of a major groove M.HpaII DPC-containing plasmid56. When this lesion is encountered by the replisome, the
DPC is first degraded by the SPRTN protease and/or the proteasome, leading to the formation of a peptide–DNA adduct that is then bypassed by TLS (Extended Data Fig. 3a)56,57. TLS across a
M.HpaII peptide adduct is a two-step mutagenic process, in which Polη inserts one or two nucleotides across the damaged base, followed by Polζ-mediated extension past the lesion15,57. In the
absence of Polη, Polζ performs both insertion and extension, but with higher mutagenesis15. The recruitment of Polκ to the DPC plasmid (Fig. 3a)56 raised the possibility that Polκ may also
have a function in the bypass of this major groove lesion. To test this, we first replicated the M.HpaII DPC plasmid (pDPC) in mock or Polκ-depleted extracts. As a control, pDPC was also
replicated in Polη-depleted or Rev1-depleted extracts (Extended Data Fig. 3b; note that Rev1 codepletes Polζ)15,22. As previously shown, while depletion of Polη had little impact on the
conversion of OC to SC molecules (Extended Data Fig. 3c, lanes 6–10), depletion of Rev1 stabilized OC molecules for up to 240 min, consistent with no bypass of the peptide–DNA adduct
(Extended Data Fig. 3c, lanes 16–20, red arrows)15,57. Strikingly, in the absence of Polκ, conversion of OC to SC molecules was also impaired, suggesting the absence of TLS (Extended Data
Fig. 3c, lanes 11–15, red arrows). To understand how Polκ depletion affects replication across the DPC, nascent leading strands were analyzed on a denaturing gel (Fig. 3b). During
replication of pDPC, nascent-strand synthesis first stalls ~30–40 nt upstream of the DPC (Fig. 3c, lane 1). Following CMG bypass of the DPC, the nascent strand advances to the lesion site
where it stalls at the −1, 0 and +1 positions (Fig. 3c, lanes 2 and 3). The nascent strand is then extended past the lesion and extension products appear by 120 min (Fig. 3c, lanes 3–5, top
radiograph). While insertion across the lesion was dependent on Polη (Fig. 3c, lanes 6–10), extension past the lesion required Rev1–Polζ (Fig. 3c, lanes 16–20), as previously described15.
Surprisingly, in the absence of Polκ, nascent strands reached the crosslink with normal kinetics but also permanently stalled at −1, 0 and +1, mimicking Rev1–Polζ depletion (Fig. 3c, lanes
11–15). This was not because of codepletion of Rev1–Polζ with the anti-Polκ antibody (Extended Data Fig. 3b). Similarly, Polκ was not codepleted upon Rev1–Polζ depletion (Extended Data Fig.
3b). Collectively, these results indicate that Polκ has an essential role in TLS extension past M.HpaII DPC lesions. Next, we investigated whether the Polκ–Rev1 interaction is needed for
this process. To this end, we performed rescue experiments with WT and RIR mutant Polκ. While the addition of WT Polκ restored TLS, addition of RIR mutant Polκ failed to do so (Fig. 3d,e).
Thus, in contrast to Polκ-mediated TLS of the minor groove adduct, the Polκ–Rev1 interaction through Polκ’s RIR is needed for the bypass of a major groove DPC. POLΚ’S NONCATALYTIC FUNCTION
DURING DPC BYPASS Next, we investigated whether Polκ activity stimulates extension past the peptide–DNA adduct and performed rescue experiments with WT or CD Polκ. Strikingly, addition of WT
or CD Polκ rescued the extension defect caused by Polκ depletion (Fig. 4a,b), indicating that Polκ has a noncatalytic function in stimulating TLS across the DPC. Consistent with this, when
Rev3, the catalytic subunit of Polζ, was depleted from extracts (Extended Data Fig. 3b), extension was also abolished (Extended Data Fig. 3d), confirming that Polζ provides the extension
activity across the DPC substrate. Identical results were obtained if pDPC was pretreated with proteinase K to digest the DPC into a short peptide adduct57, indicating that the role of Polκ
in pDPC replication is independent of DPC proteolysis (Extended Data Fig. 4a–c). Moreover, this noncatalytic function of Polκ was also observed when the DPC was placed on the leading or
lagging strand template (Extended Data Fig. 4d,e)57. Together, these results indicate a noncatalytic function of Polκ in stimulating Rev1–Polζ-mediated extension across a major groove DPC
lesion. To further validate this noncatalytic function, we purified a Polκ C-terminal fragment devoid of the catalytic domain (Polκ C-ter) (Fig. 4c). Addition of Polκ C-ter restored
extension past the lesion, albeit not as efficiently as the full-length protein (Fig. 4d,e, lanes 11–15 versus lanes 16–20). Thus, through its long disordered C-terminal region, Polκ can
stimulate Polζ-mediated extension across the DPC. Interestingly, addition of PIP1 mutant but not PIP2 mutant Polκ restored efficient TLS across DPCs (Extended Data Fig. 4f). Moreover,
addition of UBZ1 mutant but not UBZ2 mutant Polκ rescued the TLS defect (Extended Data Figs. 4g and 3e). Thus, in contrast to Polκ’s catalytic function, which is independent of Rev1, Polκ’s
noncatalytic function in DPC bypass requires binding to Rev1 (through RIR), ubiquitin (through UBZ2) and PCNA (through PIP2) (Discussion). POLΚ STIMULATES POLΖ EXTENSION ACROSS DIFFERENT DNA
LESIONS We next investigated whether the noncatalytic function of Polκ is specific to M.HpaII DPCs or whether it could be extended to other DPCs, such as those generated endogenously by
HMCES crosslinking58. It was recently shown that HMCES crosslinks to AP sites in single-stranded DNA (ssDNA) to protect them from nucleophilic attack and double-strand breaks (DSBs)58. HMCES
DPCs are endogenous intermediate lesions formed during the repair of interstrand crosslinks formed at an AP site (AP-ICL)59. During this process, the NEIL3 glycosylase is recruited to
stalled forks and unhooks the AP-ICL by cleaving the _N_-glycosyl bond of the crosslinked site. This generates an AP site on one of the daughter molecules (Fig. 5a, i and ii), which is
quickly protected by HMCES59,60. HMCES DPCs are then degraded by SPRTN and the resulting peptide–DNA adduct is bypassed by Rev1–Polζ (Fig. 5a, iii)59,60. To address whether Polκ also assists
Polζ during the bypass of HMCES DPCs, we replicated the AP-ICL plasmid in mock or Polκ-depleted extracts. In the absence of Polκ, permanent stalling at −1 was observed for the
leftward-moving fork, indicating that Polκ is required to bypass HMCES peptide–DNA adducts (Fig. 5b, lanes 1–5 versus lanes 6–10, and Extended Data Fig. 5a). This effect was rescued by the
addition of both WT and CD Polκ (Fig. 5b, lanes 11–20, and Extended Data Fig. 5a). Thus, Polκ’s noncatalytic function is not restricted to the bypass of M.HpaII DPCs but also occurs on other
DPCs, such as endogenous DPCs generated by HMCES crosslinking to AP sites. We next addressed whether Polκ is also able to assist Rev1–Polζ across another type of physiologically relevant
DNA lesion. AP sites are intermediate lesions of AP-ICL repair and dependent on Polζ for their bypass61. Thus, to stabilize the AP site, we replicated the AP-ICL plasmid in the absence of
HMCES (Extended Data Fig. 5b,c)59. In this setting, Polκ depletion again resulted in permanent stalling at −1 (Fig. 5c, lanes 5–8, and Extended Data Fig. 5d), which was again rescued by the
addition of both WT and CD Polκ (Fig. 5c, lanes 9–16, and Extended Data Fig. 5d). Thus, the noncatalytic function of Polκ extends beyond DPCs to other types of Rev1–Polζ-dependent DNA
lesions such as AP sites. POLΚ AND REV1–POLΖ FORM A COMPLEX ON DAMAGED CHROMATIN Next, we explored the mechanism by which Polκ promotes Polζ-mediated extension. We first hypothesized that
Polκ could promote polymerase switching between Polη and Polζ by stimulating the removal of Polη. To test this, we monitored Polκ’s function during pDPC replication in the absence of Polη.
Unlike Polη depletion, which is permissive to Polζ-mediated bypass (Fig. 6a, lanes 6–10, and Extended Data Fig. 6a)15, the absence of both Polη and Polκ inhibited TLS across the lesion and
nascent strands now permanently stalled at −1 (Fig. 6a, lanes 11–15). This was rescued with WT or CD Polκ (Fig. 6a, lanes 16–25, and Extended Data Fig. 6a), indicating that Polκ assists Polζ
catalysis, even in the absence of Polη. These results suggest that Polκ directly stimulates Polζ recruitment and/or activity during TLS and that this function is independent of the prior
engagement of another TLS polymerase at the lesion site. Moreover, it indicates that Polκ can assist Polζ not only during the extension but also during the insertion step across a damaged
nucleotide. We previously showed that, upon high-dose UV-C damage, Polκ is de-enriched from damaged chromatin when Rev1–Polζ is depleted from extracts (Fig. 6b)15, according to chromatin
mass spectrometry (CHROMASS) experiments performed in the absence of DNA replication. Because Polκ and Rev1 do not codeplete each other (Extended Data Fig. 3b), this suggests that Polκ may
form a complex with Rev1–Polζ on damaged chromatin. To assess the potential interdependency between Polκ and Rev1–Polζ localization to damaged chromatin, we performed a similar CHROMASS
experiment to identify proteins whose recruitment depend on either Polκ or Rev1 (ref. 62). Briefly, sperm chromatin was treated with a high dose of UV-C, followed by its incubation in mock,
Polκ-depleted or Rev1-depleted egg extracts and analysis by quantitative mass spectrometry (MS) (Fig. 6c, top). This experiment was also performed in the absence of DNA replication to
minimize effects arising from uncoupled DNA replication fork structures. Notably, when DNA is irradiated with a high UV-C dose, closely spaced opposing lesions are generated, which trigger
the recruitment of TLS polymerases following a first round of nucleotide excision63. Accordingly, we observe PCNA ubiquitylation and a robust enrichment of TLS polymerases in the absence of
DNA replication (Extended Data Fig. 6b)15,64. Consistent with Rev1–Polζ and Polκ forming a complex on damaged DNA, Rev1 depletion led to a significant de-enrichment of Polκ from damaged
chromatin (Fig. 6c, _y_ axis, Extended Data Fig. 6c and Supplementary Table 2). Similarly, Polκ depletion also led to the de-enrichment of Rev1–Polζ (Fig. 6c, _x_ axis, Extended Data Fig. 6d
and Supplementary Table 2). Note that Scai, which associates with Rev1–Polζ64, was also significantly de-enriched in both conditions (Fig. 6c). In addition to Polζ, we noted that many
Fanconi anemia (FA) proteins were significantly de-enriched from Polκ-depleted samples (Extended Data Fig. 6d). This was likely caused by a codepletion of the FA complex with the anti-Polκ
antibody (Extended Data Fig. 6e,f). Conversely, upon depletion of either Rev1 or Polκ, we observed the enrichment of Polη and its interactor Wrnip1 (refs. 65,66), as well as proteins
involved in DSB repair, such as Rnf168 and 53bp1 (Fig. 6c). This suggests that, in the absence of either Polκ or Rev1–Polζ, gap-filling synthesis across certain UV lesions is disrupted (for
example, UV-induced 6–4 photoproducts), which leads to the accumulation of Polη on chromatin and the formation of DSBs. Alternatively, the accumulation of 53bp1 could be linked to the
formation of large ssDNA gaps that form when TLS is inhibited and the described role of 53bp1 in regulating the balance between TLS and TS67. To validate our CHROMASS results, we assessed
protein recruitment to UV-damaged sperm chromatin by immunoblotting upon Rev1 or Polκ depletion. As controls, we also depleted Polη or Rfwd3, which regulates PCNA ubiquitylation and TLS
polymerase recruitment to lesion sites15. Consistent with this, Rfwd3 depletion led to impaired PCNA ubiquitylation and de-enrichment of Polη, Rev1–Polζ, and Polκ to UV-treated chromatin
compared to the control reaction (Fig. 6d, lanes 4–8 versus lanes 10–14). Confirming the CHROMASS results, the enrichment of Rev1–Polζ was also impaired in the absence of Polκ (Fig. 6d,
lanes 4 and 5 versus lanes 10 and 11). Similarly, the recruitment of Polκ was reduced in the absence of Rev1 (Fig. 6d, lanes 4–7 versus lanes 10–13). The interdependency between Polκ and
Rev1–Polζ was also observed during DNA replication following the treatment of sperm chromatin with low-dose UV-C (Extended Data Fig. 6g) and when monitoring protein recruitment to pDPC by
plasmid pulldown (Extended Data Fig. 6h). Moreover, although Rev1–Polζ is not involved in the bypass of the minor groove adduct (Fig. 2c), we could still detect its enrichment on p3d-Phen-A,
which was also partially dependent on Polκ (Extended Data Fig. 6i). Thus, the Rev1–Polζ–Polκ complex appears to form during TLS irrespectively of the DNA lesion. In contrast, Polη
enrichment to damaged chromatin was independent of either Rev1 or Polκ; conversely, the absence of Polη did not impact Rev1–Polζ or Polκ recruitment (Fig. 6d). Rev1–Polζ enrichment could be
restored by the addition of WT or CD Polκ to Polκ-depleted extracts (Fig. 6e). Together, these results suggest that Polκ and Rev1–Polζ form a stable complex on damaged DNA, independently of
Polη. CATALYTIC AND NONCATALYTIC FUNCTIONS OF POLΚ IN HUMAN CELLS To assess the functional importance of Polκ in human cells, we constructed a knockout (KO) and complementation system in
U2OS Flp-In T-REx cells. This system allows for stable doxycycline-inducible expression from the _FRT_ locus. We generated U2OS _POLK-_KO cells (Extended Data Fig. 7a, clone 1.10) and stably
complemented the cells with Venus-tagged WT or CD Polκ (Extended Data Fig. 7b). As expected, U2OS _POLK-_KO cells were hypersensitive to both illudin S and cisplatin as shown by a colony
formation assay (Fig. 7a–d)34,35,36,38. The illudin S hypersensitivity was significantly suppressed by the expression of WT but not CD Polκ (Fig. 7a,b), supporting the requirement of Polκ’s
catalytic activity for the bypass of illudin S-induced minor groove lesions. In contrast, while the cisplatin sensitivity observed in _POLK-_KO cells was only partially suppressed by the
expression of Polκ, it was suppressed equally well by WT or CD Polκ (Fig. 7c,d). This observation supports Polκ’s function in cisplatin tolerance that is independent of its catalytic
activity. DISCUSSION Polκ has the distinct ability among eukaryotic polymerases to bypass minor groove DNA adducts. Here, we study for the first time the bypass mechanism of a minor groove
DNA lesion using an extract system that recapitulates DNA replication and TLS. We demonstrate that bypass of a minor groove DNA lesion is exquisitely dependent on Polκ’s catalytic activity,
validating previous observations made with purified enzymes. Strikingly, we also uncover a noncatalytic function of Polκ during TLS that is required for Polζ-mediated extension past AP sites
and major groove DNA lesions. Whereas Polκ’s noncatalytic function in TLS requires an interaction with Rev1, Polκ’s catalytic function is independent of Rev1. Thus, Polκ serves multiple
functions during TLS, which are differentially regulated through its C-terminal disordered region. POLΚ AND MINOR GROOVE DNA LESIONS Polκ is the only vertebrate DNA polymerase shown to
synthesize across minor groove DNA lesions in vitro but direct evidence of this process in a physiological context was lacking. Using _Xenopus_ egg extracts, we show that Polκ is essential
to bypass p3d-Phen-A (Fig. 1). In the absence of Polκ, nascent strands remained stalled at the lesion (Fig. 1i), indicating that no other DNA polymerase could compensate for Polκ’s absence.
This is consistent with the structural features of Polκ, where the unique conformation of the PAD opens its catalytic core toward the minor groove of DNA28. Moreover, Polκ contains a unique
N-clasp, which contributes to stabilizing the incoming template DNA. In accordance, our base-editor screen suggested that point substitutions in the N-clasp and PAD increase cellular
sensitivity to illudin S (Fig. 1b). This is likely because of the impaired ability of these mutants in bypassing minor groove DNA adducts induced by illudin S. We further provide the
regulatory mechanisms underlying Polκ’s catalytic function across the minor groove DNA lesion. Previous in vitro studies reported that Polκ’s interaction with PCNA through its PIP1 domain
was required for Polκ’s catalytic activity, whereas PIP2 was dispensable54,55. Our base-editor screen showed that gRNAs targeting PIP1 but not PIP2 increased the sensitivity to illudin S,
reinforcing that PIP1 mediates the primary Polκ–PCNA interaction required for DNA synthesis (Fig. 1b). However, our work with extracts suggests that PIP2 also contributes to Polκ’s function.
We found that while PIP1 or PIP2 mutants exhibited mildly affected catalytic function, PIP1 and PIP2 double mutants exhibited severely impaired lesion bypass (Fig. 2d), implying that PIP1
and PIP2 can partially compensate for each other during catalysis. Consistent with our data, a composite model of the catalytic complex suggests that both PIPs are capable of binding
DNA-loaded PCNA simultaneously (Fig. 7e), which may increase Polκ’s association with PCNA during lesion bypass. In this scenario, we envision that each PIP binds successively; PIP2 initially
binds a free PCNA molecule within the trimer, followed by PIP1 binding to a second PCNA molecule, effectively locking Polκ’s catalytic site on the damaged template. In addition to
PCNA-binding domains, Polκ contains an RIR53,68. Rev1 was shown to act as a scaffolding protein that recruits Y-family TLS polymerases to damaged sites68. However, we show that
Polκ-dependent bypass of the minor groove adduct does not require Rev1 or Rev1 binding (Fig. 2b,c). Instead, Polκ’s interaction with ubiquitylated PCNA (through PIP1, PIP2 and UBZ2) appears
to be sufficient for Polκ-mediated synthesis (Fig. 7e). Together, our work supports the notion that Polκ is the vertebrate translesion polymerase specialized in bypassing minor groove DNA
lesions. POLΚ’S NONCATALYTIC FUNCTION IN TLS We show that Polκ, through its RIR, stimulates Polζ-mediated extension across different DNA lesions in a process independent of Polκ’s catalytic
activity (Figs. 3–5). In fact, even a truncated Polκ lacking its catalytic domain was able to stimulate Polζ-mediated bypass, albeit not as efficiently as the full-length protein (Fig.
4d,e). This suggests that most of the stimulatory functions are driven by Polκ’s C-terminal domains, which interact with ubiquitylated PCNA and Rev1 (Fig. 7f). The reduced rescue efficiency
by the Polκ C-terminal fragment could be attributed to its highly disordered structure, which may reduce the stability of the protein fragment compared to full-length Polκ. By superimposing
AlphaFold2-generated molecular models of _Xenopus_ Rev1–Polζ onto the known Rev1–Polζ structure from yeast69 and modeling the interactions between Polκ and this complex, we find that the
Polκ–Rev1 interaction through the RIR domain is likely mutually exclusive with a PIP1 interaction with PCNA (Fig. 7f). Consistent with this, Polκ’s noncatalytic function requires Polκ RIR
and PIP2 but is independent of PIP1 (Fig. 3e and Extended Data Fig. 4f). On the basis of our findings that Rev1–Polζ and Polκ recruitment to damaged DNA are reciprocally dependent on each
other (Fig. 6), we propose that Polκ uses its multiple interaction domains to stabilize the Rev1–Polζ complex on DNA, promoting Polζ-mediated extension beyond various DNA lesions (Fig. 7f).
This complex may serve as the last-resort TLS complex, functioning only after failed attempts by Y-family polymerases to bypass a lesion (that is, Polη, Polκ, Rev1 or Polι). These lesions
could include those across which Y-family polymerases can insert but not extend (for example, 6–4 photoproducts or M.HpaII DPCs), as well as lesions unsuited for Y-family polymerase
insertion. Notably, our results build upon a previous structural study that showed the formation of a stable Rev1–Rev3–Rev7–Polκ quaternary complex in vitro47, now highlighting the
functional relevance of this complex. Interestingly, in addition to its catalytic function across minor groove DNA lesions, Polκ possesses extension activity past mismatched bases in
vitro70,71. The formation of a Rev1–Polζ–Polκ complex would enable the recruitment of two distinct polymerases with extension activity to the same DNA lesion. Although we only observe the
stimulation of Polζ by Polκ, it is tempting to speculate that the opposite might also occur on DNA lesions that Polζ is unable to bypass. This arrangement would provide a safeguarding
mechanism, ensuring that, if one polymerase is unable to extend past the DNA lesion, the other polymerase could quickly replace it and extend beyond the damage. Together, our data reveal
both catalytic and noncatalytic functions of Polκ during TLS and demonstrate the specific roles of its different interaction domains for each function. POLΚ’S ROLE IN NER Polκ was shown to
participate in the DNA synthesis step of NER, both in human cells irradiated with UV-C37,41 and in _Xenopus_ egg extracts during the repair of a trimethylene ICL36. A recent study showed
that the participation of TLS polymerases during NER also includes Rev1, Polη and Polι when closely spaced opposing lesions are generated following high doses of UV-C63. Accordingly, we show
that Polη, Rev1–Polζ and Polκ are all recruited to chromatin treated with high-dose UV-C in the absence of DNA replication (Fig. 6). Ultimately, the nature of the lesion dictates which TLS
polymerase is used for its bypass. A previous study showed that the bypass of a trymethylene ICL during NER required Polκ’s catalytic function, whereas Polκ’s binding to Rev1 was
dispensable36. In light of our findings, we propose that this is attributed to Polκ’s catalytic role in bypassing the minor groove trimethylene adduct, rather than to a specific regulation
of Polκ by NER. ROLE OF REV1 IN Y-FAMILY TLS REGULATION Y-family TLS polymerases feature long, flexible C-terminal ends that harbor multiple interaction domains with PCNA, ubiquitin and
Rev1. Existing models propose that PCNA ubiquitylation facilitates the recruitment of TLS polymerases to sites of DNA lesions6,7,8,72. Alternatively, Rev1 acts as a scaffold, enabling the
recruitment of TLS polymerases to damaged sites regardless of PCNA ubiquitylation status19,68. Thus, two modes of TLS polymerase recruitment coexist but the precise interplay and conditions
determining the relevance of each pathway remain unknown. In our study, we showed that binding of Polκ to ubiquitylated PCNA is likely sufficient for targeting its catalytic function (Fig.
2). In contrast, Rev1 regulates Polκ’s noncatalytic function in stimulating Polζ extension (Fig. 3d,e). Thus, rather than acting as a general recruitment platform for Y-family polymerases,
Rev1 appears to govern the assembly of specialized TLS subcomplexes. We envision that these complexes may form on DNA irrespectively of the DNA lesion but the lesion type ultimately dictates
the TLS complex used to bypass it. Like Polκ, Polη and Polι also contain RIRs of unclear importance, prompting speculation that Rev1 may similarly regulate unknown functions of these
polymerases. POLΚ’S FUNCTION IN MAMMALIAN CELLS Consistent with Polκ’s unique function in replicating across a minor groove DNA adduct, _POLK_-KO cells exhibit severe sensitivity to
minor-groove-inducing agents35,36, which we show here is dependent on Polκ’s catalytic activity (Fig. 7a,b). Intriguingly, _POLK_-KO cells are also sensitive to other DNA-damaging agents,
such as UV and cisplatin, which induce lesions on the major groove of DNA that require Polζ-mediated extension for bypass20,22,35,37. Our study revealed that the sensitivity to cisplatin
observed in the absence of Polκ could be partially restored to the same extent with WT or CD Polκ (Fig. 7c,d). Hence, we propose that the sensitivity of _POLK_-KO cells to UV and cisplatin
is attributed to the noncatalytic function of Polκ, facilitating Rev1–Polζ-dependent TLS. Notably, _POLK_-KO cells have also been reported to be sensitive to oxidative agents, such as
potassium bromate, which generates AP sites and HMCES DPCs35. This sensitivity may also be linked to Polκ’s noncatalytic role across AP sites or HMCES DPCs, as we report here (Fig. 5).
Unlike _POLK-_KO cells, _REV3_−/− mouse embryonic stem cells are not viable73, which suggests that Polζ possesses functions independent of Polκ. Interestingly, Rev3 also has a role beyond
TLS by facilitating DNA replication through heterochromatic regions74, which could account for its essential role. Emerging strategies in cancer therapy include the development of TLS
inhibitors, which may enhance tumor sensitivity to first-line chemotherapeutics75,76. Notably, our results emphasize the potential benefit of targeting specific functional domains within
Y-family polymerases, such as the Polκ–Rev1 interaction. This targeted approach may selectively sensitize cancer cells to genotoxic agents. METHODS _XENOPUS_ EGG EXTRACTS AND DNA REPLICATION
REACTIONS _Xenopus_ egg extracts were prepared as described previously81. All experiments involving animals were approved by the Danish Animal Experiments Inspectorate and conform to
relevant regulatory standards and European guidelines. For plasmid DNA replication, plasmids were licensed in high-speed supernatant (HSS) at a final concentration of 7.5 ng µl−1 for 30 min
at room temperature (RT). Replication was initiated by adding two volumes of nucleoplasmic egg extract (NPE). Gap-filling reactions were performed in nonlicensing extracts (extracts that do
not support MCM2–MCM7 licensing), where one volume of HSS was premixed with two volumes of NPE before the addition of plasmid DNA (final concentration of 10 ng µl−1). For replication in the
presence of LacI, plasmid DNA (150 ng µl−1) was incubated with an equal volume of 12 µM LacI for 1 h before licensing57. The ubiquitin E1 inhibitor MLN-7243 (Active Biochem) was supplemented
to NPE at a final concentration of 200 µM 10 min before initiating the reaction. To visualize DNA replication intermediates, replication reactions were supplemented with [α-32P]dATP (Perkin
Elmer). For each time point, 1 µl of the reaction mixture was added to 5 µl of stop buffer (5% SDS, 80 mM Tris pH 8.0, 0.13% phosphoric acid and 10% Ficoll), followed by the addition of 1
µl of proteinase K (20 mg ml−1) (Roche). The samples were incubated at 37 °C for 1 h and subsequently separated using 0.9% native agarose gel electrophoresis; results were visualized using a
phosphor imager. The radioactive signal was quantified using ImageJ (National Institutes of Health). PREPARATION OF DNA CONSTRUCTS pDPC and pDPC2×Lead were prepared as previously
described56. Additionally, pDPCPK and pDPCssDNA-PK were prepared as previously described15 as pMHPK and pMHssDNA-PK, respectively. Moreover, pDPCLead and pDPCLag were prepared as previously
described57 as pDPC-LTop (Lead) and pDPC-LBot (Lag), respectively. To generate a plasmid containing p3d-Phen-A, we first removed the LacO array from pJSL3 (ref. 82) using the complementary
overhangs of the BsrG1 and BsiWI restriction sites. Subsequently, the two Nb.BsrdI nicking sites were removed using mutagenesis. An A located at position 1557 of the plasmid was mutated to a
C to remove the first Nb.BsrDI site using the following primers: 5′-CCACGATGCCTGTAGCCATGGCAACAACGTTGC-3′ and 5′- GCAACGTTGTTGCCATGGCTACAGGCATCGTGG-3′. Secondly, a T located at position 1740
of the plasmid was mutated to a C to remove the second Nb.BsrDI site using the following primers: 5′-GGTCTCGCGGTATCATCGCAGCACTGGGGCCAG-3′ and 5′-CTGGCCCCAGTGCTGCGATGATACCGCGAGACC-3′.
Afterward, we used the PciI and BsaX1 restriction sites to clone in the oligo 5′-CATGGCTCTTCNACCTCAACTACTTGACCCTCCTCATTCATTGCTTG-3′ to introduce Nt.BspQ1 and Nb.BsrD1 nicking sites. Finally,
to generate p3d-Phen-A, the vector was nicked using Nt.BspQ1 and Nb.BsrD1 and ligated with an excess of the following oligo containing 3d-Phen-A at position 15:
5′-ACCTCAACTACTTGACCCTCCTCATT-3′ (ref. 31). pAP-ICL was generated as previously described60. ANTIBODIES AND IMMUNODEPLETIONS Antibodies to Rev1 (Rev1-N and Rev1-C)22, Rfwd3 (ref. 15), Polη15
and HMCES59 were described previously. Antibodies to Polκ, Rev3 and FancA 2 were raised by New England Peptide by immunizing rabbits with Ac-CPASKKSKPNSSKNTIDRFFK-OH, Ac-CLADLSIPQLD-OH and
Ac-CSFKAPDDYDDLFFEPVF-OH, respectively. The antibody to FancA 1 was a kind gift of A. Sobeck83. To immunodeplete Rev1 from _Xenopus_ egg extracts, an equal volume of Protein A Sepharose fast
flow (PAS) (GE Health Care) beads was bound to anti-Rev-N or anti-Rev1-C antibodies overnight at 4 °C. The beads were then washed twice with 500 µl of PBS, once with ELB (10 mM HEPES pH
7.7, 50 mM KCl, 2.5 mM MgCl2 and 250 mM sucrose), twice with ELB supplemented with 0.5 M NaCl and twice with ELB. One volume of precleared HSS or NPE was then depleted by mixing with 0.2
volumes of antibody-bound beads and then incubated at RT for 15 min, before being isolated. For HSS, the depletion procedure was performed once with Rev1-N coupled beads and once with Rev1-C
coupled beads. For NPE, the depletion procedure was performed twice with Rev1-N coupled beads and once with Rev1-C coupled beads. To immunodeplete Polκ, Polη or Rfwd3 from _Xenopus_ egg
extracts, one volume of PAS beads was bound to five volumes of affinity-purified antibody (1 mg ml−1). The beads were washed as described above and one volume of precleared HSS or NPE was
then depleted by mixing with 0.2 volumes of antibody-bound beads for 15 min at RT. The depletion procedure was performed once for HSS and three times for NPE. For HMCES and Polκ combined
depletion, one volume of beads was bound to eight volumes of each affinity-purified antibody (1 mg ml−1). The beads were washed and depletion was performed as described for Polκ
immunodepletion. IMMUNOPRECIPITATIONS For the FancA and Polκ immunoprecipitation experiments, 5 μl of PAS beads were incubated with 10 μg of the respective affinity-purified antibody for 1 h
at RT. The Sepharose beads were subsequently washed twice with PBS and three times with IP buffer (10 mM HEPES pH 7.7, 50 mM KCl, 2.5 mM MgCl2 and 0.25% NP-40). Next, 5 μl of NPE was
diluted with 20 μl of IP buffer and incubated with antibody-prebound beads for 1 h at RT. The beads were then washed three times with IP buffer and resuspended in 50 μl of 2× Laemmli sample
buffer before analysis by western blotting. NASCENT LEADING-STRAND ANALYSIS For nascent leading-strand analysis, 3–4 µl of replication reaction was added to ten volumes of transparent stop
buffer (50 mM Tris-HCl pH 7.5, 0.5% SDS and 25 mM EDTA). The replication intermediates were purified as previously described84,85. The DNA was digested with the indicated restriction enzymes
and subsequently supplemented with 0.5 volumes of denaturing PAGE gel loading buffer II (Life technologies). The digested DNA products were separated on a 6% polyacrylamide sequencing gel.
PROTEIN EXPRESSION AND PURIFICATION Full-length _Xenopus laevis_ Polκ with an N-terminal 6xHis-tag was amplified from pCMV-Sport.ccdb-Polκ36 and cloned into pET28b (Novagen) using primers A
and B and restriction enzymes BamHI and XhoI. _Xenopus_ Polκ C-ter with an N-terminal 6xHis-tag was cloned into pET28b using primers B and C and restriction enzymes BamHI and XhoI. Polκ
amino acid substitutions were introduced by Quikchange mutagenesis and confirmed by Sanger sequencing. Plasmids containing WT Polκ, mutant Polκ or Polκ C-ter were transformed into BL21
_Escherichia coli_ competent cells. Cells were grown at 37 °C to an optical density of 0.6–0.8 in Luria–Bertani broth and were subsequently induced with 0.5 mM IPTG for 4 h. Bacteria were
harvested by centrifugation and resuspended in 20 ml of lysis buffer (50 mM Tris pH 7.5, 300 mM NaCl, 2 mM MgCl2, 1 mM DTT and 1× Roche EDTA-free cOmplete protease inhibitor cocktail).
Suspensions were sonicated and cleared by high-speed centrifugation at 15,000 r.p.m. in a F15-8x50cy rotor for 1 h at 4 °C. The soluble fraction was collected and incubated with 2 ml of
Ni-NTA Superflow affinity resin (Qiagen), previously equilibrated with lysis buffer, for 2 h at 4 °C. The resin was then washed three times with 20 ml of wash buffer (50 mM Tris pH 7.5, 300
mM NaCl, 2 mM MgCl2, 1 mM DTT, 0,1% Triton-X and 10 mM imidazole). Then, 6xHis-tagged Polκ was eluted with elution buffer (50 mM Tris pH 7.5, 300 mM NaCl, 2 mM MgCl2, 1 mM DTT, 10% glycerol
and 10 mM imidazole). Elution fractions containing the target proteins were pooled and dialyzed against dialysis buffer (50 mM Tris pH 7.5, 300 mM NaCl, 2 mM MgCl2, 1 mM DTT and 10%
glycerol) at 4 °C overnight. After dialysis, protein fractions were concentrated to 100 μl using centrifugal filters with a molecular weight cutoff of 30,000 (Amicon) and subsequently
aliquoted, flash-frozen in liquid nitrogen and stored at −80 °C. Primer A: 5′-ATGCGGATCCAATGGATAACAAGCAAGAAGCAGAG-3′ Primer B: 5′-ATGCCTCGAGCTACTTGAAGAATCTGTCGATGGTG-3′ Primer C:
5′-ATGCGGATCCAAAACATCACCAGAAGAGCATTACTAG-3′ Plasmids for expressing _X._ _laevis_ WT and CD Polκ in rabbit reticulocytes were kind gifts from J. Gautier36. Briefly, 2 μg of pCMV-Sport-Polκ
was incubated with 100 µl of TnT Sp6 Quick master mix (Promega) supplemented with 4 μl of 1 mM methionine for 90 min at 30 °C. The reaction volume was subsequently adjusted to 400 μl with
PBS and DNA was precipitated by the addition of 0.06% polymin-P and incubation for 30 min at 4 °C with rotation. The mixture was then centrifuged at 14,000_g_ for 30 min and the proteins in
the supernatant were precipitated with saturated ammonium sulfate to a final concentration of 55% for 30 min at 4 °C with rotation, followed by centrifugation at 16,000_g_ for 30 min. The
protein pellet was subsequently resuspended in 15 μl of ELB, dialyzed for 3 h at 4 °C in ELB. As a negative control, a reaction without DNA was performed. The Polκ protein preparations
obtained using this method were used for gap-filling synthesis experiments (Extended Data Fig. 1d). PLASMID PULLDOWN Plasmid pulldowns were performed as described previously22. Briefly, 6 μl
of streptavidin-coupled magnetic beads (Dynabead M-280, Invitrogen) per pulldown reaction were equilibrated with wash buffer 1 (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA pH 8 and 0.02%
Tween-20) and then incubated with 12 pmol of biotinylated LacI at RT for 40 min. The beads were washed four times with pulldown buffer 1 (10 mM HEPES pH 7.7, 50 mM KCl, 2.5 mM MgCl2, 250 mM
sucrose, 0.25 mg ml−1 BSA and 0.02% Tween-20), resuspended in 40 μl and stored on ice until used. At the indicated time points, 10 μl of reaction was added to the beads and rotated for 30
min at 4 °C. The beads were subsequently washed twice in wash buffer 2 (10 mM HEPES pH 7.7, 50 mM KCl, 2.5 mM MgCl2, 0.25 mg ml−1 BSA and 0.03% Tween-20) and resuspended in 40 μl of 2×
Laemmli sample buffer. CHROMATIN SPIN DOWN Demembranated _Xenopus_ sperm chromatin was prepared as described previously86 and stored at −80 °C at a concentration of 100,000 sperm chromatin
per µl (320 ng µl−1). For analysis of UV-damaged chromatin, sperm chromatin was diluted to 25,000 sperm chromatin per µl in ELB, deposited on parafilm and irradiated with 2,000 J m−2 (for
nonreplicating reactions) or 20 J m−2 (for replicating reactions) of UV-C. For nonreplicating reactions, HSS and NPE were premixed at a 1:2 ratio to block licensing. Subsequently, undamaged
or UV-damaged sperm chromatin was added at a final concentration of 16 ng µl−1. For replicating reactions, sperm chromatin was licensed in one volume of HSS for 30 min followed by the
addition of two volumes of NPE. At the indicated time points, 8 µl of replication reaction was stopped with 60 µl of ELB supplemented with 0.2% Triton-X. The mixture was carefully layered on
top of a sucrose cushion (10 mM HEPES pH 7.7, 50 mM KCl, 2.5 mM MgCl2 and 500 mM sucrose) and spun for 1 min at 6,800_g_ in a swing-bucket centrifuge at 4 °C. The chromatin pellet was
carefully washed twice with 200 µl of ice-cold ELB and resuspended in 2× Laemmli buffer. ALPHAFOLD MODEL GENERATION Molecular models were predicted using AlphaPulldown 0.30.0 (ref. 77),
running AlphaFold 2.3.1 (ref. 78). AlphaPulldown parameters were as follows: cycles = 3, models = 5 and predictions per model = 1. Structure predictions were generated for _X._ _laevis_
Q6DFE4 (POLK), P18248 (PCNA), Q6NRK6 (REV1), D0VEW8 (REV3), Q8QFR4 (REV7), O93610 (POLD2), Q76LD3 (POLD3) and P62972 (UBIQP), individually and as complexes of either full-length proteins or
protein fragments. Models were evaluated on their predicted local distance difference test78, interface predicted template modeling77,87, predicted template modeling78 and predicted aligned
error88 scores. From each prediction, the best model as determined by AlphaPulldown was selected for inclusion in the final complex models. Model building was performed using UCSF
ChimeraX89,90. The catalytic complex was modeled on a scaffold of human Polκ holoenzyme with Ub-PCNA (Protein Data Bank (PDB) 7NV1 (ref. 55)). The noncatalytic complex was modeled on a
scaffold of the yeast Polζ (PDB 6V93 (ref. 69)). To establish the relative position of Polζ to PCNA, a structure of processive human Polδ holoenzyme was used (PDB 6TNY (ref. 91)). The
monoubiquitinated PCNA and scaffold DNA attached to the polymerase complex was modeled on a structure of monoubiquitinated PCNA (PDB 3TBL (ref. 80)). CHROMASS CHROMASS experiments were
performed as previously described62. Briefly, isolated sperm chromatin was either untreated or treated with 2,000 J m−2 of UV-C. Each reaction was performed in quadruplicate. The sperm
chromatin was then incubated at a final concentration of 16 ng µl−1 in nonlicensing extracts that were mock-treated, Polκ-depleted or Rev1-depleted. Reactions were stopped after 45 min.
Specifically, 10 µl of replication reaction was stopped with 60 µl of ELB supplemented with 0.2% Triton-X and chromatin spin down performed as described above. The chromatin pellet was then
resuspended in 100 µl of denaturation buffer (9 M urea and 100 mM Tris-HCl pH 8) and transferred to a new low-binding tube. Cysteines were reduced (1 mM DTT for 15 min at RT) and alkylated
(0.55 M chloroacetamide for 40 min at RT protected from light). Proteins were first digested with 0.5 µg of LysC (2.5 h at RT) and then with 0.5 µg of trypsin at 30 °C overnight. Peptides
were acidified with 10% trifluoroacetic acid (pH < 4), followed by the addition of 400 mM NaCl, and purified by StageTip (C18 material). For this, StageTips were first activated in 100%
methanol, then equilibrated in 80% acetonitrile in 0.1% formic acid and finally washed twice in 0.1% formic acid. Samples were loaded onto the equilibrated stage tips and washed twice with
50 µl of 0.1% formic acid. StageTip elution was performed with 80 μl of 25% acetonitrile in 0.1% formic acid; eluted samples were dried to completion in a SpeedVac at 60 °C, dissolved in 10
μL 0.1% formic acid and stored at −20 °C until MS analysis. MS DATA ACQUISITION All MS samples were analyzed on an EASY-nLC 1200 system (Thermo) coupled to an Orbitrap Exploris 480 MS
instrument (Thermo). Of the _n_ = 4 biochemical replicates, 50% were analyzed per run (R1–R4). Afterward, an additional _n_ = 4 technical replicates were performed by mixing 25%:25% of R1:R2
(R5), R2:R3 (R6), R3:R4 (R7) and R4:R1 (R8), totaling _n_ = 8 technical replicates. Separation of peptides was performed using 20-cm columns (75-μm internal diameter) packed in house with
ReproSil-Pur 120 C18-AQ 1.9-μm beads (Dr. Maisch). Elution of peptides from the column was achieved using a gradient ranging from buffer A (0.1% formic acid) to buffer B (80% acetonitrile in
0.1% formic acid), at a flow of 250 nl min−1. The gradient length was 80 min per sample, including ramp up and wash out, with an analytical gradient of 58 min ranging from 7% B to 34% B.
Analytical columns were heated to 40 °C using a column oven and ionization was achieved using a NanoSpray Flex NG ion source. Spray voltage was set to 2 kV, ion transfer tube temperature was
set to 275 °C and RF funnel level was set to 40%. The full scan range was set to 300–1,300 _m_/_z_, MS1 resolution was set to 120,000, MS1 automated gain control (AGC) target was set to
‘200’ (2,000,000 charges) and MS1 maximum injection time was set to ‘auto’. Precursors with charges 2–6 were selected for fragmentation using an isolation width of 1.3 _m_/_z_ and fragmented
using higher-energy collision disassociation with a normalized collision energy of 25. Precursors were excluded from resequencing by setting a dynamic exclusion of 80 s. The MS2 AGC target
was set to ‘200’ (200,000 charges), intensity threshold was set to 360,000 charges per second, MS2 maximum injection time was set to ‘auto’, MS2 resolution was set to 30,000 and number of
dependent scans was set to 13. MS DATA ANALYSIS All MS RAW data were analyzed using the freely available MaxQuant software (version 1.5.3.30)92 in a single computational run. Default
MaxQuant settings were used, with exceptions specified below. For the generation of theoretical spectral libraries, the _X._ _laevis_ FASTA database was downloaded from UniProt on October, 3
2022. In silico digestion of proteins to generate theoretical peptides was performed with trypsin, allowing up to three missed cleavages. The minimum peptide length was set to six and
maximum peptide mass was set to 6,000 Da. Allowed variable modifications were oxidation of methionine (default), protein N-terminal acetylation (default), deamidation of asparagine and
glutamine, peptide N-terminal glutamine to pyroglutamate conversion, dioxidation of tryptophan and replacement of three protons by iron (cation Fe(III)) on aspartate and glutamate. These
variable modifications were determined by an initial analysis of the RAW data using pFind version 3.1.6 in ‘open search’ mode89 to unbiasedly determine any known modifications (from the
Unimod database) affecting >0.5% of peptide–spectrum matches (PSMs) across all samples. The maximum number of variable modifications per peptide was set to three. Label-free
quantification (LFQ) using MaxLFQ was enabled93 with ‘fast LFQ’ disabled. Matching between runs was enabled, with an alignment window of 20 min and a match time window of 1 min. A stringent
MaxQuant 1% false discovery rate (FDR) control was applied at the PSM, protein and site-decoy levels (default). MS DATA ANNOTATION AND QUANTIFICATION The _X._ _laevis_ FASTA databases
downloaded from UniProt lacked comprehensive gene name annotations. Missing or uninformative gene names were, when possible, semiautomatically curated, as described previously15.
Quantification of the MaxQuant output files (‘proteinGroups.txt’) and all statistical handling were performed using Perseus software (version 1.5.5.3)94. In total, _n_ = 8 technical
replicates (derived from _n_ = 4 biochemical replicates) were analyzed. For quantification purposes, all LFQ-normalized protein intensity values were log2-transformed and filtered for
presence in eight of eight replicates in at least one experimental condition. Missing values were inputted below the global experimental detection limit at a downshift of 1.8 and a
randomized width of 0.15 (in log2 space). The statistical significance of differences was in all cases tested using two-tailed Student’s two-sample _t_-testing, with permutation-based FDR
control applied to ensure a corrected _P_ value (that is, _q_ value) of <1%. Proteins not enriched over the no-DNA control in at least one CHROMASS condition (FDR < 1%, s0 = 1 and
2,500 rounds of randomization) were removed from the analysis, after which previously inputted values were reinputted on the basis of the new total matrix. Final biological differences were
determined using two-tailed Student’s two-sample _t_-testing (FDR < 1%, s0 = 0.5 and 2,500 rounds of randomization) on the remaining CHROMASS-enriched proteins. CELL CULTURE Cells were
cultured in high-glucose DMEM with glutaMAX Supplement and pyruvate (Gibco) supplemented with 10% FBS (Gibco) and 100 U per ml of penicillin–streptomycin (Gibco) at 37 °C with 5% CO2.
GENERATION OF U2OS FLP-IN T-REX _POLK_-KO CELLS U2OS Flp-In T-REx cells were a kind gift from H. Piwnica-Worms. Four different gRNAs targeting different regions of _POLK_
(5′-TAGGTTCAACACACCTGACG-3′, 5′-ATACATATAGATACCTCGTC-3′, 5′-ATACCGAGCTGTGAGTAAAG-3′ and 5′-AGGACAGGAAACACCAACAA-3′) were cloned into pSpCas9(BB)-2A-Puro (PX459) V2.0 (Addgene, 62988).
sgRNA-containing plasmids were transfected into U2OS Flp-In T-REx cells using Dharmacon 1 (Horizon Discovert T-2005-01) transfection reagent according to the manufacturer’s protocol. After
24 h of incubation, transfected cells were selected with 1 µM puromycin for 48 h and plated sparsely to isolate single colonies. Single colonies were screened by qPCR for a lack of _POLK_
mRNA using a primer pair (forward, 5′-TTGGGTCTAGGTTCAACACACC-3′; reverse, 5′-GCAAGCTCACTGCAAAGTTCT-3′). To perform the qPCR, total RNA was extracted using Qiagen RNeasy Mini (Qiagen 74104)
according to the manufacturer’s instructions. Complementary DNA (cDNA) was synthesized from total RNA using the iScript cDNA synthesis kit (BioRad, 1708890) according to the manufacturer’s
instructions. qPCR was performed in 96-well plates using the mentioned primers and Brilliant III ultrafast SYBR green qPCR master mix (Agilent, 600882) in a Stratagene Mx3005P machine using
standard thermocycling conditions. CLONING OF PCDNA5/FRT/TO/VENUS–_POLK_ CONSTRUCTS Human WT and CD _POLK_ (harboring D198A and E199A substitutions) cDNA sequences were a kind gift from O.
Scharer. BamHI and NotI restriction sites were added using PCR (forward primer, 5′-ATGCGGATCCATG GATAGCACAAAGGAGAAGTGTGAC-3′; reverse primer,
5′-TATAGCGGCCGCTTACTTAAAAAATATATCAAGGGTATGTTTGGG-3′) and cloned into pcDNA5/FRT/TO-Venus. Constructs were sequence-verified. GENERATION OF _POLK_-KO CELLS STABLY EXPRESSING VENUS–POLΚ To
generate stable cell lines in the Flp-In system, U2OS Flp-In T-REx _POLK_-KO cells were cotransfected with the Flp recombinase-encoding plasmid pOG44 (Invitrogen) and a pcDNA5/FRT/TO plasmid
encoding Venus–WT Polκ or Venus–CD Polκ at a 10:1 ratio using the jetOPTIMUS transfection reagent (Polyplus). Then, 48 h after transfection, cells were selected in medium supplemented with
5 μg ml−1 blasticidin S HCl and 200 μg ml−1 hygromycin B (Gibco) for 2–3 weeks. Expression from the Tet-ON inducible promoter in U2OS Flp-In T-REx cells was induced with 20 ng ml−1
doxycycline. COLONY FORMATION ASSAYS The cells were trypsinized, resuspended in medium and counted. A total of 200 cells were seeded per well in six-well plates with three wells per
condition. Venus–Polκ-expressing Flp-In T-REx cells were induced with 20 ng ml−1 doxycycline. Then, 24 h after seeding, cells were treated with the indicated compound (31.25 pg ml−1 illudin
S or 1 μM cisplatin) or left untreated. After seven additional days of growth, formed colonies were fixed and stained in a methyl violet solution (0.5% methyl violet and 25% methanol) and
the number of colonies was quantified on a GelCount (Oxford Optronix). The survival after treatment with a given compound was calculated as the average number of colonies after treatment
divided by the average number of colonies in the untreated condition multiplied by 100%. The experiments were performed three times independently and analyzed in PRISM (GraphPad). One-way
analyses of variance (ANOVAs) with Tukey’s multiple-comparisons tests were performed to test for statistical significance. WESTERN BLOT ANALYSIS OF CELL LYSATES Cells were harvested by
trypsinization, lyzed on ice in radioimmunoprecipitation assay buffer (10 mM Tris pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.5% sodium deoxycholate and 0.1% SDS) supplemented with 1 mM DTT
and cOmplete protease inhibitor cocktail (Roche) and sonicated with Bioruptor Plus (Diagenode). The lysate was cleared by centrifugation at 20,000_g_ at 4 °C for 30 min and a BCA assay
(Pierce) was used to measure protein concentrations. Samples were analyzed by SDS–PAGE and western blotting using anti-Polκ (Bethyl laboratories, A301-975A) and anti-tubulin (Abcam, ab6160)
antibodies. BASE-EDITOR TILING SCREEN AND ANALYSIS An sgRNA library targeting the coding sequence of Polκ was designed. The gRNA oligonucleotide pool was synthesized by GenScript as an 83-nt
oligonucleotide sequence, following a previously published design48. The oligonucleotide sequence consisted of primer sites for amplification with overhang sequences with Esp3I recognition
sites and the gRNA: 5′-[forward primer (20 nt)]CGTCTCACACCG[sgRNA (20 nt)]GTTTCGAGACG[reverse primer (20 nt)]. The gRNA oligonucleotide pool was amplified using NEBNext Ultra II Q5 master
mix (New England BioLabs) and the primers (forward, 5′-GTGTAACCCGTAGGGCACCT-3′; reverse, 5′-GTCGAGAGCAGTCCTTCGAC-3′). Amplicons were cloned into the Abe8e-Cas9-SpG lentiviral vector pRDA_479
(ref. 48) using Golden Gate cloning with Esp3I and T7 ligase. pRDA_479 was a gift from J. Doench and D. Root (Addgene, plasmid 179099). The ligated plasmid library was purified by PCR
(NucleoSpin gel and PCR Clean‑Up, Macherey-Nagel) and isopropanol precipitation and electroporated into Endura electrocompetent cells (Lucigen), which were grown at 30 °C for 16 h on agar
with 100 μg ml−1 carbenicillin. Plasmid DNA was prepared from the colonies on the plates (NucleoBond Xtra Maxi, Macherey-Nagel). To confirm library representation, the gRNA inserts were
amplified from the plasmid library using NEBNext Ultra II Q5 master mix and the primers D506_F and D702_R_PAGE_BE (Supplementary Table 3). Gel-purified amplicons were sequenced on a
NextSeq2000 (Illumina). The Polκ tiling library was part of a larger adenosine base editing (ABE) and cytosine base editing (CBE) tiling library, for which only Polκ with ABE is analyzed
here. Note that some CBE guides also score, most likely because of low-frequency editing beyond the optimal 4–8-nt editing window. A lentiviral library was produced by cotransfection of
HEK293T/17 cells (American Type Culture Collection, CRL-11268) with the sgRNA plasmid library and lentiviral packaging plasmids pMD2.G and psPAX2 using Lipofectamine 3000 (Invitrogen) in
Opti-MEM medium (Gibco). pMD2.G (Addgene, plasmid 12259) and psPAX2 (Addgene, plasmid 12260) were gifts from D. Trono. Then, 6 h after transfection, the medium was exchanged for DMEM
GlutaMax supplemented with 10% FBS, 100 U per ml of penicillin–streptomycin and 1% BSA. Next, 48 h after transfection, the lentiviral supernatant was collected and filtered through a 0.45-μm
syringe filter before storing at −80 °C. RPE1-hTERT _p53_−/− cells (a kind gift from D. Durocher) were cultured in DMEM GlutaMax supplemented with 10% FBS and 100 U per ml of
penicillin–streptomycin and passaged every 3 days. The screen was performed as a duplicate (two separate transductions) at a coverage of >500-fold sgRNA representation, which was
maintained throughout the screen. Cells were transduced with the lentiviral library at a low multiplicity of infection (0.3–0.4) and transductions were caried out by treating cells with 8 μg
ml−1 polybrene and lentiviral supernatant for 24 h. Transduced cells were selected by treatment with 20 μg ml−1 puromycin for 24 h followed by trypsinization and reseeding in the same
plates with 20 μg ml−1 puromycin for another 24 h. After selection, cells were passaged for 6 days before splitting into untreated or illudin-S-treated fractions, where they were passaged
for an additional 12 days in medium with or without a low dose of illudin S (1.4 ng ml−1). The dose of illudin S corresponds to a 20% reduction in cell numbers (LD20) compared to the
untreated condition in uninfected cells, which was determined in a titration experiment. Genomic DNA was extracted from cell pellets harvested after selection, which we consider the start of
the screen (_t_0) and at the final time point (_t_18). The genomic DNA region containing the integrated sgRNA was amplified by PCR using NEBNext Ultra II Q5 master mix with the LCV2_forward
and LCV2_reverse primers (Supplementary Table 3). A second PCR reaction introduced i5 and i7 multiplexing barcodes (Supplementary Table 3) and gel-purified PCR products were sequenced on
Illumina NextSeq2000. Sequencing data of _t_0, untreated _t_18 and illudin-S-treated _t_18 samples were converted to gRNA sequencing counts by MAGeCK95 and mapping was performed to gRNAs
tiling _POLK_ and control gRNAs (essential splice sites, nontargeting and intergenic)48. Low-abundance gRNAs were removed (counts < 30) and raw sequencing counts were normalized per
condition replicate to log2 transcripts per million (log2TPM). The log2TPM values were compared using limma96 for three sample pairs (_t_0 versus untreated _t_18, _t_0 versus
illudin-S-treated _t_18 and untreated _t_18 versus illudin-S-treated _t_18) and the fold change, _P_ value and _q_ value were collected for each gRNA. gRNA editing outcomes were predicted on
the basis of the editing of all adenines within the editing window of positions 4–8 (ref. 97). REPRODUCIBILITY A minimum of two independent experiments were conducted for each experimental
result shown in this manuscript. REPORTING SUMMARY Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article. DATA AVAILABILITY The
MS proteomics data were deposited to the ProteomeXchange Consortium through the PRIDE98 partner repository with the dataset identifier PXD044258. Source data are provided with this paper.
REFERENCES * Ling, H., Boudsocq, F., Woodgate, R. & Yang, W. Crystal structure of a Y-family DNA polymerase in action: a mechanism for error-prone and lesion-bypass replication. _Cell_
107, 91–102 (2001). CAS PubMed Google Scholar * Goodman, M. F. & Woodgate, R. Translesion DNA polymerases. _Cold Spring Harb. Perspect. Biol._ 5, a010363 (2013). PubMed PubMed
Central Google Scholar * Cortez, D. Replication-coupled DNA repair. _Mol. Cell_ 74, 866–876 (2019). CAS PubMed PubMed Central Google Scholar * Saldivar, J. C., Cortez, D. &
Cimprich, K. A. The essential kinase ATR: ensuring faithful duplication of a challenging genome. _Nat. Rev. Mol. Cell Biol._ 18, 622–636 (2017). CAS PubMed PubMed Central Google Scholar
* Friedberg, E. C., Wagner, R. & Radman, M. Specialized DNA polymerases, cellular survival, and the genesis of mutations. _Science_ 296, 1627–1630 (2002). CAS PubMed Google Scholar *
Hoege, C., Pfander, B., Moldovan, G.-L., Pyrowolakis, G. & Jentsch, S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. _Nature_ 419, 135–141 (2002).
CAS PubMed Google Scholar * Stelter, P. & Ulrich, H. D. Control of spontaneous and damage-induced mutagenesis by SUMO and ubiquitin conjugation. _Nature_ 425, 188–191 (2003). CAS
PubMed Google Scholar * Watanabe, K. et al. Rad18 guides Polη to replication stalling sites through physical interaction and PCNA monoubiquitination. _EMBO J._ 23, 3886–3896 (2004). CAS
PubMed PubMed Central Google Scholar * Sale, J. E., Lehmann, A. R. & Woodgate, R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. _Nat. Rev. Mol. Cell
Biol._ 13, 141–152 (2012). CAS PubMed PubMed Central Google Scholar * Johnson, R. E., Prakash, S. & Prakash, L. Efficient bypass of a thymine–thymine dimer by yeast DNA polymerase,
Polη. _Science_ 283, 1001–1004 (1999). CAS PubMed Google Scholar * Masutani, C., Kusumoto, R., Iwai, S. & Hanaoka, F. Mechanisms of accurate translesion synthesis by human DNA
polymerase η. _EMBO J._ 19, 3100–3109 (2000). CAS PubMed PubMed Central Google Scholar * McCulloch, S. D. et al. Preferential _cis_–_syn_ thymine dimer bypass by DNA polymerase η occurs
with biased fidelity. _Nature_ 428, 97–100 (2004). CAS PubMed Google Scholar * Johnson, R. E., Washington, M. T., Haracska, L., Prakash, S. & Prakash, L. Eukaryotic polymerases ι and
ζ act sequentially to bypass DNA lesions. _Nature_ 406, 1015–1019 (2000). CAS PubMed Google Scholar * Lee, Y.-S., Gregory, M. T. & Yang, W. Human Pol ζ purified with accessory
subunits is active in translesion DNA synthesis and complements Pol η in cisplatin bypass. _Proc. Natl Acad. Sci. USA_ 111, 2954–2959 (2014). CAS PubMed PubMed Central Google Scholar *
Gallina, I. et al. The ubiquitin ligase RFWD3 is required for translesion DNA synthesis. _Mol. Cell_ 81, 442–458 (2021). CAS PubMed PubMed Central Google Scholar * Gibbs, P. E. M.,
McDonald, J., Woodgate, R. & Lawrence, C. W. The relative roles in vivo of _Saccharomyces cerevisiae_ Pol η, Pol ζ, Rev1 protein and Pol32 in the bypass and mutation induction of an
abasic site, T–T (6–4) photoadduct and T–T _cis_–_syn_ cyclobutane dimer. _Genetics_ 169, 575–582 (2005). CAS PubMed PubMed Central Google Scholar * Hicks, J. K. et al. Differential
roles for DNA polymerases η, ζ, and REV1 in lesion bypass of intrastrand versus interstrand DNA cross-links. _Mol. Cell. Biol._ 30, 1217–1230 (2010). CAS PubMed Google Scholar * Yoon,
J.-H., Prakash, L. & Prakash, S. Error-free replicative bypass of (6–4) photoproducts by DNA polymerase ζ in mouse and human cells. _Genes Dev._ 24, 123–128 (2010). CAS PubMed PubMed
Central Google Scholar * Guo, C. et al. REV1 protein interacts with PCNA: significance of the REV1 BRCT domain in vitro and in vivo. _Mol. Cell_ 23, 265–271 (2006). CAS PubMed Google
Scholar * Martin, S. K. & Wood, R. D. DNA polymerase ζ in DNA replication and repair. _Nucleic Acids Res._ 47, 8348–8361 (2019). CAS PubMed PubMed Central Google Scholar * Acharya,
N., Johnson, R. E., Pagès, V., Prakash, L. & Prakash, S. Yeast Rev1 protein promotes complex formation of DNA polymerase ζ with Pol32 subunit of DNA polymerase δ. _Proc. Natl Acad. Sci.
USA_ 106, 9631–9636 (2009). CAS PubMed PubMed Central Google Scholar * Budzowska, M., Graham, T. G. W., Sobeck, A., Waga, S. & Walter, J. C. Regulation of the Rev1–Pol ζ complex
during bypass of a DNA interstrand cross-link. _EMBO J._ 34, 1971–1985 (2015). CAS PubMed PubMed Central Google Scholar * Edmunds, C. E., Simpson, L. J. & Sale, J. E. PCNA
ubiquitination and REV1 define temporally distinct mechanisms for controlling translesion synthesis in the avian cell line DT40. _Mol. Cell_ 30, 519–529 (2008). CAS PubMed Google Scholar
* Gerlach, V. L., Feaver, W. J., Fischhaber, P. L. & Friedberg, E. C. Purification and characterization of Polκ, a DNA polymerase encoded by the human _DINB1_ gene. _J. Biol. Chem._ 276,
92–98 (2001). CAS PubMed Google Scholar * Ogi, T., Kato, T. Jr, Kato, T. & Ohmori, H. Mutation enhancement by DINB1, a mammalian homologue of the _Escherichia coli_ mutagenesis
protein dinB. _Genes Cells_ 4, 607–618 (1999). CAS PubMed Google Scholar * Choi, J.-Y., Angel, K. C. & Guengerich, F. P. Translesion synthesis across bulky _N_2-alkyl guanine DNA
adducts by human DNA polymerase κ. _J. Biol. Chem._ 281, 21062–21072 (2006). CAS PubMed Google Scholar * Jarosz, D. F., Godoy, V. G., Delaney, J. C., Essigmann, J. M. & Walker, G. C.
A single amino acid governs enhanced activity of DinB DNA polymerases on damaged templates. _Nature_ 439, 225–228 (2006). PubMed Google Scholar * Jha, V., Bian, C., Xing, G. & Ling, H.
Structure and mechanism of error-free replication past the major benzo[a]pyrene adduct by human DNA polymerase κ. _Nucleic Acids Res._ 44, 4957–4967 (2016). CAS PubMed PubMed Central
Google Scholar * Jha, V. & Ling, H. 2.0 Å resolution crystal structure of human Polκ reveals a new catalytic function of N-clasp in DNA replication. _Sci Rep._ 8, 15125 (2018). PubMed
PubMed Central Google Scholar * Ogi, T., Shinkai, Y., Tanaka, K. & Ohmori, H. Polκ protects mammalian cells against the lethal and mutagenic effects of benzo[a]pyrene. _Proc. Natl
Acad. Sci. USA_ 99, 15548–15553 (2002). CAS PubMed PubMed Central Google Scholar * Malvezzi, S. et al. Mechanism of RNA polymerase II stalling by DNA alkylation. _Proc. Natl Acad. Sci.
USA_ 114, 12172–12177 (2017). CAS PubMed PubMed Central Google Scholar * Takeiri, A. et al. In vivo evidence that DNA polymerase κ is responsible for error-free bypass across DNA
cross-links induced by mitomycin C. _DNA Repair_ 24, 113–121 (2014). CAS PubMed Google Scholar * Tanasova, M. & Sturla, S. J. Chemistry and biology of acylfulvenes:
sesquiterpene-derived antitumor agents. _Chem. Rev._ 112, 3578–3610 (2012). CAS PubMed Google Scholar * Casimir, L., Zimmer, S., Racine-Brassard, F., Jacques, P.-É. & Maréchal, A. The
mutational impact of illudin S on human cells. _DNA Repair_ 122, 103433 (2023). CAS PubMed Google Scholar * Olivieri, M. et al. A genetic map of the response to DNA damage in human
cells. _Cell_ 182, 481–496 (2020). CAS PubMed PubMed Central Google Scholar * Williams, H. L., Gottesman, M. E. & Gautier, J. Replication-independent repair of DNA interstrand
crosslinks. _Mol. Cell_ 47, 140–147 (2012). CAS PubMed PubMed Central Google Scholar * Ogi, T. & Lehmann, A. R. The Y-family DNA polymerase κ (Pol κ) functions in mammalian
nucleotide-excision repair. _Nat. Cell Biol._ 8, 640–642 (2006). CAS PubMed Google Scholar * Spanjaard, A. et al. Division of labor within the DNA damage tolerance system reveals
non-epistatic and clinically actionable targets for precision cancer medicine. _Nucleic Acids Res._ 50, 7420–7435 (2022). CAS PubMed PubMed Central Google Scholar * Johnson, R. E.,
Prakash, S. & Prakash, L. The human _DINB1_ gene encodes the DNA polymerase Polθ. _Proc. Natl Acad. Sci. USA_ 97, 3838–3843 (2000). CAS PubMed PubMed Central Google Scholar *
Kanemaru, Y. et al. Catalytic and non-catalytic roles of DNA polymerase κ in the protection of human cells against genotoxic stresses. _Environ. Mol. Mutagen._ 56, 650–662 (2015). CAS
PubMed Google Scholar * Ogi, T. et al. Three DNA polymerases, recruited by different mechanisms, carry out NER repair synthesis in human cells. _Mol. Cell_ 37, 714–727 (2010). CAS PubMed
Google Scholar * Bétous, R. et al. DNA polymerase κ-dependent DNA synthesis at stalled replication forks is important for CHK1 activation. _EMBO J._ 32, 2172–2185 (2013). PubMed PubMed
Central Google Scholar * Dall’Osto, M., Pierini, L., Valery, N., Hoffmann, J.-S. & Pillaire, M.-J. A catalytically independent function of human DNA polymerase κ controls the stability
and abundance of checkpoint kinase 1. _Mol. Cell. Biol._ 41, e0009021 (2021). PubMed Google Scholar * Tonzi, P., Yin, Y., Lee, C. W. T., Rothenberg, E. & Huang, T. T. Translesion
polymerase κ-dependent DNA synthesis underlies replication fork recovery. _eLife_ 7, e41426 (2018). PubMed PubMed Central Google Scholar * Hishiki, A. et al. Structural basis for novel
interactions between human translesion synthesis polymerases and proliferating cell nuclear antigen. _J. Biol. Chem._ 284, 10552–10560 (2009). PubMed PubMed Central Google Scholar *
Vaisman, A. & Woodgate, R. Translesion DNA polymerases in eukaryotes: what makes them tick? _Crit. Rev. Biochem. Mol. Biol._ 52, 274–303 (2017). CAS PubMed PubMed Central Google
Scholar * Xie, W., Yang, X., Xu, M. & Jiang, T. Structural insights into the assembly of human translesion polymerase complexes. _Protein Cell_ 3, 864–874 (2012). CAS PubMed PubMed
Central Google Scholar * Sangree, A. K. et al. Benchmarking of SpCas9 variants enables deeper base editor screens of _BRCA1_ and _BCL2_. _Nat. Commun._ 13, 1318 (2022). CAS PubMed PubMed
Central Google Scholar * Boudsocq, F. et al. Investigating the role of the little finger domain of Y-family DNA polymerases in low fidelity synthesis and translesion replication. _J.
Biol. Chem._ 279, 32932–32940 (2004). CAS PubMed Google Scholar * Walter, J., Sun, L. & Newport, J. Regulated chromosomal DNA replication in the absence of a nucleus. _Mol. Cell_ 1,
519–529 (1998). CAS PubMed Google Scholar * Ogi, T., Kannouche, P. & Lehmann, A. R. Localisation of human Y-family DNA polymerase κ: relationship to PCNA foci. _J. Cell Sci._ 118,
129–136 (2005). CAS PubMed Google Scholar * Guo, C., Tang, T.-S., Bienko, M., Dikic, I. & Friedberg, E. C. Requirements for the interaction of mouse Polκ with ubiquitin and its
biological significance. _J. Biol. Chem._ 283, 4658–4664 (2008). CAS PubMed Google Scholar * Ohashi, E. et al. Identification of a novel REV1-interacting motif necessary for DNA
polymerase κ function. _Genes Cells_ 14, 101–111 (2009). CAS PubMed PubMed Central Google Scholar * Masuda, Y. et al. Different types of interaction between PCNA and PIP boxes contribute
to distinct cellular functions of Y-family DNA polymerases. _Nucleic Acids Res._ 43, 7898–7910 (2015). CAS PubMed PubMed Central Google Scholar * Lancey, C. et al. Cryo-EM structure of
human Pol κ bound to DNA and mono-ubiquitylated PCNA. _Nat. Commun._ 12, 6095 (2021). CAS PubMed PubMed Central Google Scholar * Larsen, N. B. et al. Replication-coupled DNA–protein
crosslink repair by SPRTN and the proteasome in _Xenopus_ egg extracts. _Mol. Cell_ 73, 574–588 (2019). CAS PubMed PubMed Central Google Scholar * Duxin, J. P., Dewar, J. M., Yardimci,
H. & Walter, J. C. Repair of a DNA–protein crosslink by replication-coupled proteolysis. _Cell_ 159, 346–357 (2014). CAS PubMed PubMed Central Google Scholar * Mohni, K. N. et al.
HMCES maintains genome integrity by shielding abasic sites in single-strand DNA. _Cell_ 176, 144–153 (2019). CAS PubMed Google Scholar * Semlow, D. R., MacKrell, V. A. & Walter, J. C.
The HMCES DNA–protein cross-link functions as an intermediate in DNA interstrand cross-link repair. _Nat. Struct. Mol. Biol._ 29, 451–462 (2022). CAS PubMed PubMed Central Google Scholar
* Semlow, D. R., Zhang, J., Budzowska, M., Drohat, A. C. & Walter, J. C. Replication-dependent unhooking of DNA interstrand cross-links by the NEIL3 glycosylase. _Cell_ 167, 498–511
(2016). CAS PubMed PubMed Central Google Scholar * Haracska, L. et al. Roles of yeast DNA polymerases δ and ζ and of Rev1 in the bypass of abasic sites. _Genes Dev._ 15, 945–954 (2001).
CAS PubMed PubMed Central Google Scholar * Räschle, M. et al. Proteomics reveals dynamic assembly of repair complexes during bypass of DNA cross-links. _Science_ 348, 1253671 (2015).
PubMed PubMed Central Google Scholar * Sertic, S. et al. Coordinated activity of Y family TLS polymerases and EXO1 protects non-S phase cells from UV-induced cytotoxic lesions. _Mol.
Cell_ 70, 34–47 (2018). CAS PubMed Google Scholar * Schubert, L. et al. SCAI promotes error-free repair of DNA interstrand crosslinks via the Fanconi anemia pathway. _EMBO Rep._ 23,
e53639 (2022). CAS PubMed PubMed Central Google Scholar * Yoshimura, A., Kobayashi, Y., Tada, S., Seki, M. & Enomoto, T. WRNIP1 functions upstream of DNA polymerase η in the
UV-induced DNA damage response. _Biochem. Biophys. Res. Commun._ 452, 48–52 (2014). CAS PubMed Google Scholar * Yuasa, M. S. et al. A human DNA polymerase η complex containing Rad18, Rad6
and Rev1; proteomic analysis and targeting of the complex to the chromatin-bound fraction of cells undergoing replication fork arrest. _Genes Cells_ 11, 731–744 (2006). CAS PubMed Google
Scholar * Chen, D. et al. _BRCA1_ deficiency specific base substitution mutagenesis is dependent on translesion synthesis and regulated by 53BP1. _Nat. Commun._ 13, 226 (2022). CAS PubMed
PubMed Central Google Scholar * Guo, C. et al. Mouse Rev1 protein interacts with multiple DNA polymerases involved in translesion DNA synthesis. _EMBO J._ 22, 6621–6630 (2003). CAS
PubMed PubMed Central Google Scholar * Malik, R. et al. Structure and mechanism of B-family DNA polymerase ζ specialized for translesion DNA synthesis. _Nat. Struct. Mol. Biol._ 27,
913–924 (2020). CAS PubMed PubMed Central Google Scholar * Haracska, L., Prakash, L. & Prakash, S. Role of human DNA polymerase κ as an extender in translesion synthesis. _Proc. Natl
Acad. Sci. USA_ 99, 16000–16005 (2002). CAS PubMed PubMed Central Google Scholar * Washington, M. T., Johnson, R. E., Prakash, L. & Prakash, S. Human _DINB1_-encoded DNA polymerase
κ is a promiscuous extender of mispaired primer termini. _Proc. Natl Acad. Sci. USA_ 99, 1910–1914 (2002). CAS PubMed PubMed Central Google Scholar * Kannouche, P. L., Wing, J. &
Lehmann, A. R. Interaction of human DNA polymerase η with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. _Mol. Cell_ 14, 491–500 (2004).
CAS PubMed Google Scholar * Lange, S. S., Wittschieben, J. P. & Wood, R. D. DNA polymerase ζ is required for proliferation of normal mammalian cells. _Nucleic Acids Res._ 40,
4473–4482 (2012). CAS PubMed PubMed Central Google Scholar * Ben Yamin, B. et al. DNA polymerase ζ contributes to heterochromatin replication to prevent genome instability. _EMBO J._ 40,
e104543 (2021). PubMed PubMed Central Google Scholar * Wojtaszek, J. L. et al. A small molecule targeting mutagenic translesion synthesis improves chemotherapy. _Cell_ 178, 152–159
(2019). CAS PubMed PubMed Central Google Scholar * Yamanaka, K., Chatterjee, N., Hemann, M. T. & Walker, G. C. Inhibition of mutagenic translesion synthesis: a possible strategy for
improving chemotherapy? _PLoS Genet._ 13, e1006842 (2017). PubMed PubMed Central Google Scholar * Yu, D., Chojnowski, G., Rosenthal, M. & Kosinski, J. AlphaPulldown—a python package
for protein–protein interaction screens using AlphaFold-Multimer. _Bioinformatics_ 39, btac749 (2023). CAS PubMed Google Scholar * Jumper, J. et al. Highly accurate protein structure
prediction with AlphaFold. _Nature_ 596, 583–589 (2021). CAS PubMed PubMed Central Google Scholar * Yockey, O. P. et al. Mechanism of error-free DNA replication past lucidin-derived DNA
damage by human DNA polymerase κ. _Chem. Res. Toxicol._ 30, 2023–2032 (2017). CAS PubMed PubMed Central Google Scholar * Zhang, Z. et al. Structure of monoubiquitinated PCNA:
implications for DNA polymerase switching and Okazaki fragment maturation. _Cell Cycle_ 11, 2128–2136 (2012). CAS PubMed PubMed Central Google Scholar * Lebofsky, R., Takahashi, T. &
Walter, J. C. DNA replication in nucleus-free _Xenopus_ egg extracts. _Methods Mol. Biol._ 521, 229–252 (2009). CAS PubMed Google Scholar * Sparks, J. L. et al. The CMG helicase bypasses
DNA–protein cross-links to facilitate their repair. _Cell_ 176, 167–181 (2019). CAS PubMed Google Scholar * Sobeck, A. et al. Fanconi anemia proteins are required to prevent accumulation
of replication-associated DNA double-strand breaks. _Mol. Cell. Biol._ 26, 425–437 (2006). CAS PubMed PubMed Central Google Scholar * Knipscheer, P. et al. The Fanconi anemia pathway
promotes replication-dependent DNA interstrand cross-link repair. _Science_ 326, 1698–1701 (2009). CAS PubMed PubMed Central Google Scholar * Räschle, M. et al. Mechanism of
replication-coupled DNA interstrand crosslink repair. _Cell_ 134, 969–980 (2008). PubMed PubMed Central Google Scholar * Sparks, J. & Walter, J. C. Extracts for analysis of DNA
replication in a nucleus-free system. _Cold Spring Harb. Protoc._ 2019, prot097154 (2019). Google Scholar * Evans, R. et al. Protein complex prediction with AlphaFold-Multimer. Preprint at
_bioRxiv_ https://doi.org/10.1101/2021.10.04.463034 (2022). * Varadi, M. et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space
with high-accuracy models. _Nucleic Acids Res._ 50, D439–D444 (2022). CAS PubMed Google Scholar * Goddard, T. D. et al. UCSF ChimeraX: meeting modern challenges in visualization and
analysis. _Protein Sci._ 27, 14–25 (2018). CAS PubMed Google Scholar * Pettersen, E. F. et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. _Protein
Sci._ 30, 70–82 (2021). CAS PubMed Google Scholar * Lancey, C. et al. Structure of the processive human Pol δ holoenzyme. _Nat. Commun._ 11, 1109 (2020). CAS PubMed PubMed Central
Google Scholar * Cox, J. & Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. _Nat.
Biotechnol._ 26, 1367–1372 (2008). CAS PubMed Google Scholar * Cox, J. et al. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio
extraction, termed MaxLFQ. _Mol. Cell. Proteomics_ 13, 2513–2526 (2014). CAS PubMed PubMed Central Google Scholar * Tyanova, S. et al. The Perseus computational platform for
comprehensive analysis of (prote)omics data. _Nat. Methods_ 13, 731–740 (2016). CAS PubMed Google Scholar * Li, W. et al. MAGeCK enables robust identification of essential genes from
genome-scale CRISPR/Cas9 knockout screens. _Genome Biol._ 15, 554 (2014). PubMed PubMed Central Google Scholar * Ritchie, M. E. et al. limma powers differential expression analyses for
RNA-sequencing and microarray studies. _Nucleic Acids Res._ 43, e47 (2015). PubMed PubMed Central Google Scholar * Richter, M. F. et al. Phage-assisted evolution of an adenine base editor
with improved Cas domain compatibility and activity. _Nat. Biotechnol._ 38, 883–891 (2020). CAS PubMed PubMed Central Google Scholar * Perez-Riverol, Y. et al. The PRIDE database
resources in 2022: a hub for mass spectrometry-based proteomics evidences. _Nucleic Acids Res._ 50, D543–D552 (2022). CAS PubMed Google Scholar Download references ACKNOWLEDGEMENTS We
thank J. Gautier (Columbia University) for sharing Polκ-expressing constructs, D. Durocher (University of Toronto) for the RPE1-hTERT _p53_−/− cells and staff of the CPR/ReNew Genomics
Platform for support: H. Wollmann, M. Michaut and A. Kalvisa. We also thank members of the Duxin laboratory and J. Walter for feedback on the manuscript. The Novo Nordisk Foundation Center
for Protein Research is supported financially by the Novo Nordisk Foundation (grant agreement NNF14CC0001). This project has received funding from the European Research Council under the
European Union’s Horizon 2020 research and innovation program (grant agreement 715975) and from the Novo Nordisk Foundation (grant NNF22OC0074140). B.B. is supported by the European
Molecular Biology Organization (grant agreement no. ALTF 1149-2020). Work in J.N.’s lab was supported by a Novo Nordisk Fonden distinguished investigator grant (NNF23OC0082227) and a grant
from the Danish Cancer Society (R269-A15586-B71). T.C.R.M. was supported by a Novo Nordisk Fonden Hallas-Møller emerging investigator grant (NNF22OC0073571), the Danish National Research
Foundation (DNRF115) and the Carlsberg Foundation (CF21-0571). J.P.D. is part of the European doctoral network (Replifate, grant agreement #101072903). We also extend our gratitude to J.
Lukas for his exceptional leadership, which has elevated the Center for Protein Research to the forefront of biological research. AUTHOR INFORMATION Author notes * These authors contributed
equally: Sara M. Ambjørn, Alberto Carli, Ivo A. Hendriks. AUTHORS AND AFFILIATIONS * Novo Nordisk Foundation Center for Protein Research, University of Copenhagen, Copenhagen, Denmark Selene
Sellés-Baiget, Sara M. Ambjørn, Ivo A. Hendriks, Irene Gallina, Bente Benedict, Alessandra Zarantonello, Sampath A. Gadi, Bob Meeusen, Emil P. T. Hertz, Michael L. Nielsen, Jakob Nilsson
& Julien P. Duxin * Center for Chromosome Stability, Department of Cellular and Molecular Medicine, Faculty of Health and Medical Sciences, University of Copenhagen, Copenhagen, Denmark
Alberto Carli & Thomas C. R. Miller * Department of Molecular Medicine, University of Padua, Padua, Italy Irene Gallina * Division of Cancer Biology, The Institute of Cancer Research,
London, UK Norman E. Davey * Biotech Research and Innovation Centre, Faculty of Health and Medical Sciences, University of Copenhagen, Copenhagen, Denmark Bente Benedict, Alessandra
Zarantonello, Sampath A. Gadi & Julien P. Duxin * Department of Health Sciences and Technology, ETH Zurich, Zurich, Switzerland Laura Slappendel & Shana Sturla * Division of
Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, CA, USA Daniel Semlow Authors * Selene Sellés-Baiget View author publications You can also search for this
author inPubMed Google Scholar * Sara M. Ambjørn View author publications You can also search for this author inPubMed Google Scholar * Alberto Carli View author publications You can also
search for this author inPubMed Google Scholar * Ivo A. Hendriks View author publications You can also search for this author inPubMed Google Scholar * Irene Gallina View author publications
You can also search for this author inPubMed Google Scholar * Norman E. Davey View author publications You can also search for this author inPubMed Google Scholar * Bente Benedict View
author publications You can also search for this author inPubMed Google Scholar * Alessandra Zarantonello View author publications You can also search for this author inPubMed Google Scholar
* Sampath A. Gadi View author publications You can also search for this author inPubMed Google Scholar * Bob Meeusen View author publications You can also search for this author inPubMed
Google Scholar * Emil P. T. Hertz View author publications You can also search for this author inPubMed Google Scholar * Laura Slappendel View author publications You can also search for
this author inPubMed Google Scholar * Daniel Semlow View author publications You can also search for this author inPubMed Google Scholar * Shana Sturla View author publications You can also
search for this author inPubMed Google Scholar * Michael L. Nielsen View author publications You can also search for this author inPubMed Google Scholar * Jakob Nilsson View author
publications You can also search for this author inPubMed Google Scholar * Thomas C. R. Miller View author publications You can also search for this author inPubMed Google Scholar * Julien
P. Duxin View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS S.S.B. performed all experiments unless stated otherwise. A.C. generated the
AlphaFold models in Figs. 1c and 7e,f and Extended Data Figs. 1b,c and 7c under the supervision of T.M. S.M.A. and B.M. performed the CRISPR base-editor tiling screen (Fig. 1b and Extended
Data Fig. 1a) under the supervision of J.N. E.P.T.H. provided technical advice on the base-editor screen. N.D. designed the CRISPR library and performed statistical analysis of the
sequencing data. I.A.H. performed MS analysis on CHROMASS samples (Fig. 6 and Extended Data Fig. 6) under the supervision of M.L.N. L.S and B.B. generated the p3d-Phen-A minor groove DNA
substrate under the supervision of S.S. and J.P.D. I.G. performed the experiment in Fig. 6d and preliminary experiments for this project. D.S. provided the AP-ICL-containing plasmid. S.A.G.
generated the _POLK_-KO cells. S.M.A. performed the colony assay experiments (Fig. 7a–d). A.Z. assisted with recombinant Polκ protein purification. S.S.B. and J.P.D. designed and analyzed
the experiments. S.S.B. and J.P.D. prepared the manuscript with feedback and input from all authors of the manuscript. CORRESPONDING AUTHOR Correspondence to Julien P. Duxin. ETHICS
DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Nature Structural & Molecular Biology_ thanks Vincent Pagès and the
other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available. Primary Handling Editor: Dimitris Typas, in collaboration with the
_Nature Structural & Molecular Biology_ team. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and
institutional affiliations. EXTENDED DATA EXTENDED DATA FIG. 1 A) Dot plot showing the results of the Polκ CRISPR base editor tiling screen comparing t0 and t18 of the untreated condition.
Each guide is shown as a dot. X-axis represents amino acid position in Polκ. Y-axis represents Log2 fold changes between t18 and t0 of the untreated condition. Larger dots represent guides
which are significantly changing between the two conditions (p-value ≤ 0.01). The various domains of Polκ are indicated. The significance and fold change was derived from limma (see Methods)
B) Composite molecular model of human Polκ. Dashed lines represent disordered regions that are not present in the model. The model was generated as described in Fig. 1c. C) Composite
molecular model of human Polκ with point mutations derived from the base editor tiling screen. Dashed lines represent disordered regions that are not present in the model. Point mutations
are highlighted in red. The left panel is a duplicate of Fig. 1c. D) pCTRL and p3d-Phen-A were replicated in the presence or absence of ubiquitin E1 inhibitor. Reaction samples were analyzed
as in Fig. 1h. E) Schematic diagram illustrating the replication intermediates generated during the replication of p3d-Phen-A. F) Western blot of the immunoprecipitation of Polκ using our
generated _Xenopus_ Polκ antibody. Sup., supernatant; IP, immunoprecipitation. Polκ #1 and Polκ #2 denotes two different affinity purifications generated from the same rabbits. G) Coomassie
blue staining of recombinant Polκ WT and Polκ CD purified from _E.coli_. * denotes a contaminant not recognized by the anti-HIS antibody (right immunoblot). H) Schematic representation of
gap filling synthesis (left scheme) when pDPCssDNA is incubated with Polκ WT, Polκ CD, or whole egg cytosolic egg extract (HSS) in the presence of [α-32P]dATP. Note the absence of DNA
synthesis in the Polκ CD reaction (lanes 6-9). Source Data EXTENDED DATA FIG. 2 A) Polκ-depleted extracts were supplemented with either buffer (+Buffer), wild-type Polκ (+WT), or RIR (RIR*)
Polκ mutant. Samples were blotted with the indicated antibodies (top immunoblots). To ensure that the same amount of protein was added back to the extracts, the different Polκ preparations
used in this study were titrated, and equal amounts were confirmed via immunoblotting (via HIS antibody, bottom immunoblot). 1x corresponds to the amount needed to add back roughly
endogenous levels of Polκ (when added to 10% F.C in NPE). We unfortunately lack the Coomassie stain of various mutants. B) Extracts from (A) were used to replicate p3d-Phen-A. Samples were
analyzed as in Fig. 1h. C) Polκ-depleted extracts were supplemented with either buffer (+Buffer), wild-type Polκ (+WT), PIP1 (PIP1*), PIP2 (PIP2*) or PIP1 and PIP2 (PIP1 + 2*) Polκ mutants.
Samples were blotted with the indicated antibodies. Note that the Polκ antibody was generated against Polκ C-terminal end which encompasses PIP2. This antibody exhibits lower affinity for
Polκ PIP2* compared to Polκ WT and Polκ PIP1*. The 6xHis tag of the recombinant protein is located on the N-terminal end. D) Extracts from (C) were used to replicate p3d-Phen-A. Samples were
analyzed as in Fig. 1h. E) Polκ-depleted extracts were supplemented with either buffer (+Buffer), wild-type Polκ (+WT), UBZ (UBZ1*), UBZ (UBZ2*) or UBZ1 and UBZ2 (UBZ1 + 2*) Polκ mutants.
Samples were blotted with the indicated antibodies. F) Extracts from (E) were used to replicate 3d-Phen-A. Samples were analyzed as in Fig. 1h. Source Data EXTENDED DATA FIG. 3 A)
Replication intermediates generated during replication of pDPC57. B) Mock-, Polη-, Polκ-, Rev1-, or Rev3-depleted extracts were blotted with the indicated antibodies. C) pDPC was replicated
in egg extracts in mock-, Polη-, Polκ-, or Rev1-depleted extracts. Reaction samples were analyzed as in Fig. 1h. D) pDPC was replicated in Polκ- or Rev1-, or Rev3-depleted egg extracts.
Samples were digested, separated on a denaturing polyacrylamide gel and analyzed as in Fig. 3c. Source Data EXTENDED DATA FIG. 4 A) Generation of pDPCPK56. B) Mock- and Polκ-depleted egg
extracts were used to replicate pDPCPK. Polκ-depleted extracts were supplemented with either buffer (+Buffer), wild-type (+WT), or catalytically inactive (+CD) Polκ. Samples were analyzed as
in Fig. 1h. C) Samples from (B) were digested and analyzed as in Fig. 3c. D) pDPCLead was replicated in either mock- or Polκ-depleted egg extracts in the presence of LacI57. Polκ-depleted
extracts were supplemented with either buffer (+Buffer), wild-type Polκ (+WT), or catalytically inactive Polκ (+CD). Samples were digested with AatII, separated on a denaturing
polyacrylamide gel and analyzed as in Fig. 3c. E) Extracts from (D) were used to replicate pDPCLag57. Samples were digested with BssHII, separated on a denaturing polyacrylamide gel and
analyzed as in Fig. 3c. F) Polκ-depleted egg extracts were supplemented with either buffer (+Buffer), recombinant PIP1 (+PIP1*), PIP2 (+PIP2*) or PIP1 and PIP2 (PIP1 + 2*) Polκ mutants.
Extracts were then used to replicate pDPC. Samples were digested and analyzed as in Fig. 3c. G) Polκ-depleted egg extracts were supplemented with either buffer (+Buffer), UBZ1(+UBZ1*), UBZ2
(+UBZ2*), or UBZ1 and UBZ2 (+UBZ1 + 2*) Polκ mutants. Extracts were then used to replicate pDPC. Samples were analyzed as in Fig. 3c. Source Data EXTENDED DATA FIG. 5 A) Mock- and
Polκ-depleted egg extracts were used to replicate pAP-ICL. Polκ-depleted extracts were supplemented with either buffer (+Buffer), wild-type Polκ (+WT), or catalytically inactive Polκ (+CD).
The samples were analyzed as in Fig. 1h. B) Western blot of the immunoprecipitation of HMCES. Sup., supernatant; IP, immunoprecipitation. C) Mock- and HMCES-extracts were blotted with the
indicated antibodies. * indicates a non-specific band. D) HMCES-depleted extracts were either mock- or Polκ-depleted and were subsequently used to replicate pAP-ICL. Polκ-depleted extracts
were supplemented with either buffer (+Buffer), wild-type Polκ (+WT), or catalytically inactive Polκ (+CD). The samples were analyzed as in Fig. 1h. Source Data EXTENDED DATA FIG. 6 A)
Mock-, Polη-, or double Polη- and Polκ-depleted egg extracts were used to replicate pDPC. Polκ-depleted extracts were supplemented with either buffer (+Buffer), wild-type Polκ (+WT), or
catalytically inactive Polκ (+CD). Samples were analyzed as in Fig. 1h. B) MS analysis of protein recruitment to UV-treated compared to untreated sperm chromatin. The volcano plot shows the
difference in abundance of proteins between the two sample conditions (x-axis), plotted against the _p_-value resulting from two- tailed Student’s two-sample t-testing (y-axis). Proteins
significantly down- or up-regulated (FDR < 5%) upon UV treatment are represented in red or blue, respectively. TLS polymerases are highlighted in orange. n = 4 biochemical and n = 8
technical replicates, significance was determined via two-tailed Student’s two-sample t-testing, with permutation-based FDR control (s0 = 0.5) to ensure an adjusted p-value (that is q-value)
of <0.05 in all cases. The significance line is drawn at q = 0.01. C) Same experiment as in (B) but comparing mock- to Rev1-depleted extracts. Small red dots, 1% < FDR < 5%; large
red dots, FDR < 1%. Polκ and Rev1 are highlighted in purple and black, respectively. D) Same experiment as in (B) but comparing mock- to Polκ-depleted extracts. Small red dots, 1% <
FDR < 5%; large red dots, FDR < 1%. Polκ and Rev1 are highlighted in purple and black, respectively. E) Western blot of FancA immunoprecipiates. Sup., supernatant; IP,
immunoprecipitation. FancA #183 and was used to validate that our FancA antibody (FancA #2) immunoprecipitates the same protein. F) Western blot of the co-immunoprecipitation of Polκ with
FancA. Sup., supernatant; IP, immunoprecipitation. G) Sperm chromatin was either untreated or treated with 20 J/m2 of UV-C and then added to replicating mock-, Polκ- or Rev1-depleted
extracts. Chromatin samples of duplicated reactions were isolated at 60 min, and the proteins associated were blotted with the indicated antibodies. H) pDPC was replicated in either mock,
Polκ or Rev1-depleted extracts. Reactions were subjected to plasmid pull-down and samples were blotted with the indicated antibodies. I) p3d-Phen-A was replicated in either mock or
Polκ-depleted extracts. Reactions were subjected to plasmid pull-down and samples were blotted with the indicated antibodies. Source Data EXTENDED DATA FIG. 7 A) Knockout of _POLK_ in U2OS
cells was validated by qPCR. Bar graphs represent _POLK_ mRNA (cDNA) expression levels relative to WT parental cells (E1) and normalised to GAPDH expression levels. Error bars represent
standard deviation from the mean of a technical triplicate PCR reaction (one representative experiment of two biological replicates is shown). Clones 1.10 was selected for further studies.
B) Western blot analysis of whole cell extracts of the indicated cell lines treated with doxycycline where indicated. Note that we were unable to detect endogenous Polκ with three
independent antibodies that were tested (A301-975A (Bethyl); A301-977A (Bethyl); sc-166667 (Santa Cruz). * indicates a non-specific band. C) AlphaPulldown predictions of protein complexes
were utilised to construct the composite molecular models presented in Fig. 7. Each panel is divided into three sections: The upper panel displays the Predicted Aligned Error (PAE) plots
associated with specific protein/fragment combinations generated by AlphaPulldown77. The PAE score in AlphaFold estimates the distance errors for residue pairs. It is represented by plots
composed by diagonal squares for inner-correlation and cross-correlation areas for protein/fragment interactions. They assess prediction confidence, with low values being reliable and high
values unreliable. The middle panel shows the predicted structures. Lastly, the lower panel includes the names of the predicted protein combinations, along with their corresponding Interface
Predicted Template Modelling (IPTM), the Predicted Template Modelling (PTM) scores and the Ranking of the model (R0). D) Summary tables featuring the protein structures used as scaffolds
for model building, along with their respective PDB codes. Single protein predictions obtained from AlphaPulldown with related Predicted Local Distance Difference Test scores (pLDDT).
Uniprot accession codes for each of the proteins used for the complex protein modelling. Source Data SUPPLEMENTARY INFORMATION REPORTING SUMMARY PEER REVIEW FILE SUPPLEMENTARY TABLES 1–3
Supplementary Table 1. _POLK_ base-editor tiling screen results and analysis (Fig. 1 and Extended Data Fig. 1). Note that the library was generated for both ABE and CBE but only ABE was
used. The significance and fold change were derived from limma (Methods). Supplementary Table 2. MS analysis of protein recruitment to UV-treated sperm chromatin in mock, Rev1-depleted or
Polκ-depleted extracts (Fig. 6 and Extended Data Fig. 6). ΔMock, mock extracts incubated in undamaged chromatin; ΔMock + UV, mock extracts incubated with UV-treated chromatin; ΔRev1 + UV,
Rev1-depleted extracts incubated with UV-treated chromatin; ΔPolκ + UV, Polκ-depleted extracts incubated with UV-treated chromatin. For quantification purposes, all LFQ-normalized protein
intensity values were log2-transformed and filtered for presence in eight of eight replicates in at least one experimental condition. Missing values were inputted below the global
experimental detection limit at a downshift of 1.8 and a randomized width of 0.15 (in log2 space). Statistical significance of differences was, in all cases, tested using two-tailed
Student’s two-sample _t_-testing with a _q_ value of <1%. Proteins not enriched over the no-DNA control in at least one CHROMASS condition (FDR < 1%, s0 = 1 and 2,500 rounds of
randomization) were removed from the analysis, after which previously inputted values were reinputted on the basis of the new total matrix. Final biological differences were determined using
two-tailed Student’s two-sample _t_-testing (FDR < 1%, s0 = 0.5 and 2,500 rounds of randomization) on the remaining CHROMASS-enriched proteins. Supplementary Table 3. PCR primers used
during base-editor tiling screen (Fig. 1, Extended Data Fig. 1 and Methods). SOURCE DATA SOURCE DATA FIGS. 1–6, EXTENDED DATA FIGS. 1–7 All source data are combined into one file. SOURCE
DATA FIG. 7 AND EXTENDED DATA FIG. 7 Statistical counts. RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0
International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s)
and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material
derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a
credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted
use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/. Reprints and permissions
ABOUT THIS ARTICLE CITE THIS ARTICLE Sellés-Baiget, S., Ambjørn, S.M., Carli, A. _et al._ Catalytic and noncatalytic functions of DNA polymerase κ in translesion DNA synthesis. _Nat Struct
Mol Biol_ 32, 300–314 (2025). https://doi.org/10.1038/s41594-024-01395-3 Download citation * Received: 01 September 2023 * Accepted: 28 August 2024 * Published: 19 September 2024 * Issue
Date: February 2025 * DOI: https://doi.org/10.1038/s41594-024-01395-3 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry,
a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative