Play all audios:
ABSTRACT Coral reefs are under existential threat from climate change and anthropogenic impacts. Genomic studies have enhanced our knowledge of resilience and responses of some coral species
to environmental stress, but reference genomes are lacking for many coral species. The blue coral _Heliopora_ is the only reef-building octocoral genus and exhibits optimal growth at a
temperature close to the bleaching threshold of scleractinian corals. Local and high-latitude expansions of _Heliopora coerulea_ were reported in the last decade, but little is known about
the molecular mechanisms underlying its thermal resistance. We generated a draft genome of _H. coerulea_ with an assembled size of 429.9 Mb, scaffold N50 of 1.42 Mb and BUSCO completeness of
94.9%. The genome contains 239.1 Mb repetitive sequences, 27,108 protein coding genes, 6,225 lncRNAs, and 79 miRNAs. This reference genome provides a valuable resource for in-depth studies
on the adaptive mechanisms of corals under climate change and the evolution of skeleton in cnidarian. SIMILAR CONTENT BEING VIEWED BY OTHERS A DRAFT GENOME ASSEMBLY OF THE REEF-BUILDING
CORAL _ACROPORA HEMPRICHII_ FROM THE CENTRAL RED SEA Article Open access 26 November 2024 HIGH-QUALITY GENOME ASSEMBLY OF THE AZOOXANTHELLATE CORAL _TUBASTRAEA COCCINEA_ (LESSON, 1829)
Article Open access 26 March 2025 THE EARLIEST DIVERGING EXTANT SCLERACTINIAN CORALS RECOVERED BY MITOCHONDRIAL GENOMES Article Open access 26 November 2020 BACKGROUND & SUMMARY Coral
reefs are one of the most diverse and productive ecosystems, which support more than one-quarter of marine life with less than 2% of the ocean floor1. In recent decades, reef-building corals
are threatened by anthropogenic climate change such as ocean warming and acidification2,3, as well as local stressors such as overfishing, pollution, and coastal development4,5,6. The world
has lost almost 50% coral coverage since the 1950s7. With projected continued degradation of coral reefs, 90% of coral reefs may disappear in the next few decades8,9,10. The blue corals
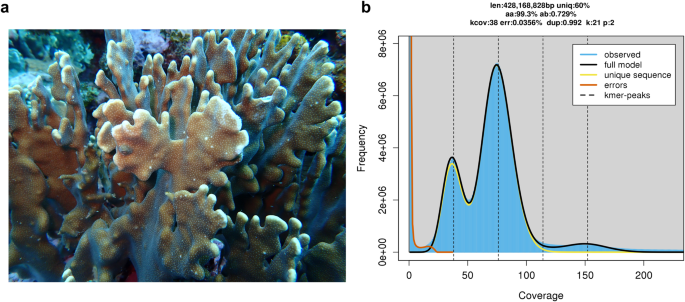
(_Heliopora_) are the only genus of octocorals that form a massive hard skeleton and symbiosis with zooxanthellae like scleractinian corals11 (Fig. 1a). Due to their massive reef structure,
blue corals are an important reef-building species in the Indo-West Pacific11,12,13,14. _H. coerulea_, with a characteristic blue skeleton, had long been regarded as the only extant member
of the family Helioporidae, until the recent description of _H. hiberniana_ (with white skeleton) in northwestern Australia15. Recent studies based on RAD-seq and Genotyping by sequencing in
blue corals revealed there are also two distinct lineages of _H. coerulea_ in the Kuroshio Current region16,17. Based on fossil records, the genus _Heliopora_ were once widely distributed
throughout the warm shallow oceans in the early Cretaceous11,18 (<120 million years ago, MYA). To date, _H. coerulea_ is distributed in the shallow warm waters of the Indo-Pacific
oceans11,17. _Heliopora coerulea_ is known to survive through bleaching events better than most scleractinian corals15,19,20. Recently, this species has been reported to expand from the
tropics to the high-latitude Tsukazaki, Japan21. A shift of dominant taxa from scleractinian corals to _H. coerulea_ has been reported in reefs of Ishigaki island, Japan22 and the South
China Sea side of the Philippines14,23. In addition, laboratory experiments showed that _H. coerulea_ had a higher growth rate when exposed at 31 °C – a temperature that would usually
trigger the bleaching of scleractinian corals7,8,9 – than at 26 °C24. To facilitate molecular studies of blue corals to understand their thermal resistance, here, we report a draft genome
assembly of _H. coerulea_ generated using long-read PacBio HiFi sequencing (Tables 1, 2). The assembled genome size of _H. coerulea_ is 429.9 Mb, consisting of 769 contigs with an N50 of
1.42 Mb, GC content of 37.4%, and 55.6% repeat elements (Fig. 2). The genome contains a total of 27,108 protein-coding genes with 95.7% functional annotated by BLASTp search against the
published protein databases. In addition, RNA sequencing shows that the _H. coerulea_ genome contains 6,225 lncRNAs and 79 miRNAs. METHODS SAMPLE COLLECTION The blue coral was collected by
SCUBA at 5 m depth from Green Island, Taiwan (22°40′37′′N 121°28′23′′E) in April 2018. Coral fragments were transported in seawater to Biodiversity Research Center, Academia Sinica, Taipei,
where they were kept in a 5 L aerated aquarium. To avoid contamination by bacteria or algae in the water, the coral fragments were rinsed several times in Milli-Q water immediately prior to
DNA and RNA sampling. Coral fragments were immediately fixed in liquid nitrogen for DNA extraction and genome sequencing, whilst tissues were fixed in RNA_later_ (Invitrogen, CA, USA) for
RNA sequencing. All samples were stored at −80 °C in a freezer until subjected to extraction. GENOMIC SEQUENCING Genomic DNA was extracted from the coral tissue using the CTAB method25. DNA
quality and quantity was measured using agarose gel electrophoresis and a Qubit fluorometer (Thermo Fisher Scientific, MA, USA), respectively. DNA samples were submitted to Novogene
(Beijing, China) for library preparation and whole genome sequencing (Table 1). Briefly, 1 µg DNA was used to construct two libraries with 350-bp and 500-bp insert sizes using the NEBNext
DNA Library Prep Kit (New England Biolabs, MA, USA), and sequenced on an Illumina HiSeq X Ten sequencer to generate 122.4 Gb paired-end reads with a read length of 150 bp. In addition, 10 µg
DNA was used to construct a HiFi SMRTbell library using the SMRTbell Express Template Prep Kit 2.0, and sequenced on a PacBio Sequel II sequencer. Total of 31.8 Gb high-quality HiFi reads
were produced using the circular consensus sequencing (CCS) mode on the PacBio long-read platform. RNA SEQUENCING Total RNA was extracted from the coral tissue using TRIzol reagent (Thermo
Fisher Scientific, MA, USA) by following the manufacturer’s protocol. The quality of the RNA samples was determined with agarose gel electrophoresis and the quantity was determined using a
Qubit fluorometer (Thermo Fisher Scientific, MA, USA). RNA samples were submitted to Novogene (Beijing, China) for mRNA, long non-coding RNA (lncRNA), and microRNA (miRNA) sequencing (Table
1). mRNA library was constructed using Illumina NEBNext Ultra RNA Library Prep Kit (New England Biolabs, MA, USA) and sequenced using an Illumina HiSeq X Ten sequencer to produce 150-bp
paired-end reads. For lncRNA, ribosomal RNA was depleted from total RNA using Epicentre Ribo-Zero rRNA Removal Kit (Epicentre, WI, USA). The cDNA libraries were prepared using the NEBNext
Ultra RNA Library Prep Kit (New England Biolabs, MA, USA), and sequenced on an Illumina NovaSeq platform under the paired-end mode to produce 150-bp reads. In addition, miRNA libraries were
prepared using the NEBNext Multiplex Small RNA Library Prep Kit (Illumina, CA, USA) and sequenced on an Illumina NovaSeq platform to produce 50-bp single-end reads. ESTIMATION OF GENOME SIZE
The genome size of _H. coerulea_ was estimated using GenomeScope v2.0 with Illumina data26. Adaptors and low-quality reads (quality score <30, length <40 bp) of the Illumina data were
trimmed with Trimmomatic v0.3827. To eliminate the zooxanthellae and prokaryotic reads, Illumina data were further filtered using bbmap.sh v39.01 (https://sourceforge.net/projects/bbmap/)
against the Symbiodiniaceae genomes (_Symbiodinium minutum_, _S. microadriaticum_, _S. kawagutii_, and _S goreaui_) from ReefGenomics database (http://reefgenomics.org/) and NCBI Prokaryotic
Refseq genomes with default settings. A total of 88.7 Gb Illumina reads were returned after quality filtering, and 77.9 Gb (87.8%) of them were from coral host. The clean Illumina data were
used to generate a 21-kmer histogram using jellyfish v2.2.028, and then characterized using GenomeScope v2.0, which predicted the genome size of 428.2 Mb and heterozygosity of 0.73% at a
k-mer size of 21 (Fig. 1b). GENOME ASSEMBLY _De novo_ assembly of HiFi reads (N50 of 14.0 kb and mean length of 13.5 kb; Table 1) were performed using nextDenovo v2.5.0
(https://github.com/Nextomics/NextDenovo) under default settings. Algal and microbial sequences were removed by binning genome assembly with MetaBAT2 v2.1529, and BLASTn v2.11.0 + search
against the 14 cnidarian genomes in Table 4, four Symbiodiniaceae genomes from ReefGenomics database (http://reefgenomics.org/), and NCBI Prokaryotic Refseq genomes with an E-value threshold
of 1e-20. The initial assembly generated 1,309.7 Mb metagenome sequences (Table 2). After binning, a total of 170 bins were identified and the “Bin167” with 600.2 Mb and >100X coverage
of Illumina data was selected (Table 2 and S1). BLASTn analysis filtered the potential symbiont sequence and resulted in the 586.0 Mb genome with 2,248 contigs. Possible alternative
heterozygous contigs were further eliminated using Purge Haplotigs v1.1.23030 (Table 2). The completeness of the final genome assembly was assessed by analyzing the Benchmarking Universal
Single-Copy Orthologs (BUSCO) v5.4.5 scores against the databases eukaryota_odb10 and eukaryota_odb10 under the genome mode31. QUAST v5.2 was used to assess the assembly statistics32. The
total assembled size of the genome is 429.9 Mb in length and the N50 is 1.42 Mb (Table 3; Fig. 2). In addition, the mitogenome of _H. coerulea_ was assembled with Illumina clean reads using
Norgal v1.0 under the default settings33, and annotated using MITOS2 online34 and tBLASTn v2.11.0 + search against the published _H. coerulea_ MT genome (GenBank: OL616236). The _H.
coerulea_ mitogenome is 18,957 bp in length with 14 protein-coding genes (Fig. 3), which is 100% identical with OL616236 in GenBank. MRNA ANNOTATION The protein coding genes of the _H.
coerulea_ genome were predicted using MAKER v3.0 pipeline35 according to Ip _et al_.36. In brief, repeat contents in the genome were identified using RepeatMasker v4.1.2-p1
(http://www.repeatmasker.org/; settings: “-e rmblast -s -gff”) with RepBase library version 2018102637 and species-specific repeat libraries in RepeatModeler v2.0.338 under the “LTRStruct”
option and the default setting for other parameters. A total of 239.1 Mb (55.6%) of the _H. coerulea_ genome consists of repetitive sequences, including 30.6% transposable elements, 21.8%
unclassified repeats, and 3.1% simple repeats and low complexity sequences (Table 3 and Fig. 2). Raw mRNA reads were trimmed using Trimmomatic v0.3827 (quality score <30, length <40
bp). The clean reads were _de novo_ and genome-guided assembled using Trinity v2.5.139 under the default settings. Cnidaria protein sequences from UniProt database were used as protein
evidence. Augustus v3.440 and SNAP v2006-07-2841 were used for _ab initio_ gene prediction. All predicted gene models were integrated into a consensus weighted annotation with
EVidenceModeler v1.1.142 under the default settings in Maker3. In addition, PASA v2.4.1 was used to improve the Maker result using the _de novo_ transcriptome43. Finally, we obtained 27,108
predicted protein-coding genes with an N50 of 1,754 bp (Table 3). The BUSCO completeness of predicted gene models was assessed against eukaryota_odb10 and metazoa_odb10 datasets31 under the
protein mode. The predicted genes were functionally annotated using Diamond v2.0.13.151 BLASTp44 against UniProt and Swissport databases under the “ultra-sensitive” option and an E-value
threshold of 1e-5. Gene functional annotation was conducted using eggNOG-mapper v245 for Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways and Pfam domain. LNCRNA
ANNOTATION The raw lncRNA reads were filtered to remove adapter and low-quality reads (quality score <30, length <40 bp) using Trimmomatic v0.3827. The clean lncRNA reads were mapped
to the _H. coerulea_ genome using HISAT2 v2.1.046 under the default settings. The resulting bam files were then assembled into transcript models using StringTie v1.3.4d47 under the default
settings. The assembled transcripts were processed through FlExible Extraction of LncRNAs (FEELnc) v0.2.148 for lncRNA identification and classification. Briefly, the script FEELnc filter.pl
was used to remove transcripts with one exon, a size < 200 bp, and overlapping with predicted protein-coding regions. The coding potential score of each candidate transcript was
calculated using the script FELLnc_codpot.pl under the shuffle mode. Finally, the FEELnc_classifier.pl was used to classify potential lncRNA with respect to the localization and the
direction of transcription of nearby protein-coding genes. A total of 6,225 lncRNA genes were predicted in the _H. coerulea_ genome (Tables S2, S3). MIRNA ANNOTATION miRNA analysis was
conducted according to Ip _et al_.36. Briefly, raw miRNA reads were trimmed with fastp v0.20.049 under the settings of length_required = 18, max_length = 35, unqualified_percent_limit = 30,
n_base_limit = 0. The clean reads were then combined and mapped to the genome using the mapper.pl script in miRDeep2 v2.0.1.250 using bowtie v1.2.251. miRNAs were predicted using the
miRDeep2.pl script in miRDeep2 with the Cnidaria mature miRNAs from miRBase v22.152. The predicted miRNAs were filtered with a miRDeep2 score ≥ 4, star (complementary) and mature read count
≥ 5, and a significant Randfold _p_-value. The target genes of miRNAs were predicted using miRanda v3.3a53 with a miRanda score ≥ 140, a dimer binding free energy < −5 kcalmol−1, and
strict 5′ seed pairing. In total, we detected 79 miRNA candidates ranging from 20 to 24 nt in length, and 10,636 mRNAs were predicted as their potential targets (Tables S4, S5). PHYLOGENY,
DIVERGENCE, AND GENE FAMILY ANALYSES Orthologous groups among _H. coerulea_ and 13 anthozoans with the outgroup species _Hydra vulgaris_ (details in Table 4 and Table S6) were identified
using OrthoFinder v2.5.4 under the “diamond_ultra_sens” option54. A total of 407 single-copy genes were aligned using MUSCLE v3.8.3155 and trimmed using TrimAL v1.456. The aligned sequences
with 91,426 amino acid positions and 1.1–13.9% gaps were concatenated for phylogenetic analysis using a maximum-likelihood method implemented in IQ-TREE v2.1357, with the best model of
Q.insect + F + I + G4 and 1000 bootstrapping replicates. MCMCtree implemented in PAML v4.9h58 was used to estimate divergence times using the burn-in, sample frequency and number of samples
of 10000000, 1000 and 10000, respectively. The node calibration among cnidarians was based on fossil records (i.e., ~55 MYA for _Acropora_59, ~145 MYA for Helioporacea18, ~540 MYA for
Hexacorallia60) and TIMETREE database61 (i.e., Edwardsiidae for 280 – 490 MYA, Anthozoa for 520 – 740 MYA). Using the orthologous results, we performed the gene family expansion and
contraction for each node using CAFÉ v4.262. These analyses revealed that _H. coerulea_ is sister to the soft coral _Dendronephthya gigantea_, which split during Triassic (~216 MYA, 95%
confidence interval of 157–301 MYA; Fig. 4). This _D. gigantea_ + _H. coerulea_ clade is then sister to the Hexacorallia clade, consistent with a previous phylogenetic analysis of 234
anthozoans63. Gene family analysis detected 167 expanded and 61 contracted gene families in _H. coerulea_ (Fig. 4; Table S7). DATA RECORDS The Illumina, PacBio HiFi, and RNAseq data have
been deposited in NCBI Sequence Read Archive with accession number SRR2353002364, SRR2353002465, SRR2353002566, SRR2353002667, SRR2353002768, SRR2353002869, SRR2353002970, SRR2353003071, and
SRR2353003172, under Bioproject accession number PRJNA936655. The genome assembly has been deposited at GenBank with accession number JASJOG00000000073. The genome annotation
(“Hco_maker_PASA_Final.gff”) and predicted genes (“Hco_v1.transcript.fasta” and “Hco_v1.protein.fasta”), lncRNA (“Hco_lncRNA.fasta”), and miRNA (“Hco_miRNA_mature.fasta”) has been deposited
in the Figshare database74. TECHNICAL VALIDATION The quality of _H. coerulea_ genome assembly was assessed by several approaches: (i) comparison with the estimated genome size, which is also
~430 Mb in total length (Figs. 1b, 2); (ii) obtaining the complete mitogenome, which is 100% identical in size and gene order with a published mitogenome of the same species (GenBank:
OL616236; Fig. 3); (iii) conducting QUAST analysis, which showed that the assembly statistics of _H. coerulea_ is comparable with published cnidarian genomes (Table 4); (iv) conducting BUSCO
analysis, which identified 98.4% eukaryotic BUSCOs and 94.4% metazoan BUSCOs in the _H. coerulea_ genome, and 98.4% eukaryotic BUSCOs and 95.3% metazoan BUSCOs in its predicted gene models
(Table 4); (v) conducting the analysis of genome coverage using SAMtools v1.15.175, which showed 100% genome coverage and 91.4% mapping rate of PacBio HiFi reads, and 94.8% genome coverage
and 88.4% mapping rate of Illumina short reads (Table 3). These results indicated the _H. coerulea_ assembly is of high-quality. CODE AVAILABILITY All bioinformatic tools used in this study
were executed according to the corresponding manual and protocols. The version and code and parameters of the main bioinformatic tools are described below. (1) Trimmomatic v0.38, parameters
used: “PE -phred33 ILLUMINACLIP:TruSeq. 3-PE.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:30 MINLEN:40”. (2) jellyfish v2.2.0, parameters used: “-C -m 21”. (3) GenomeScope v.2.0,
parameters used: ploidy 2 and kmer_length 21. (4) nextDenovo v2.5.0, parameters used: default. (5) Purge Haplotigs v1.1.2, parameters used: default. (5) MetaBAT v 2.12.1, parameters used:
default. (6) BLASTn v2.11.0+, parameters used: “-evalue 1e-20 -max_target_seqs. 1”. (8) BUSCO v5.4.5, parameters used: lineage_dataset eukaryota_odb10 (255 BUSCOs) and metazoa_odb10 (954
BUSCOs). (9) Norgal v1.0, parameters used: default. (10) MAKER v3.0, parameters used: default. (11) RepeatMasker v4.1.2-p1, parameters used: “-e rmblast -s -gff”, Database: Dfam v3.1 and
RepBaseRepeatMaskerEdition-20181026. (12) RepeatModeler v 2.0.3, parameters used: “-LTRStruct”. (13) Trinity v2.5.1, parameters used: default. (14) Augustus, version 3.4.0, parameters used:
species = Database trained with BUSCO. (15) SNAP v2006-07-28, parameters used: default. (16) EVidenceModeler v1.1.1, parameters used: default settings in Maker3. (17) PASA v2.4.1, parameters
used: “-C -R -T–ALIGNERS blat”. Augustus, version 3.4.0, parameters used: species = Database trained with BUSCO, alternatives-from-evidence = true, hintsfile = Output of RepeatMasker. (18)
Diamond v2.0.13.151 BLASTp, parameters used: “-ultra-sensitive -max-target-seqs. 1 -evalue 1e-5”. (19) HISAT2 v2.1.0, parameters used: default. (20) StringTie v1.3.4d, parameters used:
default. (21) FEELnc v0.2.1, parameters used: default. (22) fastp v0.20.0, parameters used: “length_required = 18, max_length = 35, unqualified_percent_limit = 30, n_base_limit = 0”. (23)
miRDeep2 v2.0.1.2, parameters used: default. (24) miRanda v3.3a, parameters used: “-sc 140 -en -5 -strict”. (25) OrthoFinder v2.5.4, parameters used: “-S diamond_ultra_sens”. (26) IQ-TREE
v2.1.3, parameters used: “-m TEST -bb 1000”. (27) MCMCtree implemented in PAML v4.9 h, parameters used: Tree topology from IQ-TREE result, fossil records in Fig. 4, burn-in: 10000000, sample
frequency: 1000, and number of samples: 10000. (28) CAFÉ v4.2, parameters used: default. (29) QUAST v5.2, parameters used: default. (30) bbmap v39.01, parameters used: bbsplit.sh and
mapPacBio.sh with default settings. (31) SAMtools v1.15.1, parameters used: command = coverage, depth, with default settings. REFERENCES * Knowlton, N. _et al_. Coral reef biodiversity. in
_Life in the World’s Oceans: Diversity, Distribution, And Abundance_ (ed. Mclntyre, A.) Ch. 4 (Wiley-Blackwell, 2010). * Hoegh-Guldberg, O., Poloczanska, E. S., Skirving, W. & Dove, S.
Coral reef ecosystems under climate change and ocean acidification. _Front. Mar. Sci._ 4, 158 (2017). Article Google Scholar * Anthony, K. R. _et al_. Ocean acidification and warming will
lower coral reef resilience. _Glob. Chang. Biol._ 17, 1798–808 (2011). Article ADS PubMed Central Google Scholar * Brodie, J. E. _et al_. Terrestrial pollutant runoff to the great
barrier reef: an update of issues, priorities and management responses. _Mar. Pollut. Bull._ 65, 81–100 (2012). Article CAS PubMed Google Scholar * Baum, G., Januar, H. I., Ferse, S. C.
& Kunzmann, A. Local and regional impacts of pollution on coral reefs along the Thousand Islands north of the megacity Jakarta, Indonesia. _PLoS One_ 10, e0138271 (2015). Article PubMed
PubMed Central Google Scholar * Magesh, N. S. & Krishnakumar, S. The Gulf of Mannar marine biosphere reserve, southern India. In _World seas: an environmental evaluation_ (ed.
Sheppard, C.) Ch. 8 (Cambridge: Academic Press, 2019). * Eddy, T. D. _et al_. Global decline in capacity of coral reefs to provide ecosystem services. _One Earth_ 4, 1278–1285 (2021).
Article ADS Google Scholar * Hoegh-Guldberg, O. _et al_. Impacts of 1.5 C global warming on natural and human systems. _Global warming of 1.5 °C_ (IPCC Special Report, 2018). *
Hoegh-Guldberg, O., Kennedy, E. V., Beyer, H. L., McClennen, C. & Possingham, H. P. Securing a long-term future for coral reefs. _Trends Ecol. Evol._ 33, 936–944 (2018). Article PubMed
Google Scholar * Hughes, T. P. _et al_. Spatial and temporal patterns of mass bleaching of corals in the Anthropocene. _Science_ 359, 80–83 (2018). Article ADS CAS PubMed Google
Scholar * Zann, L. P. & Bolton, L. The distribution, abundance and ecology of the blue coral _Heliopora coerulea_ (Pallas) in the Pacific. _Coral reefs_ 4, 125–134 (1985). Article ADS
Google Scholar * Abe, M. _et al_. Report of the Survey of _Heliopora coerulea_ Communities in Oura Bay, Okinawa (in Japanese) (2008). * Takino, T. _et al_. Discovery of a large population
of _Heliopora coerulea_ at Akaishi reef, Ishigaki Island, southwest Japan. _Galaxea J. Coral Reef Stud._ 12, 85–86 (2010). Article Google Scholar * Atrigenio, M. P., Conaco, C., Guzman,
C., Yap, H. T. & Aliño, P. M. Distribution and abundance of _Heliopora coerulea_ (Cnidaria: Coenothecalia) and notes on its aggressive behavior against scleractinian corals: Temperature
mediated? _Reg. Stud. Mar. Sci._ 40, 101502 (2020). Google Scholar * Richards, Z. T. _et al_. Integrated evidence reveals a new species in the ancient blue coral genus _Heliopora_
(Octocorallia). _Sci. Rep._ 8, 15875 (2018). Article ADS PubMed PubMed Central Google Scholar * Iguchi, A. _et al_. RADseq population genomics confirms divergence across closely related
species in blue coral (_Heliopora coerulea_). _BMC Evol. Biol._ 19, 1–7 (2019). Article CAS Google Scholar * Taninaka, H. _et al_. Phylogeography of blue corals (genus _Heliopora_)
across the Indo-West Pacific. _Front. Mar. Sci._ 8, 926 (2021). Article Google Scholar * Eguchi, M. Fossil Helioporidae from Japan and the South Sea Islands. _J. Paleontol_. 362–364
(1948). * Harii, S., Kayanne, H., Takigawa, H., Hayashibara, T. & Yamamoto, M. Larval survivorship, competency periods and settlement of two brooding corals, _Heliopora coerulea_ and
_Pocillopora damicornis_. _Mar. Biol._ 141, 39–46 (2002). Article Google Scholar * Kayanne, H., Harii, S., Ide, Y. & Akimoto, F. Recovery of coral populations after the 1998 bleaching
on Shiraho Reef, in the southern Ryukyus, NW Pacific. _Mar. Ecol. Prog. Ser._ 239, 93–103 (2002). Article ADS Google Scholar * Nakabayashi, A., Matsumoto, T., Kitano, Y. F., Nagai, S.
& Yasuda, N. Discovery of the northernmost habitat of the blue coral _Heliopora coerulea_: possible range expansion due to climate change? _Galaxea J. Coral Reef Stud._ 19, 1–2 (2017).
Article Google Scholar * Harii, S., Hongo, C., Ishihara, M., Ide, Y. & Kayanne, H. Impacts of multiple disturbances on coral communities at Ishigaki Island, Okinawa, Japan, during a 15
year survey. _Mar. Ecol. Prog. Ser._ 509, 171–180 (2014). Article ADS Google Scholar * Atrigenio, M., Aliño, P. & Conaco, C. Influence of the Blue coral _Heliopora coerulea_ on
scleractinian coral larval recruitment. _J. Mar. Biol._ 2017, 1–5 (2017). Article Google Scholar * Guzman, C., Atrigenio, M., Shinzato, C., Aliño, P. & Conaco, C. Warm seawater
temperature promotes substrate colonization by the blue coral, _Heliopora coerulea_. _PeerJ_ 7, e7785 (2019). Article PubMed PubMed Central Google Scholar * Porebski, S., Bailey, L. G.
& Baum, B. R. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. _Plant Mol. Biol. Rep._ 15, 8–15 (1997). Article CAS
Google Scholar * Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. _Nat. Commun._ 11, 1432 (2020).
Article ADS CAS PubMed PubMed Central Google Scholar * Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. _Bioinformatics_ 30,
2114–2120 (2014). Article CAS PubMed PubMed Central Google Scholar * Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers.
_Bioinformatics_ 27, 764–770 (2011). Article PubMed PubMed Central Google Scholar * Kang, D. D. _et al_. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome
reconstruction from metagenome assemblies. _PeerJ_ 7, e7359 (2019). Article PubMed PubMed Central Google Scholar * Roach, M. J., Schmidt, S. A. & Borneman, A. R. Purge Haplotigs:
allelic contig reassignment for third-gen diploid genome assemblies. _BMC Bioinformatics_ 19, 1–10 (2018). Article Google Scholar * Simão, F. A., Waterhouse, R. M., Ioannidis, P.,
Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. _Bioinformatics_ 31, 3210–3212 (2015). Article PubMed
Google Scholar * Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: quality assessment tool for genome assemblies. _Bioinformatics_ 29, 1072–1075 (2013). Article CAS PubMed
PubMed Central Google Scholar * Al-Nakeeb, K., Petersen, T. N. & Sicheritz-Pontén, T. Norgal: extraction and de novo assembly of mitochondrial DNA from whole-genome sequencing data.
_BMC Bioinformatics_ 18, 1–7 (2017). Article Google Scholar * Donath, A. _et al_. Improved annotation of protein-coding genes boundaries in metazoan mitochondrial genomes. _Nucleic Acids
Res._ 47, 10543–10552 (2019). Article CAS PubMed PubMed Central Google Scholar * Cantarel, B. L. _et al_. MAKER: an easy-to-use annotation pipeline designed for emerging model organism
genomes. _Genome Res._ 18, 188–196 (2008). Article CAS PubMed PubMed Central Google Scholar * Ip, J. C. H. _et al_. Host-Endosymbiont Genome Integration in a Deep-Sea Chemosymbiotic
Clam. _Mol. Biol. Evol._ 38, 502–518 (2021). Article CAS PubMed Google Scholar * Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic
genomes. _Mob. DNA._ 6, 11 (2015). Article PubMed PubMed Central Google Scholar * Flynn, J. M. _et al_. RepeatModeler2 for automated genomic discovery of transposable element families.
_Proc. Natl. Acad. Sci. USA_ 117, 9451–9457 (2020). Article ADS CAS PubMed PubMed Central Google Scholar * Haas, B. J. _et al_. De novo transcript sequence reconstruction from RNA-seq
using the Trinity platform for reference generation and analysis. _Nat. Protoc._ 8, 1494–1512 (2013). Article CAS PubMed Google Scholar * Stanke, M. & Morgenstern, B. AUGUSTUS: a web
server for gene prediction in eukaryotes that allows user-defined constraints. _Nucleic Acids Res._ 33, W465–W467 (2005). Article CAS PubMed PubMed Central Google Scholar * Korf, I.
Gene finding in novel genomes. _BMC Bioinformatics_ 5, 59 (2004). Article PubMed PubMed Central Google Scholar * Haas, B. J. _et al_. Automated eukaryotic gene structure annotation using
EVidenceModeler and the Program to Assemble Spliced Alignments. _Genome Biol._ 9, R7 (2008). Article PubMed PubMed Central Google Scholar * Haas, B. J. _et al_. Improving the
Arabidopsis genome annotation using maximal transcript alignment assemblies. _Nucleic Acids Res._ 31, 5654–5666 (2003). Article CAS PubMed PubMed Central Google Scholar * Buchfink, B.,
Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. _Nat. Methods_ 12, 59–60 (2015). Article CAS PubMed Google Scholar * Huerta-Cepas, J. _et al_. Fast
genome-wide functional annotation through orthology assignment by eggNOG-mapper. _Mol. Biol. Evol._ 34, 2115–2122 (2017). Article CAS PubMed PubMed Central Google Scholar * Kim, D.,
Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. _Nat. Methods_ 12, 357 (2015). Article CAS PubMed PubMed Central Google Scholar * Pertea,
M. _et al_. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. _Nat. Biotechnol._ 33, 290–295 (2015). Article CAS PubMed PubMed Central Google Scholar *
Wucher, V. _et al_. FEELnc: a tool for long non-coding RNA annotation and its application to the dog transcriptome. _Nucleic Acids Res._ 45, e57–e57 (2017). ADS CAS PubMed PubMed Central
Google Scholar * Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. _Bioinformatics_ 34, i884–i890 (2018). Article PubMed PubMed Central
Google Scholar * Friedländer, M. R., Mackowiak, S. D., Li, N., Chen, W. & Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades.
_Nucleic Acids Res._ 40, 37–52 (2011). Article PubMed PubMed Central Google Scholar * Langmead, B. Aligning short sequencing reads with Bowtie. _Curr. Protoc. Bioinformatics_ 32, 11.17.
11–11.17. 14 (2010). Article Google Scholar * Kozomara, A., Birgaoanu, M. & Griffiths-Jones, S. miRBase: from microRNA sequences to function. _Nucleic Acids Res._ 47, D155–D162 (2018).
Article PubMed Central Google Scholar * Enright, A. _et al_. MicroRNA targets in _Drosophila_. _Genome Biol._ 4, 1–27 (2003). Article Google Scholar * Emms, D. M. & Kelly, S.
OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. _Genome Biol._ 16, 157 (2015). Article PubMed PubMed Central
Google Scholar * Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. _Nucleic Acids Res._ 32, 1792–1797 (2004). Article CAS PubMed PubMed Central
Google Scholar * Capella-Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. _Bioinformatics_ 25,
1972–1973 (2009). Article PubMed PubMed Central Google Scholar * Nguyen, L. T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for
estimating maximum-likelihood phylogenies. _Mol. Biol. Evol._ 32, 268–274 (2014). Article PubMed PubMed Central Google Scholar * Yang, Z. PAML 4: phylogenetic analysis by maximum
likelihood. _Mol. Biol. Evol._ 24, 1586–1591 (2007). Article CAS PubMed Google Scholar * Medina, M., Collins, A. G., Takaoka, T. L., Kuehl, J. V. & Boore, J. L. Naked corals:
skeleton loss in Scleractinia. _Proc. Natl. Acad. Sci. USA_ 103, 9096–100 (2006). Article ADS CAS PubMed PubMed Central Google Scholar * Han, J. _et al_. Tiny sea anemone from the
Lower Cambrian of China. _PLoS One_ 5, e13276 (2010). Article ADS PubMed PubMed Central Google Scholar * Hedges, S. B., Dudley, J. & Kumar, S. TimeTree: a public knowledge-base of
divergence times among organisms. _Bioinformatics_ 22, 2971–2972 (2006). Article CAS PubMed Google Scholar * Han, M. V., Thomas, G. W., Lugo-Martinez, J. & Hahn, M. W. Estimating
gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. _Mol. Biol. Evol._ 30, 1987–1997 (2013). Article CAS PubMed Google Scholar * Quattrini,
A. M. _et al_. Palaeoclimate ocean conditions shaped the evolution of corals and their skeletons through deep time. _Nat. Ecol. Evol._ 4, 1531–1538 (2020). Article PubMed Google Scholar *
_NCBI Sequence Read Archive_ https://identifiers.org/ncbi/insdc.sra:SRR23530023 (2023). * _NCBI Sequence Read Archive_ https://identifiers.org/ncbi/insdc.sra:SRR23530024 (2023). * _NCBI
Sequence Read Archive_ https://identifiers.org/ncbi/insdc.sra:SRR23530025 (2023). * _NCBI Sequence Read Archive_ https://identifiers.org/ncbi/insdc.sra:SRR23530026 (2023). * _NCBI Sequence
Read Archive_ https://identifiers.org/ncbi/insdc.sra:SRR23530027 (2023). * _NCBI Sequence Read Archive_ https://identifiers.org/ncbi/insdc.sra:SRR23530028 (2023). * _NCBI Sequence Read
Archive_ https://identifiers.org/ncbi/insdc.sra:SRR23530029 (2023). * _NCBI Sequence Read Archive_ https://identifiers.org/ncbi/insdc.sra:SRR23530030 (2023). * _NCBI Sequence Read Archive_
https://identifiers.org/ncbi/insdc.sra:SRR23530031 (2023). * _NCBI GenBank_ https://identifiers.org/nucleotide:JASJOG000000000 (2023). * Ip, J. _et al_. A draft genome assembly of
reef-buliding octocoral _Heliopora coerulea_. _Figshare_ https://doi.org/10.6084/m9.figshare.22093037 (2023). * Li, H. _et al_. The sequence alignment/map format and SAMtools.
_Bioinformatics_ 25, 2078–2079 (2009). Article PubMed PubMed Central Google Scholar * Jeon, Y. _et al_. The draft genome of an octocoral, _Dendronephthya gigantea_. _Genome Biol. Evol._
11, 949–953 (2019). Article CAS PubMed PubMed Central Google Scholar * Stephens, T. G. _et al_. High-quality genome assembles from key Hawaiian coral species. _GigaScience_ 11, giac098
(2022). Article PubMed PubMed Central Google Scholar * Shinzato, C. _et al_. Eighteen coral genomes reveal the evolutionary origin of _Acropora_ strategies to accommodate environmental
changes. _Mol. Biol. Evol._ 1, 16–30 (2021). Article Google Scholar Download references ACKNOWLEDGEMENTS This work was supported by Hong Kong Baptist University’s Start-up Grant for New
Academics (162780), Environmental and Conservation Fund of Hong Kong SAR (122/2022), the Key Special Project for Introduced Talents Team of Southern Marine Science and Engineering Guangdong
Laboratory (Guangzhou) (GML2019ZD0404), and the General Research Fund of Hong Kong SAR Government’s University Grants Committee (12102018). B.K.K.C. was supported by a grant for the Senior
Investigator Award, Academia Sinica, Taiwan (AS-IA-105-L03). AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Biology, Hong Kong Baptist University, Kowloon Tong, Hong Kong Jack
Chi-Ho Ip & Jian-Wen Qiu * Biodiversity Research Center, Academia Sinica, Taipei, Taiwan Ming-Hay Ho & Benny K. K. Chan Authors * Jack Chi-Ho Ip View author publications You can also
search for this author inPubMed Google Scholar * Ming-Hay Ho View author publications You can also search for this author inPubMed Google Scholar * Benny K. K. Chan View author publications
You can also search for this author inPubMed Google Scholar * Jian-Wen Qiu View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS J.C.H.I. and
J.W.Q. designed research. B.K.K.C. and M.J.H. collected the samples and cultured them in the laboratory. J.C.H.I. conducted genomic extraction, assembled, annotated genome, and data
analyses. J.C.H.I., J.W.Q. and B.K.K.C. drafted the manuscript. All authors edited the manuscript and approved the submission. CORRESPONDING AUTHORS Correspondence to Jack Chi-Ho Ip or
Jian-Wen Qiu. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to
jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under
a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article
are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and
your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this
license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Ip, J.CH., Ho, MH., Chan, B.K.K. _et al._ A draft genome assembly of
reef-building octocoral _Heliopora coerulea_. _Sci Data_ 10, 381 (2023). https://doi.org/10.1038/s41597-023-02291-z Download citation * Received: 02 March 2023 * Accepted: 31 May 2023 *
Published: 14 June 2023 * DOI: https://doi.org/10.1038/s41597-023-02291-z SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link
Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative