Play all audios:
ABSTRACT _Slavum lentiscoides_ and _Chaetogeoica ovagalla_ are two aphid species from the subtribe Fordina of Fordini within the subfamily Eriosomatinae, and they produce galls on their
primary host plants _Pistacia_. We assembled chromosome-level genomes of these two species using Nanopore long-read sequencing and Hi-C technology. A 332 Mb genome assembly of _S.
lentiscoides_ with a scaffold N50 of 19.77 Mb, including 11,747 genes, and a 289 Mb genome assembly of _C. ovagalla_ with a scaffold N50 of 11.85 Mb, containing 14,492 genes, were obtained.
The Benchmarking Universal Single-Copy Orthologs (BUSCO) benchmark of the two genome assemblies reached 93.7% (91.9% single-copy) and 97.0% (95.3% single-copy), respectively. The
high-quality genome assemblies in our study provide valuable resources for future genomic research of galling aphids. SIMILAR CONTENT BEING VIEWED BY OTHERS THE CHROMOSOME-LEVEL GENOME
ASSEMBLY OF _APHIDOLETES APHIDIMYZA_ RONDANI (DIPTERA: CECIDOMYIIDAE) Article Open access 17 July 2024 A CHROMOSOME-LEVEL GENOME ASSEMBLY OF THE APHID _SEMIAPHIS HERACLEI_ (TAKAHASHI)
Article Open access 10 May 2025 A CHROMOSOME-LEVEL GENOME ASSEMBLY OF THE GALL MAKER PEST INQUILINE, _DIOMORUS AIOLOMORPHI_ KAMIJO (HYMENOPTERA: TORYMIDAE) Article Open access 29 August 2024
BACKGROUND & SUMMARY Some insects can induce abnormal growth and development of host plants and form highly specialized structures termed galls. Galls benefit insect inducers and their
offspring by providing abundant nutrition and protection against natural enemies and adverse abiotic factors1. Gall formation at the early stage is generally induced by insect stimuli,
including feeding and oviposition1. The molecular mechanisms of gall induction and development have been found to be inseparable from insect effectors2,3 and phytohormones4,5,6. Aphids
(Hemiptera: Aphidoidea) are an important group of plant-sapping insects that comprise over 5,000 species, 10–20% of which can induce galls on their primary host plants7,8. Galling aphid
species mainly belong to Adelgidae, Phylloxeridae, and several subfamilies of Aphididae, including Eriosomatinae, Hormaphidinae, Tamaliinae, Thelaxinae, and Aphidinae9,10. To date, reference
genomes are available for more than 40 aphid species. Among them, only seven true gall-forming species have been sequenced and assembled11,12,13,14,15,16. The lack of genomic resources for
galling aphids has greatly hindered our understanding of the genetic basis for adaptive evolution of gall induction in aphids. The tribe Fordini of subfamily Eriosomatinae is a typical
lineage of true gall-inducing aphids. Species from one subtribe, Melaphidina, induce galls on _Rhus_ (Anacardiaceae). The _Rhus_ galls usually contain high concentrations of tannins and have
economic and medicinal values17. The genome of one representative species, _Schlechtendalia chinensis_, has been reported recently15. Aphids within the other subtribe, Fordina, induce galls
on _Pistacia_ (Anacardiaceae), which is an economically significant genus of plants. _Pistacia vera_ (cultivated pistachio) produces edible nuts that are of great commercial importance.
Seeds of other wild _Pistacia_ species also possess economic value and can be utilized for local consumption, oil extraction, soap production, and most importantly, as rootstock sources for
pistachio trees18. Extracts from _Pistacia_ galls exhibit anti-inflammatory and antioxidant activities19,20. However, no genomic data are available for this galling group. Producing more
high-quality genome assemblies encompassing different lineages is foundational to the genomic study of galling aphids. In this study, we generated chromosome-level genome assemblies of two
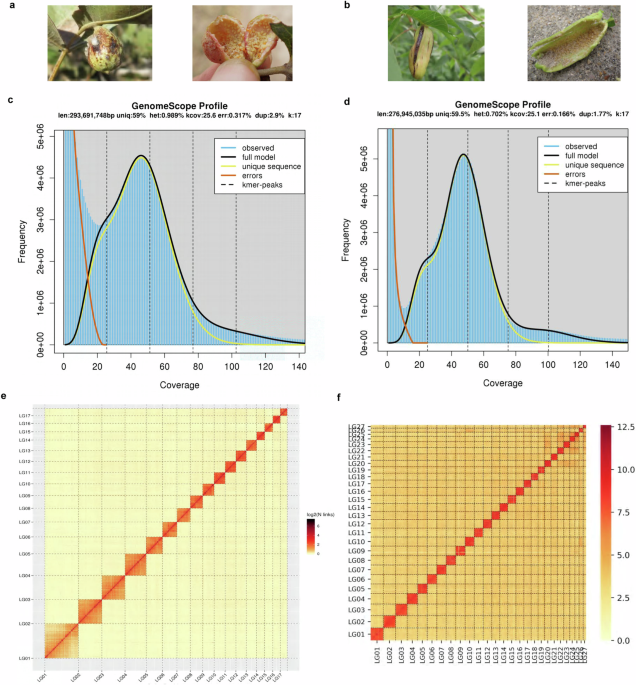
galling species from Fordina, _Slavum lentiscoides_ Mordvilko and _Chaetogeoica ovagalla_ (Zhang). Both species induce closed bag-like galls on the main veins of _Pistacia_ spp. leaves8
(Fig. 1a,b). A total of 332.26 Mb and 289.72 Mb assembled sequences were anchored to 17 and 27 pseudo-chromosomes for _S. lentiscoides_ and _C. ovagalla_, with 11,747 and 14,492 predicted
protein-coding genes, respectively. METHODS SAMPLE COLLECTION _S. lentiscoides_ samples within galls were collected on a pistachio tree (_P. vera_) from Andijan, Uzbekistan (40.719°N,
72.436°E) in September 2019. The galls of _C. ovagalla_ were obtained on _Pistacia chinensis_ from Qingdao, China (36.193°N, 120.573°E) in July 2018. The aphids from fresh galls were rapidly
frozen and stored in liquid nitrogen at the National Animal Collection Resource Center, Institute of Zoology, Chinese Academy of Sciences, Beijing, China. GENOME SEQUENCING Total genomic
DNA was extracted from over 0.2 grams of aphids within a single gall using a standard CTAB method. For Oxford Nanopore sequencing, the concentration and quality of DNA were checked using 1%
agarose gel electrophoresis, NanoDrop spectrophotometry (Thermo Fisher Scientific, Lafayette, USA), and Qubit fluorometry (Invitrogen, Darmstadt, Germany). Large-sized segment libraries were
selected (≥30 kb) with the BluePippinTM System and processed by the ONT Template Prep Kit (SQK-LSK109, Oxford Nanopore Technologies [ONT], Oxford, UK) protocol. DNA fragments were
end-repaired and 3′-adenylated using the NEB Next FFPE DNA Repair Mix kit (New England Biolabs [NEB], Ipswich, USA). The Nanopore sequencing adapters were ligated using the NEBNext Quick
Ligation Module (E6056) (NEB). The final library was sequenced on R9 flow cells using a PromethION DNA sequencer (ONT) with an ONT sequencing reagent kit (EXP-FLP001.PRO.6). The raw signal
data were called, and the FAST5 files were converted into FASTQ files using MinKNOW software v2.0 (ONT). Short reads (<2kb) and reads with low-quality bases and adapter sequences were
removed. Shotgun sequencing reads were used to estimate genome size and to correct the genome assembly. Paired-end libraries were prepared following the instructions for sequencing with the
NovaSeq 6000 Reagent Kit on a NovaSeq 6000 platform, with an insert size of 350 bp. For Hi-C sequencing, nuclear DNA from tissue cells was cross-linked and enzymatically digested with DpnII.
The DNA fragments with interaction relationships were captured with streptavidin beads and prepared for sequencing. Hi-C libraries with 300–700 bp insert sizes were constructed on the PE150
Illumina platform. Total RNA was extracted from the same colony for RNA extraction and sequencing. RNA-seq libraries were constructed using the NEBNext Ultra RNA Library Prep Kit for
Illumina (NEB) and were then sequenced on the NovaSeq 6000 platform with a 150-bp paired-end output. We finally generated for the first time chromosome-level genome assemblies of _S.
lentiscoides_ and _C. ovagalla_ using a combination of Nanopore long reads (44.14 Gb and 55.02 Gb, respectively), Illumina short reads (58.13 Gb and 36.40 Gb, respectively), and Hi-C
sequencing data (40.35 Gb and 45.54 Gb, respectively). KARYOTYPE ANALYSIS We dissected young embryos of aphids on slides. The embryos were kept in 0.7% sodium citrate for 30 min and then
fixed in Carnoy’s fixative for 15 min. After fixation, a coverslip was placed on the slide and vertically pressed down to disperse the cell mass as much as possible. The treated slide was
frozen at −80 °C for 10 min, followed by air drying. The air-dried slide was stained in 5% Giemsa solution for 15 min and then washed with a tiny stream of distilled water. The slide was
observed and subjected to photomicrography using a Leica DM6B photomicroscope (camera magnification 100 × 10). The result showed that _S. lentiscoides_ has a diploid chromosome number of 2n
= 34 (Fig. S1), while the karyotype of _C. ovagalla_ was not obtained due to sample limitations. GENOME ASSEMBLY Before genome assembly, we extracted Illumina paired sequencing reads with
approximately 50× coverage to estimate genome size and heterozygosity. The 17 k-mer frequency spectra obtained with Jellyfish v1.1.1021 and GenomeScope22 suggested that the heterozygosity of
the _S. lentiscoides_ and _C. ovagalla_ genomes was 0.99% and 0.70%, the estimated genome sizes were 293.69 Mb and 276.95 Mb, and the repeat sequence content was 41.0% and 40.5%,
respectively (Table 1, Fig. 1c,d). Different assembly strategies were used to assemble the genomes of these two aphid species. For _S. lentiscoides_, the Nanopore long reads were initially
cleaned using Canu v1.523 for sequence error correction and were then assembled from the error-corrected reads with the parameters “useGrid = false, genomeSize = 300 m, minReadLength = 2000,
minOverlapLength = 500, corOutCoverage = 135, corMinCoverage = 2”. We used the _Redundans_24 pipeline to remove bubble contigs from the draft assembly. For _C. ovagalla_, the long reads
were analyzed using Nextdenovo v2.125 for correction with the parameter “-seed_cutoff = 30k”, and then the corrected reads were assembled by SMARTdenovo
(https://github.com/ruanjue/smartdenovo) with the parameters “-k 21 -J 5000 -t 20”. We found that the Canu assembly pipeline run obviously more slowly than the latter methods. These two
draft aphid genome assemblies were subjected to two rounds of error correction using RACON26 with Nanopore reads, followed by three rounds of additional polishing using Pilon27 with Illumina
sequencing data. We further removed a few contigs with microbial contamination by BLAST searches against the NCBI nt database, and no contigs related to the mitochondrial genome were found.
After assembly and correction, we produced draft genome assemblies of _S. lentiscoides_ and _C. ovagalla_, comprising a total of 336.27 Mb and 293.08 Mb of sequences, with 313 and 128
contigs, in which the contig N50 lengths were 6.79 Mb and 5.38 Mb, respectively (Table 1). The Hi-C paired-end reads were mapped to the Nanopore assembly using BWA aln v0.7.10-r78928. The
unique read pairs around the DpnII site were determined. Then, we applied LACHESIS software29 to cluster, order, and orient contigs from the draft assembly. Invalid read pairs were filtered
with HiC-Pro30 using default settings. The predicted chromosomal genome was divided into bins of equal length (500 kb), and the number of valid Hi-C read pairs between bins was then used to
represent the interaction signals between bins. Finally, we constructed a heatmap based on these interaction signals by R. For _C. ovagalla_, considering the variability in chromosome
numbers observed in Aphididae31, we initially established a range of 10 to 30 for the cluster number. After careful consideration and analysis, we determined that a cluster number of 27 was
the most appropriate choice. As a result, 40.35 Gb and 45.54 Gb of Hi-C clean reads were generated to build chromosome-level assemblies. After clustering, ordering, and orientation of the
contigs, we obtained two final assemblies with chromosome-anchored sizes of 332.26 Mb and 289.72 Mb and scaffold N50 lengths of 19.77 Mb and 11.85 Mb for _S. lentiscoides_ and _C. ovagalla_,
respectively (Table 1). Therein, 128 and 86 contigs could be anchored to 17 and 27 linkage groups, with 98.81% and 98.85% anchoring rates, respectively (Table 1, Fig. 1e,f). The chromosome
circus plots were generated using Circos v0.69-932 (Fig. 2). GENOME ANNOTATION Before gene prediction, a de novo repeat library was built by using LTR_FINDER33 and RepeatScout34, and the
library was classified by PASTEClassifier35. Transposable element sequences in the genome were predicted with RepeatMasker36 using the de novo library and Repbase library37. Consequently, we
identified 87.71 Mb and 61.25 Mb of repeat sequences, accounting for 25.94% and 20.90% of the genome assemblies of _S. lentiscoides_ and _C. ovagalla_, respectively (Table 1). The
protein-coding genes were predicted by integrating the evidence generated via the de novo, homology-based, and RNA-seq-based methods. For ab initio prediction, we used Augustus v2.438 to
generate the de novo predictions with the default parameters. For the homology-based analysis, GeMoMa v1.3.139 was used to query the protein sequences against a database of four species
(_Myzus persicae_, _Aphis gossypii_, _Rhopalosiphum maidis_, and _Acyrthosiphon pisum_). For RNA-seq annotation, RNA-seq reads were aligned to the genome assembly using HISAT2 v2.0.440 and
StringTie v1.2.341 with the default parameters. TransDecoder v2.042, GeneMarkS-T v5.143, and PASA v2.0.244 were used to generate transcript predictions. Finally, we integrated three types of
evidence for gene prediction by EVidenceModeler (EVM) v 1.1.145. By integrating _ab initio_-based, homology-based, and transcriptome-based evidence, we predicted 11,747 genes in the _S.
lentiscoides_ genome, and 14,492 genes in the genome of _C. ovagalla_. The predicted genes were then searched for homology-based functions by BLAST searches against the NCBI non-redundant
protein sequences (nr), Eukaryotic Orthologous Groups (KOG), TrEMBL, Kyoto Encyclopedia of Genes and Genomes (KEGG), and the Gene Ontology (GO) databases with BLASTP v2.2.3146 (-evalue
1e-5). The GO terms were assigned using blast2go v5.047. A total of 99.22% (11,656 genes) and 89.73% (13,004 genes) of the predicted genes of these two genomes were supported by functional
annotation from the above five protein databases. Among these genes of _S. lentiscoides_ and _C. ovagalla_, 98.11% and 89.15% showed homology to proteins in NCBI nr, 98.89% and 67.84% in
TrEMBL, 67.34% and 57.00% in KOG, 48.76% and 43.27% in KEGG, and 38.11% and 43.17% in GO, respectively (Table 2). Three types of noncoding RNAs (ncRNAs) were identified. The microRNA (miRNA)
and ribosomal RNA (rRNA) genes were predicted by BLASTN searches against the Rfam database48. The transfer RNA (tRNA) genes were identified using tRNAscan-SE49. Finally, we identified 27
and 31 miRNAs, 66 and 68 rRNAs, and 200 and 172 tRNAs within the genomes of _S. lentiscoides_ and _C. ovagalla_, respectively (Table 1). DATA RECORDS The genome raw data, RNA sequencing
data, and genome assemblies are available at the National Center for Biotechnology Information (NCBI) under the BioProject accession numbers PRJNA765394 and PRJNA832539. The Illumina WGS
data was archived with the accession numbers SRR1604696350 and SRR2399932551. The Nanopore WGS data was deposited with the accession numbers SRR1604696452 and SRR2399932653. The RNA-Seq data
was archived with the accession numbers SRR1604696154 and SRR2399932355. The Hi-C data was archived under the accession numbers SRR1604696256 and SRR2399932457. The genome assemblies have
been deposited at GenBank under the accession numbers GCA_032441835.158 and GCA_032441825.159. The genome annotation files have been deposited at the Figshare60. TECHNICAL VALIDATION The
quality and completeness of draft assemblies were assessed by three methods as follows. The Illumina reads were mapped to the draft assembly with BWA-MEM v0.7.10-r78961, and the mapping
ratio was calculated with SAMTOOLS v1.362. In addition, the Core Eukaryotic Genes Mapping Approach (CEGMA v2.563) and BUSCO v3.1.064 were used to assess the completeness of the genome
assembly. The _S. lentiscoides_ and _C. ovagalla_ genomes contained 96.77% and 97.58% of highly conserved Core Eukaryotic Genes (CEGs), respectively. They showed a high representation of
conserved complete Benchmarking Universal Single-Copy Orthologs (BUSCOs) (_S. lentiscoides_: 93.70% complete BUSCOs of insecta_odb10; _C. ovagalla_: 97.00% complete BUSCOs of insecta_odb10),
mapped with 96.78% and 97.19% of Illumina short reads, respectively, which indicated that these genome assemblies were of high quality and near-complete. CODE AVAILABILITY This paper does
not report original code. If no detailed parameters were mentioned for the software, default parameters were used according to the software introduction. REFERENCES * Stone, G. N. &
Schönrogge, K. The adaptive significance of insect gall morphology. _Trends Ecol. Evol._ 18, 512–522 (2003). Article Google Scholar * Thorpe, P., Cock, P. J. & Bos, J. Comparative
transcriptomics and proteomics of three different aphid species identifies core and diverse effector sets. _BMC Genom._ 17, 1–18 (2016). Article Google Scholar * Cambier, S. _et al_. Gall
wasp transcriptomes unravel potential effectors involved in molecular dialogues with oak and rose. _Front. Physiol._ 10, 926 (2019). Article PubMed PubMed Central Google Scholar *
Yamaguchi, H. _et al_. Phytohormones and willow gall induction by a gall-inducing sawfly. _New Phytol._ 196, 586–595 (2012). Article CAS PubMed Google Scholar * Tooker, J. F. &
Helms, A. M. Phytohormone dynamics associated with gall insects, and their potential role in the evolution of the gall-inducing habit. _J. Chem. Ecol._ 40, 742–753 (2014). Article CAS
PubMed Google Scholar * Hirano, T. _et al_. Reprogramming of the developmental program of _Rhus javanica_ during initial stage of gall induction by _Schlechtendalia chinensis_. _Front.
Plant Sci._ 11, 471 (2020). Article PubMed PubMed Central Google Scholar * Chakrabarti, S. Diversity and biosystematics of gall-inducing aphids (Hemiptera: Aphididae) and their galls in
the Himalaya. _Orient. Insects_ 41, 35–54 (2007). Article Google Scholar * Blackman, R. L. & Eastop, V. F. Aphids on the world’s plants: an online identification and information guide.
http://www.aphidsonworldsplants.info/ (2024). * Wool, D. Galling aphids: specialization, biological complexity, and variation. _Annu. Rev. Entomol._ 49, 175–192 (2004). Article CAS PubMed
Google Scholar * Chen, J. & Qiao, G. X. Galling aphids (Hemiptera: Aphidoidea) in China: diversity and host specificity. _Psyche_ 2012, 621934 (2012). Google Scholar * Dial, D. T.
_et al_. Whole-genome sequence of the Cooley spruce gall adelgid, _Adelges cooleyi_ (Hemiptera: Sternorrhyncha: Adelgidae). _G3-Genes Genomes Genet._ 14, jkad224 (2023). Article Google
Scholar * Rispe, C. _et al_. The genome sequence of the grape phylloxera provides insights into the evolution, adaptation, and invasion routes of an iconic pest. _BMC Biol._ 18, 1–25
(2020). Google Scholar * Stern, D. L. & Han, C. Gene structure-based homology search identifies highly divergent putative effector gene family. _Genome Biol. Evol._ 14, evac069 (2022).
Article PubMed PubMed Central Google Scholar * Smith, T. E. _et al_. Elucidation of host and symbiont contributions to peptidoglycan metabolism based on comparative genomics of eight
aphid subfamilies and their _Buchnera_. _PLoS Genet._ 18, e1010195 (2022). Article CAS PubMed PubMed Central Google Scholar * Wei, H. Y. _et al_. Chromosome-level genome assembly for
the horned-gall aphid provides insights into interactions between gall-making insect and its host plant. _Ecol. Evol._ 12, e8815 (2022). Article PubMed PubMed Central Google Scholar *
Korgaonkar, A. _et al_. A novel family of secreted insect proteins linked to plant gall development. _Curr. Biol._ 31, 1836–1849 (2021). Article CAS PubMed PubMed Central Google Scholar
* Zhang, C. X., Tang, X. D. & Cheng, J. A. The utilization and industrialization of insect resources in China. _Entomol. Res._ 38, S38–S47 (2008). Article Google Scholar * Kafkas,
S., Kafkas, E. & Perl-Treves, R. Morphological diversity and a germplasm survey of three wild _Pistacia_ species in Turkey. _Genet. Resour. Crop Ev._ 49, 261–270 (2002). Article Google
Scholar * Ahmed, Z. B. _et al_. Study of the antioxidant activity of _Pistacia atlantica_ Desf. gall extracts and evaluation of the responsible compounds. _Biochem. Syst. Ecol._ 100, 104358
(2022). Article Google Scholar * Giner-Larza, E. M. _et al_. Anti-inflammatory triterpenes from _Pistacia terebinthus_ galls. _Planta Medica_ 68, 311–315 (2002). Article CAS PubMed
Google Scholar * Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. _Bioinformatics_ 27, 764–770 (2011). Article PubMed
PubMed Central Google Scholar * Vurture, G. W. _et al_. GenomeScope: fast reference-free genome profiling from short reads. _Bioinformatics_ 33, 2202–2204 (2017). Article CAS PubMed
PubMed Central Google Scholar * Koren, S. _et al_. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. _Genome Res._ 27, 722–736 (2017).
Article CAS PubMed PubMed Central Google Scholar * Pryszcz, L. P. & Gabaldón, T. Redundans: an assembly pipeline for highly heterozygous genomes. _Nucleic Acids Res._ 44, e113
(2016). Article PubMed PubMed Central Google Scholar * Hu, J. _et al_. An efficient error correction and accurate assembly tool for noisy long reads. Preprint at
https://doi.org/10.1101/2023.03.09.531669 (2023). * Vaser, R., Sović, I., Nagarajan, N. & Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. _Genome Res._
27, 737–746 (2017). Article CAS PubMed PubMed Central Google Scholar * Walker, B. J. _et al_. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly
improvement. _PLoS One_ 9, e112963 (2014). Article ADS PubMed PubMed Central Google Scholar * Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler
transform. _Bioinformatics_ 25, 1754–1760 (2009). Article CAS PubMed PubMed Central Google Scholar * Burton, J. N. _et al_. Chromosome-scale scaffolding of de novo genome assemblies
based on chromatin interactions. _Nat. Biotechnol._ 31, 1119–1125 (2013). Article CAS PubMed PubMed Central Google Scholar * Servant, N. _et al_. HiC-Pro: an optimized and flexible
pipeline for Hi-C data processing. _Genome Biol._ 16, 1–11 (2015). Article Google Scholar * Gavrilov-Zimin, I. A., Stekolshchikov, A. V. & Gautam, D. C. General trends of chromosomal
evolution in Aphidococca (Insecta, Homoptera, Aphidinea+Coccinea). _Comp. Cytogenet._ 9, 335–422 (2015). Article PubMed PubMed Central Google Scholar * Krzywinski, M. _et al_. Circos: An
information aesthetic for comparative genomics. _Genome Res._ 19, 1639–1645 (2009). Article CAS PubMed PubMed Central Google Scholar * Xu, Z. & Wang, H. LTR_FINDER: an efficient
tool for the prediction of full-length LTR retrotransposons. _Nucleic Acids Res._ 35, W265–W268 (2007). Article PubMed PubMed Central Google Scholar * Price, A. L., Jones, N. C. &
Pevzner, P. A. De novo identification of repeat families in large genomes. _Bioinformatics_ 21, i351–i358 (2005). Article CAS PubMed Google Scholar * Hoede, C. _et al_. PASTEC: an
automatic transposable element classification tool. _PLoS One_ 9, e91929 (2014). Article ADS PubMed PubMed Central Google Scholar * Chen, N. Using Repeat Masker to identify repetitive
elements in genomic sequences. _Curr. Protoc. Bioinformatics_ 5, 4.10.11–14.10.14 (2004). Article Google Scholar * Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of
repetitive elements in eukaryotic genomes. _Mobile_. _DNA_ 6, 11 (2015). Google Scholar * Stanke, M. & Waack, S. Gene prediction with a hidden Markov model and a new intron submodel.
_Bioinformatics_ 19, ii215–ii225 (2003). Article PubMed Google Scholar * Keilwagen, J. _et al_. Using intron position conservation for homology-based gene prediction. _Nucleic Acids Res._
44, e89 (2016). Article PubMed PubMed Central Google Scholar * Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. _Nat. Methods_ 12,
357–360 (2015). Article CAS PubMed PubMed Central Google Scholar * Pertea, M. _et al_. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. _Nat.
Biotechnol._ 33, 290–295 (2015). Article CAS PubMed PubMed Central Google Scholar * Haas, B. & Papanicolaou, A. TransDecoder (find coding regions within transcripts). _Google
Scholar_ (2016). * Tang, S., Lomsadze, A. & Borodovsky, M. Identification of protein coding regions in RNA transcripts. _Nucleic Acids Res._ 43, e78 (2015). Article PubMed PubMed
Central Google Scholar * Haas, B. J. _et al_. Improving the _Arabidopsis_ genome annotation using maximal transcript alignment assemblies. _Nucleic Acids Res._ 31, 5654–5666 (2003).
Article CAS PubMed PubMed Central Google Scholar * Haas, B. J. _et al_. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced
Alignments. _Genome Biol._ 9, R7 (2008). Article PubMed PubMed Central Google Scholar * Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment
search tool. _J. Mol. Biol._ 215, 403–410 (1990). Article CAS PubMed Google Scholar * Conesa, A. _et al_. Blast2GO: a universal tool for annotation, visualization and analysis in
functional genomics research. _Bioinformatics_ 21, 3674–3676 (2005). Article CAS PubMed Google Scholar * Griffiths-Jones, S. _et al_. Rfam: annotating non-coding RNAs in complete
genomes. _Nucleic Acids Res._ 33, D121–D124 (2005). Article CAS PubMed Google Scholar * Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes
in genomic sequence. _Nucleic Acids Res._ 25, 955–964 (1997). Article CAS PubMed PubMed Central Google Scholar * _NCBI Sequence Read Archive_
https://identifiers.org/ncbi/insdc.sra:SRR16046963 (2023). * _NCBI Sequence Read Archive_ https://identifiers.org/ncbi/insdc.sra:SRR23999325 (2023). * _NCBI Sequence Read Archive_
https://identifiers.org/ncbi/insdc.sra:SRR16046964 (2023). * _NCBI Sequence Read Archive_ https://identifiers.org/ncbi/insdc.sra:SRR23999326 (2023). * _NCBI Sequence Read Archive_
https://identifiers.org/ncbi/insdc.sra:SRR16046961 (2023). * _NCBI Sequence Read Archive_ https://identifiers.org/ncbi/insdc.sra:SRR23999323 (2023). * _NCBI Sequence Read Archive_
https://identifiers.org/ncbi/insdc.sra:SRR16046962 (2023). * _NCBI Sequence Read Archive_ https://identifiers.org/ncbi/insdc.sra:SRR23999324 (2023). * Institute of Zoology, Chinese Academy
of Sciences. _GenBank_ https://identifiers.org/insdc.gca:GCA_032441835.1 (2023). * Institute of Zoology, Chinese Academy of Sciences. _GenBank_
https://identifiers.org/insdc.gca:GCA_032441825.1 (2023). * Xu, S. The genome annotation files of galling aphids _Slavum lentiscoides_ and _Chaetogeoica ovagalla_. _Figshare._
https://doi.org/10.6084/m9.figshare.25602348.v1 (2024). * Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at
https://doi.org/10.48550/arXiv.1303.3997 (2013). * Li, H. _et al_. The sequence alignment/map format and SAMtools. _Bioinformatics_ 25, 2078–2079 (2009). Article PubMed PubMed Central
Google Scholar * Parra, G., Bradnam, K. & Korf, I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. _Bioinformatics_ 23, 1061–1067 (2007). Article CAS PubMed
Google Scholar * Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy
orthologs. _Bioinformatics_ 31, 3210–3212 (2015). Article PubMed Google Scholar Download references ACKNOWLEDGEMENTS All authors thank Baocheng Guo for his suggestion on data mining. The
work was supported by the National Natural Science Foundation of China (No. 32030014), the Youth Innovation Promotion Association of Chinese Academy of Sciences (No. 2020087), and the Key
Collaborative Research Program of the Alliance of International Science Organizations (Grant No. ANSO-CR-KP-2020-04). AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Key Laboratory of
Zoological Systematics and Evolution, Institute of Zoology, Chinese Academy of Sciences, Beijing, China Shifen Xu, Liyun Jiang, Zhengting Zou, Ming Zou, Gexia Qiao & Jing Chen * College
of Life Sciences, University of Chinese Academy of Sciences, Beijing, China Gexia Qiao Authors * Shifen Xu View author publications You can also search for this author inPubMed Google
Scholar * Liyun Jiang View author publications You can also search for this author inPubMed Google Scholar * Zhengting Zou View author publications You can also search for this author
inPubMed Google Scholar * Ming Zou View author publications You can also search for this author inPubMed Google Scholar * Gexia Qiao View author publications You can also search for this
author inPubMed Google Scholar * Jing Chen View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS J.C., G.Q. and L.J. designed the research. G.Q.
and L.J. identified voucher specimens. S.X. conducted karyotype experiments and all data analyses. Z.Z. and M.Z. assisted in genome assembly and annotation. J.C. guided all data analyses.
S.X. and J.C. wrote the manuscript and all authors contributed to revisions. CORRESPONDING AUTHORS Correspondence to Gexia Qiao or Jing Chen. ETHICS DECLARATIONS COMPETING INTERESTS The
authors declare no competing interests. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional
affiliations. SUPPLEMENTARY INFORMATION FIGURE S1 RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits
use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the
Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated
otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds
the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and
permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Xu, S., Jiang, L., Zou, Z. _et al._ Two chromosome-level genome assemblies of galling aphids _Slavum lentiscoides_ and _Chaetogeoica
ovagalla_. _Sci Data_ 11, 803 (2024). https://doi.org/10.1038/s41597-024-03653-x Download citation * Received: 26 April 2024 * Accepted: 15 July 2024 * Published: 20 July 2024 * DOI:
https://doi.org/10.1038/s41597-024-03653-x SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative