Play all audios:
ABSTRACT Nonalcoholic fatty liver disease (NAFLD) is a global health problem that is associated with various metabolic disorders. Telmisartan is a potential treatment for NAFLD due to its
ability to improve insulin sensitivity and decrease hepatic fat accumulation via modulation of PPARγ, and to suppress hepatic fibrosis by blocking angiotensin II receptors. However, the
underlying mechanisms of action of telmisartan have yet to be fully elucidated. In the present study, diabetic nonalcoholic steatohepatitis (NASH) mice (STAM mice) received daily
administrations of telmisartan for 6 weeks to assess the improvements in NASH. Hepatic transcriptome analyses revealed that the amelioration of NASH likely occurred through the regulation of
inflammatory- and fibrosis-related gene responses. An integrated network analysis including transcriptional and non-transcriptional genes regulated by telmisartan showed that the NAFLD
pathway is interconnected with the dysregulated RAS-PPAR-NFκB pathways. The downstream targets of PPARα, PPARδ, and RELA in this network significantly overlapped with telmisartan-induced
differentially expressed genes (DEGs), which were verified in palmitate-treated Hepa1c1c7 cell line. This transcriptome approach accompanied with cell-based molecular analyses provided the
opportunity to understand the fundamental molecular mechanisms underpinning the therapeutic effects of telmisartan, and will contribute to the establishment of a novel pharmacological
treatment for NASH patients. SIMILAR CONTENT BEING VIEWED BY OTHERS NETWORK PHARMACOLOGY-BASED INVESTIGATION OF THE PHARMACOLOGICAL MECHANISMS OF DIOSGENIN IN NONALCOHOLIC STEATOHEPATITIS
Article Open access 26 March 2025 FLUOROFENIDONE ATTENUATES CHOLINE-DEFICIENT, L-AMINO ACID-DEFINED, HIGH-FAT DIET-INDUCED METABOLIC DYSFUNCTION-ASSOCIATED STEATOHEPATITIS IN MICE Article
Open access 21 March 2025 A GUT MICROBIAL METABOLITE OF LINOLEIC ACID AMELIORATES LIVER FIBROSIS BY INHIBITING TGF-Β SIGNALING IN HEPATIC STELLATE CELLS Article Open access 03 November 2023
INTRODUCTION NAFLD is a global health problem with a prevalence of approximately 30% in Western countries1, and a rapidly increasing prevalence (with a trend towards a younger onset) in
Asian countries2. NAFLD is highly associated with metabolic disorders such as obesity, insulin resistance, type 2 diabetes mellitus, dyslipidemia, and hypertension3. Additionally, NAFLD
covers a broad spectrum of pathological abnormalities ranging from simple steatosis and NASH to advanced fibrosis and cirrhosis4. Furthermore, NASH is recognized as a significant risk factor
for hepatocellular carcinoma (HCC)5,6. A decade ago, it was proposed that NASH developed due to hepatic steatosis followed by the production of gut-derived endotoxins7. More recently, it
was proposed that numerous factors act in concert to induce NASH, including genetic predisposition, abnormal lipid metabolism, oxidative stress, lipotoxicity, mitochondrial dysfunction,
altered production of cytokines and adipokines, gut dysbiosis, and endoplasmic reticulum stress3. However, the pathogenesis of NASH has yet to be fully elucidated. Transcriptional profiling
studies with cohorts stratified based on histological liver parameters have demonstrated that several genes involved in the Wnt pathway, metabolism, cellular proliferation and extracellular
matrix (ECM) organization are dysregulated during the progression of NAFLD8,9. Additionally, an elegant study by Lefebvre _et al_.4, which investigated NASH disease activity using whole
genome profiling, revealed that gastric bypass, which is a surgical procedure that effectively improves NASH, significantly normalizes ECM homeostasis-associated genes. Thus, transcriptomic
investigations have elucidated the genetic contributors to NAFLD progression, and also provided an opportunity to establish novel pharmacological and/or medical treatment options.
Pharmacological agents, such as PPARγ activators, lipid-lowering agents, cytoprotective agents, and antioxidants have been used to treat NASH patients10. However, no optimal therapeutic
strategy has yet been established; thus, there is a need for novel NASH treatment modalities. Previous studies have suggested that the renin-angiotensin system (RAS) may play a critical role
in the progression of NAFLD, because activation of this system potentiates the accumulation of triglycerides, decreases hepatic fatty acid oxidation, alters very low-density lipoprotein
secretion, and increases _de novo_ lipogenesis in the liver11. Additionally, the RAS-mediated activation of hepatic stellate cells results in the acquisition of a myofibroblast-like
phenotype12. Taken together, these findings indicate that suppression of the RAS may be a potentially effective treatment for NAFLD. Telmisartan is an angiotensin II receptor (AGTR1)
antagonist used for the management of hypertension, which is the principle effector of RAS. Recently, it was demonstrated that telmisartan is a bifunctional molecule that activates PPARγ and
blocks angiotensin II receptors13. This unique feature allows telmisartan to improve insulin sensitivity and decrease hepatic fat accumulation via the modulation of PPARγ, as well as
suppress hepatic fibrosis by blocking angiotensin II receptors14,15. Clinical trials have shown that telmisartan improves fibrosis and the NAFLD activity score (NAS) in patients with NASH or
NAFLD, and thus has beneficial effects on fatty liver patients16,17. However, the molecular mechanisms of telmisartan, and the interaction between the RAS and PPAR, have yet to be fully
investigated. In the present study, telmisartan efficiently prevented the development of NASH in STAM mice. Additionally, hepatic transcriptomic analyses revealed that the amelioration of
NASH likely occurred via regulation of inflammatory- and fibrosis-related responses, and an integrated analysis of transcriptional and non-transcriptional genes regulated by telmisartan
identified cross-talk between angiotensin-PPAR-NFκB pathways that could contribute to the effects of telmisartan on NASH. This alternative approach to assessing the transcriptome accompanied
with the cell-based molecular analyses provided the opportunity to elucidate the underlying molecular mechanisms of the therapeutic effects of telmisartan and will contribute to the
establishment of novel pharmacological treatments for NASH patients. RESULTS TELMISARTAN-INDUCED AMELIORATION OF NASH IN STAM MICE The pharmacological effects of telmisartan were evaluated
in STAM mice from the steatosis stage (6 weeks of age) to the fibrosis stage (12 weeks of age). After 6 weeks of treatment, the bodyweights of the vehicle and telmisartan-treated mice did
not differ significantly (19.4 ± 3.2 and 19.5 ± 2.3 g, respectively; _p_ = 0.4963). In blood chemistry analyses, hypertension-related parameters including plasma triglyceride (TG) and
low-density lipoprotein (LDL) were significantly reduced (_p_ < 0.05), and high-density lipoprotein (HDL) was significantly increased (_p_ = 0.0007) by telmisartan (Table 1). The
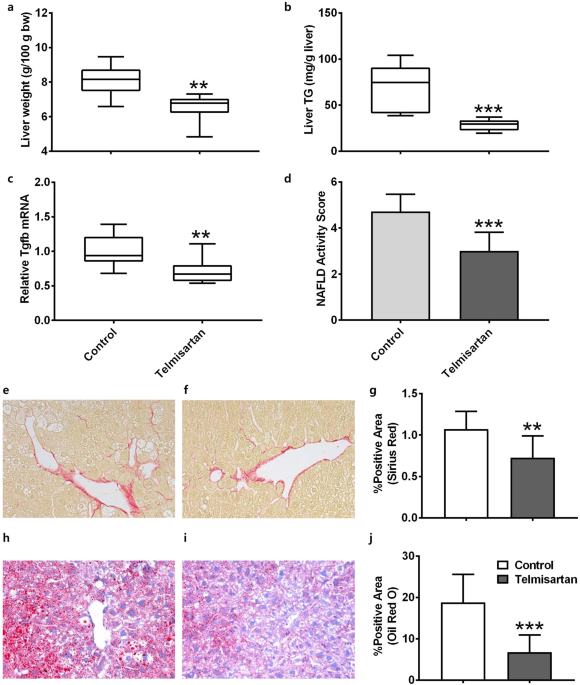
liver/bodyweight ratios of the vehicle and telmisartan-treated mice were 8.07 ± 0.92 and 6.49 ± 0.83 g/100 g bodyweight, respectively, which indicates that telmisartan significantly reduced
relative liver weight compared to the vehicle (_p_ = 0.027; Fig. 1a). Consistent with the liver/bodyweight ratio, the liver TG levels of the vehicle and telmisartan-treated mice were 67.6 ±
25.8 and 28.1 ± 6.2 mg/g liver, respectively, which shows that liver TG levels were significantly reduced by telmisartan compared to vehicle (_p_ = 0.0003; Fig. 1b). The relative mRNA levels
of fibrosis-related _Tgfb_ in the vehicle and telmisartan groups were 1.00 ± 0.23 and 0.72 ± 0.19, respectively, which indicates that telmisartan significantly decreased _Tgfb_ expression
(_p_ = 0.015; Fig. 1c). Histological examinations revealed that the telmisartan-treated mice exhibited reduction of liver steatosis, lobular inflammation as well as hepatocyte ballooning
compared to vehicle control. The NAS values in the vehicle and telmisartan groups were 4.71 ± 0.76 and 3.00 ± 0.82, respectively, which indicates that telmisartan significantly reduced the
NAS relative to the vehicle (_p_ = 0.0008; Fig. 1d, Supplementary Table 1). Furthermore, the percentages of Sirius red-positive areas in the vehicle and telmisartan groups were 1.07 ± 0.22
and 0.73 ± 0.26, respectively, which shows that telmisartan significantly reduced the degree of liver fibrosis compared to vehicle (_p_ = 0.01; Fig. 1e–g). Oil red O-positive areas in the
vehicle and telmisartan groups were 18.77 ± 6.81% and 6.76 ± 4.17%, respectively, which indicates that the presence of vesicular fat in telmisartan-treated liver tissues was significantly
reduced (_p_ = 0.0009; Fig. 1h–j). Taken together, these findings indicate that 6 weeks of daily treatment with telmisartan efficiently prevented fibrosis and lipid accumulation in the liver
and ameliorated NASH in STAM mice. TRANSCRIPTIONAL GENES-REGULATED BY TELMISARTAN Transcriptomic analyses of the liver tissues from randomly selected mice (three per group) were performed
to identify the differentially expressed transcripts due to telmisartan. After normalization, a total of 31,873 probes with signals common to all samples were subjected to hierarchical
clustering; the liver transcriptomes of the vehicle and telmisartan-treated groups exhibited distinct clusters (Fig. 2a, Supplementary Table 2). Overall, 69 DEGs exhibited a significant
change of at least 1.2-fold up- or down-regulation due to telmisartan compared to vehicle (_p_ < 0.05). Of these DEGs, 21 were up-regulated and 48 were down-regulated by telmisartan (Fig.
2b, Supplementary Table 3). To validate the microarray result, the expression levels of the down-regulated genes by telmisartan were verified by quantitative RT-PCR (qRT-PCR), those
included ankyrin repeat and SOCS box containing 13 (_Asb13_), intercellular adhesion molecule 1 (_Icam1_), _Jun_, monoacylglycerol O-acyltransferase 1 (_Mogat1_), polo like kinase 3
(_Plk3_), and Serglycin (_Srgn_). qRT-PCR confirmed the consistent reduction of the mRNA levels of these genes by telmisartan compared to vehicle control in the liver tissues (Fig. 2c). The
up- and down-regulated genes were separately applied to functional enrichment using Gene Set Enrichment Analysis [GSEA; false discovery rate (FDR) _q_ < 0.05] to clarify whether the
molecular functions were activated or inhibited by telmisartan. There were no functions enriched with up-regulated genes. In the contrary, several inflammatory- and fibrosis-related
functions were down-regulated by telmisartan, included TNFα signaling via NFκB (_q_ = 4.99E-06), allograft rejection (_q_ = 1.65E-03), the IFNγ response (_q_ = 3.93E-02), and
epithelial-mesenchymal transition (EMT; _q_ = 3.94E-02; Fig. 2d, Supplementary Table 4). A total of 15 down-regulated genes were enriched in these functions (Fig. 2e), and these genes were
considered to be the differentially expressed as well as functionally enriched genes that were transcriptionally regulated by telmisartan. Subsequently, these genes were used as
transcriptional genes regulated by telmisartan to construct the regulatory network. NON-TRANSCRIPTIONAL GENES REGULATED BY TELMISARTAN The identification of transcriptional-regulated genes
due to the perturbation of non-transcription factors is challenging and could have a serious negative impact on the construction of a precise pathway and/or network for understanding the
molecular mechanisms of drugs. To identify non-transcriptional-regulated genes associated with telmisartan, 40 telmisartan-induced DEGs were queried in the Connectivity Map (CMap). CMap
analyses were conducted using the Touchstone signature dataset, which was generated by pharmacological perturbation for identification of drugs and their target genes, as well as by paired
genetic perturbation through knockdown or over-expression. As shown in Table 2, CMap analyses revealed that the telmisartan-induced DEGs were connected with irbesartan (angiotensin receptor
antagonist, connectivity score: 99.98), benazepril (angiotensin converting enzyme inhibitor, connectivity score: 99.96), clofibrate (PPAR receptor agonist, connectivity score: 99.96),
parthenolide (NFκB pathway inhibitor and adiponectin receptor agonist, connectivity score: 99.88), etomoxir (carnitine palmitoyltransferase inhibitor, connectivity score: 99.87), and
carbacyclin (PPARδ receptor activator, connectivity score: 99.85) (Fig. 3a, Supplementary Table 5). The associated protein targets of the drugs retrieved from CMap were as follows: AGTR1 and
JUN for irbesartan, ACE for benazepril, PPARα and LPL for clofibrate, IκBKB and RELA for parthenolide, CPT1A and CPT1B for etomoxir, and PPARδ for carbacyclin. Next, the paired genetic
perturbagens that were transcriptionally similar to the telmisartan-induced DEGs in CMap were investigated (Fig. 3b, Supplementary Table 6); the strongly paired genes included _FOXP3_
(connectivity score by knockdown/over-expression: 98.92/−97.31), _CCL2_ (connectivity score: 91.16/−98.32), _ADRB2_ (connectivity score: 99.73/−97.86), and _BCL10_ (connectivity score:
97.23/−96.73). This approach identified 11 target genes of pharmacological perturbagens and 10 genetic perturbagens; these genes were regarded as non-transcriptional-regulated genes by
telmisartan. Subsequently, these genes were used to construct the regulatory network of telmisartan in combination with the transcriptional-regulated genes. TELMISARTAN-INDUCED REGULATORY
NETWORK FOR IMPROVEMENT OF NASH The transcriptional and non-transcriptional genes regulated by telmisartan were assessed using STRING and a protein-protein interaction (PPI) network was
constructed. A PPI network was generated with 19 protein nodes encoded by telmisartan-regulated genes (Fig. 4a). Of the nodes in the network, six were functionally enriched in the NAFLD
pathway (_q_ = 2.81E-07), which was located at the center of this network and interconnected with the PPAR signaling pathway (_q_ = 2.81E-07), TNFα signaling pathway (_q_ = 6.18E-08), and
angiotensin signaling pathway. To understand whether this network influenced the expression of telmisartan-induced DEGs, the associations of the transcription factors in the network with the
DEGs were investigated using ChIP-X enrichment analysis (ChEA). Interestingly, PPARα, PPARδ, and RELA were significantly associated with 6, 14, and 7 telmisartan-induced DEGs, respectively
(Fig. 4b, Supplementary Table 7), which implies that these genes were down-regulated by PPARα, PPARδ, and RELA binding, respectively. Therefore, these three transcription factors appeared to
play the essential role as network regulators, exerting an influence on the telmisartan-induced STRING network. To verify whether the telmisartan modulates PPARα, PPARδ and RELA to
influence NAFLD network, the protein levels of these transcription factors and the mRNA levels of their downstream target genes were evaluated in Hepa1c1c7 cells. As shown in Fig. 5a,
telmisartan alone did not change the protein levels of PPARα, PPARδ and RELA in Hepa1c1c7 cells. In the contrary, palmitate slightly decreased PPARα and PPARδ; furthermore, telmisartan in
palmitate-treated Hepa1c1c7 cells significantly increased the levels of PPARα and PPARδ (_p_ < 0.05), and decreased RELA (_p_ < 0.01; Supplementary Fig. 1). Coincidently, the mRNA
levels of the downstream target genes of these transcription factors were inversely correlated with the protein levels of the transcription factors (Fig. 5b). The mRNA levels of _Asb13_,
_Icam1_ and _Jun_ genes, which were predicted to be down-regulated by PPARα and/or PPARδ in ChEA, were significantly increased by palmitate (_p_ < 0.05); however, as PPARα and PPARδ
increased by co-treatment of telmisartan, the mRNA levels of these genes were significantly decreased (_p_ < 0.05). In contrast, palmitate-induced increase of _Srgn_ mRNA was
significantly reduced (_p_ < 0.01) as the RELA decreased by telmisartan. Taken together, the findings of the constructed regulatory network in conjunction with the transcriptional and
non-transcriptional genes identified as being regulated by telmisartan indicate that the AGTR1-mediated angiotensin pathway interacted with the PPAR-NFκB signaling pathway, and that the
NAFLD pathway was down-regulated through PPARα, PPARδ, and RELA as transcriptional regulators to ameliorate NASH in STAM mice. DISCUSSION Activation of the PPARγ signaling pathway improves
insulin resistance, dyslipidemia, adipokine secretion, inflammation, cell proliferation and hepatic steatosis18,19. Blockade of the RAS pathway improves oxidative stress, inflammation, and
cell proliferation, and also leads to improvements in hepatic fibrosis20. Thus, modulation of the PPARγ and RAS pathways would likely confer significant advantages for NASH patients. Based
on previous research and the present findings, the bifunctional pharmacological activities of telmisartan, as an angiotensin II receptor antagonist and PPARγ partial agonist, significantly
ameliorate NAFLD activity, alter hepatic fat accumulation, and improve hepatic fibrosis. Other PPARγ agonists, such as pioglitazone, also attenuate hepatic steatosis, inflammation, and
fibrosis to a degree similar to that of telmisartan, but also affect systemic characteristics such as lipid metabolism and body weight such that rats treated with pioglitazone exhibit
increases in body weight and subcutaneous fat. In contrast, telmisartan is associated with mild loss of body weight accompanied by marked decreases in subcutaneous inguinal and epididymal
visceral fat18. These features differentiate telmisartan and pioglitazone in terms of therapeutic efficacy for NASH patients. Telmisartan also exerts dissociable effects on hepatic steatosis
and energy expenditure to those of ordinary angiotensin II receptor antagonists, such as valsartan13. These differences may be due to differences in chemical structure. Conventional
angiotensin II receptor antagonists in clinical use today are biphenyl tetrazole derivatives, whereas telmisartan is a non-tetrazole derivative that resembles pioglitazone21. This unique
structural feature appears to grant telmisartan the ability to regulate both carbohydrate and lipid metabolism, which led to improvements in fatty liver and reductions in triglyceride
levels, without weight gain, in the present study. Gene expression profiling analysis at different stages of various diseases represents a sensitive method for elucidating the molecular
processes that underlie pathological states. The reversal of NASH and fibrosis by telmisartan seen in the present study suggests that at least some of the transcriptomic alterations were
reversible, which allowed for the identification of putative target genes that may potentially be effective against pathological processes. The unsupervised hierarchical clustering of the
present hepatic transcriptome data revealed a clear dissociation between the vehicle and telmisartan treatments. Telmisartan induced subtle changes in global gene expression levels, and this
may have been due to the adaptive nature of the pathological response to gene expression4. However, it is noteworthy that telmisartan appeared to reduce the activities of essential genes
that are associated with the inflammatory response and hepatic fibrosis. Of these genes, _Icam1_ is important in the inflammatory process of livers with NASH, and thus may be a useful marker
for the diagnosis of NASH22,23. Increased expression of _Irs2_ is associated with steatohepatitis in obese individuals24 and seems to be a critical regulator of the synthesis and oxidation
of fatty acids in the livers of rats with NASH25. Additionally, _Onecut1_ and _Cd74_ were down-regulated in telmisartan-treated liver tissues but, in contrast to the present findings, these
genes were inhibited during hepatic steatosis induction26,27. Thus, whether the activities of _Onecut1_ and _Cd74_ are regulated differentially depending on the NASH induction conditions
needs to be clarified. It is well known that gene activity is regulated by transcription, RNA processing, post-translational modification, and/or PPIs. In the present study, several
telmisartan-regulated genes were identified at the transcription level and their cellular and molecular functions were shown to attenuate NASH progression. However, it was challenging to
understand the therapeutic mechanisms underlying the effects of telmisartan on NASH, and the interaction between angiotensin II receptors and the PPAR signaling pathway, using only
transcriptional-regulated genes. The target genes of transcription factors can be efficiently revealed by knockout expression profiling because transcriptional-regulated genes would be
directly regulated by perturbations in transcription factors28. However, it would be extremely difficult to investigate the activity of non-transcriptional-regulated genes by gene expression
profiling29. Thus, CMap provides a significant opportunity to elucidate disease-drug and/or drug-drug connections at the transcription level due to massive pharmacological or genetic
perturbations that, in turn, may aid in the identification of the modes of action of certain candidate drugs, and repurpose existing drugs for alternative indications30,31. Furthermore,
pharmacological or genetic perturbation expression profiles in CMap could provide insight into the transcriptional responses of genes that are regulated in a non-transcriptional manner. To
investigate this hypothesis, the transcriptional genes that were dysregulated by telmisartan were queried in CMap. Surprisingly, CMap revealed that well-known telmisartan associated
signaling molecules such as AGTR1, ACE, PPARα, and PPARδ exhibited a pharmacological connection, while other interacting molecules had a genetic connection to telmisartan-induced DEGs; these
had never been previously identified by conventional gene expression analyses. Thus, in combination with the transcriptional genes regulated by telmisartan, the telmisartan network that
ameliorated NASH in the livers of STAM mice, and which harbored the NAFLD pathway that was interconnected with the RAS and the PPAR-NFκB signaling pathways, was successfully generated.
Furthermore, retrospective ChEAs of transcription factors in this network implied that PPARα, PPARδ, and RELA likely play important role in differentially control of target gene activity
during the reversal of NASH by telmisartan. AGTR1 has been known to activate the NFκB machinery through MAPK/ERK pathway32,33. RelA, p65 subunit of NFκB, also has been known to down-regulate
the PPARα34 and PPARδ35 activity by inhibitory binding. It implies that RelA would be a master regulator of the core transcriptional circuit by telmisartan and mediate RAS-PPAR pathway.
Interestingly, PPARγ was not identified among the transcriptional and non-transcriptional genes regulated by telmisartan. PPAR isoforms display tissue-specific expressions. For example,
PPARγ is dominant in adipose tissue, whereas PPARδ is found in various tissues and has been identified in high levels in skeletal muscle36. The potential agonism of PPARγ by telmisartan was
suggested by PPRE-dependent transcription in cells that were similar to pioglitazone-treated cells14,18. However, telmisartan also induces anti-fibrotic and anti-obesity effects through
PPARδ-dependent pathways37,38 and enacts anti-hepatic fibrosis and anti-dyslipidemic effects through PPARα-dependent pathways4,39. Therefore, telmisartan appears to inhibit NASH progression
by PPARγ activation as well as by partial activation of PPARα and PPARδ through AGTR1 antagonism, resulting in down-regulation of genes related with inflammation and fibrosis in STAM mice.
There are some potential pitfalls to consider. First, STAM mice used in this study represent diabetic, male NASH in human, and it does not explain non-diabetic NASH or female NASH patients.
Long-term HFD without streptozotocin (STZ) treatment could be an alternative model for non-diabetic NASH with variable onset and characteristics to improve the clinical relevance of the
study. Moreover, STAM mice are known as hypertension insensitive and maybe inappropriate to evaluate the anti-hypertensive effects by telmisartan40; however, telmisartan effectively
controlled the strong hypertension predictors including plasma TG, LDL and HDL at dose level used in this study. Second, there was no comparison between wild type and STAM mice to evaluate
the effect of telmisartan. However, the _in vitro_ experiment with Hepa1c1c7 cells demonstrated that telmisartan-induced amelioration of NASH would be steatosis/steatochepatitis-specific and
there would be low possibility to observe the transcriptional effect of telmisartan in wild type animals. Third, CMap has limited coverage of perturbagens. Although the coverage has been
dramatically increased in the next generation CMap with L1000 platform30, it is still retrospective and novel targets which do not have matched perturbagens would be difficult to be
connected with biological states. This limitation needs to be improved by expanding the coverage of genetic perturbagens with appropriate test system. In conclusion, telmisartan efficiently
prevented the development of NASH in STAM mice. Additionally, hepatic transcriptomic analyses revealed that the amelioration of NASH possibly occurred via the regulation of inflammatory- and
fibrosis-related responses. Integrated analyses of transcriptional and non-transcriptional genes regulated by telmisartan identified cross-talk between the angiotensin-PPAR-NFκB signaling
pathways, which could have contributed to the pharmacological effects of telmisartan on NASH. This alternative transcriptomic approach accompanied with the cell-based molecular analyses
provided the opportunity to understand the fundamental molecular mechanisms underlying the therapeutic effects of telmisartan, and will contribute to the establishment of novel
pharmacological treatments for patients with NASH. METHODS ANIMAL EXPERIMENT NASH was induced in C57BL/6 J male mice, as described previously41. Briefly, at 2 days after birth, the mice
received a single subcutaneous injection of 200 μg STZ (Sigma, St. Louis, MO, USA). Then, after 4 weeks of age, they received 60 kcal% fat HFD32 chow (CLEA Japan Inc., Tokyo, Japan) _ad
libitum_ and were assigned to receive either vehicle or telmisartan. Telmisartan (Sigma) was dissolved in 0.5% (v/v) carboxymethyl cellulose (Sigma) and orally administered (5 mg/kg/day) to
the mice from 6 to 12 weeks of age (n = 7 per group). At termination, liver tissues were obtained from the mice and stored until further analysis. This study was approved by the
Institutional Animal Care and Use Committees of Seoul National University (SNU-170912-22) and was conducted in accordance with the approved guidelines. BIOCHEMICAL ANALYSIS Blood chemistry
was analyzed using automated chemistry analyzer (Hitachi, Tokyo, Japan), those included ALT, TBIL, plasma TG, TCHO, HDL, LDL, BUM, CREA, and GLU according to Park _et al_.42. Total lipid in
the liver tissues was extracted using a 2:1 chloroform:methanol solution (v/v) and the TG contents were measured with a Triglyceride E-test kit (Wako, Osaka, Japan) according to the
manufacturer’s instructions. QRT-PCR To measure the expression levels of _Tgfb_ and ribosomal protein lateral stalk subunit P0 (_36B4_) genes, total RNA was extracted from the liver tissues
using RNAiso (Takara, Tokyo, Japan) and cDNA was prepared with Moloney murine leukemia virus reverse transcriptase (Invitrogen, Carlsbad, CA, USA). cDNA was amplified with the ABI 7700
sequence‐detector system (Applied Biosystems, Foster City, CA, USA) using a set of primers and probes that corresponded to _Tgfb_ and _36B4_ (endogenous control)41. To measure the expression
levels of _Asb13_, _Icam1_, _Jun_, _Mfge_, _Mogat1_, _Plk3_, and _Srgn_ genes, total RNA from the liver tissues (six per group) or cells was extracted using a RNeasy Mini kit (Qiagen)
according to the manufacturer’s instructions. cDNA was prepared using SuperScript III Reverse Transcriptase (Invitrogen) and qRT-PCR was performed on a StepOnePlus Real-Time PCR System
(Applied Biosystems) with Power SYBR Green PCR Master Mix (Applied Biosystems). Gene expression levels were analyzed by ΔΔCT method using _Gapdh_ as an internal control. Primers referred
from PrimerBank (https://pga.mgh.harvard.edu/primerbank/index.html)43 were summarized in Supplementary Table 8. HISTOLOGICAL ANALYSES Water-soluble glycol and resin-embedded liver sections
were cut at a thickness of 5 μm, air-dried, fixed in acetone, and then stained with a hematoxylin and eosin solution (Wako). The NAS was evaluated semi-quantitatively, as described
previously44, the degree of liver fibrosis was assessed with Sirius-red staining, and the presence of vesicular fat in the liver tissues was confirmed using oil red O. MICROARRAY ANALYSES
Total RNA was extracted from the frozen liver tissues with a RNeasy mini kit (Qiagen). Following quantitative and qualitative evaluations performed with BioAnalyzer (Agilent, Santa Clara,
CA, USA), RNA samples with an RNA integrity number (RIN) ≥ 6.7 and A260/A280 values ≥ 1.88 were subjected to cDNA synthesis, performed with the GeneChip WT cDNA synthesis and amplification
kit (Applied Biosystems). Next, the cDNA was fragmented and biotin-labeled using GeneChip WT terminal labeling kit (Applied Biosystems), and approximately 5.5 μg of labeled cDNA was
hybridized to the Affymetrix GeneChip Mouse Gene 2.0 ST Array (Affymetrix, Santa Clara, CA, USA) at 45 °C for 16 h. The hybridized arrays were scanned on a GCS3000 Scanner (Affymetrix) and
all data analyses were performed with the GeneChip Command Console Software (Affymetrix). All data were normalized using the robust multi-array average (RMA) approach and hierarchical
clustering of the expressed probes was performed using GenePattern (https://genepattern.broadinstitute.org). The distance between clusters was computed with Pearson correlations and global
gene expression profiling was conducted in triplicate for the vehicle control and telmisartan treatment groups. TELMISARTAN GENE SIGNATURE Telmisartan-induced DEGs in the liver tissues were
identified using a fold change cutoff of 1.2; these genes were compared to the vehicle control group using independent _t_-tests with a _p_-value of 0.05 taken to indicate statistical
significance. Next, the telmisartan-induced DEGs were used as seeds to generate a telmisartan gene signature. First, the DEGs were analyzed using GSEA software
(http://software.broadinstitute.org/gsea)45 to determine the molecular mechanisms of action of telmisartan. Next, the telmisartan-induced DEGs were computationally overlapped with the
Molecular Signature Database (MSigDb) using a FDR _q_-value cutoff of 0.05; DEGs enriched in a certain molecular signature were regarded as transcriptional-regulated genes by telmisartan.
Next, the transcriptional connections between telmisartan-induced DEGs and chemical and genetic perturbagens were assessed with CMap (https://clue.io)30, which is a catalog of
transcriptional responses following pharmacological or genetic (knock-down by shRNA or over-expression by transgenesis) perturbations of cell lines; the pharmacological (and their target
genes) or genetic perturbagens associated with telmisartan-induced DEGs were considered non-transcriptional-regulated genes by telmisartan. These transcriptional- and
non-transcriptional-regulated gene sets were used as the telmisartan gene signature. REGULATORY NETWORKS A system-wide understanding of the cellular functions induced by telmisartan was
obtained using the STRING protein-protein association network database (https://string-db.org)46. A telmisartan gene signature generated by GSEA and CMap analyses was used to construct both
the experimental and predicted interactions of the signature molecules using a confidence level of 0.7. To verify the experimental relevance of the network, transcription factors in the
constructed network were associated with telmisartan-induced DEGs using the ChEA gene set library47, which is a comprehensive resource for targets of transcription factors in various cell
types, mammalian organisms, and microarray platforms, as determined by ChIP-seq. CELL EXPERIMENT Hepa1c1c7 murine hepatoma cells (Korean Cell Line Bank, Korea) were maintained in αMEM medium
without nucleosides (ThermoFisher Scientific, Waltham, MA, USA) with 10% FBS (ThermoFisher Scientific) at 37 °C, 5% CO2. After reaching 70% confluency, Hepa1c1c7 cells were treated with 0.2
mM palmitate (Sigma) for 9 h to induce lipotoxicity. The concentration of palmitate was determined by cell viability assay which did not cause significant cell death. Telmisartan at 10 µM
was treated for 24 h after palmitate treatment according to Li _et al_.36. WESTERN BLOTTING Total protein collected from cells was subject to dodecyl sulfate-poly acrylamide electrophoresis,
and then transferred to nitrocellulose membranes. Membranes were incubated with anti-PPARδ, PPARα (1:500) and anti-RELA (1:250) antibodies (ThermoFisher Scientific). Expression levels of
proteins were analyzed by ImageJ (https://imagej.nih.gov/ij/) using ACTB as an internal control. STATISTICAL ANALYSES All animal data were analyzed with Student _t_-tests or Mann-Whitney U
tests depending on the homogeneity of variance of the data. All statistical analyses were performed using Prism software (ver. 7.03; GraphPad Software, Inc., San Diego, CA, USA) with
_p_-values < 0.05 considered to indicate statistical significance. All measurements are reported as means ± standard deviation (SD). DATA AVAILABILITY The dataset generated during the
current study are available in the Gene Expression Omnibus repository, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE120937. REFERENCES * Musso, G., Gambino, R. & Cassader, M.
Non-alcoholic fatty liver disease from pathogenesis to management: an update. _Obesity reviews: an official journal of the International Association for the Study of Obesity_ 11, 430–445,
https://doi.org/10.1111/j.1467-789X.2009.00657.x (2010). Article CAS Google Scholar * Fan, J. G. & Farrell, G. C. Epidemiology of non-alcoholic fatty liver disease in China. _Journal
of hepatology_ 50, 204–210, https://doi.org/10.1016/j.jhep.2008.10.010 (2009). Article PubMed Google Scholar * Caligiuri, A., Gentilini, A. & Marra, F. Molecular Pathogenesis of NASH.
_International journal of molecular sciences_ 17, https://doi.org/10.3390/ijms17091575 (2016). * Lefebvre, P. _et al_. Interspecies NASH disease activity whole-genome profiling identifies a
fibrogenic role of PPARalpha-regulated dermatopontin. _JCI insight_ 2, https://doi.org/10.1172/jci.insight.92264 (2017). * Angulo, P. Long-term mortality in nonalcoholic fatty liver
disease: is liver histology of any prognostic significance? _Hepatology_ 51, 373–375, https://doi.org/10.1002/hep.23521 (2010). Article PubMed PubMed Central Google Scholar * Adams, L.
A. _et al_. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. _Gastroenterology_ 129, 113–121 (2005). Article Google Scholar * Day, C. P. &
James, O. F. Steatohepatitis: a tale of two “hits”? _Gastroenterology_ 114, 842–845 (1998). Article CAS Google Scholar * Moylan, C. A. _et al_. Hepatic gene expression profiles
differentiate presymptomatic patients with mild versus severe nonalcoholic fatty liver disease. _Hepatology_ 59, 471–482, https://doi.org/10.1002/hep.26661 (2014). Article CAS PubMed
Google Scholar * Murphy, S. K. _et al_. Relationship between methylome and transcriptome in patients with nonalcoholic fatty liver disease. _Gastroenterology_ 145, 1076–1087,
https://doi.org/10.1053/j.gastro.2013.07.047 (2013). Article CAS PubMed PubMed Central Google Scholar * Mummadi, R. R., Kasturi, K. S., Chennareddygari, S. & Sood, G. K. Effect of
bariatric surgery on nonalcoholic fatty liver disease: systematic review and meta-analysis. _Clinical gastroenterology and hepatology: the official clinical practice journal of the American
Gastroenterological Association_ 6, 1396–1402, https://doi.org/10.1016/j.cgh.2008.08.012 (2008). Article Google Scholar * Wu, Y. _et al_. Lipid disorder and intrahepatic renin-angiotensin
system activation synergistically contribute to non-alcoholic fatty liver disease. _Liver international: official journal of the International Association for the Study of the Liver_ 36,
1525–1534, https://doi.org/10.1111/liv.13131 (2016). Article CAS Google Scholar * Pereira, R. M., dos Santos, R. A., da Costa Dias, F. L., Teixeira, M. M. & Simoes e Silva, A. C.
Renin-angiotensin system in the pathogenesis of liver fibrosis. _World journal of gastroenterology_ 15, 2579–2586 (2009). Article CAS Google Scholar * Sugimoto, K. _et al_. Telmisartan
but not valsartan increases caloric expenditure and protects against weight gain and hepatic steatosis. _Hypertension_ 47, 1003–1009, https://doi.org/10.1161/01.HYP.0000215181.60228.f7
(2006). Article CAS PubMed Google Scholar * Benson, S. C. _et al_. Identification of telmisartan as a unique angiotensin II receptor antagonist with selective PPARgamma-modulating
activity. _Hypertension_ 43, 993–1002, https://doi.org/10.1161/01.HYP.0000123072.34629.57 (2004). Article CAS PubMed Google Scholar * Yokohama, S. _et al_. Inhibitory effect of
angiotensin II receptor antagonist on hepatic stellate cell activation in non-alcoholic steatohepatitis. _World journal of gastroenterology_ 12, 322–326 (2006). Article CAS Google Scholar
* Hirata, T. _et al_. Effect of Telmisartan or Losartan for Treatment of Nonalcoholic Fatty Liver Disease: Fatty Liver Protection Trial by Telmisartan or Losartan Study (FANTASY).
_International journal of endocrinology_ 2013, 587140, https://doi.org/10.1155/2013/587140 (2013). Article CAS PubMed PubMed Central Google Scholar * Alam, S. _et al_. Effect of
telmisartan on histological activity and fibrosis of non-alcoholic steatohepatitis: A 1-year randomized control trial. _Saudi journal of gastroenterology: official journal of the Saudi
Gastroenterology Association_ 22, 69–76, https://doi.org/10.4103/1319-3767.173762 (2016). Article Google Scholar * Fujita, K. _et al_. Telmisartan, an angiotensin II type 1 receptor
blocker, controls progress of nonalcoholic steatohepatitis in rats. _Digestive diseases and sciences_ 52, 3455–3464, https://doi.org/10.1007/s10620-007-9741-4 (2007). Article CAS PubMed
Google Scholar * Fuentes, E., Guzman-Jofre, L., Moore-Carrasco, R. & Palomo, I. Role of PPARs in inflammatory processes associated with metabolic syndrome (Review). _Molecular medicine
reports_ 8, 1611–1616, https://doi.org/10.3892/mmr.2013.1714 (2013). Article CAS PubMed Google Scholar * Schupp, M., Janke, J., Clasen, R., Unger, T. & Kintscher, U. Angiotensin type
1 receptor blockers induce peroxisome proliferator-activated receptor-gamma activity. _Circulation_ 109, 2054–2057, https://doi.org/10.1161/01.CIR.0000127955.36250.65 (2004). Article CAS
PubMed Google Scholar * Kurtz, T. W. & Pravenec, M. Antidiabetic mechanisms of angiotensin-converting enzyme inhibitors and angiotensin II receptor antagonists: beyond the
renin-angiotensin system. _Journal of hypertension_ 22, 2253–2261 (2004). Article CAS Google Scholar * Soderberg, C. _et al_. Microvesicular fat, inter cellular adhesion molecule-1 and
regulatory T-lymphocytes are of importance for the inflammatory process in livers with non-alcoholic steatohepatitis. _APMIS: acta pathologica, microbiologica, et immunologica Scandinavica_
119, 412–420, https://doi.org/10.1111/j.1600-0463.2011.02746.x (2011). Article PubMed Google Scholar * Ito, S., Yukawa, T., Uetake, S. & Yamauchi, M. Serum intercellular adhesion
molecule-1 in patients with nonalcoholic steatohepatitis: comparison with alcoholic hepatitis. _Alcoholism, clinical and experimental research_ 31, S83–87,
https://doi.org/10.1111/j.1530-0277.2006.00292.x (2007). Article PubMed Google Scholar * Rametta, R. _et al_. Increased insulin receptor substrate 2 expression is associated with
steatohepatitis and altered lipid metabolism in obese subjects. _International journal of obesity_ 37, 986–992, https://doi.org/10.1038/ijo.2012.181 (2013). Article CAS PubMed Google
Scholar * Matsunami, T. _et al_. Regulation of synthesis and oxidation of fatty acids by adiponectin receptors (AdipoR1/R2) and insulin receptor substrate isoforms (IRS-1/−2) of the liver
in a nonalcoholic steatohepatitis animal model. _Metabolism: clinical and experimental_ 60, 805–814, https://doi.org/10.1016/j.metabol.2010.07.032 (2011). Article CAS Google Scholar * van
Breda, S. G. J. _et al_. Integrative omics data analyses of repeated dose toxicity of valproic acid _in vitro_ reveal new mechanisms of steatosis induction. _Toxicology_ 393, 160–170,
https://doi.org/10.1016/j.tox.2017.11.013 (2018). Article CAS PubMed Google Scholar * Heinrichs, D. _et al_. Protective role of macrophage migration inhibitory factor in nonalcoholic
steatohepatitis. _FASEB journal: official publication of the Federation of American Societies for Experimental Biology_ 28, 5136–5147, https://doi.org/10.1096/fj.14-256776 (2014). Article
CAS Google Scholar * Reimand, J., Vaquerizas, J. M., Todd, A. E., Vilo, J. & Luscombe, N. M. Comprehensive reanalysis of transcription factor knockout expression data in Saccharomyces
cerevisiae reveals many new targets. _Nucleic acids research_ 38, 4768–4777, https://doi.org/10.1093/nar/gkq232 (2010). Article CAS PubMed PubMed Central Google Scholar * Ray, J. C.,
Tabor, J. J. & Igoshin, O. A. Non-transcriptional regulatory processes shape transcriptional network dynamics. _Nature reviews. Microbiology_ 9, 817–828,
https://doi.org/10.1038/nrmicro2667 (2011). Article CAS PubMed PubMed Central Google Scholar * Subramanian, A. _et al_. A Next Generation Connectivity Map: L1000 Platform and the First
1,000,000 Profiles. _Cell_ 171, 1437–1452 e1417, https://doi.org/10.1016/j.cell.2017.10.049 (2017). Article CAS PubMed PubMed Central Google Scholar * Shin, E., Lee, Y. C., Kim, S. R.,
Kim, S. H. & Park, J. Drug Signature-based Finding of Additional Clinical Use of LC28-0126 for Neutrophilic Bronchial Asthma. _Scientific reports_ 5, 17784,
https://doi.org/10.1038/srep17784 (2015). Article ADS CAS PubMed PubMed Central Google Scholar * Van Beek, M. _et al_. Bcl10 links saturated fat overnutrition with hepatocellular NF-kB
activation and insulin resistance. _Cell reports_ 1, 444–452, https://doi.org/10.1016/j.celrep.2012.04.006 (2012). Article CAS PubMed PubMed Central Google Scholar * Capettini, L. S.
_et al_. Role of renin-angiotensin system in inflammation, immunity and aging. _Current pharmaceutical design_ 18, 963–970 (2012). Article CAS Google Scholar * Rual, J. F. _et al_.
Towards a proteome-scale map of the human protein-protein interaction network. _Nature_ 437, 1173–1178, https://doi.org/10.1038/nature04209 (2005). Article ADS CAS PubMed Google Scholar
* Jove, M., Laguna, J. C. & Vazquez-Carrera, M. Agonist-induced activation releases peroxisome proliferator-activated receptor beta/delta from its inhibition by palmitate-induced
nuclear factor-kappaB in skeletal muscle cells. _Biochimica et biophysica acta_ 1734, 52–61, https://doi.org/10.1016/j.bbalip.2005.02.002 (2005). Article CAS PubMed Google Scholar * Li,
L. _et al_. Telmisartan improves insulin resistance of skeletal muscle through peroxisome proliferator-activated receptor-delta activation. _Diabetes_ 62, 762–774,
https://doi.org/10.2337/db12-0570 (2013). Article CAS PubMed PubMed Central Google Scholar * Mikami, D. _et al_. Telmisartan activates endogenous peroxisome proliferator-activated
receptor-delta and may have anti-fibrotic effects in human mesangial cells. _Hypertension research: official journal of the Japanese Society of Hypertension_ 37, 422–431,
https://doi.org/10.1038/hr.2013.157 (2014). Article CAS Google Scholar * He, H. _et al_. Telmisartan prevents weight gain and obesity through activation of peroxisome
proliferator-activated receptor-delta-dependent pathways. _Hypertension_ 55, 869–879, https://doi.org/10.1161/HYPERTENSIONAHA.109.143958 (2010). Article CAS PubMed Google Scholar * Yin,
S. N. _et al_. Telmisartan increases lipoprotein lipase expression via peroxisome proliferator-activated receptor-alpha in HepG2 cells. _Endocrine research_ 39, 66–72,
https://doi.org/10.3109/07435800.2013.828741 (2014). Article CAS PubMed Google Scholar * Yoshimine, Y. _et al_. Hepatic expression of the Sptlc3 subunit of serine palmitoyltransferase is
associated with the development of hepatocellular carcinoma in a mouse model of nonalcoholic steatohepatitis. _Oncology reports_ 33, 1657–1666, https://doi.org/10.3892/or.2015.3745 (2015).
Article CAS PubMed Google Scholar * Fujii, M. _et al_. A murine model for non-alcoholic steatohepatitis showing evidence of association between diabetes and hepatocellular carcinoma.
_Medical molecular morphology_ 46, 141–152, https://doi.org/10.1007/s00795-013-0016-1 (2013). Article CAS PubMed Google Scholar * Park, Y. J., Rim, J. H., Yim, J., Lee, S. G. & Kim,
J. H. Effects of two types of medical contrast media on routine chemistry results by three automated chemistry analyzers. _Clinical biochemistry_ 50, 719–725,
https://doi.org/10.1016/j.clinbiochem.2017.02.023 (2017). Article CAS PubMed Google Scholar * Spandidos, A., Wang, X., Wang, H. & Seed, B. PrimerBank: a resource of human and mouse
PCR primer pairs for gene expression detection and quantification. _Nucleic acids research_ 38, D792–799, https://doi.org/10.1093/nar/gkp1005 (2010). Article CAS PubMed Google Scholar *
Kleiner, D. E. _et al_. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. _Hepatology_ 41, 1313–1321, https://doi.org/10.1002/hep.20701 (2005).
Article PubMed Google Scholar * Subramanian, A. _et al_. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. _Proceedings of the
National Academy of Sciences of the United States of America_ 102, 15545–15550, https://doi.org/10.1073/pnas.0506580102 (2005). Article ADS CAS PubMed PubMed Central Google Scholar *
Szklarczyk, D. _et al_. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. _Nucleic acids research_ 45, D362–D368,
https://doi.org/10.1093/nar/gkw937 (2017). Article CAS PubMed Google Scholar * Kuleshov, M. V. _et al_. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update.
_Nucleic acids research_ 44, W90–97, https://doi.org/10.1093/nar/gkw377 (2016). Article CAS PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS This study was
supported by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI) funded by the Ministry for Health and Welfare of Korea
(HI14C1135 to C.S.C. and J.P.) and LG Chem Inc. (1403-20170093 to J.P.). AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * LG Chem R&D Campus, Daejeon, Korea Jung Gyu Park & Hee Dong
Park * Graduate School of International Agricultural Technology, Seoul National University, Seoul, Korea Jong Soo Mok, Tae Sub Park & Joonghoon Park * Institute of Green Bio Science and
Technology, Seoul National University, Seoul, Korea Young In Han, Tae Sub Park & Joonghoon Park * College of pharmacy, Seoul National University, Seoul, Korea Keon Wook Kang * Korea
mouse metabolic phenotyping center, Lee Gil Ya cancer and diabetes institute, Gachon University School of Medicine, Seongnam-si, Republic of Korea Cheol Soo Choi * Endocrinology, Internal
Medicine, Gachon University Gil Medical Center, Seongnam-si, Republic of Korea Cheol Soo Choi Authors * Jung Gyu Park View author publications You can also search for this author inPubMed
Google Scholar * Jong Soo Mok View author publications You can also search for this author inPubMed Google Scholar * Young In Han View author publications You can also search for this author
inPubMed Google Scholar * Tae Sub Park View author publications You can also search for this author inPubMed Google Scholar * Keon Wook Kang View author publications You can also search for
this author inPubMed Google Scholar * Cheol Soo Choi View author publications You can also search for this author inPubMed Google Scholar * Hee Dong Park View author publications You can
also search for this author inPubMed Google Scholar * Joonghoon Park View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS J.G.P., H.D.P. and
J.P. conceived the study. J.G.P., J.S.M., Y.I.H., T.S.P., K.W.K., C.S.C. and J.P. designed and conducted the experiments. J.P. wrote the manuscript with input from other authors.
CORRESPONDING AUTHOR Correspondence to Joonghoon Park. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION PUBLISHER’S NOTE: Springer
Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION RIGHTS AND PERMISSIONS OPEN
ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format,
as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third
party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the
article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright
holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Park, J.G., Mok, J.S., Han, Y.I. _et
al._ Connectivity mapping of angiotensin-PPAR interactions involved in the amelioration of non-alcoholic steatohepatitis by Telmisartan. _Sci Rep_ 9, 4003 (2019).
https://doi.org/10.1038/s41598-019-40322-1 Download citation * Received: 18 July 2018 * Accepted: 30 January 2019 * Published: 08 March 2019 * DOI: https://doi.org/10.1038/s41598-019-40322-1
SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy
to clipboard Provided by the Springer Nature SharedIt content-sharing initiative