Play all audios:
ABSTRACT Hyperoxia-induced lung injury plays a key role in the development of bronchopulmonary dysplasia (BPD), characterized by inflammatory injury and impaired lung development in preterm
infants. Although BPD is a predictor of poor neurodevelopmental outcomes, currently it is uncertain how lung injury contributes to brain injury in preterm infants. Extracellular vesicles
(EVs) are a heterogeneous group of cell-derived membranous structures that regulate intercellular and inter-organ communications. Gasdermin D (GSDMD) has emerged as a key executor of
inflammasome-mediated cell death and inflammation. In this study, we utilized a neonatal rat model of BPD to assess if hyperoxia stimulates lung release of circulating EVs and if these EVs
induce lung and brain injury. We found that hyperoxia-exposed rats had elevated numbers of plasma-derived EVs compared to rats maintained in room air. These EVs also had increased cargos of
surfactant protein C, a marker of type II alveolar epithelial cells (AEC), and the active (p30) form of GSDMD. When these EVs were adoptively transferred into normal newborn rats via
intravenous injection, they were taken up both by lung and brain tissues. Moreover, EVs from hyperoxic animals induced not only the pathological hallmarks of BPD, but also brain inflammatory
injury in recipient rats, as well as inducing cell death in cultured pulmonary vascular endothelial cells and neural stem cells (NSC). Similarly, hyperoxia-exposed cultured AEC-like cells
released EVs that also contained increased GSDMD-p30 and these EVs induced pyroptotic cell death in NSC. Overall, these data indicate that hyperoxia-activated circulating EVs mediate a lung
to brain crosstalk resulting in brain injury and suggest a mechanism that links lung injury and neurodevelopmental impairment in BPD infants. SIMILAR CONTENT BEING VIEWED BY OTHERS UMBILICAL
CORD BLOOD EXOSOMES FROM VERY PRETERM INFANTS WITH BRONCHOPULMONARY DYSPLASIA AGGRAVATE LUNG INJURY IN MICE Article Open access 27 May 2023 STEM CELL-DERIVED EXTRACELLULAR VESICLES: A
POTENTIAL INTERVENTION FOR BRONCHOPULMONARY DYSPLASIA Article Open access 09 September 2024 ADIPONECTIN AMELIORATES HYPEROXIA-INDUCED LUNG ENDOTHELIAL DYSFUNCTION AND PROMOTES ANGIOGENESIS
IN NEONATAL MICE Article 25 March 2021 INTRODUCTION Extremely premature infants born at less than 28 weeks of gestational age (GA) are at great risk of having multi-organ injury and
developmental abnormalities that predominately involve the lung and brain1,2. The lungs of these infants are immature at birth, which predisposes them to respiratory failure and in need of
oxygen therapy1,3. However, life sustaining oxygen therapy can cause lung inflammation that leads to lung structure damage and ultimately to bronchopulmonary dysplasia (BPD)1, 3. The brains
of these infants are also immature and are prone to injurious stimuli such as oxygen toxicity and inflammation. Consequently, these infants are at greater risk of developing both short-term
and long-term neurological complications2. Thus, BPD survivors not only suffer from pulmonary dysfunction but are also often complicated with long-term neurodevelopmental impairment (NDI).
There is mounting clinical evidence that even in the absence of catastrophic brain injuries, severe BPD is an independent risk factor for adverse neurodevelopmental outcomes4,5,6. However,
in spite of recent advances in neonatal intensive care and extensive research, the extent to which BPD contributes to NDI is uncertain, and there is no effective therapy for either the
prevention or treatment of these conditions. Extracellular vesicles (EVs) are a heterogeneous group of cell-derived nano-sized membranous structures, including exosomes and microvesicles,
that are increasingly recognized as signal mediators of intercellular as well as inter-organ communications, in health and disease7,8. EVs are released by a variety of living cells in
response to inflammation, oxidative stress, and cell activation or damage and have been isolated from most bodily fluids including bronchoalveolar lavage fluid (BALF)9 and blood and
cerebrospinal fluid (CSF)10. EVs carry complex cargos of proteins, lipids and nucleic acids, and their cargo composition is highly dependent on the biological function of the parental
cells7,8,9,10. Being membranous, EVs protect their cargo from the extracellular environment, thus allowing for safe transport and delivery of their intact cargo to near or distant target
cells, resulting in modification of the target cell’s gene expression, signaling pathways and overall function7,8,9,10. Recently, lung epithelial cell-secreted EVs isolated from BALF have
been shown to play a role in regulating inflammatory responses in adult lung diseases11,12. In preterm infants with severe BPD, increased numbers of EVs were detected in their tracheal
aspirates, and interestingly, the majority of these EVs were found to be of epithelial origin13. These data suggest that lung epithelial cells can release bioactive EVs into airspace fluid
upon inflammatory injury. Similarly, EVs contribute to a number of adult central nervous system disorders, and they can bidirectionally cross the blood brain barrier (BBB)14,15. Moreover, we
have recently reported that traumatic brain injury in adult patients and mice causes the release of EVs into the circulation that travel to the lung to cause acute lung injury, confirming
EV BBB transit and suggesting a brain to lung crosstalk16. However, to date, there are no clinical or pre-clinical studies that report lung epithelial cells releasing EVs into the
circulation that can cross the BBB and induce injury in the developing brain. Gasdermin D (GSDMD), a 53-kilodalton (kDa) cytosolic protein, was recently found to be a key executer of
pyroptosis, a form of inflammasome-mediated cell death17,18,19. Inflammasome activation by pathogens or host-derived danger signals leads to the activation of caspase-1 and
caspase-1-mediated cleavage of GSDMD releases the GSDMD’s 30-kDa N-terminal domain (p30) which oligomerizes in the cell membrane to form pores causing cellular swelling, membrane rupture and
cell death17,18,19. The pores additionally allow rapid release of active interleukin (IL)-1β and IL-18, resulting in secondary inflammation17,18,19. We have previously demonstrated a
critical role for the inflammasome pathway in experimental models of BPD20,21,22, and importantly, inhibition of GSDMD activation attenuates hyperoxia-induced lung and brain injury22.
Moreover, GSDMD-p30 has also recently been detected in serum exosomal microparticles from adult patients with sepsis and acute lung injury23, but it is presently unknown if exosomal GSDMD
plays a mechanistic role in the pathogenesis of BPD or brain injury in preterm infants. In the present study, we tested the hypothesis that hyperoxia-induced neonatal lung and brain injury
is mediated by circulating GSDMD-laden EVs released by alveolar type II epithelial cells (AEC) after their uptake by lung and brain tissues. We found supporting evidence for this hypothesis
using a combination of in vivo adoptive EV transfer experiments in neonatal rats and in vitro studies using cultured AEC, pulmonary vascular endothelial cells (PVEC) and neural stem cells
(NSC). RESULTS HYPEROXIA EXPOSURE STIMULATES THE RELEASE OF EVS WITH AN INCREASED CARGO OF SPC/GSDMD INTO THE CIRCULATION OF NEONATAL RATS To determine if hyperoxia stimulates lung
epithelial cells to release GSDMD-containing EVs into the circulation, we analyzed EV isolates from the plasma of neonatal rats maintained in room air (RA-EVs) or hyperoxia (O2-EVs) for 14
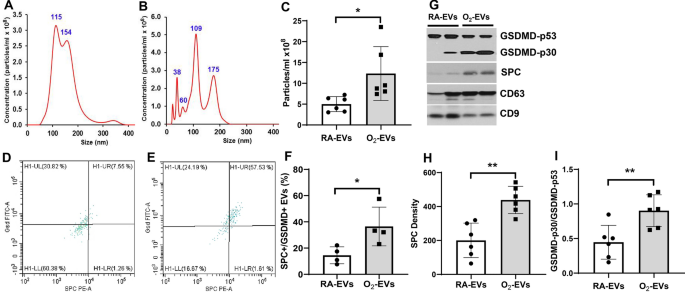
days. Nanosight tracking assay revealed that both RA-EVs (Fig. 1A) and O2-EVs (Fig. 1B) contained nanoparticles that were primarily of a 30–150 nm exosome size, with both peaking at 100–150
nm in diameter, but the nanoparticle concentrations of O2-EVs were 2.4-fold higher than RA-EVs (Fig. 1C, _P_ < 0.05). FACS analysis after capture on anti-tetraspanin (CD9, CD63 and CD81)
beads confirmed the exosome nature of the EVs7, 8 and demonstrated that the RA-EVs (Fig. 1D) contained a smaller population of SPC + /GSDMD + EVs compared to O2-EVs (Fig. 1E) (Fig. 1F,
RA-EVs 14.5 ± 6.5% vs. O2-EVs 36.5 ± 14.8%, _P_ < 0.05). Western blot analysis confirmed that O2-EVs contained twofold increased levels of SPC (Fig. 1G, H, O2-EVs 438.7 ± 80.5 vs. RA-EVs
200.3 ± 101.8, _P_ < 0.01) and activated GSDMD-p30 compared to RA-EVs, which contained primarily inactive GSDMD-p53 (Fig. 1G, I, O2-EVs 0.90 ± 0.23 vs. RA-EVs 0.45 ± 0.24, _P_ < 0.01)
(Supplemental Fig. 1G). Western blot analysis also confirmed that both RA-EVs and O2-EVs contained relatively equal amounts of exosomal markers, CD63 and CD9 tetraspanins, when the proteins
were loaded in equal amounts/sample (Fig. 1G). Thus, hyperoxia stimulates AEC to release circulatory EVs, with an increased cargo of active GSDMD which are characteristically exosomes based
on their size and high levels of multiple tetraspanin expression, although the presence of other nano or microparticles with similar characteristics cannot be ruled out. ADOPTIVELY
TRANSFERRED CIRCULATING EVS TRAVEL TO THE LUNG AND BRAIN OF NORMAL NEONATAL RATS As a prelude to determining if circulating EVs from hyperoxia-exposed rats can cause lung and brain injury,
we performed adoptive transfer experiments and examined their potential to traffic not only to the lung but also to the brain. RA-EVs or O2-EVs were labeled with tracking dyes and adoptively
transferred into normal neonatal rats by intravenous injection. As illustrated in Fig. 2A–C, both Exo-Glow labelled RA-EVs and O2-EVs rapidly distributed throughout the body and were
localized in the lungs and brain at 1 and 4 h after tail vein injection. Their uptake by the brain (Fig. 2D) and lung (Fig. 2E) tissues was further confirmed by ex vivo imaging at 4 h. To
determine if they are present in brain tissue for longer than 4 h, we similarly injected Dil-dye-labeled EVs and isolated EVs from the CSF and examined brain tissue sections 24 h later. We
detected fluorescent Dil signals in the brain tissue sections from both RA-EVs and O2-EVs injected rats but not from sham animals (Fig. 2F–H), and when compared to sham animals (Fig. 2I,
4.95 ± 0.57 × 107, n = 4 pooled of 3 CSF) animals that received either RA-EVs (Fig. 2J, 17.55 ± 1.9 × 107, n = 2 pooled of 3 CSF, _P_ < 0.01) or O2-EVs (Fig. 2K, 26.7 ± 16.6 × 107, n = 2
pooled of 3 CSF, _P_ < 0.05) had much higher concentrations of CSF EVs. Overall, these results confirm that hyperoxia-induced circulating EVs can cross the BBB and be taken up by brain
cells. ADOPTIVE TRANSFER OF CIRCULATING EVS FROM HYPEROXIA EXPOSED RATS INDUCES LUNG INFLAMMATORY INJURY IN NORMAL NEONATAL RATS We next investigated if adoptive transfer of circulating EVs
with increased cargo of GSDMD-p30 could cause inflammatory lung injury in normal neonatal rats. We found that when compared to RA-EVs injected rats (Fig. 3A), rats that received O2-EVs (Fig.
3B) had increased numbers of total leukocytes in their BALF (Fig. 3C, O2-EVs 57.7 ± 14.6 × 104 vs. RA-EVs 27.0 ± 13.8 × 104, _P_ < 0.01). Differential counts of macrophages, lymphocytes,
and neutrophils were similarly elevated in rats receiving O2-EVs when compared to rats receiving RA-EVs, accordingly, macrophages (Fig. 3D, O2-EVs 49.6 ± 17.1 × 104 vs. RA-EVs 25.9 ± 14.5 ×
104, _P_ < 0.05); lymphocytes (Fig. 3E, O2-EVs 330.2 ± 228.5 vs. RA-EVs 26.1 ± 13.7, _P_ < 0.01); and neutrophils (Fig. 3F, O2-EVs 4456.1 ± 383.7 vs. RA- EVs 385.7 ± 312.9, _P_ <
0.05). We further analyzed lung inflammation by measuring lung tissue gene expression of several inflammation-associated factors by qRT-PCR (Fig. 3G). The lungs of rats that received O2-EVs
had increased expression of the inflammatory mediators IL-18 (_P_ < 0.05), TNF-α (_P_ < 0.05), chemokine CXCL1 (_P_ < 0.01), and profibrotic factors CTGF (_P_ < 0.05) and TGF-β
(_P_ < 0.05). Thus, O2-EVs induce lung inflammatory injury. CIRCULATING EVS FROM HYPEROXIC RATS INHIBIT ALVEOLARIZATION AND VASCULAR DEVELOPMENT IN NORMAL NEONATAL RATS IN VIVO AND
DECREASE CELL PROLIFERATION AS WELL AS INCREASE CELL DEATH IN PVEC IN VITRO Impaired alveolarization and vascular development are hallmarks of BPD. Thus, we next evaluated alveolar and
vascular development in neonatal rats that received adoptive transfer of RA-EVs and O2-EVs. Microscopy of H&E stained lung tissue sections showed that rats injected with O2-EVs had
larger and more simplified alveolar structures as well as significantly decreased radial alveolar count (RAC) in comparison to rats that received RA-EVs (Fig. 4A–C, O2-EVs 17.6 ± 1.4 vs.
RA-EVs 20.7 ± 1.7, _P_ < 0.01). Moreover, vascular density was significantly decreased in rats injected with O2-EVs (Fig. 4D–F, O2-EVs 7.1 ± 0.7 vs. RA- EVs 8.8 ± 0.7, _P_ < 0.01).
These results confirm that hyperoxia causes the release of EVs capable of inducing BPD-like pathology. To further examine the effects of circulating EVs on lung vasculature, we treated
cultures of PVEC with RA-EVs and O2-EVs and evaluated cell proliferation by Ki67 immunofluorescent staining and cell death by TUNEL. We found that PVEC treated with O2-EVs had markedly
decreased proliferation indices compared to PVEC treated with RA-EVs (Fig. 4G–I, O2-EVs 10.8 ± 3.6% vs. RA-EVs 41.8 ± 9.9%, _P_ < 0.001). This inhibitory effect can at least be partially
attributed to increased cell death as we also observed significantly more TUNEL positive cells in PVEC cultures treated with O2-EVs than with RA-EVs (Fig. 4J–L, O2-EVs 10.9 ± 2.0% vs. RA-EVs
3.3 ± 1.3%, _P_ < 0.001). Thus O2-EVs exhibit inhibitory effects on the growth of lung vasculature in vivo and in vitro. ADOPTIVELY TRANSFERRED CIRCULATING EVS FROM HYPEROXIA EXPOSED
RATS INDUCE BRAIN INFLAMMATORY INJURY IN NORMAL NEONATAL RATS We further investigated if circulating EVs from hyperoxia-exposed rats can also cause brain inflammatory injury when adoptively
transferred into normal neonatal rats. First, we performed immunostaining for allograft-inflammatory-factor-1 (AIF-1), a marker for microglial cells, on brain tissue sections and found that
when compared to RA-EVs, O2-EVs greatly increased the presence of enlarged activated inflammatory microglial cells in the SVZ (Fig. 5A, B), SGZ (Fig. 5C, D) and cortex (Fig. 5E, F) of
recipient rats. We also performed qRT-PCR to assess gene expression of inflammatory mediators involved in microglial cell activation, namely: IL-1α, IL-18, TNF-α, fibronectin (FN1) and
platelet derived growth factor receptor beta (PDGFRβ), and we found that all of these factors were significantly higher in rats that received O2-EVs when compared to rats that received
RA-EVs (Fig. 5G, _P_ < 0.05). Moreover, when we further examined the brain sections for evidence of neural cell death by TUNEL assay we found a 3.3 fold increase in dead cells in the SVZ
of rats who received O2-EVs , compared to rats who received RA-EVs (Fig. 5H–J, O2-EVs 2.7 ± 1.29% vs. RA-EVs 0.8 ± 0.45%, _P_ < 0.01). These novel findings indicate that O2-EVs can cause
brain damage. CIRCULATING EVS FROM HYPEROXIA EXPOSED RATS DECREASE NSC PROLIFERATION AND INCREASE CELL DEATH IN VITRO To examine the effects circulating EVs had on the brain at a cellular
level, we treated cultures of NSC with RA-EVs and O2-EVs and found that O2-EVs decreased NSC proliferation by more than two-fold compared to RA-EVs (Fig. 6A–C, O2-EVs 18.5 ± 3.0% vs. RA-EVs
41.9 ± 5.4%, _P_ < 0.001). Furthermore, the observed growth inhibition may be mediated by cell death since NSC had increased cell death indices when cultured with O2-EVs, compared to
cells cultured with RA- EVs (Fig. 6D–F, O2-EVs 16.2 ± 3.9% vs. RA-EVs 3.2 ± 1.9%, _P_ < 0.001). These results suggest that the O2-EVs induced brain cell death observed in our in vivo
studies may be primarily occurring in NSC. HYPEROXIA STIMULATES MLE-12 CELLS TO RELEASE EVS WITH AN INCREASED CARGO OF GSDMD-P30 WHICH INDUCE NSC CELL DEATH IN VITRO In order to confirm our
in vivo finding that hyperoxia stimulated AEC to release EVs that contain an increased cargo of activated GSDMD, we examined the effect of hyperoxia on EV release from cultured MLE-12 cells,
an AEC-like cell line. We found EVs isolated from the supernatant media of cells cultured under room air (RA-MEVs) or hyperoxia (O2-MEVs) conditions were comparably sized, with peak
distributions of 100–150 nm, which is similar to our neonatal rat plasma EVs (Fig. 7A, B), but in contrast to our in vivo results, RA-MEVs and O2-MEVs had similar EV concentrations (data not
shown). However, like circulating EVs induced by hyperoxia, O2-MEVs when compared to RA-MEVs had minimal full-length GSDMD-p53 (Fig. 7C, D, O2-MEVs 17.3 ± 2.5 vs. RA-MEVs 255.2 ± 89.0, _P_
< 0.01) but contained an increase cargo of activated GSDMD-p30 (Fig. 7C, E, O2-MEVs 397.8 ± 17.4 vs. RA-MEVs 111.9 ± 41.8, _P_ < 0.001), and both RA-MEVs and O2-MEVs contained SPC as
well as EV exosomal marker CD9 (Fig. 7C), confirming their AEC lineage and EV nature. These results reinforce our in vivo conclusion that hyperoxia activation of AEC GSDMD results in AEC
release of GSDMD-p30 rich EVs into the circulation. To determine if O2-MEVs also induced increased NSC inflammasome-mediated cell death, as rat O2-EVs had, we treated NSC with MLE-12 cell
EVs. We found this to be the case, as when compared to RA-MEVs exposed cells (Fig. 7F), O2-MEVs increased pyroptotic cell death as observed appearance of “blebbed” cells (Fig. 7G), and as
measured an increased number of TUNEL positive cells (Fig. 7H–J, O2-MEVs 32.3 ± 2.7% vs. RA-MEVs 9.3 ± 3.7%, _P_ < 0.001) and an increased number of propidium iodide (PI) positive cells
(Fig. 7K–M, O2-MEVs 7.25 ± 1.25 vs. RA-MEVs 3.25 ± 0.95, _P_ < 0.01). These results suggest that circulatory GSDMD-rich EVs are responsible for in vivo neural cell death that leads to
brain injury in hyperoxia-exposed rodents. DISCUSSION BPD, characterized by inflammatory injury and impaired lung development, is the most common morbidity complicating preterm birth1,3, and
a predictor of poor neurodevelopmental outcomes4,5,6. Currently, it is unclear to what extent lung injury contributes to brain injury in preterm infants, and there are no effective
therapies for these two conditions. EVs have recently emerged as key regulators of cellular crosstalk by shuttling functional and signaling molecules to both neighboring and distal cells,
affecting physiological functions or pathological responses of the recipient cells7,8. Our previous studies have shown that the inflammasome-GSDMD cascade plays a critical role in a
hyperoxia-induced mouse model of BPD and brain injury22. In the present study, we have isolated nanoparticles from the plasma of neonatal rats that are considered to be EVs by MISEV2018
standards7 due to their extracellular nature, isolation method (commercial polymer precipitation kit), nanoparticle size, lipid membrane structure (Dil-dye labelling), general protein and
non-lipid content (BCA assay and ExoGlow-Vivo-EV labelling), and the presence of EV tetraspanin membrane protein markers characteristic of exosomes by both Western blot (CD9 and CD63) and
capture by anti-tetraspanin (CD9, CD63, and CD81)-conjugated magnetic beads prior to FACS analysis. Moreover, we demonstrated that hyperoxia-exposed neonatal rats had elevated levels of
plasma EVs containing an increased cargo of both the AEC marker, SPC, and GSDMD-p30 compared to room air-maintained controls. We also found that these EVs traveled not only to the lung but
also to the brain when adoptively transferred into the peripheral circulation of normal newborn rats. Importantly, adoptive transfer of O2-EVs induced not only the pathological hallmarks of
BPD but also led to brain inflammatory injury. We further showed that O2-EVs induced cell death in cultured PVEC and NSC. We also confirmed in vitro that hyperoxia stimulated AEC to release
GSDMD-p30-containing EVs into their culture media, and these EVs induced NSC pyroptotic death. Overall, these findings support a novel mechanism through which hyperoxia-activated circulating
EVs induces lung and brain injury and suggest that targeting this mechanism may have therapeutic value in preventing BPD-associated brain injury. Moreover, if elevated levels of circulating
SPC+/GSDMD+ EVs also occur in preterm infants, they could be used as a predictive biomarker of BPD and possibly poor neurodevelopmental outcome. One of the key findings of this study is the
discovery of an increased number of circulatory AEC-derived EVs in hyperoxia-exposed neonatal rats. While there are previously published studies showing the presence of hyperoxia-induced
EVs in BALF and serum of adult rodents, this report is the first linking circulating AEC-derived EVs to a hyperoxia-induced neonatal rat model of BPD. For example, two previous studies in
adult rodent models of hyperoxia-induced acute lung injury have shown that lung epithelial cells can release EVs and microvesicles into BALF or serum11,24. Another recent study found
tracheal aspirates from infants with severe BPD had increased numbers of, but smaller, exosomes compared with term controls13. This study also found increased exosomes in BALF from a
hyperoxia-induced mouse model of BPD. Interestingly, the majority of these exosomes were epithelial in origin, but this study did not evaluate whether there were increased plasma exosomes in
either the clinical or experimental BPD arm of the study. Thus, our study extends this report to show that hyperoxia stimulates AEC to release EVs with some exosomal characteristics into
the circulation using a rat BPD model. Additionally it also suggests that if elevated levels of SPC-positive AEC-derived EVs could be used as a novel biomarker of BPD, they would be easily
obtainable from plasma as opposed to invasive BALF sampling. Our results also demonstrated that the circulating EVs from hyperoxia-exposed neonatal rats had increased cargos of GSDMD,
particularly its pyroptosis-inducing inflammasome-activated GSDMD-p30 form. Our FACS data further showed that the increased GSDMD was primarily present in SPC+/GSDMD+ EVs, indicating that
the majority of the hyperoxia-induced EV GSDMD originated in AEC. Moreover, they suggest a novel mechanism for our recent observations that GSDMD activation is a part of the inflammasome
pathway activated by hyperoxia in the lung and brain of neonatal mice22. To our knowledge, this is the first study reporting increased lung epithelial-derived active GSDMD in circulating EVs
in experimental models of BPD. When combined with previous reports of microparticle GSDMD release by activated monocytes in vitro25 and GSDMD plasma microparticles in adult septic
patients23, they suggest that the activation of cellular GSDMD by inflammatory injury and its release in circulating EVs/microparticles may be common to many inflammatory diseases. While the
autocrine-like effect of intracellular inflammasome-activated GSDMD to induce cell death and secondary inflammation is certain17,18,19, whether EV GSDMD can function in a paracrine-like or
endocrine-like manner to induce neighboring cell or distant organ injury is currently unclear. However, such bystander signaling mechanisms, although not directly attributed to GSDMD, are
well documented for both localized and systemic EV-mediated radiation-induced cellular death26. But, our adoptive transfer experiments do suggest that endocrine-like mechanisms are possible
as they revealed that intravenously injected RA-EVs and O2-EVs were both taken up by the lung and brain tissues of recipient normal neonatal rats. More importantly, we found that adoptive
transfer of O2-EVs into normal newborn rats produced lung changes similar to those seen in hyperoxia-induced rodent models of BPD20,21,22. These changes were not seen in their counterparts
who received RA-EVs. Histologically, the changes seen in O2-EVs recipients were decreased alveolarization and vascular density, two hallmarks of BPD, suggesting the adoptive transfer of
O2-EVs was responsible for the creation of a BPD phenotype in these otherwise normal newborn rats. These histological changes were accompanied by evidence of BPD-characteristic lung
inflammation as increased counts for total leukocytes, lymphocytes, macrophages and neutrophils were found in BALF, and increased gene expression of the BPD-associated cytokines IL-18,
TNF-α, CXCL1, CTGF and TGF-β1 was found in lung tissue samples. IL-18 and TNF-α are pro-inflammatory cytokines previously reported to be associated with BPD or pulmonary hypertension in
preterm infants or experimental models of BPD27,28,29,30. Interestingly, IL18 is activated by the inflammasome, and its secretion is dependent on GSDMD-p30 pore formation17,18,19. CXCL1 is a
pivotal chemokine that regulates neutrophil recruitment during infection31 and chronic inflammatory responses characterized by increased CXCL1 and TNF-α have been observed in hyperoxia
models of BPD32 as well as in neonatal lung injury induced by bromine33. While CTGF and TGF-β are fibrosis-inducing cytokines previously shown by us and others to play a causal role in
inducing abnormal alveolarization and fibrosis in the developing lungs in general and in hyperoxia-induced rodent models of BPD in particular34,35,36. Taken together, these results suggest
that upregulation of these important inflammatory and fibrotic mediators contributes to the induction of BPD pathology by O2-EVs in the receipt rats. Although many cell types in the lung
could be the targets of circulating EVs, we focused our investigation on PVEC because PVEC would have direct contact with the circulating nanoparticles, and their injury can lead to abnormal
vascular development in BPD37. Utilizing in vitro PVEC cultures, we demonstrated that O2-EVs decreased cell proliferation and increased cell death. We speculate that these in vitro vascular
endothelial effects may translate into poor vascular growth as well as endothelial cell damage, leading to vascular leakage that would allow inflammatory cells to infiltrate into the
alveolar airspaces, as noted in our in vivo results. Although it has been long known that BPD is an independent risk factor for adverse neurological outcomes in preterm infants4,5,6, the
underlining mechanisms connecting lung injury to brain injury are poorly understood. However, our previous findings show that inflammasome-activated GSDMD plays a critical role in the brain
injury seen in a mouse model of BPD22, and circulating EVs are known to play a key role in inter-organ communications. Thus, we additionally investigated if adoptive transfer of
hyperoxia-activated circulating EVs could induce inflammatory injury in the brain of normal neonatal rats, as they did to the lung. Our investigation yielded pivotal data that support a
novel endocrine-like mechanism in which O2-EVs induce the same brain inflammatory injury seen in hyperoxia-exposed mice. We first demonstrated that intravenous injection of both labeled
RA-EVs and O2-EVs can be detected in the brains of recipient rats by in vivo and ex vivo imaging. Furthermore, we detected increased CSF EVs in both RA-EVs and O2-EVs injected rats. These
results are not surprising since previous reports show that circulating EVs can cross the BBB and are taken up by brain cells14,15,38. Next, and more importantly, we discovered that O2-EVs
are biologically active as they induced brain inflammatory injury. O2-EVs activated microglial cells, and this activation was accompanied, as we had seen in the lung, by increased expression
of inflammatory mediators, including IL-1α, IL-18, TNF-α, fibronectin and PDGFRβ. Previous studies have shown that IL-1α, IL-18 and TNF-α are important mediators of neonatal inflammatory
brain injury39,40, and fibronectin has been shown to promote several pro-inflammatory functions of microglia41,42. In the brain, PDGFRβ expressing cells are pericytes, the mural cells of
blood microvessels, and these cells play an important role in relaying inflammatory signals from the circulatory system to neurons43. We further found that O2-EVs induced cell death in the
SVZ of recipient rats, which with the SGZ are the main sites of postnatal neurogenesis in mammalian brains44,45,46, both of which we previously found to be sites of cell death in
hyperoxia-exposed mice22. The developing SVZ and SGZ are enriched in NSC that have the ability to proliferate and generate transient neural progenitor cell clusters (NPC), which then further
differentiate into astrocytes, oligodendrocytes, and neurons44,45,46. Since the NPC lie in close proximity to blood vessels, and circulating mediators are known to greatly affect their
biological functions47,48,49, they would be logical O2-EVs targets. Our in vitro results support this idea as both O2-EVs and O2-MEVs induced cell death in cultured NSC. Moreover, the O2-EVs
and O2-MEVs induced NSC cell death was found to be pyroptotic in nature and thus likely mediated by EV GSDMD-p30. Collectively, these data reveal a novel in vivo paradigm that links
hyperoxic lung injury, lung-derived circulating EVs, and inflammatory brain injury together. Our study has limitations. One of the limitations is that we did not investigate whether other
cell types, such as inflammatory cells and vascular endothelial cells contribute to increased circulating EV GSDMD levels seen in our hyperoxia-induced rat model of BPD. Thus, future studies
are needed to determine if any of the circulating GSDMD positive EVs found in hyperoxia-injured neonatal rats are of inflammatory cell-derived or endothelial cell-derived. Another
limitation of this study is that we did not explore whether the EV-mediated effects we observed on the lung and brain are totally GSDMD-dependent. Thus, we plan to utilize recently described
GSDMD knockout mice50 and pharmacologic GSDMD inhibitory approaches similar to those we have previously used22 to definitively prove that GSDMD is required for circulating EV-induced lung
and brain injury in neonatal rats. Alternatively, studies directed at inhibiting EV formation or cellular uptake will also be important in understanding the mechanisms by which EV GSDMD
plays a pivotal role in BPD-associated brain injury. In conclusion, in this study we demonstrate that hyperoxia stimulates AEC to release GSDMD-p30 laden EVs into the circulation of neonatal
rats, and that adoptive transfer of these circulating EVs into normal neonatal rats induces the pathological hallmarks of BPD in the lung. More importantly, these EVs are also capable of
crossing the BBB and inducing inflammatory brain injury. We speculate that targeting EV trafficking may provide novel strategies to prevent and treat BPD and its associated brain injury in
preterm infants. MATERIALS AND METHODS MATERIALS Pregnant Sprague-Dawley rats were purchased from Charles River Laboratory (Wilmington, MA). The following antibodies were used for
immunostaining, immunofluorescence staining, Western blot analysis and FACS analysis: rabbit anti-GSDMD and mouse anti-AIF-1 from Novusbio (Littleton, CO); rabbit anti-pro-SPC and mouse
anti-CD63 from EDMillipore (Temecula, CA); mouse anti-CD9 antibody from ThermoFisher (Waltham, MA); rabbit anti-Ki67 from Abcam (Cambridge, MA); mouse anti-vonWillebrand factor (vWF) from
Dako (Carpinteria, CA); rabbit anti-SPC-PE from Biossusa (Woburn, MA). Total Exosome Isolation Kit was obtained from ThermoFisher. Exo-FLOW Exosome Purification Kit and ExoGlow-Vivo EV
Labeling Kit were obtained from Systems Biosciences (Palo Alto, CA). The lipophilic fluorescent dye, 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarocyanine perchlorate (DiI) was purchased
from Sigma (Louis, MO). MLE-12 cells were obtained from ATCC (Manassas, VA), pulmonary vascular endothelial cells (PVEC) were obtained from Lonza (Walkersville, MD), and rat fetal neural
stem cells (NSC) were obtained from ThermoFisher. The primers for all the qRT-PCR were obtained from ThermoFisher. ANIMAL MODELS All animal study protocols (16-030, 2016; 19-030-LF, 2019)
were approved by the University of Miami Animal Care and Use Committee with pregnant Sprague-Dawley rats and their pups treated according to NIH guidelines for laboratory animals and the
ARRIVE guidelines. IN VIVO EXPERIMENT 1: NEONATAL HYPEROXIA MODEL, EV ISOLATION AND CHARACTERIZATION Newborn rats were randomized on postnatal day 1 (P1) into two groups to receive RA or
hyperoxia, 85% O2 as previously described21,22. On P14, pups were anesthetized, blood samples were collected by right ventricular puncture, and plasmas were obtained. The EVs from an equal
volume of plasmas from room air-maintained and hyperoxia-exposed rats were isolated using the appropriate Total Exosome Isolation Kit (ThermoFisher) and resuspended in an equal volume of
PBS. A 4 µl from each EV sample were analyzed for particle numbers and size distributions by nanoparticle tracking assay using the Nanosight NS300 system (Malvern Instruments, Malvern, UK)
as previously described16. Nanoparticle concentrations were expressed per ml of plasma. Expression of GSDMD, SPC, CD9 and CD63, in 20 µg of EVs was determined by Western blot analysis as
previously described22. For FACS analyses 25 µg samples of plasma EVs were captured on anti-tetraspanin (CD9, 63 and 81)-conjugated magnetic beads (Exo-FLOW Exosome Purification Kit, Systems
Biosciences), stained sequentially with an anti-GSDMD antibody and a FITC-labeled secondary antibody, followed by an PE-labeled anti-SPC antibody, and then analyzed using a flow cytometer
(CytoFLEX, Beckman Coulter, Brea, CA). IN VIVO EXPERIMENT 2: TRACKING OF INJECTED EVS Plasma RA-EVs and O2-EVs obtained in Experiment 1 were labeled with non-lipophilic near IR dye
(ExoGlow-Vivo EV Labeling Kit) as instructed by the manufacturer (Systems Biosciences)51. The labeled EVs (50 µg/sample) or sham-labeled normal saline (NS) negative control were injected via
the tail vein into normal neonatal rats on P7. Whole body imaging was done in vivo at 15 min, 1 h and 4 h after injection, and brain and lung tissues were dissected at 4 h for ex vivo
imaging using an In Vivo Imaging system (PerkinElmer, Hopkinton, MA). In a separate experiment, RA-EVs and O2-EVs were also labeled with DiI dye (Sigma, St. Louis, MO), and these EVs and
sham-labeled NS were injected via the tail vein to a different sets of normal neonatal rats at P7. Brain tissues were collected 24 h later and tissue sections were examined by fluorescent
microscopy for Dil signals. CSF was collected by tapping the cisternal magna and EVs were isolated from pooled CSF in each condition and analyzed by nanoparticle tracking. IN VIVO EXPERIMENT
3: ASSESSMENT OF EV EFFECTS Normal neonatal rats were randomized to receive adoptive transferring of plasma RA-EVs (pooled from 10 animals) or O2-EVs (pooled from 10 animals) isolated in
Experiment 1 by temporal vein injection at a dose of 50 µg/animal on P3, and again on P7 via tail vein injection. These rats were maintained in room air, and on P17 they were sacrificed, and
lung and brain tissues were collected. ASSESSMENT OF LUNG INFLAMMATION Rat pups were sedated with 1% isoflurane, tracheotomized with a 22-gauge angiocatheter which was secured in place with
a suture. For BAL, ice cold normal saline (0.5 ml) was instilled into the airway and gently withdrawn for 4 times. The lavage was repeated four times to recover a total volume of 1.5–2 ml.
The cells were stained with trypan blue and total live cell counts were performed with a hemocytometer. Cytospin (Cytospin 2; Shandon, Waltham, MA) slides were prepared from the BALF and
were then fixed and stained using Neat Stain Hematology Kit (Polysciences, Inc., Warrington, PA). A total of 100 cells/slide were viewed and counted for differential leukocyte analysis52.
Gene expression of inflammatory mediators in lung tissues was assessed by qRT-PCR53. ASSESSMENT OF LUNG MORPHOMETRY AND VASCULARIZATION RAC was analyzed by identifying 10 terminal
respiratory bronchioles under the 10 × magnification on each H&E stained tissue section. The number of distal air sacs that were transected by a line drawn from a terminal respiratory
bronchiole to the nearest pleural surface was calculated and RAC was determined as the average number of distal air sacs from each lung tissue sections22. To determine vascular density,
immunofluorescence staining for vWF, an endothelial marker, was performed. Ten random images were taken under the 20 × magnification on each lung section and the average number of vWF
stained vessels (< 50 μm in diameter) was calculated22. ASSESSMENT OF BRAIN INFLAMMATION AND CELL DEATH The presence of neuroinflammatory microglial cells was assessed by immunohistology
for AIF-1. Gene expression of inflammatory mediators was assessed by qRT-PCR53. Cell death was determined by TUNEL assay22. IN VITRO HYPEROXIA EXPOSURE OF CULTURED MLE-12 CELLS MLE-12 cells
were cultured in room air or hyperoxia (95% O2) in media supplemented with 2% EV-free FBS for 48 h. EVs were isolated from supernatant media with a Total Exosome Isolation Kit
(ThermofFisher) and analyzed by Nanosight tracking assay and Western blot. IN VITRO ASSESSMENT OF CELL PROLIFERATION AND DEATH Room air cultured PVEC and NSC were treated for 48 h with
RA-MEVs or O2-MEVs and cell proliferation was assessed by immunofluorescent staining for Ki67, cell death was determined by TUNEL and pyroptosis was measured by uptake of PI. WESTERN BLOT
ANALYSIS EV concentrations were measured by BCA protein assay using a commercial kit from Pierce Biotechnology Inc (Rockford, IL). Total proteins (20 µg/sample) were fractionated by SDS-PAGE
on 4–12% Tris–glycine precast gradient gels (ThermoFisher) and then transferred to nitrocellulose membranes (Amersham, Piscataway, NJ). The membranes were incubated overnight at 4 °C with
respective primary antibodies and then incubated for 1 h at room temperature with HRP-conjugated secondary antibodies. Antibody bound proteins were detected using ECL chemiluminescence
methodology (Amersham). The intensities of protein bands were quantified by Quantity One Imaging Analysis Program (Bio-Rad, Hercules, CA). RNA ISOLATION AND QUANTITATIVE QRT-PCR Total RNA
was isolated from frozen lung and brain tissues and treated with DNase to remove possible DNA contamination as described53. One µg of total RNA was reverse-transcribed in a 20 µl reaction by
using a first-strand cDNA synthesis kit according to supplier’s protocol (Invitrogen). The Real-time qRT-PCR was performed on an ABI Fast 7500 System (Applied Biosystems, Foster City, CA).
Each reaction included diluted first-strand cDNA, target gene primers, or 18S rRNA gene primers and master mix containing TaqMan probes according to the supplier’s instruction (Applied
Biosystems). qRT-PCR conditions were 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 30 s. RNase-free water was used as a negative control. The expression levels of
target genes were normalized to 18S rRNA. STATISTICAL ANALYSIS Data were expressed as mean ± SD and comparisons were performed by Student t-test. _P_ values < 0.05 were considered
statistically significant. CHANGE HISTORY * _ 06 OCTOBER 2021 A Correction to this paper has been published: https://doi.org/10.1038/s41598-021-99450-2 _ REFERENCES * Jobe, A. H. &
Bancalari, E. Bronchopulmonary dysplasia. _Am. J. Respir. Crit. Care Med._ 163, 1723–1729 (2001). CAS PubMed Google Scholar * Volpe, J. J. Brain injury in premature infants: a complex
amalgam of destructive and developmental disturbances. _Lancet Neurol._ 8, 110–124 (2009). PubMed PubMed Central Google Scholar * Higgins, R. D. _et al._ Bronchopulmonary dysplasia:
executive summary of a workshop. _J. Pediatr._ 197, 300–308 (2018). PubMed PubMed Central Google Scholar * Anderson, P. J. & Doyle, L. W. Neurodevelopmental outcome of
bronchopulmonary dysplasia. _Semin. Perinatol._ 30, 227–232 (2006). PubMed Google Scholar * Schmidt, B. _et al._ Impact of bronchopulmonary dysplasia, brain injury, and severe retinopathy
on the outcome of extremely low-birth-weight infants at 18 months: results from the trial of indomethacin prophylaxis in preterms. _JAMA_ 289, 1124–1129 (2003). PubMed Google Scholar *
Sriram, S. _et al._ Congnitive development and quality of life associated with BPD in 10-year-olds born preterm. _Pediatrics_ 141(6), e20172719 (2018). PubMed Google Scholar * Thery, C.
_et al._ Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014
guidelines. _J. Extracell. Vesicles_ 7, 1–111 (2018). Google Scholar * Thery, C., Ostrowski, M. & Segura, E. Membrane vesicles as conveyors of immune responses. _Nat. Rev. Immunol._ 9,
581–593 (2009). CAS PubMed Google Scholar * Wahlund, C. J. E., Eklund, A., Grunewald, J. & Gabrielsson, S. Pulmonary extracellular vesicles as mediators of local and systemic
Inflammation. _Front. Cell Dev. Biol._ 5, 39 (2017). PubMed PubMed Central Google Scholar * Thompson, A. G. _et al._ Extracellular vesicles in neurodegenerative disease pathogenesis to
biomarkers. _Nat. Rev. Neurol._ 12, 346–357 (2016). CAS PubMed Google Scholar * Moon, H. G. _et al._ Lung epithelial cell-derived extracellular vesicles activate macrophage-mediated
inflammatory responses _via_ ROCK1 pathway. _Cell Death Dis._ 6, 2016 (2015). Google Scholar * Kulshreshtha, A., Ahmad, T., Agrawal, A. & Ghosh, B. Proinflammatory role of epithelial
cell-derived exoosmes in allergic airway inflammation. _J. Allergy Clin. Immunol._ 131, 1194–1203 (2013). CAS PubMed Google Scholar * Lal, C. V. _et al._ Exosomal microRNA predicts and
protects against severe bronchopulmonary dysplasia in extremely premature infants. _JCI Insight_ 3, 093994 (2018). Google Scholar * Ridder, K. _et al._ Extracellular vesicle-mediated
transfer of genetic information between the hematopoitic system and the brain in response to inflammation. _PLoS Biol._ 12, e1001874 (2014). PubMed PubMed Central Google Scholar * Osier,
N. _et al._ Exosomes in acquired neurological disorders: new insights into pathophysiology and treatment. _Mol. Neurobiol._ 55, 9280–9293 (2018). CAS PubMed Google Scholar * Kerr, N. A.
_et al._ Traumatic brain injury-induced acute lung injury: evidence for activation and inhibition of a neural—respiratory-inflammasome axis. _J. Neurotrauma_ 35, 2067–2976 (2018). PubMed
PubMed Central Google Scholar * Shi, J. _et al._ Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. _Nature_ 526, 660–665 (2015). ADS CAS PubMed Google Scholar
* Kayagaki, N. _et al._ Caspase-11 cleaves gasdermin D for non-canonical inflammasome signaling. _Nature_ 526, 666–671 (2015). ADS CAS PubMed Google Scholar * He, W. T. _et al._
Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. _Cell Res._ 25, 1285–1298 (2015). CAS PubMed PubMed Central Google Scholar * Hummler, J. K. _et
al._ Inhibition of Rac1 signaling downregulates inflammasome activation and attenuates lung injury in neonatal rats exposed to hyperoxia. _Neonatology_ 111, 280–288 (2017). CAS PubMed
Google Scholar * Donda, K. _et al._ Riociguat prevents hyperoxia-induced lung injury and pulmonary hypertension in neonatal rats without effects on long bone growth. _PLoS ONE_ 13, e0199927
(2018). PubMed PubMed Central Google Scholar * Dapaah-Siakwan, F. _et al._ Caspase-1 inhbition attenuates hyperoxia-induced lung and brain injury in neonatal mice. _Am. J. Respir. Cell
Mol. Biol._ 61, 341–354 (2019). CAS PubMed Google Scholar * Homsy, E. _et al._ Circulating gasdermin-D in critically ill patients. _Crit. Care Explor._ 1, e0039 (2019). PubMed PubMed
Central Google Scholar * Lee, H., Zhang, D., Zhu, A., Dela Cruz, C. S. & Jin, Y. Epithelial cell-derived microvesicles activate macrophages and promote inflammation _via_
microvesicle-containing microRNAs. _Sci. Rep._ 6, 35250 (2016). ADS CAS PubMed PubMed Central Google Scholar * Mitra, S. _et al._ Microparticlulate caspase-1 regulates GSDMD and
pulmonary vascular endothelial cell injury. _Am. J. Respir. Cell Mol. Biol._ 59, 56–64 (2018). CAS PubMed PubMed Central Google Scholar * Szatmári, T. _et al._ Extracellular vesicles
mediate radiation-induced systemic bystander signals in the bone marrow and spleen. _Front. Immunol._ 8, 347 (2017). PubMed PubMed Central Google Scholar * D’Angio, C. T. _et al._ Blood
cytokine profiles associated with distinct patterns of bronchopulmonary dysplasia among extremely low birth weight infants. _J. Pediatr._ 174, 45–51 (2016). CAS PubMed PubMed Central
Google Scholar * Ding, L. _et al._ Prediction of bronchopulmonary dysplasia in preterm infants using postnatal risk factors. _Front. Pediatr._ 8, 349 (2020). ADS PubMed PubMed Central
Google Scholar * Sahni, M. _et al._ Novel biomarkers of bronchopulmonary dysplasia and bronchopulmonary dysplasia-associated pulmonary hypertension. _J. Perinatol._
https://doi.org/10.1038/s41372-020-00788-8 (2020). Article PubMed PubMed Central Google Scholar * Yue, Y. _et al._ Excessive activation of NMDA receptor inhibits the protective effect of
endogenous bone marrow mechenchymal stem cells on promoting alveolarization in bronchopulmonary dysplasia. _Am. J. Physiol. Cell Physiol._ 16, C815–C827 (2019). Google Scholar * Paudel, S.
_et al._ CXCL1 regulates neutrophil homestasis in pneumonia-derived sepsis caused by _Streptococcus pneumoniae_ serotype 3. _Blood_ 133, 1335–1345 (2019). CAS PubMed PubMed Central
Google Scholar * James, M. L., Ross, A. C., Nicola, T., Steele, C. & Ambalavanan, N. VARA attenuates hyperoxia-induced impaired alveolar development and lung function in newborn mice.
_Am. J. Physiol. Lung Cell. Mol. Physiol._ 304, L803–L812 (2013). CAS PubMed PubMed Central Google Scholar * Lilling, T. _et al._ Exposure of nenatal mice to bromine impairs their
alveolar development and lung function. _Am. J. Physiol. Lung Cell. Mol. Physiol._ 314, L137–L143 (2018). Google Scholar * Alapati, D. _et al._ CTGF antibody therapy attenuates
hyperoxia-induced lung injury in neonatal rats. _Am. J. Respir. Cell Mol. Biol._ 45(6), 1169–1177 (2011). CAS PubMed Google Scholar * Sureshbabu, A. _et al._ Conditional overexpression of
TGFβ1 promotes pulmonary inflammation, apoptosis and mortality _via_ TGFβR2 in the developing mouse lung. _Respir. Res._ 16, 4–15 (2015). PubMed PubMed Central Google Scholar *
Nakanishi, H., Sugiura, T., Streisand, J. B., Lonning, S. M. & Roberts, J. D. Jr. TGF-beta-neutralizing antibodies improve pulmonary alveologenesis and vasculogenesis in the injured
newborn lung. _Am. J. Physiol. Lung Cell. Mol. Physiol._ 293, L151-161 (2007). CAS PubMed Google Scholar * Abman, S. H., Grenolds, A. & Mourani, P. Pulmonary vascular disease in
bronchopulmonary dysplasia. _Adv. Pulm. Hypertens._ 15, 92–99 (2016). Google Scholar * Saint-Pol, J., Gosselet, F., Duban-Deweer, S., Pottiez, G. & Karamanos, Y. Targeting and crossing
the blood–brain barrier with extracellular vesicles. _Cells_ https://doi.org/10.3390/cells9040851 (2020). Article PubMed PubMed Central Google Scholar * Jin, C., Londono, I., Mallard, C.
& Lodygensky, G. A. New means to assess neonatal inflammatory brain injury. _J. Neuroinflamm._ 12, 180 (2015). Google Scholar * Dammann, O. & O’Shea, M. Cytokines and perinatal
brain damage. _Clin. Perinatol._ 35, 643 (2008). PubMed PubMed Central Google Scholar * Milner, R. _et al._ Fibronectin and vitronectin induced microglial activation and matrix
metalloproteinase-9 expression is mediated by integrins alpha5beta1 and alphavbeta5. _J. Immunol._ 178, 8158–8167 (2007). CAS PubMed Google Scholar * Summers, L., Kielty, C. &
Pinteaux, E. Adhesion to fibronectin regulates interleukin-1 beta expression in microglial cells. _Mol. Cell. Neurosci._ 41, 148–155 (2009). CAS PubMed Google Scholar * Duan, L. _et al._
PDGFRb cells rapidly relay inflammatory signal from the circulatory system to neurons _via_ chemokine CCL2. _Neuron_ 100, 183–200 (2018). CAS PubMed Google Scholar * Porzionato, A. _et
al._ Effects of postnatal hyperoxia exposure on the rat dentate gyrus and subventricular zone. _Brain Struct. Funct._ 2015(220), 229–247 (2015). Google Scholar * Yang, Z. & Levison, S.
W. Hypoxia/ischemia expands the regenerative capacity of progenitors in the perinatal subventricular zone. _Neuroscience_ 139, 555–564 (2006). CAS PubMed Google Scholar * Jin, K. _et al._
Neurogenesis in dentate subgranular zone and rostral subventricular zone after focal cerebral ischemia in the rat. _Proc. Nat. Acad. Sci._ 98, 4710–4715 (2001). ADS CAS PubMed PubMed
Central Google Scholar * Palmer, T. D., Willhoite, A. R. & Gage, F. H. Vascular niche for adult hippocampal neurogenesis. _J. Comp. Neurol._ 425, 479–494 (2000). CAS PubMed Google
Scholar * Shen, Q. _et al._ Adult SVZ stem cells lie in a vascular niche: a quantitative analysis of niche cell–cell interactions. _Cell Stem Cell_ 3, 289–300 (2008). CAS PubMed PubMed
Central Google Scholar * Tavazoie, M. _et al._ A specialized vascular niche for adult neural stem cells. _Cell Stem Cell_ 3, 279–288 (2008). CAS PubMed PubMed Central Google Scholar *
Bulek, K. _et al._ Epithelial-derived gasdermin D mediates nonlytic IL-1β release during experimental colitis. _J. Clin. Invest._ 130, 4218–4234 (2020). CAS PubMed PubMed Central Google
Scholar * Kuwajima, Y. _et al._ Trans-trigeminal transport of masseter-derived neprilysin to hippocampus. _Arch. Oral Biol._ 118, 104861 (2020). CAS PubMed Google Scholar * Hummler, S.
C. _et al._ Targeting glycogen synthase-3β to prevent hyperoxia-induced lung injury in neonatal rats. _Am. J. Respir. Cell Mol. Biol._ 48, 578–588 (2013). CAS PubMed Google Scholar *
Chen, S. _et al._ CTGF disrupts alveolarization and induces pulmonary hypertension in neonatal mice: implication in the pathogenesis of severe bronchopulmonary dysplasia. _Am. J. Physiol.
Lung Cell. Mol. Physiol._ 300, L330–L340 (2011). CAS PubMed PubMed Central Google Scholar Download references FUNDING Project Newborn (SW), Batchelor Award (SW) and Dean’s Bridge Award
(SW) from the University of Miami Miller School of Medicine, and NIH Grant R01HL156803 (SW). AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Division of Neonatology and Batchelor Children’s
Research Institute, Department of Pediatrics, University of Miami Miller School of Medicine, P. O. Box 016960, Miami, FL, 33101, USA Anum Ali, Ronald Zambrano, Matthew R. Duncan, Shaoyi
Chen, Shihua Luo, Huijun Yuan, Pingping Chen, Merline Benny, Augusto Schmidt, Karen Young & Shu Wu * Department of Neurological Surgery, Miami Project to Cure Paralysis, University of
Miami Miller School of Medicine, Miami, USA Nadine Kerr, Juan Pablo de Rivero Vaccari, Robert W. Keane & W. Dalton Dietrich * Department of Physiology and Biophysics, University of Miami
Miller School of Medicine, Miami, FL, USA Nadine Kerr, Juan Pablo de Rivero Vaccari & Robert W. Keane Authors * Anum Ali View author publications You can also search for this author
inPubMed Google Scholar * Ronald Zambrano View author publications You can also search for this author inPubMed Google Scholar * Matthew R. Duncan View author publications You can also
search for this author inPubMed Google Scholar * Shaoyi Chen View author publications You can also search for this author inPubMed Google Scholar * Shihua Luo View author publications You
can also search for this author inPubMed Google Scholar * Huijun Yuan View author publications You can also search for this author inPubMed Google Scholar * Pingping Chen View author
publications You can also search for this author inPubMed Google Scholar * Merline Benny View author publications You can also search for this author inPubMed Google Scholar * Augusto
Schmidt View author publications You can also search for this author inPubMed Google Scholar * Karen Young View author publications You can also search for this author inPubMed Google
Scholar * Nadine Kerr View author publications You can also search for this author inPubMed Google Scholar * Juan Pablo de Rivero Vaccari View author publications You can also search for
this author inPubMed Google Scholar * Robert W. Keane View author publications You can also search for this author inPubMed Google Scholar * W. Dalton Dietrich View author publications You
can also search for this author inPubMed Google Scholar * Shu Wu View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS Conception and design of
the study: A.A., N.K., R.W.K., J.P.d.R.V., W.D.D., and S.W. Acquisition, analysis and interpretation of data: A.A., R.Z., M.R.D., S.C., S.L., H.Y., P.C., M.B., A.S., K.Y., N.K., J.P.d.R.V.,
R.W.K., W.D.D., S.W. Drafting and editing of manuscript: A.A., M.R.D., J.P.d.R.V., and S.W., Reviewed the manuscript: A.A., R.Z., M.R.D., S.C., S.L., P.C., M.B., A.S., K.Y., N.K.,
J.P.d.R.V., R.W.K., W.D.D., S.W. CORRESPONDING AUTHOR Correspondence to Shu Wu. ETHICS DECLARATIONS COMPETING INTERESTS JPdRV, WDD and RWK are co-founders and managing members of
InflamaCORE, LLC and have licensed patents on inflammasome proteins as biomarkers of injury and disease as well as on targeting inflammasome proteins for therapeutic purposes. JPdRV, WDD and
RWK are Scientific Advisory Board Members of ZyVersa Therapeutics. ADDITIONAL INFORMATION PUBLISHER'S NOTE Springer Nature remains neutral with regard to jurisdictional claims in
published maps and institutional affiliations. The original online version of this Article was revised: The Competing Interests statement in the original version of this Article was
incorrect. “The authors declare no competing interests” now reads: “JPdRV, WDD and RWK are co-founders and managing members of InflamaCORE, LLC and have licensed patents on inflammasome
proteins as biomarkers of injury and disease as well as on targeting inflammasome proteins for therapeutic purposes. JPdRV, WDD and RWK are Scientific Advisory Board Members of ZyVersa
Therapeutics.” SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION. RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International
License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source,
provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative
Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not
permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Ali, A., Zambrano, R., Duncan, M.R. _et al._ Hyperoxia-activated circulating
extracellular vesicles induce lung and brain injury in neonatal rats. _Sci Rep_ 11, 8791 (2021). https://doi.org/10.1038/s41598-021-87706-w Download citation * Received: 05 November 2020 *
Accepted: 16 March 2021 * Published: 22 April 2021 * DOI: https://doi.org/10.1038/s41598-021-87706-w SHARE THIS ARTICLE Anyone you share the following link with will be able to read this
content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative